

Abstract
Oxidative stress and endoplasmic reticulum (ER) stress are related states that can occur in cells as part of normal physiology but occur frequently in diseases involving inflammation. In this article, we review recent findings relating to the role of oxidative and ER stress in the pathophysiology of acute and chronic nonmalignant diseases of the lung, including infections, cystic fibrosis, idiopathic pulmonary fibrosis and asthma. We also explore the potential of drugs targeting oxidative and ER stress pathways to alleviate disease.
Keywords: cystic fibrosis, endoplasmic reticulum stress, inflammation, lung disease, oxidative stress, protein misfolding, respiratory epithelium
Introduction to oxidative and ER stress
Oxidative stress and endoplasmic reticulum (ER) stress are related states that can occur in cells as part of normal physiology but which have been linked in the pathophysiology of many diseases, particularly diseases involving acute or chronic inflammation.1 Oxidative stress occurs when there is an imbalance between the production and degradation of reactive oxygen or nitrogen species (hereto ROS and RNS, respectively) within a cell, or when there is an excess of environmental ROS/RNS that can diffuse into the cell. One consequence of oxidative stress is disruption of the correct oxidative environment within the ER where proteins in the secretory pathway are produced, leading to misfolding of these proteins and ER stress. Interestingly, other triggers of protein misfolding in the ER result in excessive ROS production and therefore oxidative stress, further linking these states. Oxidative stress and ER stress are entwined with inflammation because inflammatory factors drive the production of ROS/RNS and because the stress activates inflammatory signalling in the affected cells, potentially setting up a forward‐feeding loop of stress and inflammation.1 In this article, we review recent findings relating to the role of oxidative and ER stress in the pathophysiology of acute and chronic nonmalignant diseases of the lung, and the potential of drugs targeting these pathways to alleviate disease. Although there is evidence for basal and therapy‐induced ER stress in lung cancer, and ER stress is being explored as a therapeutic target in this and other cancers,2 this topic will not be dealt with in this review.
ER function and protein misfolding/ER stress
The endoplasmic reticulum (ER) is responsible for the initial steps of biosynthesis of secretory pathway proteins including folding, N‐glycosylation, disulphide bond formation, other posttranslational modifications and for protein quality control, which ensures newly made secretory pathway proteins are suitable for function on the cell surface or secretion. The ER is also important for calcium homeostasis, being the major reservoir of intracellular Ca2+. A range of chaperones and enzymes resident within the ER are essential for correct protein folding and maintenance of the ER biosynthetic machinery.3 Typically, these chaperones disengage from proteins once the correct conformation is achieved allowing exit of the protein from the ER. Although some protein misfolding occurs in all cells and increases with increasing protein complexity, when an excess of unfolded protein is present, or when intracellular Ca2+ levels are disturbed, the unfolded protein response (UPR) is triggered. The UPR involves a series of signalling and transcriptional events to restore ER homeostasis via decreased translation, upregulation of ER chaperones and other molecules associated with productive folding (ER‐associated folding or ERAF), and increased degradation of misfolded proteins (ER‐associated degradation or ERAD), and this has been extensively reviewed.3, 4, 5 Briefly, there are three major arms of the UPR regulated by (1) the ER resident protein kinase RNA‐like ER kinase (PERK) which suppresses translation via eIF2α and includes transcriptional responses via activating transcription factor‐4 (ATF4) and CCAAT/enhancer‐binding protein homologous protein (CHOP), which can lead to ER stress‐induced apoptosis; (2) the endoribonucleases inositol requiring enzyme 1 (IRE1α) (ubiquitously expressed) and IRE1β (confined to mucosal secretory cells including in the lung), which splice the X‐box‐binding protein (XBP1) mRNA leading to translation of the sXBP1 transcription factor that drives expression of chaperones and other ER resident proteins required for folding and ERAD; and (3) the transcription factor ATF6, which also promotes production of proteins enhancing ER function.
Many factors can contribute to increased rates of misfolding in the ER, including increased rates of protein synthesis, missense polymorphisms in individual proteins, alterations in the oxidative environment, energy depletion, osmotic stress, viral infection and increased temperature. Prolonged ER stress can eventually lead to inflammatory signalling and premature apoptosis through several different mechanisms.6 The UPR has been linked to the pathogenesis of diabetes, inflammatory bowel disease, Alzheimer's disease, Parkinson's disease and many respiratory conditions including cystic fibrosis (CF),7, 8, 9, 10 chronic obstructive pulmonary disease (COPD),11 asthma,12 idiopathic pulmonary fibrosis (IPF)13, 14 and infection.15, 16, 17
Oxidative stress and redox balance
An endogenous factor that has been linked to ER stress and the UPR is the excess production of ROS and RNS disturbing the redox balance of the cell. ROS/RNS play a critical role in many cellular processes and can be produced in multiple organelles, including mitochondria, peroxisomes and ER. Mitochondria and peroxisomes contain enzymes involved in the production of ROS/RNS which in turn can be important for oxidation of other molecules and for metabolism and signal transduction. However, increased production of mitochondrial and peroxisomal ROS/RNS leads to cellular oxidative stress and can induce protein misfolding and ER stress. In a stressed ER, protein misfolding and particularly dysregulated disulphide bond formation and reduction may result in ROS accumulation, diffusion into the cytoplasm and thus cause cellular oxidative stress. Thus, ER stress and oxidative stress are intrinsically linked with each process triggering the other in differing scenarios encountered by the cell. Oxidative stress can lead to activation on inflammatory signalling pathways, including via activation of (1) the NFκB transcription factor that regulates inflammatory genes but also can regulate ROS/RNS production and degradation, and (2) the NRF2 transcription factor that primarily regulates the oxidative state of the cell but can also lead to inflammatory signalling.18, 19 Interpretation of studies of ROS/RNS on inflammatory activation pathways is complex as the large number of potential pathways affected by ROS/RNS means that both direct and indirect activation mechanisms occur. However, generally the oxidative stress‐mediated activation of these transcription factors can initiate the release of cytokines and chemokines, which further contribute to cellular dysfunction, local tissue damage and inflammatory pathologies in the airways.
Susceptibility to stress of cell types found in the lung
The lung is constituted by many different types of cells. Because susceptibility to oxidative and ER stress varies with cell type and their differentiation/activation status, it is important to understand how each of these cell types may be affected in disease states.20 Therefore, we now will discuss the roles of airway epithelial cells, stromal cells and immune cells in the context of oxidative and ER stress.
