

Summary
The structure of genetic interaction networks predicts that, analogous to synthetic lethal interactions between non-essential genes, combinations of compounds with latent activities may exhibit potent synergism. To test this hypothesis, we generated a chemical-genetic matrix of 195 diverse yeast deletion strains treated with 4915 compounds. This approach uncovered 1221 genotype-specific inhibitors, which we termed cryptagens. Synergism between 8128 structurally disparate cryptagen pairs was assessed experimentally and used to benchmark predictive algorithms. A model based on the chemical-genetic matrix and the global genetic interaction network failed to accurately predict synergism. However, a combined random forest and Naïve Bayesian learner that associated chemical structural features with genotype-specific growth inhibition had strong predictive power. This approach identified previously unknown compound combinations that exhibited species-selective toxicity towards human fungal pathogens. This work demonstrates that machine learning methods trained on unbiased chemical-genetic interaction data may be widely applicable for the discovery of synergistic combinations in different species.
Keywords: antifungal, combination, synergism, genetic network, bipartite graph, Bayesian analysis, machine learning, random forest, chemical-genetic interaction
Graphical abstract
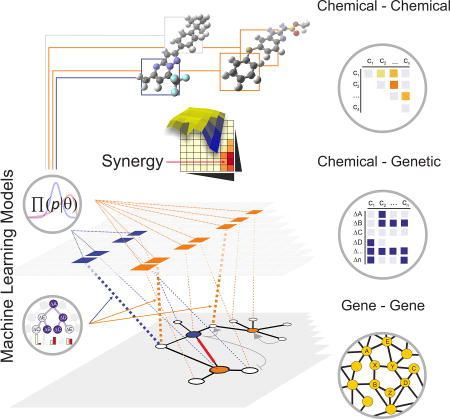
Introduction
The modern era of drug discovery has been dominated by the "magic bullet" concept developed by Ehrlich more than 100 years ago (Strebhardt and Ullrich, 2008). This concept is predicated on the notion that a pathogen, genetic mutation or physiological defect can be remedied by a single chemical agent. This approach has proven spectacularly successful in the development of anti-infective agents and specific enzyme inhibitors to treat particular conditions. However, despite investments in genome-based approaches for target identification, validation and screening, the current repertoire of approved drugs targets stands at only ~500 proteins, with at most only a further 500 targets under active exploration (Overington et al., 2006; Rask-Andersen et al., 2014). The druggable genome has been focused on a select number of enzyme and receptor target classes, suggesting that only a tiny fraction of the potential target space has been tapped to date. Cell-based phenotypic screens explore all possible targets but with the caveat that the mechanism of action can be difficult if not impossible to discern (Nijman, 2015).
It has long been evident that the development, physiology and phenotype of all organisms is controlled by complex genetics (Waddington, 1957). Recent systematic genetic screens have uncovered the depth of this genetic complexity, which manifests as network of genetic interactions. A genetic interaction between two genes is observed when a phenotype caused by a mutation in the first gene is exacerbated or ameliorated by a mutation in the second gene, such that the combined effect exceeds the sum of each individual effect. In the extreme case, the synthetic combination of two or more non-lethal mutations may result in a lethal genetic interaction (Mani et al., 2008). Systematic genetic screens in budding yeast have revealed that while only ~1000 of the ~6000 genes are essential, at least 200,000 documented genetic interactions occur between non-essential genes (Costanzo et al., 2010; Chatr-Aryamontri et al., 2015). This dense genetic architecture reflects the global hierarchy of cellular sub-systems and allows the cell to coordinate cellular activities, resist perturbation and evolve new functions (Hartman et al., 2001; Kitano, 2007; Costanzo et al., 2010). Within this context, disease is a consequence of network perturbation caused by genetic mutation, pathogen infection and/or environmental insult.
The complexity of the genetic landscape suggests that effective drug therapies may require modulation of multiple network nodes (Sharom et al., 2004; Csermely et al., 2005; Fitzgerald et al., 2006; Kitano, 2007; Lehar et al., 2009). Drug combinations can lead to increased clinical efficacy (Sharom et al., 2004; Kitano, 2007; Lehar et al., 2009) and prevent the emergence of resistance (Crystal et al., 2014; Chen et al., 2015). Different approaches have been taken to identify synergistic drug combinations. Historically, drugs have been combined in an ad hoc manner in the hope of achieving additional therapeutic benefit through synergistic interactions (Sharom et al., 2004). Similarly, the efficacy of single drugs may result from chance cooperative effects of drug action on multiple targets, an effect termed polypharmacology (Hopkins, 2008). The discovery of de novo drug interactions is confounded by the vast combinatorial chemical space that must be screened (Lehar et al., 2009), and in practice has been limited to screens with known drugs (Feala et al., 2010; Ejim et al., 2011; Spitzer et al., 2011; Tan et al., 2012; Robbins et al., 2015). Chemo-genomic sensitivity profiles in budding yeast (Ericson et al., 2008; Hillenmeyer et al., 2008; Lee et al., 2014) have been used to predict synergism but have led to different conclusions regarding the basis for drug interactions (Jansen et al., 2009; Cokol et al., 2011; Spitzer et al., 2011). Finally, network-based analysis has been used to predict synergistic drug interactions in human cells (Nelander et al., 2008; Crystal et al., 2014) but this approach is currently limited by incomplete network data and a paucity of established drug targets (Bansal et al., 2014).
By analogy to the genetic networks that underpin biology, it should be possible to identify combinations of chemicals that mimic genetic interactions, such that compounds that cause minimal phenotypes alone exhibit strong synergies when combined (Sharom et al., 2004; Fitzgerald et al., 2006; Kitano, 2007; Roemer and Boone, 2013). A barrier to the discovery of such genetically-inspired combinations is the dearth of compounds that mimic the effect of mutations in non-essential genes. Such compounds would cause little or no discernible phenotype in wild type cells yet may have strong activity in a given genetic context (Sharom et al., 2004). These chemical-genetic relationships may access a large previously hidden sector of chemical space, sometimes referred to as chemical dark matter (Wassermann et al., 2015). In this study, we identify novel combinations of compounds in yeast that alone are inert but which together potently inhibit cell growth. Diverse chemical libraries screened against a multitude of genetic backgrounds were used to generate an experimental matrix of chemical-genetic and chemical-chemical interactions, which was used to develop and test algorithmic methods for synergy prediction. Synergistic combinations did not exhibit an obvious relationship with genetic interaction networks, but could be predicted accurately by machine learning approaches applied to chemical-genetic data. These findings demonstrate the feasibility of de novo identification of synergistic chemical interactions through methods that should be applicable in many different disease contexts.
