

Abstract
Background
Historical factors, demography, reproduction and dispersal are crucial in determining the genetic structure of seabirds. In the Antarctic marine environment, penguins are a major component of the avian biomass, dominant predators and important bioindicators of ecological change. Populations of chinstrap penguins have decreased in nearly all their breeding sites, and their range is expanding throughout the Antarctic Peninsula. Population genetic structure of this species has been studied in some colonies, but not between breeding colonies in the Antarctic Peninsula or at the species’ easternmost breeding colony (Bouvetøya).
Results
Connectivity, sex-biased dispersal, diversity, genetic structure and demographic history were studied using 12 microsatellite loci and a mitochondrial DNA region (HVRI) in 12 breeding colonies in the South Shetland Islands (SSI) and the Western Antarctic Peninsula (WAP), and one previously unstudied sub-Antarctic island, 3600 km away from the WAP (Bouvetøya). High genetic diversity, evidence of female bias-dispersal and a sign of population expansion after the last glacial maximum around 10,000 mya were detected. Limited population genetic structure and lack of isolation by distance throughout the region were found, along with no differentiation between the WAP and Bouvetøya (overall microsatellite FST = 0.002, p = 0.273; mtDNA FST = − 0.004, p = 0.766), indicating long distance dispersal. Therefore, genetic assignment tests could not assign individuals to their population(s) of origin. The most differentiated location was Georges Point, one of the southernmost breeding colonies of this species in the WAP.
Conclusions
The subtle differentiation found may be explained by some combination of low natal philopatric behavior, high rates of dispersal and/or generally high mobility among colonies of chinstrap penguins compared to other Pygoscelis species.
Electronic supplementary material
The online version of this article (10.1186/s12862-018-1207-0) contains supplementary material, which is available to authorized users.
Keywords: Seabirds, Pygoscelis antarcticus, Dispersal, Gene flow, Microsatellites, D-loop, Sex-biased, Antarctica
Background
Investigating population genetic structure is important for understanding evolutionary processes [1], and establishing conservation actions for species [2]. Genetic structure is mainly determined by four processes: demography, historical factors, mating system and dispersal [3, 4]. Population demography is influenced by biotic and abiotic factors that promote variability across a species’ range. Habitat suitability, topographical barriers, resource availability and quality, and interaction with other organisms are drivers that can lead to genetic differentiation between populations [5]. Historical factors, such as the Last Glacial Maximum (LGM) and the expansion or isolation of species in different refugia also affect evolutionary processes [6, 7]. Life-history traits such as mating systems can affect genetic structure at both biparentally and maternally inherited markers, sometimes differently [1]. Finally, migratory patterns also influence genetic structure by affecting spatial patterns, species’ ranges, and environmental adaptation of the species [8]. The predisposition of an individual to return to its natal colony throughout its reproductive lifetime is known as philopatry [9]. In most birds, females have a greater tendency to leave their natal groups and disperse larger distances than males [9, 10]. These sex biases in dispersal are important to investigate, to understand its evolution [11].
Population genetic structure is typically investigated using indirect methods, such as inferring gene flow levels among colonies [12]. As levels of gene flow increase towards panmixia, the power to statistically detect distinct populations using clustering algorithms decreases [13, 14]. Additionally, population size has implications for genetic differentiation, as larger populations are more robust to the effects of genetic drift than smaller ones [15]. Thus, considering these processes, three broadly different patterns of population genetic structure can be observed: 1) absence of both genetic structure and differentiation among populations, 2) significant genetic structure, but no geographic pattern to explain it, or 3) significant genetic and geographic structured populations [5].
In the Southern Ocean, penguins are a major component of the avian biomass [16], dominant predators [17], and bioindicators of ecosystem changes [18, 19]. In the South Shetland Islands (SSI) and the Western Antarctic Peninsula (WAP), three species of Pygoscelis penguins breed sympatrically: Adélie (Pygoscelis adelie), gentoo (P. papua) and chinstrap (P. antarcticus) [20]. Population genetic structure of these penguins has recently been well documented. Microsatellite and mitochondrial data on Adélie penguins have revealed a lack of genetic differentiation between colonies around the Antarctic continent, and a sign of population expansion after the LGM [21–23]. In contrast, genetic markers employed for gentoo penguins’ analyses have revealed significant population genetic structure in Antarctica, and also evidenced divergent lineages between Antarctica and each sub-Antarctic colony studied [6, 24]. This is explained by the presence of a physical barrier (Antarctic Polar Front) and large geographical distances [24].
Unlike Adélies and gentoos, almost the entire breeding distribution of chinstrap penguins is restricted to the Antarctic Peninsula (up to approximately 64° S) and the South Shetland, South Orkney, and South Sandwich Islands in the Scotia Sea region [20, 25–29]. Additionally, small breeding populations are described on South Georgia, Bouvetøya, Heard and the Balleny Islands [20, 27]. The non-breeding range of the chinstrap penguin is extensive, with large dispersal being reported. To exemplify, Trivelpiece et al. [30] demonstrated through satellite telemetry that penguins could migrate from the South Shetland Islands to the South Orkney and South Sandwich Islands, 800 and 1300 km away, respectively. Biuw et al. [31] described a migration of 3600 km from Bouvetøya to the South Sandwich Islands for a single pre-moulting adult chinstrap penguin. Although all three Pygoscelis species show some degree of natal philopatry, chinstrap penguins are the least philopatric of the genus [32, 33]. At fine geographical scales, this species appears to show weak or even no significant population structure, with no isolation by distance [22, 34, 35]. No sex-bias has been detected for these birds using microsatellite loci [35], although the authors reported test values consistent with female bias dispersal. Currently, chinstrap penguins are listed as of Least Concern on the IUCN’s Red List of Threatened Species [36]. However, there have been reports of continuous declines at nearly all breeding sites of this species [33, 34, 37–46].
Population genetic structure of chinstrap penguins has been studied in some parts of their range [22, 34, 35]. However, this species remains the least studied of the Pygoscelis penguins, and the connectivity between breeding colonies in the WAP, or between the WAP and the easternmost breeding colony in the species’ distribution (Bouvetøya), is still unknown. Population declines reported in numerous colonies highlight the importance of investigating the connectivity of breeding colonies in terms of source and sink population dynamics, and other genetic effects these reductions might have. Indeed, in the context of conservation biology, the proper identification of population genetic structure is crucial [2]. Thus, to investigate this, we used 12 microsatellite markers and mitochondrial DNA Hypervariable Region I (HVRI) sequences of chinstrap penguins from 13 different locations to: (1) investigate the demographic history following the LGM, (2) describe patterns of distribution of genetic diversity and population structure, (3) quantify levels of connectivity among colonies in the WAP and the easternmost limit of the species’ distribution at Bouvetøya, and (4) evaluate levels of sex-biased dispersal. We proposed two hypotheses: (a) lack of or reduced genetic structure among breeding colonies of chinstrap penguins in Antarctica, as observed in P. antarcticus in a few locations and in another species of the genera with similar ecological features (P. adeliae), and (b) strong genetic structure between Antarctica and Bouvetøya, due to isolation explained by large geographical distances.
Methods
Field sampling and DNA extraction
Between January and February of 2009 and 2016 (plus Miers Bluff in 2003), a total of 251 blood samples from chinstrap penguins were collected at 13 sites, including 10 locations in the South Shetland Islands (n = 183), two in the Antarctic Peninsula (n = 45) and Bouvetøya (n = 23) (Fig. 1 and Table 1). To avoid disturbance within the breeding colonies, adult penguins were captured using hand-held nets while entering the water. Each individual was stained with bromophenol blue to avoid re-sampling. Up to 1 mL of blood was obtained from brachial or medial metatarsal veins using a 23 G needle, and stored in 96% ethanol. All procedures were done following an accepted restraining method for penguins [47].
Fig. 1.

Chinstrap penguin sampled sites during this study (total n = 251)
Table 1.