Airway epithelial cells
Airway epithelium acts as the front‐line defence for air‐borne particulate matter and pathogens to protect the lung from foreign body damage and infection. In the healthy airway, there is a thin coating of mucus that is continuously removed from the lung via the action of cilia, and if it accumulates by cough. This airway mucus is composed mainly of mucin glycoproteins and water, and provides a matrix for a wide variety of antimicrobial molecules including antibodies, defensins, protegrins, collectins, cathelicidins, lysozyme, histatins and nitric oxide21, 22, 23. When the mucus barrier has increased viscosity making it resistant to mucociliary clearance, or when the barrier is deficient or depleted, the risk of opportunistic microbial infection is increased.24 Mucins, primarily MUC5B, are tonically secreted by the nonciliated airway epithelial surface cells, known as club cells in the small airway, and can be released in large amounts in response to inhaled material by submucosal gland mucous cells, which contain large numbers of mucin granules ready for stimulated secretion.22
Under appropriate stimulation, the club cells transdifferentiate and both store and hyper‐secrete mucins, typically markedly upregulating production of the MUC5AC mucin. Mucin upregulation is controlled by the transcription factor SPDEF which also upregulates other proteins required for mucin biosynthesis, expands the size and biosynthetic capacity of the ER and modulates responses to TLR ligands.25, 26 Increased production of airway mucins by mucosal epithelial cells can be stimulated by adherence of probiotic bacteria and microbial products to assist clearance of pathogens. Expression and production of mucins can also be upregulated by inflammatory cytokines including IL‐1β,27 IL‐4,28 IL‐6,29 IL‐9,30 IL‐13,31 TNF‐α,32 nitric oxide,33 neutrophil elastase34 and other uncharacterised inflammatory factors, which might contribute to pathogenesis in human inflammatory airway disorders. IRE1β can also regulate mucin production, linking the UPR to mucin expression.35 However, if the increased production of mucin proteins is not resolved, mucus accumulation may contribute to the pathogenesis of human inflammatory airway disorders. Overproduction of airway mucus is a common problem in many airway diseases including bronchiectasis, CF and COPD where increased concentration of mucins in mucus is a key characteristic.36, 37
The secreted mucin glycoproteins are large in size, contain highly folded cysteine‐rich domains containing multiple intra‐ and intermolecular disulphide bonds formed in the ER and therefore present a substantial challenge for correct folding in the ER.22 ER stress occurs when proteins misfold during biosynthesis, and ER stress in mucin‐producing cells (known as mucous cells or goblet cells depending on the tissue) can occur during inflammation and could thus be a feature of airway diseases, especially those involving mucus hypersecretion. ER stress in mucin‐producing goblet cells has been demonstrated in a mouse model, in which aberrant mucin biosynthesis due to a protein misfolding mutation in the MUC2 mucin cells leads to goblet cell ER stress and an inflammatory bowel disease‐like phenotype.38, 39 Mediators that can induce ER stress in the gut and airway epithelial cells include oxidative stress and pro‐inflammatory cytokines. It is logical to suspect that the large demand of mucin glycoprotein production and the inflammatory environment in many mucopurulent lung diseases could result in ER stress in the airway epithelial cells, which could further lead to inflammatory signalling and chronic nonresolving inflammation. We will explore the evidence for this contention in a range of respiratory conditions in this review. In addition to the mucus‐producing cells, other epithelial cells such as the ciliated cells could be subjected to oxidative and ER stress and play an important role in the pathology of airway disease.
Stromal cells
The lung mucosa also contains stromal cells of a mesenchymal derivation that may also be exposed to oxidative and ER stress including fibroblasts, myofibroblasts and endothelial cells. We will also explore how these cells types, which are critical determinants of the fibrosis that develops in chronic lung diseases, are affected by oxidative and ER stress in acute and chronic lung disease.
Immune cells
In health, multiple populations of immune cells occupy the normal respiratory mucosa and the airways themselves, and additionally, in acute and chronic respiratory disease, there can be substantial recruitment and activation of leucocytes into the respiratory mucosa. Normal resident cells are dominated by macrophages which in the healthy lung are dominated by the alveolar macrophages. Whilst alveolar macrophages have an important role in ‘house‐keeping’ in the terminal airways, they can also become an arm of innate immunity following infection. Other important leucocytes in the respiratory mucosa include dendritic cells, both regulatory and effector T cells, innate lymphoid cells and NK cells. Conspicuous recruitment of neutrophils is also typical of many chronic inflammatory conditions in the lung. Many of these cell types produce effectors, such as inflammatory cytokines, that can induce oxidative and ER stress, or directly produce ROS/RNS into the respiratory microenvironment, as is characteristic of neutrophils and activated macrophages. Each of these immune cell types is also a potential target of ER and oxidative stress that needs to be considered in the context of assessing ER and oxidative stress in respiratory disease.
Oxidative and ER stress in infectious respiratory disease
Oxidative stress and ER stress have been reported to occur in many respiratory infections and could arise via direct effects of the pathogen on infected cells or as a consequence of the immune response to the pathogen. Because viruses hijack the secretory pathway to manufacture viral glycoproteins, they place demands on the ER which often result in substantial misfolding, ER stress and subsequent activation of the UPR. In fact, viral infection has probably shaped the evolution of the UPR such that both the UPR‐induced PERK‐mediated translational block and apoptosis can be viewed as appropriate responses to minimise viral replication. The best evidence that the UPR is important in limiting viral replication is the fact that many viruses have developed specific mechanisms to interfere with the UPR or to subvert or circumvent its mechanisms of action in order that viral replication can continue in the face of intrinsic misfolding (reviewed by Frabutt and Zheng40, Li et al.41, Verchot42). In somewhat of a contradiction, for some viruses, the action of at least some arms of the UPR actually promotes greater viral replication. The influenza A virus (IAV) is an interesting example of this phenomenon. Infection of respiratory epithelial cells with IAV activates the UPR in a distinct way with suppression of the PERK and ATF6 arms of the UPR concomitant with strong activation of IRE1α.43 The mechanism by which this is achieved, given the common activation mechanisms for these three major arms of the UPR, is not clear, but it is functionally important because boosted activation of IRE1α is necessary for continued viral replication.43 Respiratory syncytial virus (RSV) is another respiratory virus that causes ER stress (for a detailed review, see Cervantes‐Ortiz et al.44) but results in a noncanonical activation of the UPR, with in this case activation of IRE1α and ATF6, but not PERK.45 However, in complete contrast to IAV, in RSV infection, IRE1α suppresses viral replication.45
Another mechanism by which infection leads to respiratory oxidative and ER stress is via mediators of the activated immune system, such as cytokines and direct production of ROS/RNS, which are discussed below in the context of chronic inflammatory lung diseases. In this context during the infectious exacerbations that frequently occur in chronic lung diseases, it is important to consider the combined effect on the ER stress pathway of infections and pre‐existing ‘noninfectious’ inflammation. As an example of experimental evidence of this concept, concomitant RSV infection exacerbates the ER stress that occurs in the murine bleomycin‐induced model of pulmonary fibrosis.16 Figure 1 summarises the cell intrinsic and environmental factors that potentially drive both oxidative stress and ER stress in respiratory epithelial cells.
Figure 1.