Results
Generation of a chemical-genetic matrix
Based on the principle of genetic synthetic lethality, any given S. cerevisiae non-essential deletion strain should serve as a sentinel to identify compounds with cryptic bioactivities that would not be evident in a wild type context (Hartman et al., 2001). We assembled a cohort of chemical-genetic interaction profiles for many different deletion strain backgrounds, which we termed the chemical-genetic matrix (CGM), and developed a workflow to predict and test synergistic chemical interactions (Fig. 1A). To construct the CGM, we screened a total of 4915 unique compounds derived from four different chemical libraries against a panel of 195 non-essential deletion strains (Table S1, S2). This compound collection represented largely uncharacterized chemical space compared to previous large scale chemo-genomic studies (Ericson et al., 2008; Hillenmeyer et al., 2008; Lee et al., 2014) and was comprised of approximately 50% approved drugs or drug-like compounds, 40% synthetic products with drug-like properties, and 10% natural products (Fig. S1A,B). To maximize discovery of cryptic bioactivities, sentinel strains were chosen to cover a broad spectrum of biological processes across the genetic landscape (Costanzo et al., 2010) (Fig. 1B, S1C; Table S3), including genes that encode subunits of all major protein complexes (Fig. S1D).
Figure 1.

A Chemical Genetic Matrix (CGM). (A) Data generation and analysis workflow. 4915 unique molecules from 4 different chemical libraries were screened against a panel of 195 S. cerevisiae deletion strains (termed sentinels) to identify compounds that inhibit the growth of specific deletion strains (termed cryptagens). Pairwise combinations of 128 structurally diverse cryptagens from the CGM were screened in a 128×128 cryptagen matrix (CM) to identify synergistic compound pairs. The CGM dataset and chemical structural features were used to build a Naïve Bayes multi-class learner (NBL) to predict compound activity likelihoods for each sentinel strain. A graph-based algorithm was used to integrate chemical-genetic and genetic interactions to predict compound targets, based on either CGM interaction data (SONARG) or NBL likelihood scores (SONARGN). A random forest-based machine learning algorithm was used to enhance synergy prediction using the CM as training data, based on NBL likelihoods with (SONARGNR) or without (SONARNR) genetic interaction constraints. Predicted synergistic combinations were tested in S. cerevisiae, fungal pathogens and human cell lines. (B) GO SLIM categories represented by sentinel deletion strains. Color indicates significance of gene enrichment based on hypergeometric test. Numbers indicate genes in each category. (C) Heatmap of chemical-genetic interactions in the CGM. Compound activities versus sentinel strains are shown for each individual library screened in this study. (D) Compound activities (Zscore < −4) across sentinel strains. 1221 cryptagens that sensitized > 4 and < 2/3 of all deletion strains are indicated. Inset: Median growth inhibition across all sentinel screens for each compound. See also Figures S1, S2.
We carried out over 600 growth-based screens in duplicate at a compound concentration of 20 µM. In total, this large-scale experiment comprised pairwise tests of 713,000 chemical-genetic interactions, which formed the CGM (Fig. 1C). To quantify growth inhibitory effects of each compound, Z-scores were calculated and averaged for the replicate screens. Each sentinel strain was sensitized by specific subsets of compounds, consistent with previous findings that 97% of all S. cerevisiae deletion strains exhibit specific chemical sensitivities (Hillenmeyer et al., 2008). Approximately two thirds of all unique compounds exhibited activity in at least one genetic background; of these, 300 compounds were active in only a single strain background and 100 compounds were active in all strains tested (Fig. 1D). The CGM contained 1221 compounds, which we termed cryptagens, that inhibited growth of at least 4 and less than two-Wildenhain et al - Page 6 thirds of the sentinel strains compared to DMSO controls (> 3 median absolute deviation (MAD), corresponding to > 20% growth inhibition) (Fig. 1D; Table S1). This range was chosen to minimize the chances of spurious effects on single strains and to ensure a computationally tractable density of associated genetic interactions for subsequent analyses.
Contrary to our expectations, the number of genetic or protein interactions did not significantly correlate with the number of chemical-genetic interactions exhibited by the corresponding sentinel strain (Fig. S2A,B). Although chemical inhibitors may be conceptualized as mimetics of gene deletions or mutations, this correspondence is often less than precise. For example, chemical-genetic interactions of the Erg11 inhibitor fluconazole only partially overlap with the genetic interactions of an erg11#x00394; strain (Parsons et al., 2004). This imprecise overlap of chemical-gene versus gene-gene interactions may arise from partial inhibitory activity, pleiotropic protein function, neomorphic activity and compound off-target effects, all of which may further propagate and interact through the genetic network. These effects confound target and synergy prediction, as discussed below.
A cryptagen matrix (CM) enriches for synergistic chemical interactions
In order to develop unbiased computational approaches for synergy prediction, we required a large unbiased dataset of pairwise chemical combinations and growth phenotypes. We therefore used the CGM data to seed an extensive test matrix of chemical-chemical interaction data for training and evaluation of synergy prediction algorithms. We chose 128 cryptagens with distinct chemical-genetic profiles (Fig. 2A; Table S4) and maximal structural diversity based on Tanimoto similarity scores (Fig. 2B). Interactions between these 128 compounds were systematically tested in a 128×128 pairwise combination screen at a single concentration of 10 µM for each compound. Synergism was calculated using the Bliss independence model (see Supplemental Information) in which each drug is considered to act independently of the other. Analogous to genetic interactions, growth inhibition that exceeds the multiplied effect of each agent alone is termed synergism, while growth inhibition that is less than the multiplied effect is termed antagonism (Greco et al., 1995). Based on the distribution of all Bliss independence values, we defined synergistic effects with Bliss > 0.25 and antagonistic effects with Bliss < −0.18, corresponding to a 90% confidence interval of 3 MAD (Fig. 2C). We observed 730 synergistic and 118 antagonistic chemical interactions among the 8128 pairs tested. Approximately 30% of the compounds tested (45 of 128) showed specific synergistic interactions with each other and with other compounds in the CM. To our knowledge none of these combinations have been previously reported as synergistic or antagonistic. Hierarchical clustering of the Bliss scores revealed a well dispersed matrix of interactions (Fig. 2D). Only two compounds, haloperidol and echinocystic acid, were promiscuous for chemical interactions. We did not observe an obvious correlation between the number of chemical-genetic interactions and the number of synergistic chemical interactions, suggesting that cryptagens were not enriched for promiscuous compounds (Fig. 2E). Physico-chemical properties such as molecular weight, solubility and compound structure were also not predictive of synergistic interactions (Fig. S3A,B).