Summary of chinstrap penguin samples used, genetic diversity indices and neutrality test results
| HVRI | Microsatellite loci | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | Colony (abbreviation) | Coordinates | N | H | S | Hd | π | ∏ | D | Fs | N | A | Ho | He | FIS | |
| WAP – SSI | Elephant Island (EI) | 61°13’S | 55°21’W | 17 | 13 | 13 | 0.96 | 0.01 | 2.35 | −1.47 | − 10.1 | 17 | 5.55 | 0.62 | 0.62 | 0.003 |
| Penguin Island (PI) | 62°06’S | 57°56’W | 17 | 14 | 19 | 0.98 | 0.01 | 3.43 | −1.54 | −9.47 | 19 | 5.91 | 0.70 | 0.67 | −0.053 | |
| Barton Peninsula (BP) | 62°14’S | 58°46’W | 22 | 19 | 18 | 0.98 | 0.01 | 3.37 | −1.17 | −17.4 | 29 | 6.18 | 0.57 | 0.62 | 0.081 | |
| Ardley Island (AI) | 62°13’S | 58°56’W | 12 | 10 | 13 | 0.97 | 0.01 | 3.26 | −1.03 | −5.45 | 14 | 5.27 | 0.60 | 0.66 | 0.096 | |
| Greenwich Island (GI) | 62°31’S | 59°47’W | 14 | 12 | 16 | 0.98 | 0.01 | 2.97 | −1.68 | −8.57 | 14 | 5.18 | 0.61 | 0.64 | 0.048 | |
| Miers Bluff (MB) | 62°43’S | 60°26’W | 9 | 8 | 10 | 0.97 | 0.01 | 2.89 | −1.00 | −4.47 | 11 | 5.00 | 0.61 | 0.62 | 0.019 | |
| Hannah Point (HP) | 62°39’S | 54°36’W | 20 | 14 | 17 | 0.95 | 0.01 | 3.13 | −1.30 | −8.19 | 25 | 5.91 | 0.64 | 0.64 | 0.001 | |
| Cape Shirreff (CS) | 62°28’S | 60°48’W | 22 | 18 | 19 | 0.97 | 0.01 | 2.92 | −1.63 | −16.4 | 30 | 6.46 | 0.61 | 0.64 | 0.039 | |
| Baily Head (BH) | 62°58’S | 60°30’W | 8 | 7 | 9 | 0.96 | 0.01 | 2.60 | −1.21 | −3.67 | 9 | 5.10 | 0.70 | 0.71 | 0.011 | |
| Vapour Col (VC) | 63°00’S | 60°44’W | 13 | 13 | 15 | 1 | 0.01 | 3.44 | −1.21 | −11.9 | 15 | 5.46 | 0.67 | 0.65 | −0.025 | |
| WAP - AP | Kopaitic Island (KI) | 63°19’S | 57°55’W | 26 | 26 | 32 | 1 | 0.01 | 4.40 | −1.77 | −25.7 | 30 | 6.36 | 0.63 | 0.64 | 0.028 |
| Georges Point (GP) | 64°40’S | 62°39’W | 9 | 9 | 14 | 1 | 0.01 | 3.72 | −1.33 | −6.00 | 15 | 5.82 | 0.64 | 0.64 | −0.003 | |
| SAI | Bouvetøya (BI) | 54°26’S | 3°23′E | 18 | 16 | 18 | 1 | 0.01 | 3.44 | −1.32 | −13.1 | 23 | 5.73 | 0.65 | 0.65 | −0.001 |
| Total | 61°10’S | 55°00’W | 207 | 119 | 55 | 0.98 | 0.01 | 3.32 | −1.92 | −25.9 | 251 | 5.69 | 0.63 | 0.65 | 0.003 | |
Bold values are significantly different from zero after the FDR correction (p < 0.05 for D and p < 0.01 for Fs)
Geographical regions (SAI: Sub-Antarctic Islands; WAP: West Antarctic Peninsula; SSI: South Shetland Islands; AP: Antarctic Peninsula), information of localities (with abbreviations), coordinates, number of samples used from each locality for each marker (HVRI and 11 microsatellite loci), genetic diversity indices’ results (N: sample size; H: number of haplotypes; S: number of polymorphic sites; Hd: haplotype diversity; π: nucleotide diversity; ∏: pairwise difference, A: mean number of alleles per locus; Ho: mean observed heterozygosity; He: mean expected heterozygosity and FIS: inbreeding coefficient) and neutrality tests (D: Tajima’D test, Fs: Fu’s Fs test)
Total genomic DNA was extracted using a salt protocol [48], modified as follows: a lysis buffer based on TNE 1X, Tris-HCl pH 7.8 and SDS 25% in place of Tris-HCl pH 8.0, EDTA and SDS 20%. Additionally, 10 M ammonium acetate was used instead of NaCl, and tubes spun down for 20 min at 14, 000 rpm. After the extraction, DNA samples were stored in TE Buffer (Tris-EDTA; 10 mM Tris base, 0.1 mM EDTA) at − 20 °C or − 80 °C.
Amplification, sequencing and genotyping protocols
A 305 base pair (bp) of the mitochondrial DNA (mtDNA) HVRI was amplified using forward primer Forw2 (5’-ACAGTACGAGATAAGTCATGGTTCC-3′) or L-tRNAGlu (5′-CCCGCTTGGCTTYTCTCCAAGGTC-3), and reverse primer AH530 (5′- CTGATTTCACGTGAGGAGACCG-3′) [49]. The PCR conditions and amplification cycles were done following Peña et al. [6]. The mtDNA PCR products were purified and Sanger sequenced bi-directionally in Macrogen Inc. (Seoul, South Korea). All mtDNA sequences were deposited in GenBank accession numbers: MF966819 – MF966902 and MH025646 – MH025759.
Genetic diversity and population differentiation were examined at 12 tetranucleotide microsatellite loci (AP-3, AP-19, AP-26, AP-61, AP-78, AP-85, AP-90, CP-6, CP-25, GP-6, GP-15 and GP-36) isolated from the genome of three species of Pygoscelis penguin sequenced by NGS (Next Generation Sequencing), as part of another study [50]. Forward primers were synthesized using 5′-end-M13 tail-labelled fluorophores with one of three dyes (6-FAM, HEX, or NED; Applied Biosystems) to adjust simultaneous genotyping at multiple loci with overlapping size ranges. Protocols applied for primer’s sequences, PCR conditions, and amplification cycles for microsatellite loci were those described in Vianna et al. [50]. DNA samples were separated by electrophoresis through a 2% agarose gel, run for 0.5 h at 300 V. Genotyping of the obtained PCR products were performed at Macrogen Inc. (Seoul, South Korea). All PCRs were conducted in an Applied Biosystem machine, and the mixtures contained 10–100 ng of genomic DNA. The microsatellite genotypes were assigned using GeneMarker® v.1.75 (Softgenetics LLC™) software for allele size identification.
Gene diversity
The mtDNA sequences (HVRI) were aligned and edited according to the chromatogram utilizing Sequencher v.5.1 (Gene Codes, Ann Arbor, MI, USA). Polymorphic sites (S), number of haplotypes (h), haplotype diversity (Hd), average number of differences between pairs of sequences (∏), and nucleotide diversity (π) were estimated with Arlequin v.3.5.1.2 [51], applying 10,000 permutations. To create an mtDNA haplotype network, the sequence alignment was used to create a maximum parsimony tree using MEGA7 [52]. For this analysis, the program determined K2 + G + I (Kimura 2-parameter + gamma + invariable sites) as the best substitution model with a gamma parameter of 0.54. The maximum parsimony tree generated subsequently by MEGA7 and the sequence alignment were used to generate a haplotype network with Haplotype Viewer [53].
For all microsatellite data, PGDSpider v.2.1.0.1 software was used as an automated data conversion tool [54]. For these data sets, the presence of null alleles or potential genotyping errors were evaluated using Micro-Checker v.2.2.3 [55]. Arlequin v.3.5.1.2 [51] was used to study genetic diversity within samples from each chinstrap penguin colony, calculate the mean number of alleles per locus, and evaluate the observed (Ho) and expected heterozygosities (He). Expectations for Hardy-Weinberg equilibrium (HWE) were estimated as deviation of the Wright’s FIS index and these were tested for each locus, for all loci, and for each population utilizing randomization procedures using 10,000 permutations with GENETIX v.4.05.2 [56]. To test the presence of linkage disequilibrium, the same program was applied with a likelihood-ratio test and the empirical distribution generated by 10,000 permutations. Corrections for multiple testing were made using the False Discovery Rate (FDR) [57, 58].
Population genetic structure and isolation by distance
Arlequin v.3.5.1.2 [51] was utilized to calculate FST and ФST between pairwise populations on microsatellite and mtDNA sequence data using 10,000 permutations. P values were corrected with the FDR method for multiple tests [57, 58]. Employing microsatellite loci, isolation by distance was evaluated by means of the Adegenet package in R [59]. For this, Adegenet uses a Mantel test between a matrix of genetic distances, and a matrix of geographical distances [60]. Google Earth (Google, v.7.1.8.3036) was used to calculate the shortest geographical distance by sea between locations.
To determine the most likely number of clusters (K), multilocus genotypes were analyzed through Bayesian clustering methods implemented in STRUCTURE v2.3.4 [61], BAPS v.6.0 [62] and GENELAND v.3.1 [63]. The software STRUCTURE v2.3.4 was run using different models assuming (ad)mixture, (un)correlated allele frequencies both with and without a priori specification of sample locations [61, 64]. The models were run with the likely number of populations (K) set from 1 to 13. For each K, the model was run 10 times with a burn-in length of 100,000 iterations followed by 1,000,000 Markov Chain Monte Carlo (MCMC) subsequent iterations. The optimum number of clusters was inferred by deriving the posterior probability of K (LnP(D)) from each independent run. As the ΔK method of Evanno’s does not allow K = 1 to be tested [65], this method was employed when K was higher than one for log-likelihood using STRUCTURE HARVESTER [66]. To align multiple replicates of files produced by STRUCTURE, CLUMPP v1.1.2 (CLUster Matching and Permutation Program) [67] was applied. Results generated by the genetic clustering program were visualized through DISTRUCT v1.1 [68].
A Bayesian Analysis of Population Structure (BAPS v6.0) was performed using a combination of analytical and stochastic methods, based on molecular markers and geographical sampling [62]. Calculations were performed over 10,000 iterations with both spatial and non-spatial, and both a mixture and an admixture model, with the maximum number of populations possible set to 13.
An analysis of spatial structure using the R package GENELAND v3.1 was carried out to determine the most likely number of populations and to assign individuals to population clusters. This program is based on an algorithm which includes not only genotypes, but also the geographical location of all individuals to estimate the number of groups and delineate their spatial boundaries [69]. Analyses were performed under the spatial model assuming both correlated and uncorrelated allele frequency. The correlated frequency model, in comparison with the uncorrelated frequency model, might be more capable of detecting subtle differentiations. However, it could also be more sensitive to departure from model assumptions (as presence of isolation-by-distances), and more prone to algorithm instabilities [69]. Ten independent MCMC simulations were run allowing the number of populations to vary between 1 and 13, with the following parameters: 1,000,000 MCMC iterations with a thinning of 100, a maximum rate of Poisson processes fixed to 500 and a maximum number of nuclei in the Poisson-Voroni tessellation fixed to 300. The best-supported K value was determined based on the highest averaged maximum likelihood score of the models.
Additionally, a Discriminant Analysis of Principal Components (DAPC) was carried out to determine the number of clusters of genetically related individuals, using a non-Bayesian approach. DAPC uses sequential K-means and model selection to identify genetic clusters [70]. The Adegenet package in R [59] was used, retaining all principal components.
To assign or exclude individual colonies as being the origins of individuals based on genotype data, assignment testing of microsatellite loci was done using GENECLASS2 v.2.0.h [71]. Two separate analyses were performed: one employed the likelihood method based on allele frequencies [72], and the other used the Bayesian method approach [73]. The probability that each individual was assigned to a candidate population was estimated using a Monte Carlo resampling method (number of simulated individuals = 10,000; type I error = 0.01) [74]. The same program and parameters were also applied for the detection of first-generation migrants.