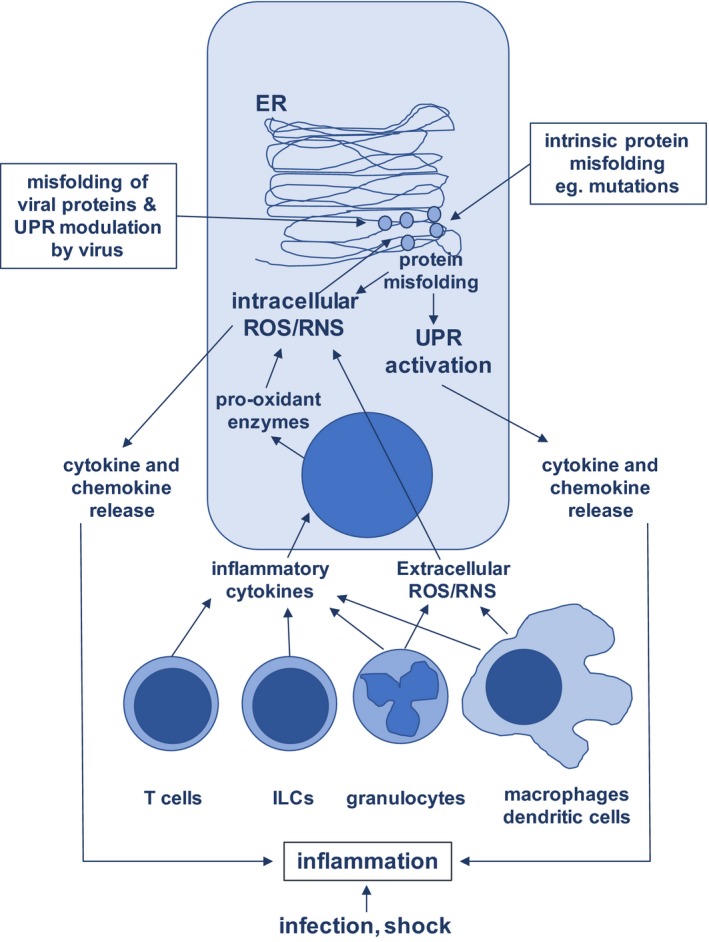
Interaction of cell intrinsic and environmental factors on the development of oxidative and ER stress in respiratory epithelial cells. ER, endoplasmic reticulum; ILC, innate lymphoid cell; ROS/RNS, reactive oxygen species, reactive nitrogen species; UPR, unfolded protein response.
Oxidative and ER stress in chronic inflammatory and mucopurulent diseases
Figure 2 summarises the influences of cell intrinsic and environmental factors that may contribute to oxidative stress and ER stress in a range of respiratory diseases, and each is dealt with in the following sections.
Figure 2.
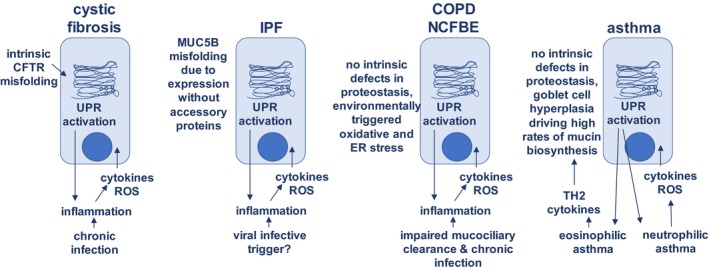
Interaction of cell intrinsic and environmental factors on the development of oxidative and ER stress in specific respiratory conditions. CFTR, cystic fibrosis conductance receptor; COPD, chronic obstructive pulmonary disease; NCFBE, noncystic fibrosis bronchiectasis; ER, endoplasmic reticulum; ROS, reactive oxygen species; TH2, T‐helper 2 immune response; UPR, unfolded protein response.
Cystic fibrosis (CF) and non‐CF bronchiectasis (NCFBE)
Cystic fibrosis is a disease where on face value one would predict the development of ER stress because of (1) intrinsic misfolding of the CFTR protein in the case of many mutations, including the common F508del mutation that results in misfolding of all the protein and complete absence of CFTR on the cell surface; (CFTR is normally expressed by a variety of different epithelial cell types but most highly in submucosal gland epithelium); (2) chronic bacterial infection and frequent exacerbations due to viral infection; (3) chronic complex inflammation with activation of both innate immunity and adaptive immunity; and (4) chronic mucus overproduction increasing the ER biosynthetic load of mucin‐secreting cells. Much of the work on ER stress in CF has been conducted by the Ribeiro group at University of North Carolina, and they have recently reviewed ER stress and UPR activation in CF.46 The most instructive data on the contribution of CFTR mutations to ER stress emanate from studies of cultured human bronchial epithelial cells (HBECs) derived from CF patients and healthy donors, separating the cells with the intrinsic defect from the inflamed and infected in vivo environment in the disease. Somewhat surprisingly, these studies indicate that the defects do not intrinsically drive increased ER stress or UPR activation. Whilst ER expansion and increased ER Ca2+ storage and signalling can be demonstrated in CF epithelial cells in short‐term cultures, these revert to normal in long‐term, nonstimulated cultures.9, 47 However, in another study, overexpression of F508del‐CFTR was reported to increase XBP1 splicing, suggesting that the mutant protein can increase ER stress.7 In considering the influence of the mutated protein, it is important to take into account that a large proportion (~60–80%) of wild‐type CFTR misfolds during biosynthesis due to the protein's complex transmembrane and nucleotide‐binding domains48; consequently, there will be a background of compensatory UPR activation in all CFTR‐expressing cells. The key question is whether the increased misfolding in the case of mutant CFTR substantially disturbs the state of proteostasis normally achieved in these respiratory epithelial cells.
Despite the lack of evidence for an intrinsic CFTR mutation driver of ER stress, there is evidence for ER stress and UPR activation in CF epithelium in vivo, and mucopurulent secretions from CF patients stimulate ER stress in healthy cultured HBECs with consequent XBP1‐dependent expansion of the ER8. Consistent with these observations, P. aeruginosa infection in mice induced lung inflammation and UPR activation as measured by splicing of XBP‐1 measured in vivo using the ERAI ER stress‐reporter transgene.49 Development of ER stress is unsurprising as CF lungs are chronically infected and the mucosa and lung secretions contain a broad array of inflammatory cytokines50, 51 many of which are known to drive oxidative stress and therefore ER stress (reviewed in Hasnain et al.1, 52).
Additionally, CF airways are characterised by pronounced neutrophil and/or macrophage accumulation and activation of these cells during infection and inflammation often results in release of ROS and RNS into the microenvironment in an attempt to control infection, increasing oxidative stress. Production of ROS is a feature of CF that has been linked with damage to the epithelium and progressive bronchiectasis and failure of lung function.53 Even in children with CF, there is a large amount of myeloperoxidase (MPO) produced by neutrophils and macrophages. MPO converts H2O2 into several damaging oxidants and MPO activity appears to be accompanied by depletion in the lung of counteracting reducing agent, glutathione, further enhancing the level of oxidative stress.54, 55 Activated neutrophils produce the superoxide ion (O2 −) as a result of activation of the NOX2 NADPH oxidase complex, whereas airway epithelial cells primarily utilise DUOX1/2 to produce ROS (reviewed in Pongnimitprasert et al.56, Lee and Yang57). In contrast, inducible nitric oxide synthase (NOS2) is reduced in CF airway epithelial cells most likely in direct response to the CFTR deficiency,58 and there is no significant difference in the natural NOS inhibitor asymmetric dimethylarginine (ADMA) in the breath condensate from children with CF,59 suggesting much of the oxidative stress is driven by ROS rather than RNS. How much the altered ROS/RNS environment in the lung also affects ER stress remains unclear; however, there is evidence that the neutrophils themselves experience ER stress in CF.60
An interesting question in CF is whether the new class of drugs designed to facilitate folding/biogenesis of CFTR, such as Lumacaftor, influence stress in the ER.61, 62 Lumacaftor is a chaperone type drug that is designed to aid folding in the ER to enhance release of mutant CFTR to the cell surface where it has partial ion channel function. Lumacaftor is typically used in combination therapy with Ivacaftor which potentiates ion channel function of CFTR. These drugs are having a significant impact in the clinic, and although the primary benefit will be via partial restoration of the appropriate ionic environment and hydration of the airway, some benefit may be derived from reducing ER stress in highly CFTR‐expressing cells in the airway.63 Alternatively, it is possible that disruption of normal ER function/quality control with these modulatory drugs increases ER stress and UPR signalling, working against the desired function of the drugs. However, there are no data exploring these possibilities.
