

Abstract
Genome Wide Association Studies (GWAS) have mapped thousands of genetic variants associated with complex disease risk and regulating quantitative traits, thus exploiting an unprecedented high-resolution genetic characterization of the human genome. A small fraction (3.7%) of the identified associations is located in untranslated regions (UTRs), and the molecular mechanism has been elucidated for few of them. Genetic variations at UTRs may modify regulatory elements affecting the interaction of the UTRs with proteins and microRNAs. The overall functional consequences include modulation of mRNA transcription, secondary structure, stability, localization, translation, and access to regulators like microRNAs (miRNAs) and RNA-binding proteins (RBPs). Alterations of these regulatory mechanisms are known to modify molecular pathways and cellular processes, potentially leading to disease processes. Here, we analyze some examples of genetic risk variants mapping in the UTR regulatory elements. We describe a recently identified genetic variant localized in the 3′UTR of the TNFSF13B gene, associated with autoimmunity risk and responsible of an increased stability and translation of TNFSF13B mRNA. We discuss how the correct use and interpretation of public GWAS repositories could lead to a better understanding of etiopathogenetic mechanisms and the generation of robust biological hypothesis as starting point for further functional studies.
Graphical Abstract
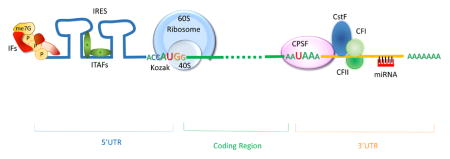
Representation of human mature mRNA with indicated the main regulatory regions in the UTRs and the corresponding trans-acting factors.
Introduction
In the last decade, Genome Wide Association Studies (GWAS) have uncovered many robust associations between genetic variants and risk of numerous complex diseases. The availability of high-quality genotyping microarrays1–3 and the advent of large-scale human genome sequencing4–7, and their integration using appropriate statistical methods, like imputation10, have provided unprecedented high-resolution genetic profiles. These advances have allowed the analysis by GWAS of millions of genetic variants of the entire human genome with deep coverage.
However, only ~4% of the total genome-wide associated variants identified result in differences in the protein product, and in even fewer cases, the link between coding variant and mechanism contributing to the disease is apparent.4
Part of this discrepancy is likely due to problems inherent in the assessment of genetic variation. Although recent technological advances have enabled large-scale whole-genome sequencing (WGS) in some studies, most large GWAS have relied on DNA microarray (chip array) analysis, which only genotypes a subset of all genetic variation. In fact, microarray construction must balance numerous factors, like allele frequency, accuracy of genotyping and marker physical position, as well as purported functional role of the polymorphisms.2 Likewise, the identification of the genetic variants themselves has not been a uniform, standardized search, but has instead been conditioned by the search method (expressed sequence tag searches, gene-region searches, etc), and also by the genotyped population, with some variants common in one human population being rare or absent in others.6,7 As a result, a GWAS can provide the most-associated variant as the “best available marker”, rather than as the “probable causal variant”. Similarly, even in the case of WGS, non-random association of specific alleles at specific positions (known as “Linkage Disequilibrium” - LD),8 complicates the problem of detecting the variant effectively associated with a disease or phenotype in a “haplotype block”.
Because of their easier interpretation, genetic variants in the coding sequence of a gene and present in the expressed coding regions (exons) have often been given priority, although it has long been clear that coding sequence variants per se were insufficient for mapping complex diseases.9 However, variants in the intervening sequences (introns) or in the untranslated regions (UTRs), although not changing the predicted protein sequence, may be pivotal in the regulation of gene expression. The UTRs are the mRNA sequences flanking the beginning and end of the coding sequence; as their name suggests, UTRs are part of the mRNA but are not translated into protein. Notably, 3.7% of the genetic variants detected in GWAS studies are located in the UTRs.11,12
By convention, a genetic variant in the DNA sequence that occurs in a population with a frequency of 1% or higher is defined as “polymorphism” while the rarer ones (frequency <1%) are defined as “mutations”.13,14 Polymorphisms and mutations can comprise one or more nucleotide changes. Interestingly, mutations predominate in 5′UTRs, while polymorphisms are more common in 3′UTRs.13,14 Gene expression is regulated at the RNA level by virtue of the presence of 5′ and 3′UTR regulatory elements such as upstream open reading frames (uORFs), internal ribosome entry sites (IRESs), as well as the UTR’s secondary structure, sequence composition, and length. The majority of regulatory elements are recognized by RBPs or by non-coding RNAs (ncRNAs) such as miRNAs. Overall, these mechanisms modulate the mRNA stability, localization, and translation.15,16
Alteration of these regulatory mechanisms can modify molecular pathways and cellular processes, thus affecting phenotype, disease onset and possibly even disease outcome. In fact, genetic variations in the UTRs have been already implicated in several diseases such as melanoma, Alzheimer’s disease, multiple myeloma, fragile X syndrome, bipolar disorder, breast cancer and other pathologies.11
In this review, we will describe the mechanisms of UTR regulation, and the role of genetic variants in modulating RNA processing and thus protein production in human disease. We will then give an overview of the GWAS results linked to UTR regions, and discuss an example of a genetic variant in the 3′UTR of TNFSF13B affecting the risk of autoimmune diseases and related immune phenotypes.17 We conclude by presenting several open questions about UTR mechanisms.