Demographic history
To evaluate deviations from Wright-Fisher equilibrium, two neutrality tests were applied: Tajima [75] and Fu [76]. Both tests were performed in Arlequin v.3.5.1.3 [51]. The historical demographic changes were inferred and reconstructed with a Bayesian approach using BEAST v.1.8 [77] and Tracer v.1.5.0 [78] programs. The coalescence model elected was Bayesian Skyline plot and the molecular clock utilized was Lognormal relaxed clock (uncorrelated). The best fit nucleotide substitution model determined with Jmodeltest v2.1.10 [79] was HKY + G + I (Hasegawa-Kishino-Yano + gamma + invariant sites). The mutational rate used was 0.55 s/s/millions of years [80]. The MCMC chain length was 50,000,000 sampled every 1000 generations.
Sex determination and sex-biased dispersal
For molecular sex identification, a region of the Chromosome-helicase-DNA binding protein (CHD1) gene was amplified, with primer pair 2550F/2718R [81]. PCRs were carried out in 25 μL volume containing 10–100 ng genomic DNA, 1X reaction buffer, 0.5 μM of each primer, 1.5 mM MgCl2, 100 μM dNTPs and 0.7 U Taq DNA polymerase (Invitrogen Life Technologies). The reactions’ conditions were as follows: an initial denaturing step at 94 °C for 5 min; followed by 45 cycles at 94 °C for 30 s, 46 °C for 45 s, and 72 °C for 25 s; and a final extension step at 72 °C for 5 min. All reactions were conducted on an Applied Biosystem machine. The amplification products were separated on 2% agarose gel for approximately 1 h at 150 V and visualized with GelRed® under UV light.
Using microsatellite data of the sex-identified individuals by molecular techniques, sex-biased dispersal was evaluated with FSTAT v.2.9.3.2 [82]. Two hypotheses were tested: a one-tailed test was done assuming males as the most philopatric group, since dispersal is female-biased in most birds [9]. Next, a two-sided test under the assumption of no differences between male and female dispersion in chinstrap penguins [35]. For both tests, differences in the inbreeding coefficient (FIS), fixation index (FST), relatedness between individuals (r), mean Assignment Index (mAIc) and variance of Assignment Indices (vAIc) between sexes were calculated. FST, r and mAIc were expected to be lower in the sex that disperses most, whereas FIS and vAIc were expected to be higher [11]. The p values of each test were estimated using 10,000 randomizations.
Results
Genetic diversity
For mtDNA HVRI results, high genetic diversity was found in all locations. A total of 119 haplotypes (n = 207, S = 55) were found, along with high haplotype diversity (Hd = 0.98, n = 207) ranging from 0.95 (HP) to 1 (VC, KI, GP and BI), and low nucleotide diversity (π = 0.01, Table 1). The number of haplotypes ranged from 7 (BH) to 26 (KI), while polymorphic sites ranged from 9 (BH) to 32 (KI).
For microsatellite data, only one locus was monomorphic (GP-6), so it was not used in further analyses. The remaining 11 microsatellite loci were polymorphic for all populations, except for AP-3 at Baily Head (BH) (Additional file 1: Table S1). The inbreeding coefficient (FIS) was low in all populations, and no significant p-values were found, indicating no significant heterozygote excess or deficiencies. Therefore, deviations from HWE were not detected in populations at the 11 loci (Table 1). Linkage disequilibrium of each pair of loci was not detected within or among populations. For microsatellite markers, overall allele numbers per locus varied between three (locus AP-3) and 12 (locus GP-15), with an average of 5.69 alleles for all sample sites. The expected heterozygosity ranged from 0.62 (Elephant Island, Barton Peninsula and Miers Bluff) to 0.71 (BH), with an average of 0.65. The observed heterozygosity exhibits a similar level of variation with an average of 0.63 over all locations. Values of allelic richness ranged between 5.00 to 6.46 per sample site. Locus-by-locus allelic richness and diversity measures for each sample location are shown in the Additional file 1: Table S1.
Population genetic structure and isolation by distance
For pairwise values using mtDNA data, no significant genetic differentiation was found between any pairwise locations (Fig. 2a, b, and Additional file 2: Table S2 and Additional file 3: Table S3). Notably, there was also an absence of population genetic structure between the WAP and Bouvetøya (FST = − 0.004, p = 0.766) (Additional file 2: Table S2 and Additional file 3: Table S3).
Fig. 2.

Distance matrices pairwise. a) pairwise FST values from mtDNA (HVRI), b) pairwise ΦST values from mtDNA (HVRI), and c) pairwise FST microsatellite values (STRs, 11 loci). Each cell of the heat plot is color-coded, illustrating relative differences. Darker colors indicate higher levels of genetic differences and white, lower. The asterisks indicate significant FST values
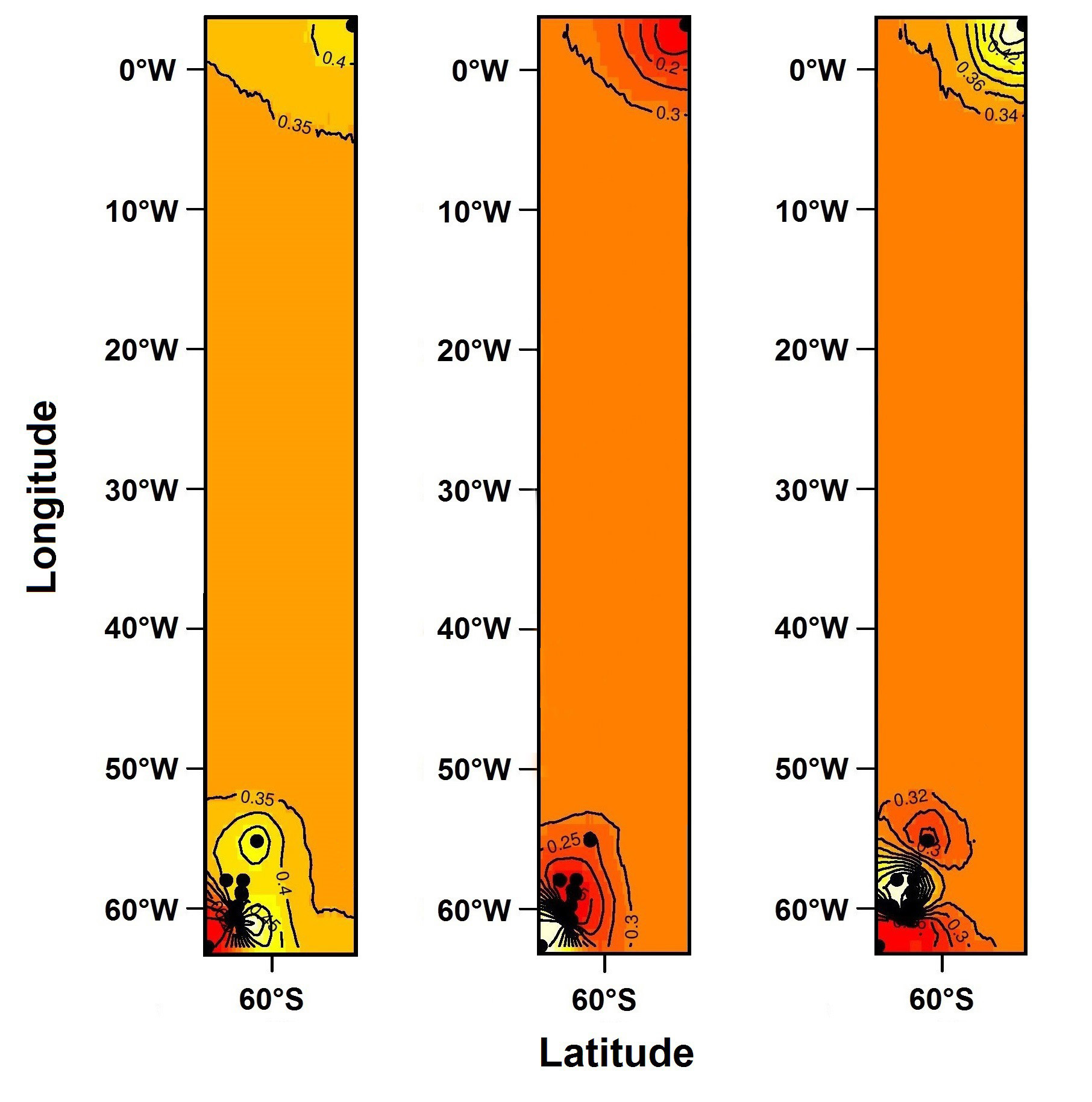
For microsatellite loci, the FST values were generally not significant (Fig. 2c). Seven of 78 pairwise FST comparisons were significantly different, all corresponding to the southernmost locality of this study: Georges Point (GP). However, statistically significant FST values varied from 0.031 to 0.054, indicating a weak differentiation between GP and other colonies (Additional file 4: Table S4). GP FST values differed significantly from seven of the 13 northernmost studied sites (EI, PI, BP, AI, CS, KI and BI, Fig. 2c). Mantel’s testing did not detect isolation by distance in microsatellite data (r = 0.05, p = 0.40). Although studies have questioned the performance of the Mantel test [83, 84], it can be an effective approach if it is used cautiously [85].