NCFBE is a chronic mucopurulent lung disease which can arise via primary ciliary dyskinesia or as a consequence of severe lung damage from infections. Although arising in the absence of any defects in CFTR, NCFBE shares many features with CF including impaired mucociliary clearance, chronic bacterial infection, frequent viral exacerbations, chronic inflammation (typically involving accumulation of activated neutrophils) and progressively declining lung function.64, 65, 66, 67 Despite these common features, our studies have failed to demonstrate any clear evidence of ER stress and UPR activation in NCFBE.
Idiopathic pulmonary fibrosis (IPF)
Idiopathic pulmonary fibrosis is a progressive interstitial lung disease arising around the distal airway/alveoli that involves epithelial cell death, inflammation and fibrosis, with few treatment options and a high mortality rate.68 Although the aetiology is unclear, there are several strong genetic links to predisposition to IPF, including rare alleles in surfactant (SFTPC, SFTPA2) and telomerase pathway (TERT, TERC, PARN and RTEL) genes, and more common alleles of MUC5B strongly associated with a less aggressive form of IPF (reviewed by Evans et al.68). ER stress has been linked with disease development by multiple lines of evidence in human IPF and in the bleomycin‐induced murine model of IPF (for recent reviews of ER stress and IPF see Zhang et al.69 and Tanjore et al.70). Evidence for ER stress in human respiratory epithelial cells, particularly in type II pneumocytes, includes high expression of CHOP,71 activation of ATF6, XBP1 and ATF4.14 Activation of the UPR has been found in both inherited and sporadic IPF and associated with viral infection.13 Additionally, fibroblasts from the lungs of IPF patients show increased ER stress in response to TGFβ.72 IPF‐linked mutations in SFTPC and SFTPA2 cause misfolding of the encoded surfactant proteins and ER stress in type II pneumocytes, providing a direct mechanistic driver for the ER stress in these forms of IPF.13, 73 Interestingly, there is substantial evidence for the importance of environmental triggers, and mice transgenic for the Δexon‐4 SFTPC mutation develop spontaneous lung disease,74 whereas those expressing L188Q SFTPC mutations only develop disease when exposed to low dose bleomycin.75 Cultured type II pneumocytes and transfected cells carrying the Δexon‐4 SFTPC mutation show accumulation of the SFTPC precursor protein, UPR activation and increased cell death when infected with RSV, suggesting viral infection could be a trigger for development of IPF in susceptible individuals.76
The common variant rs35705950 in the promoter region of MUC5B is carried by 9% of the European population and is the strongest risk factor for developing IPF accounting for 30–35% of the risk and also predicting asymptomatic mild fibrosis.68, 77, 78, 79 There are multiple potential explanations for how this polymorphism could lead to fibrosis (for a discussion, Evans et al.68), one of which is the ER stress pathway. Inappropriate expression of MUC5B in type II pneumocytes, particularly in the honeycomb cysts characteristic of this disease, is a feature of IPF,80, 81 and the MUC5B promoter polymorphism leads to enhanced MUC5B mRNA expression.77 Interestingly, this expression, potentially driven by altered transcription factor binding sites in the promoter, occurs in the absence of expression of SPDEF,80 which is a transcription factor that drives expression of mucin genes and a network of other genes involved in mucin biosynthesis and secretion.82 Taking these things together, it is inviting to propose that high expression of a complex mucin protein in a cell type lacking the appropriate ER machinery to fold and process the mucin appropriately predisposes the cell to ER stress. Perhaps this level of stress can be managed in the absence of other stressors but with ageing and accumulated environmental insults, including from viral infection, ER stress and inflammation emerge and progress with consequent fibrosis. Whilst the mucin is normally expressed only in differentiated cells, the polymorphism may drive inappropriate MUC5B expression in stem cells, as suggested by Evans et al.,68 and could lead to altered survival or function of the stem cells that are needed to appropriately renew epithelium in the terminal airways.
Animal models for IPF are imperfect, with the most commonly used being a model where fibrosis is induced by installation of bleomycin in the airway. ER stress develops in airway cells in this model and is exacerbated by viral infection, and the UPR has been associated with the differentiation status of macrophages, and intrinsic ER stress within macrophages also occurs.16, 71, 75, 83, 84, 85, 86
Asthma
Asthma is a condition involving airway hyper‐responsiveness and there is emerging evidence that both oxidative stress and ER stress are features of asthma, most prominently in the neutrophil‐dominated glucocorticoid resistant severe endotype (for a review, see Kim et al.87). Genetic predisposition has also linked asthma and ER stress via the ORMLD3 gene which encodes a protein that modulates function of the sarcoendoplasmic reticulum Ca2+ ATPase pump (SERCA) that regulates ER vs cytosolic Ca2+ concentrations, and thereby modulates protein folding and Ca2+‐signalling.12, 88, 89 Studies of ER stress in the human disease are rather limited but there is evidence of ER stress in both cells sampled by bronchiolar lavage and in peripheral blood leucocytes.90 A range of animal models of allergy and asthma have also implicated ER stress in the pathophysiology of these conditions.90, 91, 92, 93, 94, 95, 96 Airway goblet cell hyperplasia and mucus hypersecretion are characteristics of most forms of asthma and the increased biosynthetic load in these cells may be principal drivers of ER stress, as may the ROS/RNS released by phagocytes, particularly in neutrophilic disease. Furthermore, viral infection is a trigger for exacerbating asthma and has been shown to exacerbate ER stress in animal models of allergic asthma.91
Chronic obstructive pulmonary disease (COPD) and chronic bronchitis
Chronic obstructive pulmonary disease and chronic bronchitis are characterised by inflammation, oxidative stress and mucus hypersecretion, all of which, based on earlier discussion, could drive ER stress in these conditions. There is evidence for ER stress in COPD but it is based on a limited number of validated studies.97, 98, 99 However, there is reasonably clear evidence that exposure of bronchial epithelial cells to cigarette smoke (the environmental driver of COPD) increases ER stress.100, 101
α‐1‐antitrypsin (AAT) deficiency