REGULATORY ELEMENTS IN THE UNTRANSLATED REGIONS
Genetic variants at the 5′UTR
The 5′UTR is the RNA sequence immediately upstream of the coding RNA. It is generally not translated, although some exceptions, in which part of the 5′ UTR is translated, do exist.18 In eukaryotes, its length ranges from a few nucleotides (nt) to several thousand, with an average in humans of about 200 nt.16 The 5′ UTR should possess a Kozak consensus sequence (ACCAUGG), which contains the translation initiation codon. It may also contain numerous regulatory elements, like CpG sites, uORFs, IRESs, and RBP binding sites, which will be treated later. Additionally, secondary structures, such as hairpin loops, may be important in translation regulation, and often occur within the 5′UTR. Thus, genetic variants modifying these regulatory elements can have important impact on the overall production of the protein by affecting RNA transcription, stability, and translation.16,19, 20
In the next sections, we will describe the regulatory elements at the 5′UTR and give some examples of genetic variants affecting their function.
5′UTR length, CpG sites and Kozak sequence
Genes with differences in 5′UTR length are relatively common mainly due to the presence of multiple promoters21 or alternative splicing mechanisms within UTRs22 and may have clinical effects. For example, deletion in the 5′UTR of the ATPase copper transporting beta, ATP7B, reduces the activity of the ATP7B promoter resulting in less protein and increased Wilson disease predisposition.23
Besides length per se, CpG sites, repeats of cytosine followed by guanine, are often present in 5′UTRs. These regions can undergo cytosine methylation, an epigenetic modification that promotes gene silencing by recruiting proteins involved in gene repression of by inhibiting the binding of transcription factors.24 The higher the number of CpG sites in a 5′UTR, the higher the probability that the gene expression will be downregulated as a consequence of cytosine methylation. For example, fragile X syndrome, a genetic disorder characterized by several intellectual disabilities, is caused by the expansion of a CGG-repeat sequence in the 5′UTR of the FMR1 gene. Expansion to >200 copies of the CGG-repeat leads to hypermethylation of FMR1 and silencing of this gene, resulting in an insufficient amount of fragile X mental retardation protein that is pivotal for neuronal development.25
Other mutations in specific regulatory elements, such as the Kozak consensus sequence26 can have an important impact on protein production. For example, mutation in the Kozak sequence of the β-globin gene leads to a 30% reduction of the translational rate of the beta-globin gene, while not altering the transcription level.19
Open reading frames
The ORF is defined as the part of a reading frame that has the potential to be translated; it consists of a sequence of nucleotide triplets that specify an amino acid chain. While the mRNA of a gene will have a principal ORF that specifies the main polypeptide product, there may be several other ORFs, each of which modulate the overall expression of the main protein product. The ORFs located upstream to the canonical initiation codon and out-of-frame with respect to the main coding sequence are called upstream (u)ORFs and are characterized by their own upstream starting codon (uAUG) and stop codon. At least half of the human transcripts contain uORFs. They correlate significantly with reduction of protein expression (30–80%) of the downstream ORF, with only a modest impact on mRNA levels.27 The uORF-mediated translational control can occur through different mechanisms, depending on the efficiency of uAUG ribosome recognition and of uORF translational termination.28 The uORFs can be generated or disrupted by genetic variants leading to dysregulation of gene expression and increased disease risk.27,29
For example, the creation of a new uORF was observed in the SPINK1 gene. This gene encodes a trypsin inhibitor, Serine Peptidase Inhibitor, Kazal Type 1, that prevents the activation of zymogens within the pancreas.30 A mutation (C>T) at position -53 to the main AUG start codon generates a new AUG start codon and a uORF that dampens the efficiency of Spink1 translation, leading to hereditary pancreatitis.