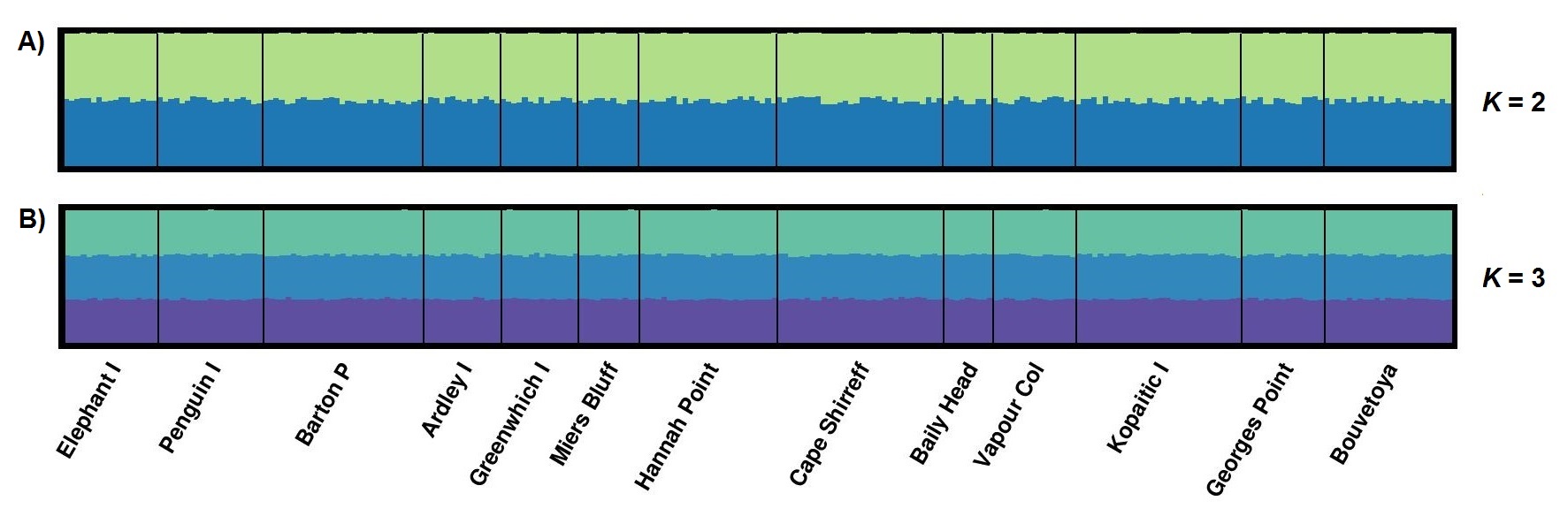
To identify the number of populations among the 13 locations, four approaches were used with microsatellite loci, yielding different optimal numbers of clusters. Using the mean log-likelihood in STRUCTURE, the analysis inferred that the number of populations (K) was one, for seven of the eight tested different model assumptions. Only one model selected K = 3, therefore it was also evaluated by Evanno’s method, which suggested K = 2. Nonetheless, when analyzing individual assignment plots, no group could be identified (See Additional file 5: Figure S1). In the BAPS analysis, the inferred number of populations was K = 1 when the spatial model was applied, despite whether mixture or admixture models were performed. On the other hand, when a non-spatial model was run, the optimal number of clusters was K = 7, but without any geographical relation (Additional file 6: Figure S2). For GENELAND, the variation of estimated number of groups depended on whether the uncorrelated or correlated frequency model was used, although in both models the 10 runs consistency converged on a single K value. When employing the uncorrelated model, the inferred number of populations K was one. Contrastingly, GENELAND estimated K = 3 clusters for chinstrap penguins when the correlated allele frequency model was employed. These clusters corresponded to three distinct populations: (1) Kopaitic Island, (2) Georges Point, and (3) northern WAP locations and Bouvetøya (Additional file 7: Figure S3). Additionally, in the pairwise FST comparison, GENELAND also identified Georges Point as the most differentiated breeding colony, however, the probabilities of cluster membership were very low (< 0.5). Models and estimated number of populations (K) for all Bayesian programs used are summarized in Table 2. The final approach, DAPC, estimated the optimal number of clusters to K = 6, however, they were geographically meaningless and overlapped extensively (Additional file 8: Figure S4). Although some analyses suggested clusters larger than one, the graphic results did not show any consistent group. Finally, for estimation of dispersal patterns, assignment tests were only successful for assigning 13.9% of the individuals (assignment threshold of 0.05) to the proper colony, and low values were again observed for the first-migrant generation, revealing high gene flow among all sampled colonies (Additional file 9: Table S5).
Table 2.
Bayesian clustering analyses and different models used to infer the optimal number of population (K)
| Bayesian clustering software | Model use | Inferred number of cluster (K) |
|---|---|---|
| BAPS | Spatial, with mixture model | K = 1 |
| Spatial, with admixture model | K = 1 | |
| Non-spatial model, with admixture model | K = 7 | |
| GENELAND | Spatial model, with uncorrelated allele frequency | K = 1 |
| Spatial model, with correlated allele frequency | K = 3 | |
| STRUCTURE | Admixture, with correlated allele frequency, using location information | K = 1 |
| Admixture, with independent allele frequency, using location information | K = 1 | |
| Admixture, with correlated allele frequency, no location information supplied | K = 1 | |
| Admixture, with independent allele frequency, no location information supplied | K = 1 | |
| No admixture, with correlated allele frequency, using location information | K = 1 | |
| No admixture, with independent allele frequency, using location information | K = 1 | |
| No admixture, with correlated allele frequency, no location information supplied | K = 1 | |
| No admixture, with independent allele frequency, no location information supplied | K = 3, K = 2 * |
*Inferred number of cluster using Evanno’s method
Demographic history
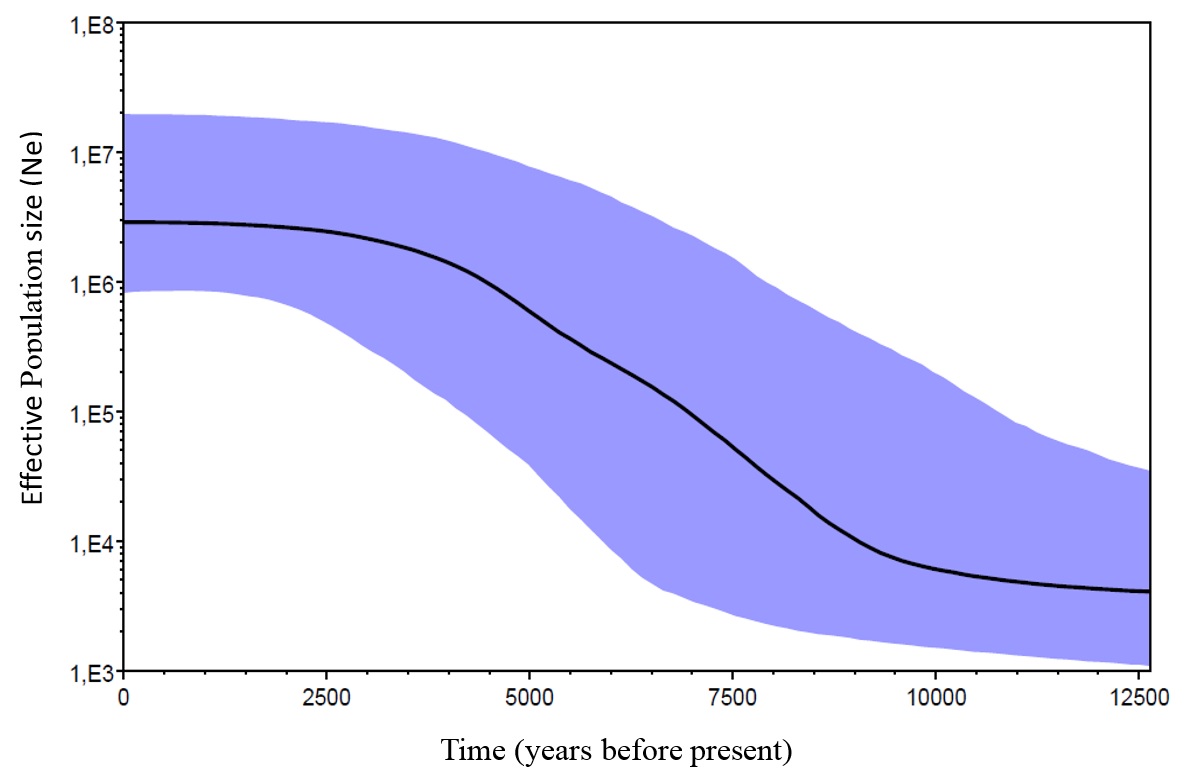
The haplotype network analysis showed high genetic diversity, lack of divergent lineages and a star-like topology, suggesting population expansion (Fig. 3). A sign of population expansion was also observed for all studied locations using a Bayesian approach (Additional file 10: Figure S5); the historical time for this expansion was around 10,000 years ago. This is supported by the negative and significant values of the Tajima test (D = − 1.92, p < 0.05, Table 1) and Fu (Fs = − 25.9, p < 0.001, Table 1) for the species, and for the majority of the locations for Fu’Fs.
Fig. 3.

Haplotype network based on the mtDNA (HVRI) haplotypes according to sampling localities. Size of circles is proportional to haplotype frequency
Sex-biased dispersal
Sex of 196 individuals was determined: 93 females and 103 males. The majority of tests do not support sex-biased dispersal, however, the mean assignment index test revealed a significant difference between males and females for one-tailed and two-sided tests, that may indicate females as the dispersing sex in chinstrap penguins (Table 3).
Table 3.
Sex-biased dispersal in chinstrap penguins. One-tailed test results, their corresponding p-values and the number (N) of females and males used for the analyses
| Assignment indices | ||||||
|---|---|---|---|---|---|---|
| N | F IS | F ST | Relatedness | Mean | Variance | |
| Females | 93 | 0.002 | 0.002 | 0.145 | −0.509 | 9.56 |
| Males | 103 | 0.042 | 0.97 | 0.171 | 0.459 | 10.43 |
| p-value | 0.111 | 0.291 | 0.397 | 0.029 | 0.70 | |
Bold values are significantly different from zero after the FDR correction (p < 0.05)
Discussion
Chinstrap penguins throughout the 13 colonies studied herein, showed high levels of genetic diversity, low levels of genetic structure between study sites and no isolation by distance. Results also suggest female biased-dispersal and a sign of population expansion since the Last Glacial Maximum.
Although chinstrap penguin populations have decreased dramatically over the last four decades, high genetic diversity for both mtDNA and microsatellites were found in all studied colonies. This could be the result of a historically large population size or simply a result of the currently large population (7,5 million pairs) [25], combined with high levels of gene flow between colonies. This study’s finding of high genetic diversity is consistent with previous reports for chinstrap penguin colonies from the WAP, South Orkney and South Sandwich Islands, using microsatellite and mtDNA data [22, 35]. High genetic diversity has also been documented through mtDNA from gentoo penguins [6, 22, 24, 86] and microsatellite markers from Adélie penguins [21]. Additionally, investigations in other penguin species have also demonstrated high genetic diversity, such as in rockhopper [87], magellanic [88] and Humboldt penguins [89].