Figure 2.

Cryptagen Matrix (CM) of Chemical-Chemical Interactions. (A) Activity heatmap of 128 structurally diverse cryptagens against 195 deletion strains. (B) Tanimoto similarity across all 4915 molecules screened. Edges represent > 30% similarity. The 128 cryptagens used in the CM are shown in red. (C) Histogram of 8128 Bliss independence values for cryptagen pairs. (D) Heatmap of cryptagen interactions. Colors are as in panel C. (E) Scatterplot of chemical-genetic interactions versus observed synergistic interactions for the 128 cryptagens. See also Figure S3.
Prediction of compound targets and synergies based on genetic interactions
Based on the assumption that chemical perturbations may mimic genetic lesions (Hartwell et al., 1997), the integration of chemical-genetic interaction data with genetic network data may enable the prediction of compound synergies. We developed a software toolset to predict chemical synergies, collectively called the Second Order Naïve Bayesian and Random Forest (SONAR) suite. We first implemented a network-based algorithm that integrates chemical-genetic data with genetic interactions to predict the target genes/proteins for each compound and concomitantly synergistic chemical interactions. In an idealized example, integration of the chemical-genetic interactions for a set of deletion strains with the corresponding genetic interaction network should allow the inference of synergistic chemical interactions (Fig. 3A,B, S3C). However, this over-simplification is likely to fail since previous experimental studies suggest that bioactive compounds typically elicit pleiotropic responses (Hillenmeyer et al., 2008), consistent with the lack of correlation between genetic and chemical-genetic interactions in our dataset (Fig. S2A,B).
Figure 3.

Synergy Prediction Based on Chemical-Genetic and Genetic Interactions. (A) Deletion strains are sensitized to specific cryptagens. (B) Underlying genetic interaction network. (C) SONARG integrates chemical-genetic and genetic interactions to predict chemical synergies. Sentinel strains sensitive to cryptagen c represent first order connections s. Second order connections t are inferred from genetic interactions of sentinel strains and ranked by interactions with sentinel strains in s. Edge weights between target spaces ti and tj are based on genetic interaction counts. See Methods for details. (D) PCA biplot of loadings for 7 SONARG parameters in comparison to Bliss independence values from the CM. Abbreviations: sgi, shared genetic interactions between deletion strains for each compound pair; pval, p-value; hs, high sum on V vertices for x and y and E edges between x and y. (E) Naïve Bayes multi-class likelihoods from the CGM. ECFP4 fingerprints for all compounds and activity probabilities for each feature are calculated for all sentinel strains. The integrated probability for compound activity across all features and classes is represented as a likelihood score. (F) Heatmap of CGM based on NBL likelihoods. (G) PCA biplot for SONARGN parameters. (H) Receiver-operator characteristics (ROC) for the single property Exy (AUC = 0.64) and for synergy scores based on SONARGNR parameters (AUC = 0.87). Inset: Precision-recall plot for SONARGNR model. (I) Distribution of SONARGNR scores for synergistic and non-synergistic pairs based on CM data. See also Figures S3–S6.
To account for the complexity of chemical effects on the genetic network, we created a bipartite representation of genetic interaction data by separation of the experimental genetic nodes s in the CGM and their known interaction partners t in the global genetic interaction network. Nodes in target space t should represent known gene/protein targets of the cryptagen compounds, and each node was ranked according to the number of connected neighbors in the genetic network, such that nodes with more shared neighbors received a higher score (Fig. 3C). This algorithm was termed SONARG, for genetic interaction network. To assess the ability of SONARG to identify compound target genes/proteins, we curated a set of 104 known chemicals from the literature (Parsons et al., 2004; Hillenmeyer et al., 2008; Jansen et al., 2009; Cokol et al., 2011), of which 27 were classified as cryptagens in this study. SONARG correctly predicted the known targets or associated target pathways within the 6 highest-ranked genes for 16 of these 27 compounds (Table S5).
Once each compound was assigned a candidate list of gene/protein targets, the genetic interactions between these targets could then be used to predict potential compound-compound interactions. We thus applied the SONARG counting algorithm to pairs of compounds in the CM to predict chemical interactions based on the genetic network (Fig. 3C, S4). Similar to previous graph-based approaches, we used enrichment of genetic interactions between chemical-genetic profiles to rank potential synergistic compound pairs (Spitzer et al., 2011). For each compound combination cx−cy, we calculated parameters based on the sum of the highest ranked target candidates hsVx and hsVy, the corresponding p-values pvalVx and pvalVy, and the inter-compound edge list hsExy, where hs indicates high sum. As more than 80% of genes in the genetic network interact via second-degree neighbors (Costanzo et al., 2010), the SONARG bipartite graph is extremely dense because each compound sensitizes a number of sentinel strains that in turn exhibit many genetic interactions. For computational tractability, we restricted the target space represented by hsExy to the 35 top ranked genes by interaction count (the procedure was robust to the actual cut-off value chosen for counts, see Methods). We also calculated Pearson correlations between chemical-genetic interactions for all compound combinations (Pxy) and the number of shared genetic interactions between the compound pair target spaces, sgi (Spitzer et al., 2011). We applied principal component analysis (PCA) to these seven parameters derived from the CGM (pvalVx, pvalVy, hsVx, hsVy, hsExy, Pxy, sgi) to identify those that might explain the observed Bliss independence. However, none of the parameters contributed strongly to the Bliss vector (Fig. 3D) nor did any parameters perform better than random as measured by the area under the curve (AUC) for receiver operating characteristic (ROC) plots (Fig. S3D). From these results, we concluded that, at least as implemented in SONARG, the genetic interaction network did not have appreciable power for prediction of synergistic chemical interactions. This poor result led us to consider other parameters in our chemical-genetic and chemical-chemical interaction datasets to improve synergy prediction.