The disruption of uORFs are also frequent in nature, for example a point mutation in the second uORF of the gene encoding the human hairless homolog (HR) causes the elimination of the ATG codon, leading to the absence of the corresponding 34 amino acid peptide, which has a negative regulatory effect on the main HR. In this way, the variant causes an increased translation of HR and Marie Unna hereditary hypotrichosis, an autosomal dominant form of genetic hair loss.31
Other mechanisms involving mutations in the uORF can affect the expression of the corresponding encoded peptide, predisposing to disease. For example in bipolar disorder, the missense mutation C178T in the uORF reduces the repressive activity of the uORF encoded peptide (P16S amino acid change), causing an increase in HT3A, a receptor subunit involved in neuronal depolarization.32
Internal Ribosome Entry Sites
Translation initiation is a complex event requiring several proteins, called initiation factors (IFs), which allow i) the formation of the ribosomal pre-initiation complex, ii) the recruitment of the 43S complex to the 5′ end of the mRNA, iii) the scanning of the 5′UTR, and iv) the recognition of the AUG codon and the 5′ cap. Capping at the 5′ end is a key process consisting in the addition of 7-methylguanosine to the 5′UTR, which confers protection to the mRNA against degradation, ultimately promoting nuclear export and translation.33
Internal ribosome entry sites (IRESs) are RNA elements that allow the translation initiation in a cap-independent manner by recruiting the ribosome to the mRNA for protein synthesis. Discovered in 1988 in the poliovirus RNA genome,34 IRES are characterized by several elements recognized by proteins involved in translation, such as IRES trans-acting factors (ITAFs), but also canonical initiation factors. IRESs are present in many viruses and in eukaryotic mRNAs involved in responses to stress conditions (hypoxia, heat shock, nutrient limitation) or in response to signals to survive, differentiate, proliferate, or undergo apoptosis.35
IRES mutations can alter protein expression and cause disease. For example, the 5′UTR of the proto-oncogene c-MYC contains an IRES and, in patients with multiple myeloma, a C>T substitution within the IRES causes increased synthesis of c-MYC protein by favouring the binding of two ITAFs, Y-box binding protein 1 (YB-1) and polypyrimidine tract-binding protein 1 (PTB-1).36 Another example is represented by a mutation in the IRES of the connexin-32 gene, which abolishes translation of the corresponding mRNA in nerve cells, leading to Charcot-Marie-Tooth disease, a neurodegenerative disorder.37
RNA-binding proteins and 5′UTR regulation
RBPs are key components of the ribonucleoproteins complexes (RNPs) which modulate gene expression by binding the mRNA molecules and may act in both the cytoplasm and the nucleus. RBPs regulate several phases of co- and post-transcriptional gene expression, such as RNA capping, splicing and polyadenylation, and mRNA export, localization, stability and translation.38
Binding of RBPs to RNA targets is mediated by a set of modular RNA-binding domains, such as the RNA recognition motif, heterogeneous nuclear RNP K-homology domain, and zinc fingers,39 while the RNA target is characterized by short, single-stranded (ss)RNA sequences, often having specific secondary structures.40
Functional genetic variants in the RNA targets can affect RBPs recruitment by generating or disrupting their binding sites, as well as by modifying the RNA secondary structure.