α‐1‐Antitrypsin is primarily caused by a variety of mutations in AAT that produce disease of varying severity including liver disease and a COPD‐like emphysema that is more prevalent and severe in smokers.102 The liver disease arises as a result of polymerisation of AAT in hepatocytes, and accumulation within the ER and subsequent ER stress, with the hepatocyte stress driving chronic hepatitis potentially progressing to malignancy.102 The aetiology of the respiratory disease is less clear, with the simplistic interpretation that AAT deficiency contributes via loss of its capacity to neutralise neutrophil elastase. However, the lung phenotype of AAT individuals with complete serum deficiency varies markedly, and there is evidence for respiratory epithelial cell ER stress in the human disease and in mice transgenic for the ZZ‐AAT genotype. The human Z allele is a deletion which results in an amino acid substitution changing the conformation of the AAT molecule, the ZZ genotype is responsible for 98% of AAT represents the most severe emphysema.103 Furthermore, in cultured AAT mutant epithelial cells, the ER stress and consequent release of inflammatory cytokines and chemokines are exacerbated by exposure to cigarette smoke, consistent with the enhanced disease in smokers.103
Oxidative and ER stress in acute respiratory conditions
Oxidative stress and ER stress have been implicated in the pathophysiology of a variety of more acute conditions affecting the lung. In septic shock and lung stress induced in animal models by exposure to LPS and other TLR ligands, both oxidative stress and ER stress occur in the damaged lung and appear to be a feature of the pathological process.104, 105, 106 In sepsis/LPS‐induced lung injury, both ER stress and autophagy (which may be a response to ER stress) occur and the systemic factor cold‐induced RNA‐binding protein (CIRP), local inflammatory cytokines such as IL‐17 and neutrophil activation have been linked with the development of ER stress and activation of the UPR.104, 105, 106 Pulmonary arterial hypertension results in chronic hypoxia, inflammation, and consequently oxidative and ER stress, and induction of autophagy, in the respiratory mucosa.107 In contrast, bronchopulmonary dysplasia (BPD) is a condition affecting ventilated premature neonates that develops as a result of hyperoxia and also appears to involve ER stress. In animal models of BPD hyperoxia in neonates drives ROS production, ER stress and ER stress‐induced apoptosis in the alveolar epithelium providing an explanation for the alveolar damage characteristic of BPD.108, 109
Therapeutic approaches to resolve oxidative and ER stress in respiratory disease
The development of oxidative and ER stress in lung diseases described above suggests that therapeutic approaches to either dampen oxidative and ER stress, or modify the UPR, may be successful. Anti‐oxidants have relieved ER stress and pathology in some animal models of respiratory disease. For example, the strong anti‐oxidant, chlorogenic acid, suppressed ER stress, apoptosis and fibrosis in bleomycin‐induced fibrosis in mice.86 However, targeting oxidative stress in humans has been largely disappointing with the most evidence coming from trials of both airway and systemically delivered N‐acetyl‐cysteine (NAC) in CF, NCFBE and COPD.110, 111 Complicating matters, the limited beneficial effects seen including reduced exacerbation frequency and improved airway function are difficult to interpret as NAC has mucolytic as well as anti‐oxidant properties, and the beneficial effects may be attributed to improved mucociliary clearance rather than resolution of oxidative stress. Other approaches trialled include NOX‐inhibitors, superoxide dismutase‐mimetics, myeloperoxidase inhibitors and NRF2 activators; however, many of these drugs have failed in clinical trials and their development has been discontinued.112 New approaches or improved agents are required to suppress oxidative stress. For example, we have found that the cytokine, IL‐22, drives a robust natural anti‐oxidant programme in secretory cells,113 including respiratory epithelial cells, potentially providing a more effective mechanism to protect cells from both environmental and intrinsically‐generated ROS/RNS.
There have been reasonably intense efforts to therapeutically manipulate ER stress and the UPR for a broad range of diseases, including primary misfolding diseases, chronic inflammatory diseases, diabetes and cancer. There are several different classes of therapeutics including (1) modulators of specific arms of the UPR, (2) chaperone modulators, (3) chemical chaperones and (4) modulators of ERAD (for a review, see Rivas et al.5). Modulation of the UPR needs to be carefully considered as it could have complex consequences. For example, suppression of an arm of the UPR could reduce downstream inflammatory signalling and protect from apoptosis, providing benefit, but at the same time may impair the production of ER molecules involved in folding thereby exacerbating misfolding and driving the other arms of the UPR with potentially adverse consequences. Use of these experimental drugs in respiratory diseases is mainly restricted to preclinical models of disease and some examples are provided below.
Salubrinal is a selective inhibitor of the phosphatase‐mediated dephosphorylation of eIF2α which is downstream of PERK and is responsible for repression of translation during ER stress. Salubrinal has been demonstrated to repress cigarette smoke‐induced airway epithelial ER stress which is relevant for COPD therapy.98 4‐phenylbutyrate (4‐PBA) and tauroursodeoxycholic acid (TUDCA) are chemical chaperones which promote correct folding in the ER that have been used widely in animal models of ER stress. 4‐PBA reduced ER stress, inflammation and fibrosis in the bleomycin model in mice,84 attenuated symptoms of asthma in the house dust mite allergen model in mice90 and also reduced ER stress and autophagy in LPS‐induced acute lung injury.104 Similarly, TUDCA has also been demonstrated to alleviate ER stress and associated pathology in the bleomycin fibrosis model and in asthma models in mice.83, 92 Whilst these studies provide proof of principle that targeting ER stress may be beneficial in human disease, we await the development of more specific, less toxic and more effective drugs, followed by clinical trials in the range of respiratory conditions that may benefit from these agents.
Summary and conclusions
Oxidative stress and ER stress are frequent and clearly linked phenomena in a wide range of acute and chronic respiratory conditions, and there is substantial evidence that they are integral components of the pathophysiology of these conditions. More often than not oxidative stress and ER stress are accompanied by inflammation, both because the stress can be a consequence of factors produced by activated leucocytes, and because the cellular pathways activated by oxidative or ER stress lead to the release of factors driving both innate immunity and adaptive immunity. This potentially sets up a forward‐feeding cycle of cellular stress and inflammation, and in each specific respiratory condition, there is a need for a deeper understanding of the relative primary importance of inflammation and oxidative/ER stress, and whether targeting individual or a combination of these pathways will best reverse the pathological processes. Such studies are hampered by the lack of sophisticated therapeutic agents to modify oxidative and ER stress, with refinement of the current agents and approaches required before benefits can be realised in the clinic.
Acknowledgments
M McGuckin is supported by National Health and Medical Research Council Principal Research Fellowship APP1059726.
Conflicts of Interest
MAM is an inventor on a patent concerning therapeutic uses of IL‐22. The authors have no further conflict of interests to declare.