Using mtDNA (HVRI) from four breeding sites, Clucas et al. [22] found weak differentiation between colonies of chinstrap penguins from the WAP, South Shetland and South Orkney Islands in relation to the South Sandwich Islands. In this current study, no significant genetic differentiation was found in breeding colonies from the WAP and South Shetland Islands using the same marker. Most clustering data analyses suggests only one genetic group for chinstrap penguins. However, the lack of consensus reached for a few microsatellite analyses (Table 2) could be explained due to that the accuracy of Bayesian analyses commonly diminishes when levels of genetic differentiation among populations decreases, performing better with FST > 0.05 [14]. Microsatellite loci showed absence or reduced population structure among chinstrap penguins breeding in the WAP. Interestingly, absence of structure between the WAP and Bouvetøya was observed (Additional file 5: Figure S1 and Additional file 6: Figure S2, and Additional file 4: Table S4). These results complement and confirm previous genetic investigations that have found little (if any) population structure between chinstrap penguins in Antarctica with the use of microsatellites. For example, limited genetic variation was found among colonies from the WAP and archipelagos within the Scotia Arc. Nonetheless, limited numbers of breeding colonies (only two in the WAP, and two in the Scotia Arc) were studied [35]. Weak genetic differences and high level of gene flow between two colonies from the South Shetland Islands were also found using amplified fragment length polymorphism (AFLP) analyses, but in populations located within a short distance of 32 km [34]. The limited genetic structure found in the present study is likely the result of recurrent and long-distance migration of individuals between sample sites, supported by the inability of assignment tests to successfully place individuals in their exact populations of origin. The slight levels of genetic differentiation reported between Georges Point and the northernmost studied locations coincide with one of the southernmost distributions of chinstrap penguins along the Antarctic Peninsula. Thus, the incipient differentiation may be explained by a founder effect from the northernmost colonies to the south. Although the source colony is expected to present higher genetic diversity values than the newer ones [90], the genetic diversity indices found here were similar in all sample sites. A similar pattern was observed in the trumpeter finch (Bucanetes githagineus) in peripheral populations [91]. Another observed pattern that could support the hypothesis of chinstrap penguins colonizing new breeding habitats is that they are currently expanding their range southward along the Antarctic Peninsula [92]. Numerous studies have reported the presence of small numbers of chinstrap penguins south of their natural breeding range [93–95]. During field work conducted in January 2017, this was also observed: two breeding pairs on Waterboat Point (Gabriel González Videla base; 64°49’S, 62°51’W), a single individual surrounded by gentoos on Doumer Island (Yelcho base; 64°65’S, 63°35’W) and another single chinstrap surrounded by Adélies on Avian Island (67°46’S, 68°54’W) (Additional file 11: Figure S6). This may suggest that chinstraps tend to prospect other colonies and breeding habitats far away from their colony of origin, similar to that observed in king penguins [96].
Dispersal has significant effects on population size (growth or reduction), species’ persistence and genetics [97]. In most birds, dispersal tends to be female-biased, however, male-biased natal dispersal has been reported for Adélie penguins [98]. In contrast, the first study which compared connectivity between males and females through genetic tools in chinstrap penguins, reported several value test results (females with higher FIS, negative mAIc and higher vAIC) pointing towards a female-biased dispersal, though none of the indexes were significant [35]. In the current study, our data suggests that females are the dispersing sex and males are the philopatric sex (Table 3). However, this should be considered with caution, as most of the sex biased tests were not conclusive. Some studies mention that penguins are not always philopatric [32, 33]. Natal philopatry evince that individuals are likely to have low rates of movement between colonies [21]. However, only a proportion of all individuals are faithful to one locality [9], and a small number of migrants could homogenize population structure easily [99]. Indeed, Adélie penguins, which exhibit strong natal philopatry, do not show strong genetic difference among colonies, potentially due to interaction between large effective population sizes in combination with some dispersal [21]. Evolutionary reasonings for sex-biased dispersal are inbreeding avoidance and evasion of intersexual competition [10]. Philopatry has several benefits, such as development of antipredator strategies, social facilitation and spatial heterogeneity of breeding and foraging habitats [32]. However, stressful environmental conditions (such as extensive sea ice or obstruction to usual migration patterns) may be driving an increase in dispersion rates, leading penguin species to have less philopatric behaviour than previously thought [100].
Finally, chinstrap penguins also show a signature of population expansion after the Last Glacial Maximum, similar to that detected for other Pygoscelis penguins in the WAP [6, 22, 24]. Potentially, the LGM may have contracted the populations at lower latitudes such as the Scotia Arc and Bouvetøya, maintaining the large population size here observed, followed by a reexpansion about 10,000 years ago. This scenario of recolonization may have contributed to the absence of population genetic structure in Antarctica, and between Antarctica and Bouvetøya. A pattern of historical divergent lineages has been described between gentoo penguin colonies throughout sub-Antarctic islands with shorter geographical distances [24] than those observed for chinstrap penguins.
Conclusions
Most of the data analyses suggests a single large population of chinstrap penguins throughout the Southern Ocean, with minimal population structure in the WAP region, and absence of genetic differentiations between the WAP and a sub-Antarctic island located 3600 km away. Georges Point, one of the southernmost breeding colonies of chinstrap penguin in the Antarctic Peninsula, was the most differentiated of all. The lack of genetic structure among distant reproductive colonies of chinstrap penguins in the Southern Ocean may be due to different factors, such as a historical large population size making it unyielding to drift, long-range gene flow between breeding colonies, stressful environmental conditions forcing penguins to increase dispersion rates and/or post-LGM recolonization between WAP and Bouvetøya.
Additional files
Figure S1. Plot of assignment probabilities from STRUCTURE. A vertical bar represents an individual, and colors represent the different clusters found. All plots were generated via running 10 replicates. Figures show the optimal number of clusters for no admixture model, with independent allele frequency and no location information supplied using A) Posterior probability of K (LnP(D)) and B) Evanno’s method. (DOCX 20 kb)
Figure S2. Plot of assignment probabilities from BAPS. Vertical lines represent each individual and the color refers to clusters found through this analysis. A) spatial with both, mixture and admixture models (K = 1) and B) non-spatial admixture model (K = 7). (DOCX 16 kb)
Figure S3. Posterior probabilities of population membership from the spatial model with correlated allele frequencies’ model. Lighter colors indicate higher probabilities of population membership. Three genetic clusters were identified using GENELAND. Left: Kopaitic Island, middle: Georges Point, and right: northern WAP locations and Bouvetøya. (DOCX 16 kb)
Figure S4. Discriminant Analysis of Principal Components (DAPC). The six genetic clusters identified by Adegenet are shown in different colors. All six groups overlapped extensively, and none of them represent a specific colony. (DOCX 16 kb)
{kind=link}
Figure S5. Skyline plot mtDNA (HVRI) for chinstrap penguins. (JPG 144 kb)
{kind=link}
Figure S6. Chinstrap penguins south of their traditional breeding range. A) Breeding pairs at Waterboat Point (Gabriel González Videla base; 64°49’S, 62°51’W), B) a single individual surrounded by gentoo colonies on Doumer Island (Yelcho base; 64°65’S, 63°35’W) and c) another single bird on Avian Island (67°46’S, 68°54’W) in the midst of Adélie penguins. (JPG 43 kb)
{kind=link}
Table S1. Allelic richness (A), expected (HE) and observed heterozigosity (HO) values for 11 microsatellite loci, for all examined populations. Study site (collection location) abbreviations correspond to EI: Elephant Island, PI: Penguin Island, BP: Barton Peninsula, AI: Ardley Island, GI: Greenwich Island, MB: Miers Bluff, HP: Hannah Point, CS: Cape Shirreff, BH: Baily Head, VC: Vapour Col, KI: Kopaitic Island, GP: Georges Point, and BI: Bouvetøya. (JPG 424 kb)
Table S2. Summary of pairwise genetic differences (ΦST) between chinstrap penguin colonies for mtDNA marker (HVRI). Bellow the diagonal are ΦST values, and their corresponding p-values above the diagonal. (TIFF 2112 kb)
Table S3. Summary of pairwise genetic differences (FST) between chinstrap penguin colonies for mtDNA marker (HVRI). Below the diagonal are FST values, and their corresponding p-values above the diagonal. (DOCX 18 kb)
{kind=link}
Table S4. Summary of pairwise genetic differences between chinstrap penguin colonies (FST) calculated from the 11 microsatellite loci. Below the diagonal are FST values, and corresponding p-values above the diagonal. (JPG 76 kb)
{kind=link}
Table S5. GeneClass2 percentage test results using microsatellite data for chinstrap penguins from 13 colonies for (a) genetic assignment using Paetkau et al. (1995) criterion and (b) first-generation migrant. Lines indicate the samples’ site collection and columns indicate the colonies to which the individuals were assigned. Colony self-assignments are in bold. (JPG 5437 kb)
Acknowledgements
We thank the Spanish polar ships “Las Palmas” and “Hespérides” for transport to the study area, the Spanish Antarctic base “Gabriel de Castilla” for their hospitality and the Unit of Marine Technology (CSIC) for logistics. We also thank Daly Noll for her support in laboratory work, and Trinidad Quito Bravo for designing the map of locations sampled in this study.
Funding
Financial support for this study was provided by the Chilean Antarctic Institute (INACH T_27–10, G_06–11, RT_12–14), FONDECYT projects (1150517), CONICYT PIA ACT172065 GAB, CONICYT-PCHA/MagísterNacional/2016–22162312, National Council for Scientific and Technological Research – CNPq (n 482501/2013–8; INCT-APA 574018/2008–5) and the Spanish Ministry of Economy and Competitiveness (CGL2004–01348, CGL2007–60369, CTM2011–24427). None of the funding body had any role in the design of the study and collection, analysis and interpretation of the data, nor in the manuscript.