Structural correlates of chemical-genetic interactions
To extract more granular information from the CGM dataset, we built a Naïve Bayes learner (NBL) to identify characteristic structural features of active versus non-active molecules for every sentinel strain (Fig. 3E). The NBL builds a Bayesian probabilistic model wherein each deletion strain represents a different class to which a likelihood score is assigned for the inhibitory effect of different compound substructures. This powerful approach has previously been used to associate chemical features with specific target classes (Keiser et al., 2009; Besnard et al., 2012) but to our knowledge has not been applied to chemical-genetic interaction data. Structural characteristics of each cryptagen were represented by Extended-Connectivity Fingerprints (ECFP4, see Methods) and combined with CGM data using a Naïve Bayes multi-class algorithm (SciTegic Pipeline Pilot, see Methods and Fig. S5A) to predict compound activities towards each sentinel strain. The NBL reliably associated structural features with good sensitivity (AUC>0.7) for more than half of all deletion strains (Table S6). The distribution of median experimental Z-scores for growth inhibition across all sentinel strains confirmed that most cryptagens exhibited specific activities against only a few sentinel strains (Fig. S5B), suggesting that each compound acts in a specific fashion. The likelihoods derived by the NBL algorithm showed a similarly centered distribution and were significantly correlated with the experimental Z-scores derived from the CGM (Pearson correlation distribution mode ≤ −0.5, Fig. S5B), demonstrating that the structural sub-features of each compound could predict genetic sensitivities. The extensive CGM dataset allowed the NBL model to co-segregate chemical structural properties with strain sensitivity, as illustrated for cyclosporine, mebendazole and chrysarobin (Fig. S5C), and many other examples (Fig. S5D). The chemical structure-based likelihood scores were agnostic to particular gene functions but accurately reflected the sensitivity of the genetic network to specific chemical perturbations. These results generalize previous structure-activity predictions for specific target classes (Keiser et al., 2009; Besnard et al., 2012), and suggested that chemical substructures may improve predictions of chemical synergism.
Integration of NBL likelihoods and genetic interactions
The NBL-derived likelihoods deconstructed the activity response for each compound over a large chemical feature space and across multiple strains, and thereby extracted more information from the CGM than simple interaction counts. We therefore tested whether re-calculation of SONAR parameters based on these likelihoods, termed SONARGN (for genetic and NBL), would improve the predictive power of one or more parameters. When based on NBL likelihoods, the vectors for sgi, Pxy and hsExy all showed the same directionality as Bliss independence, indicating that these parameters might be informative for synergy prediction (Fig. 3F). The use of the single strongest parameter, hsExy, resulted in an AUC of 0.64, whereas sgi or Pxy alone generated AUCs of 0.60 and 0.50, respectively (Fig. 3G, S3D). These weak outcomes still suggested that neither the underlying genetic network nor the correlation of chemical-genetic profiles between two compounds alone were useful predictors of synergistic interactions.
To further improve accuracy, we combined all seven SONARGN parameters using a random forest learner; this ensemble approach efficiently combines a set of weak predictors into a stronger overall strong predictor through a series of decision trees. The random forest model was trained on one-third of the CM data with 5-fold cross-validation, and optimized for tree size and variable split points (Fig. S3E). This algorithm, termed SONARGNR (for genetic, NBL and random forest), yielded synergy scores that predicted synergistic interactions in the CM with an AUC of 0.87 (Fig. 3G). The score distributions for synergistic and non-synergistic chemical pairs were well separated for a subset of synergistic combinations; however, synergy for more than half of all combinations was not predicted by SONARGNR (Fig. 3H,I, S3F). Synergistic chemical interactions bridged high-level processes in a similar overall manner as genetic interactions but with some bias towards particular processes (Fig. S6A). Many genes contributed to predicted synergistic interactions but it was not possible to trace a definitive genetic path from one compound to the other in a synergistic pair (Fig. S6B), again reflecting the manifold nature of compound action on genetic networks. Despite this complexity, the integration of chemical structural features with genetic interactions via the combined SONARGNR algorithm was able to effectively predict synergistic chemical interactions.
NBL-based random forest synergy prediction
Since the density of the genetic interaction network limits SONARGNR from the practical perspective of computational tractability, we tested the performance of a simpler prediction algorithm based entirely on the NBL likelihoods and a random forest learner. This algorithm, termed SONARNR (for NBL and random forest), did not use any information derived from the genetic interaction network (Fig. S7A). Compared to the seven SONARGN parameters derived from chemical-genetic network structure, the pure NBL model of SONARNR incorporates a much larger parameter space, namely the likelihoods corresponding to each of the 195 sentinel strains. This ensemble classifier was optimized for tree depth, forest size and split point sampling (Fig. S7B–D). SONARNR proved an even better model for synergy prediction with good separation of synergistic and non-synergistic compound pairs and an AUC of 0.91 (Fig. 4A). The predicted synergy scores from SONARNR correlated well with the experimental Bliss independence values (Pearson correlation = 0.56) but without a clear separation that would occur in an over-fitted dataset (Fig. 4B; Table S7). This lack of reliance on genetic interactions may seem counter-intuitive, but it is in accordance with current hypotheses about genetic network function (see Discussion).
Figure 4.

Random Forest-Based Learner for Synergy Prediction Based on Chemical-Genetic Interactions and Chemical Structural Features. (A) ROC for synergy prediction with SONARNR model. Inset: Precision-recall plot. (B) Scatterplot of Bliss independence values and SONARNR synergy scores. (C) Naïve Bayes classes of top-ranked deletion strains that predict synergistic interactions. Mean decrease in Gini represents the influence of variables in partitioning the data into defined classes. (D) Sentinel strains associated with synergistic interactions predicted by SONARNR. Genes are grouped by biological processes. Edge weights are determined by NBL likelihood of two genes being among the top three sensitive genes for synergistic pairs, corrected by subtraction of weights for the same graph generated from 730 non-synergistic pairs. (E) Corresponding edge weights for genetic interactions between strains for graph in panel D. See also Figures S8, S9.