References
- 1. Hasnain SZ, Lourie R, Das I et al The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol 2012; 90: 267–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marcinak SJ, Ron D. The unfolded protein response in lung disease. Proc Am Thorac Soc 2010; 7: 356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334: 1081–1086. [DOI] [PubMed] [Google Scholar]
- 4. McGuckin MA, Eri RD, Das I et al ER stress and the unfolded protein response in intestinal inflammation. Am J Physiol Gastrointest Liver Physiol 2010; 298: G820–G832. [DOI] [PubMed] [Google Scholar]
- 5. Rivas A, Vidal RL, Hetz C. Targeting the unfolded protein response for disease intervention. Expert Opin Ther Targets 2015; 19: 1203–1218. [DOI] [PubMed] [Google Scholar]
- 6. Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 2014; 21: 396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bartoszewski R, Rab A, Jurkuvenaite A et al Activation of the unfolded protein response by {Delta}F508 CFTR. Am J Respir Cell Mol Biol 2008; 39: 448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martino ME, Olsen JC, Fulcher NB et al Airway epithelial inflammation‐induced endoplasmic reticulum Ca2+ store expansion is mediated by X‐box binding protein‐1. J Biol Chem 2009; 284: 14904–14913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ribeiro CM, Paradiso AM, Schwab U et al Chronic airway infection/inflammation induces a Ca2+i‐dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J Biol Chem 2005; 280: 17798–17806. [DOI] [PubMed] [Google Scholar]
- 10. Kerbiriou M, Le Drévo M‐A, Férec C et al Coupling cystic fibrosis to endoplasmic reticulum stress: differential role of Grp78 and ATF6. Biochim Biophys Acta (BBA) 2007; 1772: 1236–1249. [DOI] [PubMed] [Google Scholar]
- 11. Malhotra D, Thimmulappa R, Vij N et al Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: the role of Nrf2‐regulated proteasomal activity. Am J Respir Crit Care Med 2009; 180: 1196–1207. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Cantero‐Recasens G, Fandos C, Rubio‐Moscardo F et al The asthma‐associated ORMDL3 gene product regulates endoplasmic reticulum‐mediated calcium signaling and cellular stress. Hum Mol Genet 2010; 19: 111–121. [DOI] [PubMed] [Google Scholar]
- 13. Lawson WE, Crossno PF, Polosukhin VV et al Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol 2008; 294: L1119–L1126. [DOI] [PubMed] [Google Scholar]
- 14. Korfei M, Ruppert C, Mahavadi P et al Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2008; 178: 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roberson EC, Tully JE, Guala AS et al Influenza induces ER stress, caspase‐12‐ dependent apoptosis and JNK mediated TGF‐β release in lung epithelial cells. Am J Respir Cell Mol Biol 2011; 46: 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L, Cheng W, Zhang Z. Respiratory syncytial virus infection accelerates lung fibrosis through the unfolded protein response in a bleomycin‐induced pulmonary fibrosis animal model. Mol Med Rep 2017; 16: 310–316. [DOI] [PubMed] [Google Scholar]
- 17. Merquiol E, Uzi D, Mueller T et al HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS ONE 2011; 6: e24660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bryan HK, Olayanju A, Goldring CE et al The Nrf2 cell defence pathway: Keap1‐dependent and ‐independent mechanisms of regulation. Biochem Pharmacol 2013; 85: 705–717. [DOI] [PubMed] [Google Scholar]
- 19. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF‐kappaB signaling. Cell Res 2011; 21: 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martinon F, Chen X, Lee AH et al TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 2010; 11: 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nicholas B, Skipp P, Mould R et al Shotgun proteomic analysis of human‐induced sputum. Proteomics 2006; 6: 4390–4401. [DOI] [PubMed] [Google Scholar]
- 22. Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Ann Rev Physiol 2008; 50: 5.1–5.28. [DOI] [PubMed] [Google Scholar]
- 23. Button B, Cai LH, Ehre C et al A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 2012; 337: 937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roy MG, Livraghi‐Butrico A, Fletcher AA et al Muc5b is required for airway defence. Nature 2014; 505: 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park KS, Korfhagen TR, Bruno MD et al SPDEF regulates goblet cell hyperplasia in the airway epithelium. J Clin Invest 2007; 117: 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Korfhagen TR, Kitzmiller J, Chen G et al SAM‐pointed domain ETS factor mediates epithelial cell‐intrinsic innate immune signaling during airway mucous metaplasia. Proc Natl Acad Sci USA 2012; 109: 16630–16635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gray T, Coakley R, Hirsh A et al Regulation of MUC5AC mucin secretion and airway surface liquid metabolism by IL‐1beta in human bronchial epithelia. Am J Physiol Lung Cell Mol Physiol 2004; 286: L320–L330. [DOI] [PubMed] [Google Scholar]
- 28. Dabbagh K, Takeyama K, Lee HM et al IL‐4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J Immunol 1999; 162: 6233–6237. [PubMed] [Google Scholar]
- 29. Neveu WA, Allard JB, Dienz O et al IL‐6 is required for airway mucus production induced by inhaled fungal allergens. J Immunol 2009; 183: 1732–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reader JR, Hyde DM, Schelegle ES et al Interleukin‐9 induces mucous cell metaplasia independent of inflammation. Am J Respir Cell Mol Biol 2003; 28: 664–672. [DOI] [PubMed] [Google Scholar]
- 31. Zhen G, Park SW, Nguyenvu LT et al IL‐13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am J Respir Cell Mol Biol 2007; 36: 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Delmotte P, Degroote S, Merten MD et al Influence of TNFalpha on the sialylation of mucins produced by a transformed cell line MM‐39 derived from human tracheal gland cells. Glycoconj J 2001; 18: 487–497. [DOI] [PubMed] [Google Scholar]
- 33. Song JS, Kang CM, Yoo MB et al Nitric oxide induces MUC5AC mucin in respiratory epithelial cells through PKC and ERK dependent pathways. Respir Res 2007; 8: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou J, Perelman JM, Kolosov VP et al Neutrophil elastase induces MUC5AC secretion via protease‐activated receptor 2. Mol Cell Biochem 2013; 377: 75–85. [DOI] [PubMed] [Google Scholar]
- 35. Martino MB, Jones L, Brighton B et al The ER stress transducer IRE1beta is required for airway epithelial mucin production. Mucosal Immunol 2012; 6: 639–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Button B, Anderson WH, Boucher RC. Mucus hyperconcentration as a unifying aspect of the chronic bronchitic phenotype. Ann Am Thorac Soc 2016; 13(Suppl 2): S156–S162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kesimer M, Ford AA, Ceppe A et al Airway mucin concentration as a marker of chronic bronchitis. N Engl J Med 2017; 377: 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heazlewood CK, Cook MC, Eri R et al Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med 2008; 5: e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eri RD, Adams RJ, Tran TV et al An intestinal epithelial defect conferring ER stress results in inflammation involving both innate and adaptive immunity. Mucosal Immunol 2011; 4: 354–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frabutt DA, Zheng YH. Arms race between enveloped viruses and the host ERAD machinery. Viruses 2016; 8: 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li S, Kong L, Yu X. The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit Rev Microbiol 2015; 41: 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Verchot J. How does the stressed out ER find relief during virus infection? Curr Opin Virol 2016; 17: 74–79. [DOI] [PubMed] [Google Scholar]