Availability of data and materials
The mitochondrial DNA sequences are available in the GenBank repository, accession numbers: MF966819 – MF966902 and MH025646 – MH025759.
Abbreviations
- ∏
Average number of differences between pairs of sequences
- A
Mean number of alleles per locus
- AI
Ardley Island
- AP
Antarctic Peninsula
- BAPS
Bayesian Analysis of Population Structure
- BH
Baily Head
- BI
Bouvetøya
- bp
base pair
- CHD1
Chromosome-helicase-DNA binding protein
- CS
Cape Shirreff
- D
Tajima’D test
- DAPC
Discriminant Analysis of Principal Components
- EI
Elephant Island
- FDR
False Discovery Rate
- FIS
Inbreeding coefficient
- Fs
Fu’s Fs test
- FST
Fixation index
- GI
Greenwich Island
- GP
Georges Point
- h
Number of haplotypes
- Hd
Haplotype diversity
- He
Expected heterozigosity
- HKY + G + I
Hasegawa-Kishino-Yano + gamma + invariant sites
- Ho
Observed heterozigosity
- HP
Hannah Point
- HVRI
Hypervariable Region I
- HWE
Hardy-Weinberg equilibrium
- K2 + G + I
Kimura 2-parameter + gamma + invariable sites
- KI
Kopaitic Island
- LGM
Last Glacial Maximum
- mAIc
mean Assignment Index
- MB
Miers Bluff
- MCMC
Markov Chain Monte Carlo
- mtDNA
mitochondrial DNA
- N
Sample size
- NGS
Next Generation Sequencing
- PI
Penguin Island
- r
Relatedness between individuals
- S
Polymorphic sites
- SAI
Sub-Antarctic Islands
- SSI
South Shetland Islands
- vAIc
variance of Assignment Indices
- VC
Vapour Col (VC)
- WAP
Western Antarctic Peninsula
- π
Nucleotide diversity
Authors’ contributions
IMJ performed, analyzed and interpreted mtDNA and microsatellite loci data, and wrote the manuscript; CP performed part of the mtDNA laboratory experiments and computer programs; EP contributed to interpretation and discussion of results; GMPD provided samples and analyzed and interpreted BAPS data; DGA, AB, MVP, KMK and AL collected and provided samples; JAV contributed to the manuscript’s design, discussion and interpretation of results. All authors read and approved the final manuscript.
Ethics approval
Samples from the WAP were collected with permits in accordance to Annex II, Article 3 of the Protocol on Environmental Protection to the Antarctic Treaty, and under regulation of the Scientific Committee on Antarctic Research (SCAR) provided by the Chilean Antarctic Authorities (INACH), the Brazilian Antarctic Authorities through PROANTAR and Environmental Ministry, and the Spanish Polar Committee. Permits incorporated authorizations for sample collection for all locations including special authorizations for Antarctic Specially Protected Areas (ASPAs). Bioethics permits were provided by Universidad de Concepción and Pontificia Universidad Católica de Chile, which were required for INACH permits. Samples from Bouvetøya were collected under Animal Ethics and sampling Permit #7001 issued by the Norwegian Food Safety Authority.
Competing interests
The authors declare they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12862-018-1207-0) contains supplementary material, which is available to authorized users.
Contributor Information
Isidora Mura-Jornet, Email: imura@uc.cl.
Carolina Pimentel, Email: caropiga@gmail.com.
Gisele P. M. Dantas, Email: dantasgpm@gmail.com
Maria Virginia Petry, Email: vpetry@unisinos.br.
Daniel González-Acuña, Email: danigonz@udec.cl.
Andrés Barbosa, Email: barbosa@mncn.csic.es.
Andrew D. Lowther, Email: andrew.lowther@npolar.no
Kit M. Kovacs, Email: kit.kovacs@npolar.no
Elie Poulin, Email: epoulin@uchile.cl.
Juliana A. Vianna, Phone: 56-2-3547210, Email: jvianna@uc.cl
References
- 1.Duminil J, Fineschi S, Hampe A, Jordano P, Salvini D, Vendramin GG, Petit RJ. Can population genetic structure be predicted from life history traits? Am Nat. 2007;169:662–672. doi: 10.1086/513490. [DOI] [PubMed] [Google Scholar]
- 2.Palsbøll PJ, Bérubé M, Allendorf FW. Identification of management units using population genetic data. Trends Ecol Evol. 2006;22:11–12. doi: 10.1016/j.tree.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Chepko-Sade BD, Halpin ZT. Mammalian dispersal patterns: the effects of social structure on population genetics. Chicago: University of Chicago Press; 1987. [Google Scholar]
- 4.Hewitt GM. Some genetic consequences of ice ages, and their role, in divergence and speciation. Biol J Linn Soc. 1996;58:247–276. doi: 10.1111/j.1095-8312.1996.tb01434.x. [DOI] [Google Scholar]
- 5.Bossart JL, Prowell DP. Genetic estimates of population structure and gene flow: limitations, lessons and new directions. Trends Ecol Evol. 1998;13:202–206. doi: 10.1016/S0169-5347(97)01284-6. [DOI] [PubMed] [Google Scholar]
- 6.Peña F, Poulin E, Dantas GPM, González-Acuña D, Petry MV, Vianna JA. Have historical climate changes affected gentoo penguin (Pygoscelis papua) populations in Antarctica? PLoS One. 2014;9:e95375. doi: 10.1371/journal.pone.0095375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritchie PA, Millar CD, Gibb GC, Baroni C, Lambert DM. Ancient DNA enables timing of the pleistocene origin and holocene expansion of two Adélie penguin lineages in Antarctica. Mol Biol Evol. 2001;21:240–248. doi: 10.1093/molbev/msh012. [DOI] [PubMed] [Google Scholar]
- 8.Lenormand T. Gene flow and the limits to natural selection. Trends Ecol Evol. 2002;17:183–189. doi: 10.1016/S0169-5347(02)02497-7. [DOI] [Google Scholar]
- 9.Greenwood PJ. Mating systems, philopatry and dispersal in birds and mammals. Anim Behav. 1980;28:1140–1162. doi: 10.1016/S0003-3472(80)80103-5. [DOI] [Google Scholar]
- 10.Pusey AE. Sex-biased dispersal and inbreeding avoidance in birds and mammals. Trends Ecol Evol. 1987;2:295–299. doi: 10.1016/0169-5347(87)90081-4. [DOI] [PubMed] [Google Scholar]
- 11.Goudet J, Perrin N, Waser P. Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol Ecol. 2002;11:1103–1114. doi: 10.1046/j.1365-294X.2002.01496.x. [DOI] [PubMed] [Google Scholar]
- 12.Bohonak AJ. Dispersal, gene flow, and population structure. Q Rev Biol. 1999;74:21–45. doi: 10.1086/392950. [DOI] [PubMed] [Google Scholar]
- 13.Waples R, Gaggiotti O. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol. 2006;15:1419–1439. doi: 10.1111/j.1365-294X.2006.02890.x. [DOI] [PubMed] [Google Scholar]
- 14.Latch EK, Dharmarajan G, Glaubitz JC, Rhodes OE. Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conserv Genet. 2006;7:295–302. doi: 10.1007/s10592-005-9098-1. [DOI] [Google Scholar]
- 15.Taylor BL, Dizon AE. The need to estimate power to link genetics and demography for conservation. Society fot Conservation Biology. 1996;10:661–664. doi: 10.1046/j.1523-1739.1996.10020661.x. [DOI] [Google Scholar]
- 16.Woehler EJ, Cooper J, Croxall JP, Fraser WR, Kooyman GL, Miller, G.D, Patterson DL, Peter HU, Ribic CA, Salwicka K, Trivelpiece WZ, Weimerskirch H. A statistical assessment of the status and trends of a statistical assessment of the status and trends of antarctic and subantarctic seabirds. SCAR. 2001. http://www.scar.org/scar_media/documents/publications/Woehler_etal_Status_of_Seabirds_2001.pdf. Accessed 13 Aug 2017.
- 17.Brooke M de L. The food consumption of the world’s seabirds. Proc R Soc Lond B Biol Sci. 2004;271(Suppl 4):S246–S248. doi: 10.1098/rsbl.2003.0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheifler R, Gauthier-Clerc M, Bohec CL, Crini N, Cœurdassier M, Badot PM, Giraudoux P, Maho YL. Mercury concentrations in king penguin (Aptenodytes patagonicus) feathers at Crozet Islands (sub-Antarctic): temporal trend between 1966–1974 and 2000–2001. Environ Toxicol Chem. 2005;24:125–128. doi: 10.1897/03-446.1. [DOI] [PubMed] [Google Scholar]
- 19.Carravieri A, Bustamante P, Curlaud CY. Penguins as bioindicators of mercury contamination in the Southern Ocean: birds from the Kerguelen Islands as a case study. Sci Total Environ. 2013;454:141–148. doi: 10.1016/j.scitotenv.2013.02.060. [DOI] [PubMed] [Google Scholar]
- 20.Borboroglu PG, Boersma PD. Penguins: natural history and conservation: University of Washington Press; 2015.