To understand the importance of each sentinel strain for synergy prediction, we investigated whether specific sentinel strains or biological processes dominated the prediction of synergy. A balanced subset of 700 compound pairs each for synergistic and non-synergistic classes was selected at random from the CM (see Methods). The contribution of each deletion strain model was assessed by Gini impurity, a measure of the importance of input variables provided to a random forest learner. A wide variety of biological processes contributed to synergy prediction, including membrane transport, DNA repair, chromatin assembly and transcription and mRNA processing (Fig. 4C). This diversity of functions continued into the long tail of Gini contributions, demonstrating that the SONARNR model draws from all aspects of the CGM dataset. The SONARNR-derived network (Fig. 4D, S8) linked high level biological processes through synergistic interactions, with more diverse connections between processes than the corresponding genetic network (Fig. 4E) or the SONARGNR-derived network. The estimated sensitivity (0.93) and specificity (0.97) for the cross-validated training data indicated that SONARNR correctly predicted non-synergistic combinations, but that half of the synergistic predictions were incorrect (Table S8; Fig S9 A,B). Nevertheless, SONARNR achieved a 6-fold enrichment of synergistic pairs compared to chance selection and for this dataset would have reduced compound space prior to screening by 3.5-fold (Table S8).
To test the SONARNR classifier against an independent compound library, we screened the Maybridge HitsKit library of 1000 synthetic compounds against a wild type S. cerevisiae strain in combination with sub-inhibitory concentrations of camptothecin, cyclosporine and fenbendazole to identify new compound synergies. Even though SONARNR was trained on a different compound set (i.e., the 128 cryptagens from the Spectrum library), it achieved an AUC of 0.72 for the 300 top-ranked combinations from the HitsKit screen (Table S8), corresponding to a reduction of chemical search space by 3-fold. Since this synthetic Maybridge library covers very different chemical space compared to the diverse natural product-like composition of the MicroSource Spectrum library, the current SONARNR classifier is relatively robust to chemical composition. This result further validates the use of NBL likelihoods derived from chemical substructures and demonstrates that SONARNR can reduce the vast chemical search space for drug interactions (Lehar et al., 2009; Feala et al., 2010).
Experimental verification of predicted synergies
The validation of predictive approaches based on the single concentration measurements in the CM dataset was subject to the caveat that one or both compounds might exhibit non-linear dosage effects that would obviate the synergy term. Extensive dose-response surfaces were therefore used to validate a subset of the SONARNR synergy predictions. Since many crytpagens had no activity even at the highest concentration tested it was not possible to estimate synergism by either Loewe additivity or FICI; instead, each pairwise concentration was evaluated for Bliss independence across the dose-response surface. We tested 98 compound combinations in total, comprised of 37 synergistic pairs and 61 non-synergistic pairs derived from the CM (Table S9). Dose-response surfaces confirmed 27 synergistic and 43 non-synergistic combinations resulting in an AUC of 0.71 (Fig. 5A; Table S8, S9, S10; Fig. S10A).
Figure 5.

Validation of Predicted Synergistic Interactions. (A) Predicted combinations between cryptagens from the Microsource Spectrum collection. Compound abbreviations are provided in the Supplemental Information. (B) Novel predicted synergistic combinations of cyclosporine, fenbendazole and camptothecin with synthetic Maybridge compounds. (C) Specific interactions between structurally diverse compounds camptothecin (Cam), cyclosporine (Cic), SEW 06533 and SP 01215. (D) De novo predicted synergistic pairs from the ChemDiv Kinase library. Assays were carried out in 8×8 matrices in a pdr1Δpdr3Δ yeast drug pump deficient strain at 0, 1, 2, 4, 8, 16, 32 and 64 µM for each compound. Data are provided in Table S8.
We also performed full dose-response assays for 7 Maybridge HitsKit compounds that exhibited synergy with camptothecin, cyclosporine or fenbendazole. From a total of 21 possible combinations, SONARNR correctly predicted 10 synergistic and 3 non-synergistic combinations with 5 false positive and 3 false negative predictions (Fig. 5B; Table S8, S10). To examine potential promiscuity between the synergizers, we also tested all possible pairwise combinations between camptothecin, cyclosporine, and two Maybridge HitsKit compounds (SP 01215 and SEW 06533). We observed a high degree of specificity in pair-wise compound effects on cell growth (Fig. 5C; Table S10).
To benchmark the SONARNR classifier against a de novo compound set, we calculated synergy scores for a 1000 × 1000 compound matrix corresponding to the ChemDiv kinase inhibitor library, using only the structural features learned in the original SONARNR model. The top 5 and bottom 5 compound combinations based on SONARNR synergy scores were evaluated experimentally in dose-response assays. 2 of the 5 top-ranked synergy score combinations from SONARNR were confirmed as synergistic, while none of the 5 bottom-ranked combinations were synergistic (Fig. 5D; Table S8, S10). The selected structures predicted by the SONARNR algorithm to exhibit synergy did not resemble any previous structures or structural combinations in the CM. The SONARNR model thus exhibited substantial predictive power across different real world compound libraries.
Selective activity of synergistic combinations against fungal pathogens
Although established anti-fungal agents are often active against different fungal species, in many instances bioactive compounds can exhibit species-specific effects (Spitzer et al., 2011; Brown et al., 2014; Robbins et al., 2015). To examine if synergistic combinations might transpose to the related genetic networks of pathogenic fungi, we tested 18 combinations, 11 of which were active towards wild type S. cerevisiae, against the human pathogens C. neoformans, C. gattii, C. albicans, C. parapsilosis and A. fumigatus (Fig. 6A,B, S10B; Table S10). Focused dose-response assays were carried out over 4 concentrations for each compound and synergism assessed using Bliss independence at 48 h and 72 h (see Supplemental Information for details on each species). We observed that 16 synergistic combinations were active against at least one pathogen with considerable species specificity: 12 combinations were active against C. neoformans, 9 against C. gattii, 7 against C. albicans, 5 against C. parapsilosis, and 2 against A. fumigatus. Furthermore, species-specific combinations were identified for C. neoformans, C. gattii and C. albicans, but not for C. parapsilosis or A. fumigatus. For example, the combination of erythromycin and mitoxanthrone was effective against C. neoformans and C. albicans; strikingly, neither compound exhibited any growth inhibition alone up to a concentration of 128 µM (Fig. 6B). Many combinations were specific to fungal pathogens since only 5 out of the 16 combinations also inhibited growth of HEK293 cells (Fig. 6A). The antibiotic erythromycin in combination with the serotonin/dopamine antagonist methiothepin showed strong synergistic effects against all pathogenic fungi tested. Of the agents tested, the antipsychotic fluphenazine and the antifungal tolnaftate exhibited the most potent synergistic activity towards C. neoformans, C. gattii, C. albicans and A. fumigatus (Fig. 6B), with little or no activity towards HEK293 or HeLa cells (Fig. 6C). These results demonstrate that synergistic combinations can be selective even for species within the same genera and that potential synergistic antifungal agents active against clinically important human pathogens can be readily identified by SONARNR based on data from the tractable model yeast S. cerevisiae.