- 43. Hassan IH, Zhang MS, Powers LS et al Influenza A viral replication is blocked by inhibition of the inositol‐requiring enzyme 1 (IRE1) stress pathway. J Biol Chem 2012; 287: 4679–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cervantes‐Ortiz SL, Zamorano Cuervo N, Grandvaux N. Respiratory syncytial virus and cellular stress responses: impact on replication and physiopathology. Viruses 2016; 8: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hassan I, Gaines KS, Hottel WJ et al Inositol‐requiring enzyme 1 inhibits respiratory syncytial virus replication. J Biol Chem 2014; 289: 7537–7546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ribeiro CM, Lubamba BA. Role of IRE1alpha/XBP‐1 in cystic fibrosis airway inflammation. Int J Mol Sci 2017; 18: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ribeiro CM, Paradiso AM, Carew MA et al Cystic fibrosis airway epithelial Ca2+ i signaling: the mechanism for the larger agonist‐mediated Ca2+ i signals in human cystic fibrosis airway epithelia. J Biol Chem 2005; 280: 10202–10209. [DOI] [PubMed] [Google Scholar]
- 48. Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol Med 2012; 18: 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith RS, Wolfgang MC, Lory S. An adenylate cyclase‐controlled signaling network regulates Pseudomonas aeruginosa virulence in a mouse model of acute pneumonia. Infect Immun 2004; 72: 1677–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tiringer K, Treis A, Fucik P et al A Th17‐ and Th2‐skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am J Respir Crit Care Med 2013; 187: 621–629. [DOI] [PubMed] [Google Scholar]
- 51. Tiringer K, Treis A, Kanolzer S et al Differential expression of IL‐33 and HMGB1 in the lungs of stable cystic fibrosis patients. Eur Respir J 2014; 44: 802–805. [DOI] [PubMed] [Google Scholar]
- 52. Hasnain SZ, Prins JB, McGuckin MA. Oxidative and endoplasmic reticulum stress in beta‐cell dysfunction in diabetes. J Mol Endocrinol 2016; 56: R33–R54. [DOI] [PubMed] [Google Scholar]
- 53. Thomson E, Brennan S, Senthilmohan R et al Identifying peroxidases and their oxidants in the early pathology of cystic fibrosis. Free Radic Biol Med 2010; 49: 1354–1360. [DOI] [PubMed] [Google Scholar]
- 54. Kettle AJ, Chan T, Osberg I et al Myeloperoxidase and protein oxidation in the airways of young children with cystic fibrosis. Am J Respir Crit Care Med 2004; 170: 1317–1323. [DOI] [PubMed] [Google Scholar]
- 55. Dickerhof N, Pearson JF, Hoskin TS et al Oxidative stress in early cystic fibrosis lung disease is exacerbated by airway glutathione deficiency. Free Radic Biol Med 2017; 113: 236–243. [DOI] [PubMed] [Google Scholar]
- 56. Pongnimitprasert N, El‐Benna J, Foglietti MJ et al Potential role of the “NADPH oxidases” (NOX/DUOX) family in cystic fibrosis. Ann Biol Clin (Paris). 2008; 66: 621–629. [DOI] [PubMed] [Google Scholar]
- 57. Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro‐inflammatory mediators‐induced airway and pulmonary diseases. Biochem Pharmacol 2012; 84: 581–590. [DOI] [PubMed] [Google Scholar]
- 58. Kelley TJ, Drumm ML. Inducible nitric oxide synthase expression is reduced in cystic fibrosis murine and human airway epithelial cells. J Clin Invest 1998; 102: 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lucca F, Da Dalt L, Ros M et al Asymmetric dimethylarginine and related metabolites in exhaled breath condensate of children with cystic fibrosis. Clin Respir J. 2018; 12: 140–148. [DOI] [PubMed] [Google Scholar]
- 60. White MM, Geraghty P, Hayes E et al Neutrophil membrane cholesterol content is a key factor in cystic fibrosis lung disease. EBioMedicine 2017; 23: 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ren HY, Grove DE, De La Rosa O et al VX‐809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane‐spanning domain 1. Mol Biol Cell 2013; 24: 3016–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Roth DM, Hutt DM, Tong J et al Modulation of the maladaptive stress response to manage diseases of protein folding. PLoS Biol 2014; 12: e1001998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bali V, Lazrak A, Guroji P et al Mechanistic approaches to improve correction of the most common disease‐causing mutation in cystic fibrosis. PLoS ONE 2016; 11: e0155882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Serisier DJ. The evidence base for non‐CF bronchiectasis is finally evolving. Respirology 2014; 19: 295–297. [DOI] [PubMed] [Google Scholar]
- 65. Chen AC, Martin ML, Lourie R et al Adult non‐cystic fibrosis bronchiectasis is characterised by airway luminal Th17 pathway activation. PLoS ONE 2015; 10: e0119325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Taylor SL, Rogers GB, Chen AC et al Matrix metalloproteinases vary with airway microbiota composition and lung function in non‐cystic fibrosis bronchiectasis. Ann Am Thorac Soc 2015; 12: 701–707. [DOI] [PubMed] [Google Scholar]
- 67. Rogers GB, van der Gast CJ, Cuthbertson L et al Clinical measures of disease in adult non‐CF bronchiectasis correlate with airway microbiota composition. Thorax 2013; 68: 731–737. [DOI] [PubMed] [Google Scholar]
- 68. Evans CM, Fingerlin TE, Schwarz MI et al Idiopathic pulmonary fibrosis: a genetic disease that involves mucociliary dysfunction of the peripheral airways. Physiol Rev 2016; 96: 1567–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang L, Wang Y, Pandupuspitasari NS et al Endoplasmic reticulum stress, a new wrestler, in the pathogenesis of idiopathic pulmonary fibrosis. Am J Transl Res 2017; 9: 722–735. [PMC free article] [PubMed] [Google Scholar]
- 70. Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2012; 302: L721–L729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yao Y, Wang Y, Zhang Z et al Chop deficiency protects mice against bleomycin‐induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther 2016; 24: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Alvarez D, Cardenes N, Sellares J et al IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol 2017; 313: L1164–L1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Maitra M, Wang Y, Gerard RD et al Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J Biol Chem 2010; 285: 22103–22113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bridges JP, Wert SE, Nogee LM et al Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J Biol Chem 2003; 278: 52739–52746. [DOI] [PubMed] [Google Scholar]
- 75. Lawson WE, Cheng DS, Degryse AL et al Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA 2011; 108: 10562–10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bridges JP, Xu Y, Na CL et al Adaptation and increased susceptibility to infection associated with constitutive expression of misfolded SP‐C. J Cell Biol 2006; 172: 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Seibold MA, Wise AL, Speer MC et al A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011; 364: 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hunninghake GM, Hatabu H, Okajima Y et al MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med 2013; 368: 2192–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Peljto AL, Zhang Y, Fingerlin TE et al Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013; 309: 2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Plantier L, Crestani B, Wert SE et al Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 2011; 66: 651–657. [DOI] [PubMed] [Google Scholar]