- 21.Roeder AD, Marshall RK, Mitchelson AJ, Visagathilagar T, Ritchie PA, Love DR, Pakai TJ, McPartland HC, Murray ND, Robinson NA, Kerry KE, Lambert DM. Gene flow on the ice: genetic differentiation among Adélie penguin colonies around Antarctica. Mol Ecol. 2001;10:1645–1656. doi: 10.1046/j.0962-1083.2001.01312.x. [DOI] [PubMed] [Google Scholar]
- 22.Clucas GV, Dunn MJ, Dyke G, Emslie SD, Levy H, Naveen R, Polito MJ, Pybus OG, Rogers AD, Hart T. A reversal of fortunes: climate change “winners” and “losers” in Antarctic peninsula penguins. Sci Rep. 2014;4:1–7. doi: 10.1038/srep05024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Younger JL, Emmerson LM, Miller KJ. The influence of historical climate changes on Southern Ocean marine predator populations: a comparative analysis. Glob Chang Biol. 2016;22:474–493. doi: 10.1111/gcb.13104. [DOI] [PubMed] [Google Scholar]
- 24.Vianna JA, Noll D, Dantas GPM, Petry MV, Barbosa A, González-Acuña D, Le Bohec C, Bonadonna F, Poulin E. Marked phylogeographic structure of Gentoo penguin reveals an ongoing diversification process along the Southern Ocean. Mol Phylogenet Evol. 2016;107:486–498. doi: 10.1016/j.ympev.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Woehler EJ. The distribution and abundance of Antarctic and Subantarctic penguins. 1993. [Google Scholar]
- 26.Jablonski B. Distribution, abundance and biomass of a summer community of birds in the region of the Admiralty Bay (king George Island, south Shetland Islands, Antarctica) Polish Polar Research. 1986;7:217–260. [Google Scholar]
- 27.Sander M, Balbão TC, Polito MJ, Costa ES, Carneiro APB. Recent decrease in chinstrap penguin (Pygoscelis antarctica) populations at two of Admiralty Bay’s islets on king George Island, south Shetland Islands, Antarctica. Polar Biol. 2007;30:659–661. doi: 10.1007/s00300-007-0259-1. [DOI] [Google Scholar]
- 28.Sierakowski K, Korczak-Abshire M, Jadwiszczak P. Changes in bird communities of Admiralty Bay, king George Island (West Antarctic): insights from monitoring data (1977–1996) Polish Polar Research. 2017;38:231–262. doi: 10.1515/popore-2017-0010. [DOI] [Google Scholar]
- 29.Croxall JP, Kirkwood ED. Balogh scientific books. 1979. The distribution of penguins on the Antarctic peninsula and islands of the Scotia Sea. [Google Scholar]
- 30.Trivelpiece WZ, Buckelew S, Reiss C, Trivelpiece SG. The winter distribution of chinstrap penguins from two breeding sites in the south Shetland Islands of Antarctica. Polar Biol. 2007;30:1231–1237. doi: 10.1007/s00300-007-0283-1. [DOI] [Google Scholar]
- 31.Biuw M, Lydersen C, De Bruyn PN, Arriola A, Hofmeyr GG, Kritzinger P, Kovacs KM. Long-range migration of a chinstrap penguin from Bouvetøya to Montagu Island, south Sandwich Islands. Antarct Sci. 2010;22:157–162. doi: 10.1017/S0954102009990605. [DOI] [Google Scholar]
- 32.Ainley DG, Nur N, Woehler EJ. Factors affecting the distribution and size of Pygoscelid penguin colonies in the Antarctic. American Ornithological Society. 1995;112:171–182. [Google Scholar]
- 33.Macdonald JA, Barton KJ, Metcalf P. Chinstrap penguins (Pygoscelis antarctica) nesting on Sabrina islet, Balleney Islands, Antarctica. Polar Biology. 2002;25:442–447. [Google Scholar]
- 34.Korczak-Abshire M, Chwedorzewska KJ, Wąsowicz P, Bednarek PT. Genetic structure of declining chinstrap penguin (Pygoscelis antarcticus) populations from south Shetland Islands (Antarctica) Polar Biol. 2012;35:1681–1689. doi: 10.1007/s00300-012-1210-7. [DOI] [Google Scholar]
- 35.Freer JJ, Mable BK, Clucas G, Rogers AD, Polito MJ, Dunn M, Naveen R, Levy H, Hart T. Limited genetic differentiation among chinstrap penguin (Pygoscelis antarctica) colonies in the scotia arc and western Antarctic peninsula. Polar Biol. 2015;38:1493–1502. doi: 10.1007/s00300-015-1711-2. [DOI] [Google Scholar]
- 36.BirdLife International . The IUCN red list of threatened species 2016: e.T22697761A93638235. 2016. Pygoscelis antarcticus. [Google Scholar]
- 37.Ciaputa P, Sierakowski K. Long-term population changes of Adélie, chinstrap, and gentoo penguins in the regions of SSSI no. 8 and SSSI no. 34, king George Island, Antarctica. Polish. Polar Res. 1999;20:355–365. [Google Scholar]
- 38.Forcada J, Trathan PN, Reid K, Murphy EJ, Croxall JP. Contrasting population changes in sympatric penguin species in association with climate warming. Glob Chang Biol. 2016;12:411–423. doi: 10.1111/j.1365-2486.2006.01108.x. [DOI] [Google Scholar]
- 39.Hinke JT, Salwicka K, Trivelpiece SG, Watters GM, Trivelpiece WZ. Divergent responses of Pygoscelis penguins reveal a common environmental driver. Oecologia. 2007;153:845–855. doi: 10.1007/s00442-007-0781-4. [DOI] [PubMed] [Google Scholar]
- 40.Sander M, Balbão TC, Costa ES, Dos Santos CR, Petry MV. Decline of the breeding population of Pygoscelis antarctica and Pygoscelis adeliae on Penguin Island, south Shetland, Antarctica. Polar Biol. 2007;30:651–654. doi: 10.1007/s00300-006-0218-2. [DOI] [Google Scholar]
- 41.Trivelpiece WZ, Hinke JT, Miller AK, Reiss CS, Trivelpiece SG, Watters GM. Variability in krill biomass links harvesting and climate warming to penguin population changes in Antarctica. Proc Natl Acad Sci. 2011;108:7625–7628. doi: 10.1073/pnas.1016560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barbosa A, Benzal J, De León A, Moreno J. Population decline of chinstrap penguins (Pygoscelis antarctica) on deception island, south Shetlands, Antarctica. Polar Biol. 2012;35:1453–1457. doi: 10.1007/s00300-012-1196-1. [DOI] [Google Scholar]
- 43.Lynch HJ, Naveen R, Trathan PN, Fagan WF. Spatially integrated assessment reveals widespread changes in penguin populations on the Antarctic peninsula. Ecology Society of America. 2012;93:1367–1377. doi: 10.1890/11-1588.1. [DOI] [PubMed] [Google Scholar]
- 44.Naveen R, Lynch HJ, Forrest S, Mueller T, Polito M. First direct, site-wide penguin survey at Deception Island, Antarctica, suggests significant declines in breeding chinstrap penguins. Polar Biol. 2012;35:1879–1888. [Google Scholar]
- 45.Dunn MJ, Jackson JA, Adlard S, Lynnes AS, Briggs DR, Fox D, Waluda CM. Population size and decadal trends of three penguin species nesting at Signy Island, south Orkney Islands. PLoS One. 2016;11:e0164025. doi: 10.1371/journal.pone.0164025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niemandt C, Kovacs KM, Lydersen C, Dyer BM, Isaksen K, Hofmeyr GG, Mehlum F, de Bruyn PJN. Chinstrap and macaroni penguin diet and demography at Nyrøysa, Bouvetøya. Antarct Sci. 2016;28:91–100. doi: 10.1017/S0954102015000504. [DOI] [Google Scholar]
- 47.Wilson RP. A method for restraining penguins. Mar Ornithol. 1997;25:72–73. [Google Scholar]
- 48.Aljanabi SM, Martinez I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 1997;25:4692–4693. doi: 10.1093/nar/25.22.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roeder AD, Ritchie PA, Lambert DM. New DNA markers for penguins. Conserv Genet. 2002;3:341–344. doi: 10.1023/A:1019982009059. [DOI] [Google Scholar]
- 50.Vianna JA, Noll D, Mura-Jornet I, Valenzuela-Guerra P, González-Acuña D, Navarro C, Loyola DE, Dantas GPM. Comparative genome-wide polymorphic microsatellite markers in Antarctic penguins through next generation sequencing. Genet Mol Biol. 2017;40:676–687. doi: 10.1590/1678-4685-gmb-2016-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 52.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ewing GB. Haplotype Viewer. In: Center for Integrative Bioinformatics Vienna, Vienna. 2012. http://www.cibiv.at/~greg/haploviewer. Accessed 15 Aug 2017.
- 54.Lischer HEL, Excoffier L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics. 2012;28:298–299. doi: 10.1093/bioinformatics/btr642. [DOI] [PubMed] [Google Scholar]
- 55.Van Oosterhout C, Hutchinson WF, Wills D, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Resour. 2004;4:535–538. doi: 10.1111/j.1471-8286.2004.00684.x. [DOI] [Google Scholar]
- 56.Belkhir K, Borsa P, Chikhi L, Raufaste N, Catch F. GENETIX 4.0. 5.2., Software under Windows™ for the genetics of the populations. University of Montpellier, Montpellier, France. 2004.
- 57.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
- 58.Pike N. Using false discovery rates for multiple comparisons in ecology and evolution. Methods in Ecology & Evolution. 2011;2:278–282. doi: 10.1111/j.2041-210X.2010.00061.x. [DOI] [Google Scholar]
- 59.Jombart T. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 60.Jombart T. An introduction to Adegenet 2.0.0. 2015. http://adegenet.r-forge.r-project.org/documentation.html. Acceded 08 jun 2017.
- 61.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics Society of America. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corander J, Marttinen P, Tang J. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics. 2008;9:1–14. doi: 10.1186/1471-2105-9-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guillot G, Mortier F, Estoup A. GENELAND: a computer package for landscape genetics. Mol Ecol Notes. 2005;5:712–715. doi: 10.1111/j.1471-8286.2005.01031.x. [DOI] [Google Scholar]
- 64.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics Society of America. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Evanno F, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 66.Earl DA, VonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4:359–361. doi: 10.1007/s12686-011-9548-7. [DOI] [Google Scholar]
- 67.Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- 68.Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Mol Ecol. 2004;4:137–138. doi: 10.1046/j.1471-8286.2003.00566.x. [DOI] [Google Scholar]
- 69.Guillot G, Santos F, Estoup A. Analysing georeferenced population genetics data with Geneland: a new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics. 2008;24:1406–1407. doi: 10.1093/bioinformatics/btn136. [DOI] [PubMed] [Google Scholar]
- 70.Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010;11:1–15. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A. GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered. 2004;95:536–539. doi: 10.1093/jhered/esh074. [DOI] [PubMed] [Google Scholar]
- 72.Paetkau D, Calvert W, Stirling I, Strobeck C. Microsatellite analysis of population structure in Canadian polar bears. Mol Ecol. 1995;4:347–354. doi: 10.1111/j.1365-294X.1995.tb00227.x. [DOI] [PubMed] [Google Scholar]
- 73.Rannala B, Mountain JL. Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci. 1997;94:9197–9201. doi: 10.1073/pnas.94.17.9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paetkau D, Slade R, Burden M, Estoup A. Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol. 2014;13:55–65. doi: 10.1046/j.1365-294X.2004.02008.x. [DOI] [PubMed] [Google Scholar]
- 75.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Am J Hum Genet. 1989;68:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics Society of America. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7 Research article. Molecular Biology & Evolution. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rambaut A, Suchard MA, Xie D, Drummond AJ. Tracer v1.6. 2014. http://beast.bio.ed.ac.uk/Tracer.