Figure 6.

Synergistic Combinations in Pathogenic Fungi and Human cells. (A) Bliss independence values for five pathogenic fungi and HEK cells. (B) Growth inhibition surface plots and corresponding Bliss independence heatmaps for 12 combinations from panel A for the S. cerevisiae pdr1Δpdr3Δ strain. Filled symbols indicate synergism, open symbols indicate lack of compound interaction. Abbreviations as in panel A. (C) Synergistic pair active against a fungal pathogen but not human cells. See also Figure S10.
Discussion
Genetic network structure suggests that combinations of compounds with little or no individual activity may offer potential for the therapeutic control of disease. However, the vast potential of synergistic chemical space has been underexplored, in part because efforts have focused heavily on known drugs (Feala et al., 2010). In this study, we used unbiased approaches to build the CGM and CM datasets, which then served as benchmark resources for the development of different predictive models for chemical synergism. Our initial expectation was that integration of chemical-genetic interaction data with the underlying genetic interaction network would enable prediction of chemical synergies (Fig. 7A). However, our SONARG graph-based model had little predictive power. To extract more information from the CGM, we developed a Naïve Bayesian model that assigned a likelihood score for genetic interactions associated with structural features represented in the CGM. Incorporation of the NBL-derived likelihoods in the SONARGN model slightly improved predictive power but combining all seven SONARGN parameters with a random forest-based machine learning algorithm to yield SONARGNR greatly augmented prediction accuracy. Unexpectedly, this effect was largely independent of genetic network information because a simpler and more computationally tractable model based only on NBL likelihoods and an RF learner, called SONARNR, performed even better. While genetic interactions must obviously underpin the network response to compound combinations, this result suggests that explicit knowledge of the genetic network is not required for accurate synergy prediction (Fig. 7B). This point is illustrated by the performance of SONARNR on unrelated chemical-genetic data and compounds that lie outside of the training datasets.
Figure 7.

Relationship Between Genetic, Chemical-Genetic and Chemical Interaction Space. (A) Conceptually, genetic synthetic lethal interactions can be integrated with chemical-genetic interactions to predict chemical synergies. The algorithm SONARG is predicated on this concept but has little predictive power. (B) In reality, genetic networks and bioactive compounds exhibit exceedingly complex interactions that defy simple interpretation (see text for details). However, machine learning approaches, such as in SONARNR, can associate chemical structural features with chemical-genetic interactions to predict chemical synergism.
Genetic interaction networks and prediction of chemical synergism
Current genetic network data has limited apparent power for prediction of chemical synergies for several reasons. First, the genotype-to-phenotype mapping problem may be too complex to solve in any general sense (Lehner, 2013; Taylor and Ehrenreich, 2015), and therefore precludes a solution to the more complex problem of chemical-to-phenotype mapping. Moreover, the still incomplete yeast binary genetic interaction network is already so dense that discrimination between different interaction paths becomes arbitrary (Costanzo et al., 2010), and context-dependent feedback loops further confound efforts to trace genetic causality (Ideker and Krogan, 2012). Second, while compound action can be idealized as genetic loss of function, in almost all cases chemical inhibition is incomplete and rife with multi-target effects that propagate through the genetic network. As demonstrated previously, chemical sub-structures often map to many hidden activity correlates and may be used to predict polypharmacological properties (Keiser et al., 2009; Besnard et al., 2012). Genetic interactions between deletion alleles may thus be a poor general proxy for chemical-chemical interactions. Third, the problem of complex chemical-genetic interaction profiles is multiplied when two compounds are co-administered.
Previous analyses of the basis for drug synergism have arrived at different conclusions. It has been suggested that compounds with similar chemogenomic profiles are likely to synergize (Jansen et al., 2009). However, this effect and other reported synergistic interactions may be explained by promiscuous (non-specific) compound actions that perturb general aspects of membrane structure or lipid metabolism (Cokol et al., 2011; Spitzer et al., 2011). We note that individual compounds that exhibited synergism in out study did not have promiscuous genetic interaction profiles. Although genetic interactions can correlate with chemical-genetic interactions (Costanzo et al., 2010), this effect is weak and has not been substantiated by other studies (Cokol et al., 2011). We similarly observed that highly connected genetic nodes are not predictive of chemical-genetic interactions. Our data do nevertheless suggest a relative enrichment for synergy between high level processes, such as for compounds that target vesicle transport and the fungal cell wall. The complexities of chemical action and genetic network density currently preclude rationalization of this synergism at the level of individual genetic interactions. We also note the difficulty in predicting synergism from genome-scale correlates that are only indirectly linked to phenotype (Bansal et al., 2014).
Genetic network differences between species suggest that synergistic combinations may be exploited to differentially target pathogens compared to benign commensal species or the host (Spitzer et al., 2011; Brown et al., 2014; Robbins et al., 2015). A number of synergistic combinations that we identified in S. cerevisiae exhibited cross-species activity, and some in fact exhibited more potent activity against pathogenic fungi. Network-specific combinatorial inhibition may prove effective in combating anti-fungal drug resistance in important human and plant fungal pathogens (Denning and Bromley, 2015; Lucas et al., 2015).
Machine learning and drug discovery
There has been tremendous interest in the efficient identification of synergistic compound interactions to improve drug efficacy, particularly in complex disease states (Feala et al., 2010). Collectively, the above arguments suggest that the complex relationship between the genetic network and chemical action may not be understandable through simple intuitive models. As shown here, focused forward chemical screens across many genetic contexts coupled to machine learning-based prediction algorithms reduce the size of the screening space for detection of chemical synergies. Thus, a moderate number of individual screens allows triangulation on potential synergistic pairs within a given collection of compounds. Analogous machine learning based approaches for synergism discovery may be feasible in other bacterial and fungal pathogens for which chemical-genetic interaction data can be readily acquired (Ejim et al., 2011; Roemer and Boone, 2013; Brown et al., 2014; Robbins et al., 2015). With the advent of robust methods for detection of genetic interactions in human cells (Shalem et al., 2015), the same strategy should be feasible in cancer and other complex diseases.