- 81. Liptzin DR, Watson AM, Murphy E et al MUC5B expression and location in surfactant protein C mutations in children. Pediatr Pulmonol 2015; 50: 1270–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chen G, Korfhagen TR, Xu Y et al SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J Clin Invest 2009; 119: 2914–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tanaka Y, Ishitsuka Y, Hayasaka M et al The exacerbating roles of CCAAT/enhancer‐binding protein homologous protein (CHOP) in the development of bleomycin‐induced pulmonary fibrosis and the preventive effects of tauroursodeoxycholic acid (TUDCA) against pulmonary fibrosis in mice. Pharmacol Res 2015; 99: 52–62. [DOI] [PubMed] [Google Scholar]
- 84. Zhao H, Qin HY, Cao LF et al Phenylbutyric acid inhibits epithelial‐mesenchymal transition during bleomycin‐induced lung fibrosis. Toxicol Lett 2015; 232: 213–220. [DOI] [PubMed] [Google Scholar]
- 85. Ayaub EA, Kolb PS, Mohammed‐Ali Z et al GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin‐induced pulmonary fibrosis. J Pathol 2016; 239: 411–425. [DOI] [PubMed] [Google Scholar]
- 86. Wang YC, Dong J, Nie J et al Amelioration of bleomycin‐induced pulmonary fibrosis by chlorogenic acid through endoplasmic reticulum stress inhibition. Apoptosis 2017; 22: 1147–1156. [DOI] [PubMed] [Google Scholar]
- 87. Kim SR, Lee YC. Endoplasmic reticulum stress and the related signaling networks in severe asthma. Allergy Asthma Immunol Res 2015; 7: 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Moffatt MF, Kabesch M, Liang L et al Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007; 448: 470–473. [DOI] [PubMed] [Google Scholar]
- 89. Hsu KJ, Turvey SE. Functional analysis of the impact of ORMDL3 expression on inflammation and activation of the unfolded protein response in human airway epithelial cells. Allergy Asthma Clin Immunol 2013; 9: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kim SR, Kim DI, Kang MR et al Endoplasmic reticulum stress influences bronchial asthma pathogenesis by modulating nuclear factor kappaB activation. J Allergy Clin Immunol 2013; 132: 1397–1408. [DOI] [PubMed] [Google Scholar]
- 91. Lu Y, Xu JY, Zhang XH et al Gu‐Ben‐Fang‐Xiao decoction attenuates sustained airway inflammation by suppressing ER stress response in a murine asthma remission model of respiratory syncytial virus infection. J Ethnopharmacol 2016; 192: 496–509. [DOI] [PubMed] [Google Scholar]
- 92. Siddesha JM, Nakada EM, Mihavics BR et al Effect of a chemical chaperone, tauroursodeoxycholic acid, on HDM‐induced allergic airway disease. Am J Physiol Lung Cell Mol Physiol 2016; 310: L1243–L1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Park SH, Gong JH, Choi YJ et al Kaempferol inhibits endoplasmic reticulum stress‐associated mucus hypersecretion in airway epithelial cells and ovalbumin‐sensitized mice. PLoS ONE 2015; 10: e0143526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Makhija L, Krishnan V, Rehman R et al Chemical chaperones mitigate experimental asthma by attenuating endoplasmic reticulum stress. Am J Respir Cell Mol Biol 2014; 50: 923–931. [DOI] [PubMed] [Google Scholar]
- 95. Hoffman SM, Tully JE, Nolin JD et al Endoplasmic reticulum stress mediates house dust mite‐induced airway epithelial apoptosis and fibrosis. Respir Res 2013; 14: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lee KS, Jeong JS, Kim SR et al Phosphoinositide 3‐kinase‐delta regulates fungus‐induced allergic lung inflammation through endoplasmic reticulum stress. Thorax 2016; 71: 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ribeiro CM, O'Neal WK. Endoplasmic reticulum stress in chronic obstructive lung diseases. Curr Mol Med 2012; 12: 872–882. [DOI] [PubMed] [Google Scholar]
- 98. Min T, Bodas M, Mazur S et al Critical role of proteostasis‐imbalance in pathogenesis of COPD and severe emphysema. J Mol Med (Berl) 2011; 89: 577–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cantin AM, Richter MV. Cigarette smoke‐induced proteostasis imbalance in obstructive lung diseases. Curr Mol Med 2012; 12: 836–849. [DOI] [PubMed] [Google Scholar]
- 100. Geraghty P, Baumlin N, Salathe MA et al Glutathione peroxidase‐1 suppresses the unfolded protein response upon cigarette smoke exposure. Mediators Inflamm 2016; 2016: 9461289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kenche H, Ye ZW, Vedagiri K et al Adverse outcomes associated with cigarette smoke radicals related to damage to protein‐disulfide isomerase. J Biol Chem 2016; 291: 4763–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Marciniak SJ, Ordonez A, Dickens JA et al New concepts in alpha‐1 antitrypsin deficiency disease mechanisms. Ann Am Thorac Soc 2016; 13(Suppl 4): S289–S296. [DOI] [PubMed] [Google Scholar]
- 103. Alam S, Li Z, Atkinson C et al Z alpha1‐antitrypsin confers a proinflammatory phenotype that contributes to chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2014; 189: 909–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zeng M, Sang W, Chen S et al 4‐PBA inhibits LPS‐induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol Lett 2017; 271: 26–37. [DOI] [PubMed] [Google Scholar]
- 105. Khan MM, Yang WL, Brenner M et al Cold‐inducible RNA‐binding protein (CIRP) causes sepsis‐associated acute lung injury via induction of endoplasmic reticulum stress. Sci Rep 2017; 7: 41363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kim SR, Kim HJ, Kim DI et al Blockade of interplay between IL‐17A and endoplasmic reticulum stress attenuates LPS‐induced lung injury. Theranostics 2015; 5: 1343–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Federti E, Matte A, Ghigo A et al Peroxiredoxin‐2 plays a pivotal role as multimodal cytoprotector in the early phase of pulmonary hypertension. Free Radic Biol Med 2017; 112: 376–386. [DOI] [PubMed] [Google Scholar]
- 108. Teng RJ, Jing X, Michalkiewicz T et al Attenuation of endoplasmic reticulum stress by caffeine ameliorates hyperoxia‐induced lung injury. Am J Physiol Lung Cell Mol Physiol 2017; 312: L586–L598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lu HY, Zhang J, Wang QX et al Activation of the endoplasmic reticulum stress pathway involving CHOP in the lungs of rats with hyperoxia‐induced bronchopulmonary dysplasia. Mol Med Rep 2015; 12: 4494–4500. [DOI] [PubMed] [Google Scholar]
- 110. Fowdar K, Chen H, He Z et al The effect of N‐acetylcysteine on exacerbations of chronic obstructive pulmonary disease: a meta‐analysis and systematic review. Heart Lung 2017; 46: 120–128. [DOI] [PubMed] [Google Scholar]
- 111. Tam J, Nash EF, Ratjen F et al Nebulized and oral thiol derivatives for pulmonary disease in cystic fibrosis. Cochrane Database Syst Rev 2013; CD007168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mannam P, Srivastava A, Sugunaraj JP et al Oxidants in acute and chronic lung disease. J Blood Lymph 2014; 4: 1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hasnain SZ, Borg DJ, Harcourt BE et al Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med 2014; 20: 1417–1426. [DOI] [PubMed] [Google Scholar]