- 79.Darriba D, Taboada GL, Doallo R, Posada D. Europe PMC funders group jModelTest 2: more models, new heuristics and high-performance computing. Nat Methods. 2015;9:6–9. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Millar CD, Dodd A, Anderson J, Gibb GC, Ritchie PA, Baroni C, Woodhams MD, Hendy MD, Lambert DM. Mutation and evolutionary rates in Adélie penguins from the Antarctic. PLoS Genet. 2008;4:1–9. doi: 10.1371/journal.pgen.1000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fridolfsson AK, Ellegren H. A simple and universal method for molecular sexing of non-ratite birds. J Avian Biol. 1999;30:116–121. doi: 10.2307/3677252. [DOI] [Google Scholar]
- 82.Goudet J. FSTAT, version 2.9.3, A program to estimate and test gene diversities and fixation indices. 2001. https://www2.unil.ch/popgen/softwares/fstat.htm.
- 83.Guillot G, Rousset F. Dismantling the mantel tests. Methods in Ecology & Evolution. 2013;4:336–344. doi: 10.1111/2041-210x.12018. [DOI] [Google Scholar]
- 84.Harmon LJ, Glor RE. Poor statistical performance of the mantel test in phylogenetic comparative analyses. Evolution. 2010;64:2173–2178. doi: 10.1111/j.1558-5646.2010.00973.x. [DOI] [PubMed] [Google Scholar]
- 85.Diniz-Filho JAF, Soares TN, Lima JS, Dobrovolski R, Landeiro VL, Telles MPD, C, Rangel TF, Bini, L M Mantel test in population genetics Genetics & Molecular Biology 2013;36: 475–485. [DOI] [PMC free article] [PubMed]
- 86.Dantas GPM, de Oliveira LR, Marasco ACM, de Araujo J, Hurtado R, Durigon EL, San Fillipo LF, Morgante JS. Demographic history of the gentoo penguin (Pygoscelis papua) and the Adélie penguin (Pygoscelis adeliae) on Admiralty Bay, king George Island, Antarctica. Waterbirds. 2014;37:410–418. doi: 10.1675/063.037.0408. [DOI] [Google Scholar]
- 87.Jouventin P, Cuthbert R, Ottvall R. Genetic isolation and divergence in sexual traits: evidence for the northern rockhopper penguin Eudyptes moseleyi being a sibling species. Mol Ecol. 2016;15:3413–3423. doi: 10.1111/j.1365-294X.2006.03028.x. [DOI] [PubMed] [Google Scholar]
- 88.Bouzat JL, Walker BG, Dee Boersma P. Regional genetic structure in the Magellanic penguin (Spheniscus magellanicus) suggests metapopulation dynamics. The American Ornithologist’s Union. 2009;126:326–334. [Google Scholar]
- 89.Schlosser JA, Dubach JM, Garner TW, Araya B, Bernal M, Simeone A, Smith KA, Wallace RS. Evidence for gene flow differs from observed dispersal patterns in the Humboldt penguin, Spheniscus humboldti. Conserv Genet. 2009;10:839–849. doi: 10.1007/s10592-008-9644-8. [DOI] [Google Scholar]
- 90.Bensch S, Hasselquist D. Phylogeographic population structure of great reed warblers: an analysis of mtDNA control region sequences. Biol J Linn Soc. 1999;66:171–185. doi: 10.1111/j.1095-8312.1999.tb01882.x. [DOI] [Google Scholar]
- 91.Barrientos R, Kvist L, Barbosa A, Valera F, López-Iborra GM, Moreno E. Colonization patterns and genetic structure of peripheral populations of the trumpeter finch (Bucanetes githagineus) from Northwest Africa, the Canary Islands and the Iberian peninsula. J Biogeogr. 2009;36:210–219. doi: 10.1111/j.1365-2699.2008.01995.x. [DOI] [Google Scholar]
- 92.Forcada J, Trathan PN. Penguin responses to climate change in the Southern Ocean. Glob Chang Biol. 2009;15:1618–1630. doi: 10.1111/j.1365-2486.2009.01909.x. [DOI] [Google Scholar]
- 93.Ainley DG, Wood RC, Sladen WJL. Bird Life at Cape Crozier. Ross Island Wilson Bull. 1978;90:492–510. [Google Scholar]
- 94.Raymond JA. Chinstrap penguin at McMurdo sound. Antarctic Journal of United States. 1975;10:275. [Google Scholar]
- 95.Spurr E. Records of chinstrap penguins in the Ross Sea region Antarctica. Notornis. 1985;32:1–4. [Google Scholar]
- 96.Clucas GV, Younger JL, Kao D, Rogers AD, Handley J, Miller GD, Jouventin P, Nolan P, Gharbi K, Miller KJ, Hart T. Dispersal in the sub-Antarctic: king penguins show remarkably little population genetic differentiation across their range. BMC Evol Biol. 2016;16:1–14. doi: 10.1186/s12862-016-0784-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lowe WH, Allendorf FW. What can genetics tell us about population connectivity? Mol Ecol. 2010;19:3038–3051. doi: 10.1111/j.1365-294X.2010.04688.x. [DOI] [PubMed] [Google Scholar]
- 98.Clarke AL, Sæther B, Røskaft E. Sex biases in avian dispersal: a reappraisal. Nordic Society Oikos. 1997;79:429–438. doi: 10.2307/3546885. [DOI] [Google Scholar]
- 99.Wright S. Evolution and the genetics of populations: Vol. 2. The theory of gene frequencies. Chicago: University of Chicago Press; 1969. [Google Scholar]
- 100.Dugger KM, Ainley DG, Lyver POB, Barton K, Ballard G. Survival differences and the effect of environmental instability on breeding dispersal in an Adélie penguin meta-population. Proc Natl Acad Sci. 2010;107:12375–123380. doi: 10.1073/pnas.1000623107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Plot of assignment probabilities from STRUCTURE. A vertical bar represents an individual, and colors represent the different clusters found. All plots were generated via running 10 replicates. Figures show the optimal number of clusters for no admixture model, with independent allele frequency and no location information supplied using A) Posterior probability of K (LnP(D)) and B) Evanno’s method. (DOCX 20 kb)
Figure S2. Plot of assignment probabilities from BAPS. Vertical lines represent each individual and the color refers to clusters found through this analysis. A) spatial with both, mixture and admixture models (K = 1) and B) non-spatial admixture model (K = 7). (DOCX 16 kb)
Figure S3. Posterior probabilities of population membership from the spatial model with correlated allele frequencies’ model. Lighter colors indicate higher probabilities of population membership. Three genetic clusters were identified using GENELAND. Left: Kopaitic Island, middle: Georges Point, and right: northern WAP locations and Bouvetøya. (DOCX 16 kb)
Figure S4. Discriminant Analysis of Principal Components (DAPC). The six genetic clusters identified by Adegenet are shown in different colors. All six groups overlapped extensively, and none of them represent a specific colony. (DOCX 16 kb)
Figure S5. Skyline plot mtDNA (HVRI) for chinstrap penguins. (JPG 144 kb)
Figure S6. Chinstrap penguins south of their traditional breeding range. A) Breeding pairs at Waterboat Point (Gabriel González Videla base; 64°49’S, 62°51’W), B) a single individual surrounded by gentoo colonies on Doumer Island (Yelcho base; 64°65’S, 63°35’W) and c) another single bird on Avian Island (67°46’S, 68°54’W) in the midst of Adélie penguins. (JPG 43 kb)
Table S1. Allelic richness (A), expected (HE) and observed heterozigosity (HO) values for 11 microsatellite loci, for all examined populations. Study site (collection location) abbreviations correspond to EI: Elephant Island, PI: Penguin Island, BP: Barton Peninsula, AI: Ardley Island, GI: Greenwich Island, MB: Miers Bluff, HP: Hannah Point, CS: Cape Shirreff, BH: Baily Head, VC: Vapour Col, KI: Kopaitic Island, GP: Georges Point, and BI: Bouvetøya. (JPG 424 kb)
Table S2. Summary of pairwise genetic differences (ΦST) between chinstrap penguin colonies for mtDNA marker (HVRI). Bellow the diagonal are ΦST values, and their corresponding p-values above the diagonal. (TIFF 2112 kb)
Table S3. Summary of pairwise genetic differences (FST) between chinstrap penguin colonies for mtDNA marker (HVRI). Below the diagonal are FST values, and their corresponding p-values above the diagonal. (DOCX 18 kb)
Table S4. Summary of pairwise genetic differences between chinstrap penguin colonies (FST) calculated from the 11 microsatellite loci. Below the diagonal are FST values, and corresponding p-values above the diagonal. (JPG 76 kb)
Table S5. GeneClass2 percentage test results using microsatellite data for chinstrap penguins from 13 colonies for (a) genetic assignment using Paetkau et al. (1995) criterion and (b) first-generation migrant. Lines indicate the samples’ site collection and columns indicate the colonies to which the individuals were assigned. Colony self-assignments are in bold. (JPG 5437 kb)
Data Availability Statement
The mitochondrial DNA sequences are available in the GenBank repository, accession numbers: MF966819 – MF966902 and MH025646 – MH025759.