Methods
Strains, cell culture and chemical screens
S. cerevisiae deletion strains were obtained from the Euroscarf deletion set and are isogenic to BY4741 (Table S3). Fungal pathogen isolates were Cryptococcus neoformans (H99), Cryptococcus gattii (R265), Candida albicans (ATCC#90028), Candida parapsilosis (ATCC#90018) and Aspergillus fumigatus (Af293). All fungal species were grown and screened in synthetic complete (SC) medium with 2 % glucose. HeLa and HEK293 cells were grown in Phenol Red-free medium with 10 % dialyzed fetal bovine serum (Gibco) at 37 °C and 5 % CO2. Compounds were from the MicroSource Spectrum, LOPAC Sigma, Maybridge Hitskit 1000 (Ryan Scientific) and a custom Bioactive Collection. Screens were conducted in duplicate in 96 well plates at 20 µM per compound, with DMSO and 10 µM cycloheximide controls in each plate. OD600 was measured after ~18 h growth or until saturation was achieved for solvent controls. OD values were normalized and Z-scores calculated. The 128×128 cryptagen matrix was generated using a pdr1Δpdr3Δ strain at 10 µM per compound in duplicate experiments. Bliss independence was calculated using the equation Exy = Ex + Ey − (ExEy). Dose-response surfaces were assessed in wild type (BY4743) and pdr1Δpdr3Δ (MT2481) strains over 2-fold serial dilutions for each compound (1 µM to 128 µM) in an 8×8 matrix. Synergism in fungal pathogens was assessed in 4×4 mini-matrix at mid-point concentrations that had minimal effect on growth. For HeLa and HEK293 cells, dilution series were added to 100 µL seed cultures at ~5000 cells per well for 48 h, followed by viability assessment with PrestoBlue (Invitrogen).
Naïve Bayes Learner (NBL)
Regression analysis was performed on active compound features f for each sentinel strain s and conditional probability p(s|f) computed assuming that features were independent. For each strain, compounds were classified as active (Z-score < −4) or non-active. For each compound, ECFP4 fingerprints based on a circular topological connectivity traverse algorithm were calculated with Pipeline Pilot 6.0 (Accelrys). Structural features were learned for each strain using a multi-class Laplacian-Modified Naïve Bayes learner and leave-one-out cross-validation (Figure S5A). All models and AUC performance values are provided in Table S4.
Synergy prediction from genetic network data
SONARG and SONARGN were based on bipartite graphs and a second-degree count measure to link compounds via genetic interactions. A graph was built between two compounds, ci and cj, and genetic interaction edges ranked between ci and cj. Sensitive deletion strains VS for each cryptagen were identified based on CGM data (Z-score ≤ −4 for SONARG) or the NBL likelihoods (using the third quartile, Q3, for SONARGN). Genetic interactions for the set of sensitive deletion strains VS were used to identify all neighbors VT that formed the target space. VS and VT were connected by genetic interaction edges based on BioGRID release number 3.076 (Chatr-Aryamontri et al., 2015). To rank the nodes in VT, each node tj was scored as the sum of all edges that link to tj and the sum of the n=35 highest used to characterize the target space of each cryptagen. The highest ranked genes in represent likely targets of molecule ci (Table S6). An interaction score between ci and cj was calculated using the target space sets and . Empirical p-values were calculated from 1,000 permutations to estimate the background distribution.
Synergy prediction using a random forest ensemble learner
SONARGNR and SONARNR used random forest learners from the R packages caret and randomForest. Data was analyzed using R and the FactomineR and ROCR packages. The SONARGNR descriptor space used for machine learning was based on the 7 parameters defined for SONARG above (see Figure S3). The CM dataset was randomly split into 1/3 training with 5-fold cross validation and 2/3 test data using 512 trees with 3 variables at each split. The RF algorithm for SONARNR used 512 trees with 17 sentinels at each split and a node size limit of minimum 14 outcomes per leaf node.
Further details for all methods, algorithms and statistical analysis are provided in the Supplemental Information. All raw and processed data is available online at http://chemgrid.org/cgm.
Supplementary Material
Acknowledgments
We thank S. Giguere, G. Giaever and L. Fischer for discussions, and M. Costanzo and C. Boone for genetic interaction data. Supported by Canada Research Chairs in Molecular Studies of Antibiotics (GDW) and in Systems and Synthetic Biology (MT), a Howard Hughes Medical Institute International Research Scholar Award (MT), a Royal Society Wolfson Research Merit Award (MT), a Scottish Universities Life Sciences Alliance Research Chair (MT), and by grants from the Canadian Institutes for Health Research (MOP 119572 to GDW and MT), the European Research Council (SCG-233457 to MT), the Wellcome Trust (085178 to MT), the National Institutes of Health (R01RR024031 to MT), an award from the Ministère de l’enseignement supérieur, de la recherche, de la science et de la technologie du Québec through Génome Québec (MT), and the US Office of the Assistant Secretary of Defence for Health Affairs through the Breast Cancer Research Program (DAMD17-03-1-0471 to DB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
JW, DB, MT designed the study and experimental approach; DB, SD, NJ, RW, MS, JW, MR, EG performed experiments; JW devised all algorithms and analyzed the data; JW, MS, GDW and MT wrote the manuscript.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- Bansal M, Yang J, Karan C, Menden MP, Costello JC, Tang H, Xiao G, Li Y, Allen J, Zhong R, et al. A community computational challenge to predict the activity of pairs of compounds. Nat Biotechnol. 2014;32:1213–1222. doi: 10.1038/nbt.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JC, Nelson J, VanderSluis B, Deshpande R, Butts A, Kagan S, Polacheck I, Krysan DJ, Myers CL, Madhani HD. Unraveling the biology of a fungal meningitis pathogen using chemical genetics. Cell. 2014;159:1168–1187. doi: 10.1016/j.cell.2014.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, Boucher L, Heinicke S, Chen D, Stark C, Breitkreutz A, Kolas N, O'Donnell L, et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015;43:D470–478. doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Mulla WA, Kucharavy A, Tsai HJ, Rubinstein B, Conkright J, McCroskey S, Bradford WD, Weems L, Haug JS, et al. Targeting the adaptability of heterogeneous aneuploids. Cell. 2015;160:771–784. doi: 10.1016/j.cell.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokol M, Chua HN, Tasan M, Mutlu B, Weinstein ZB, Suzuki Y, Nergiz ME, Costanzo M, Baryshnikova A, Giaever G, et al. Systematic exploration of synergistic drug pairs. Mol Syst Biol. 2011;7:544. doi: 10.1038/msb.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480–1486. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csermely P, Agoston V, Pongor S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci. 2005;26:178–182. doi: 10.1016/j.tips.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Denning DW, Bromley MJ. Infectious Disease. How to bolster the antifungal pipeline. Science. 2015;347:1414–1416. doi: 10.1126/science.aaa6097. [DOI] [PubMed] [Google Scholar]
- Ejim L, Farha MA, Falconer SB, Wildenhain J, Coombes BK, Tyers M, Brown ED, Wright GD. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat Chem Biol. 2011;7:348–350. doi: 10.1038/nchembio.559. [DOI] [PubMed] [Google Scholar]
- Ericson E, Gebbia M, Heisler LE, Wildenhain J, Tyers M, Giaever G, Nislow C. Off-target effects of psychoactive drugs revealed by genome-wide assays in yeast. PLoS Genet. 2008;4:e1000151. doi: 10.1371/journal.pgen.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feala JD, Cortes J, Duxbury PM, Piermarocchi C, McCulloch AD, Paternostro G. Systems approaches and algorithms for discovery of combinatorial therapies. Wiley Interdiscip Rev Syst Biol Med. 2010;2:181–193. doi: 10.1002/wsbm.51. [DOI] [PubMed] [Google Scholar]
- Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol. 2006;2:458–466. doi: 10.1038/nchembio817. [DOI] [PubMed] [Google Scholar]
- Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995;47:331–385. [PubMed] [Google Scholar]
- Hartman JLt, Garvik B, Hartwell L. Principles for the buffering of genetic variation. Science. 2001;291:1001–1004. doi: 10.1126/science.291.5506.1001. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–1068. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, et al. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science. 2008;320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol. 2012;8:565. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen G, Lee AY, Epp E, Fredette A, Surprenant J, Harcus D, Scott M, Tan E, Nishimura T, Whiteway M, et al. Chemogenomic profiling predicts antifungal synergies. Mol Syst Biol. 2009;5:338. doi: 10.1038/msb.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H. A robustness-based approach to systems-oriented drug design. Nat Rev Drug Discov. 2007;6:202–210. doi: 10.1038/nrd2195. [DOI] [PubMed] [Google Scholar]
- Lee AY, St Onge RP, Proctor MJ, Wallace IM, Nile AH, Spagnuolo PA, Jitkova Y, Gronda M, Wu Y, Kim MK, et al. Mapping the cellular response to small molecules using chemogenomic fitness signatures. Science. 2014;344:208–211. doi: 10.1126/science.1250217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehar J, Krueger AS, Zimmermann GR, Borisy AA. Therapeutic selectivity and the multi-node drug target. Discov Med. 2009;8:185–190. [PubMed] [Google Scholar]
- Lehner B. Genotype to phenotype: lessons from model organisms for human genetics. Nat Rev Genet. 2013;14:168–178. doi: 10.1038/nrg3404. [DOI] [PubMed] [Google Scholar]
- Lucas JA, Hawkins NJ, Fraaije BA. The evolution of fungicide resistance. Adv Appl Microbiol. 2015;90:29–92. doi: 10.1016/bs.aambs.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Mani R, St Onge RP, Hartman JLt, Giaever G, Roth FP. Defining genetic interaction. Proc Natl Acad Sci U S A. 2008;105:3461–3466. doi: 10.1073/pnas.0712255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelander S, Wang W, Nilsson B, She QB, Pratilas C, Rosen N, Gennemark P, Sander C. Models from experiments: combinatorial drug perturbations of cancer cells. Mol Syst Biol. 2008;4:216. doi: 10.1038/msb.2008.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SM. Functional genomics to uncover drug mechanism of action. Nat Chem Biol. 2015;11:942–948. doi: 10.1038/nchembio.1963. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol. 2004;22:62–69. doi: 10.1038/nbt919. [DOI] [PubMed] [Google Scholar]
- Rask-Andersen M, Masuram S, Schioth HB. The druggable genome: Evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu Rev Pharmacol Toxicol. 2014;54:9–26. doi: 10.1146/annurev-pharmtox-011613-135943. [DOI] [PubMed] [Google Scholar]
- Robbins N, Spitzer M, Yu T, Cerone RP, Averette AK, Bahn YS, Heitman J, Sheppard DC, Tyers M, Wright GD. An Antifungal Combination Matrix Identifies a Rich Pool of Adjuvant Molecules that Enhance Drug Activity against Diverse Fungal Pathogens. Cell Rep. 2015;13:1481–1492. doi: 10.1016/j.celrep.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer T, Boone C. Systems-level antimicrobial drug and drug synergy discovery. Nat Chem Biol. 2013;9:222–231. doi: 10.1038/nchembio.1205. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharom JR, Bellows DS, Tyers M. From large networks to small molecules. Curr Opin Chem Biol. 2004;8:81–90. doi: 10.1016/j.cbpa.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Spitzer M, Griffiths E, Blakely KM, Wildenhain J, Ejim L, Rossi L, De Pascale G, Curak J, Brown E, Tyers M, Wright GD. Cross-species discovery of syncretic drug combinations that potentiate the antifungal fluconazole. Mol Syst Biol. 2011;7:499. doi: 10.1038/msb.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strebhardt K, Ullrich A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat Rev Cancer. 2008;8:473–480. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- Tan X, Hu L, Luquette LJ, 3rd, Gao G, Liu Y, Qu H, Xi R, Lu ZJ, Park PJ, Elledge SJ. Systematic identification of synergistic drug pairs targeting HIV. Nat Biotechnol. 2012;30:1125–1130. doi: 10.1038/nbt.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MB, Ehrenreich IM. Higher-order genetic interactions and their contribution to complex traits. Trends Genet. 2015;31:34–40. doi: 10.1016/j.tig.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. The Strategy of the Genes. London: Allen & Unwin; 1957. [Google Scholar]
- Wassermann AM, Lounkine E, Hoepfner D, Le Goff G, King FJ, Studer C, Peltier JM, Grippo ML, Prindle V, Tao J, et al. Dark chemical matter as a promising starting point for drug lead discovery. Nat Chem Biol. 2015;11:958–966. doi: 10.1038/nchembio.1936. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.