Tryptophan hydroxylase (TPH) is an enzyme responsible for neuronal serotonin (5-HT) synthesis and it is encoded by two genes, TPH1 and TPH2. While TPH1 is primarily expressed in the periphery, TPH2 is predominantly expressed in the brain. Polymorphisms in TPH2 are associated with a range of behavioural traits and psychiatric disorders.41,42 Chen and colleagues demonstrated that the 90 A/G polymorphism at the 5′UTR of the THP2 mRNA alters the mRNA structure and/or RNA–protein interaction, thus affecting TPH2 gene expression at the post-transcriptional level.43
The amyotrophic lateral sclerosis-associated RNA-binding protein (TDP-43) was linked to the pathogenesis of fragile X-associated tremor/ataxia syndrome (FXTAS). In FXTAS, a CGG repeat expansion in the 5′UTR of the FMR1 gene caused a progressive neurodegeneration in human patients. In a drosophila model of FXTAS, He and colleagues identified TDP-43 as the suppressor of the CGG-induced toxicity, although it required two heterogeneous nuclear ribonucleoproteins (Hrb87F and Hrb98DE) for its activity. In fact, deletions in TDP-43 that prevented the interaction with the two ribonucleoproteins nullified the beneficial effect of TDP-43 function of CGG-repeat toxicity. These results suggest a model in which the repeat expansion of CGG at the 5′UTR and the modified interaction with a RBP are implicated in neurodegenerative disease.44
Introns at 5′UTR
Approximately the 35% of human genes contain introns in the 5′UTR.45 These 5′ introns are less common than introns within coding regions, but are, on average, longer.46 By analysing the expression profiles of genes with 5′UTR introns, Cenik and colleagues found that the most highly expressed genes reveal a strong enrichment of short 5′UTR introns with respect to long or absent 5′UTR introns.18 No relationship was found between length and expression level for genes with intermediate or long 5′ introns. Considering that expression depends on production and degradation rates of mRNAs, Cenik’s results suggest that short 5′UTR introns tend to increase transcription or stabilize mature mRNAs.
Genetic variants at the 3′UTR
The 3′UTR is located downstream of the coding sequence, and it is involved in regulatory processes, including RNA stability, mRNA translation and localization. The 3′ UTR is characterized by binding sites for RBPs and miRNAs, and thus any variation in the 3′UTR length and sequence may change the binding for miRNAs and RBPs, leading to change in gene expression.
3′UTR length and alteration of the polyadenylation signal
The importance of 3′UTR length in mRNA stability, translation, tissue-specific expression, timing and function is demonstrated by several studies in health47–49 and disease.50,51 For example, in a recent work Romo and colleagues showed that the huntingtin gene (HTT) is characterized by three mRNA isoforms, two of which had different 3′UTR lengths. The amount of the two 3′UTR isoforms differed between Huntington disease patients and controls; moreover, while the longer isoform is more represented in the neuronal precursor cells, breast and ovary, the shorter isoform is more prevalent in testes, B cells and muscle, and the abundance of HTT isoforms changes in a tissue-specific manner in Huntington patients.52
One important mechanism causing alteration in the 3′UTR length is the modification of the polyadenylation signal. Like the 5′ cap on mature mRNA, the stability, nuclear export and translation is also regulated by a stretch of adenine nucleotides that are added at the 3′end of the mRNA by specialized enzymes. The cleavage and polyadenylation specificity factor (CPSF) recognizes the specific sequence AAUAAA on the pre-mRNA and cuts 3′UTR about 10–30 nucleotides downstream of its binding site. Other proteins, such as the cleavage stimulation factor (CstF) and cleavage factors (CFI, CFII) work in concert creating, together with CPSF, the RNA-cleavage complex. Once the RNA is cut, the polyadenylate polymerase adds adenosine monophosphates creating the poly-A tail.47
Several genes contain multiple polyadenylation sites which, by changing the length of the 3′ untranslated regions, may alter the number of binding sites for miRNAs and RBPs, thus modifying protein expression patterns and influencing disease. An example is represented by a point mutation in the canonical poly-A signal (AAUAAA→AAUGAA) of the forkhead box P3 gene (FOXP3), highly expressed in regulatory T cells. The mutation reduces the protein expression and causes a rare autoimmune disease, named immune dysfunctions polyendocrinopathy enteropathy X-linked, also known as IPEX syndrome.53 Similarly, mutations in the poly-A signal of α- and β-globin genes cause a decreased production of the corresponding proteins resulting in thalassemia.54–56 Conversely, the introduction of a novel alternative poly-A signal can dysregulate the protein production, leading to increased risk for disease. An example is represented by the recently discovered variant, associated to both multiple sclerosis and systemic lupus erythemathosus, which will be described below.17
MicroRNAs and miRNA binding sites
MicroRNAs are small non-coding RNAs acting as regulatory elements in gene expression.57 miRNAs are transcribed as primary miRNA (pri-miRNA) and then cleaved by a nuclear complex, including the Drosha and Pasha proteins, resulting in the production of a precursor miRNA (pre-miRNA).58–60 Pre-miRNAs are then exported to the cytoplasm by exportin-5 and cleaved by the Dicer enzyme, yielding a double-stranded (ds) miRNA.61 Finally, miRNAs are loaded in the RNA-induced silencing complex (RISC) to suppress stability and/or translation of the mRNA target.62 miRNAs recognize and bind miRNA Regulatory Elements mostly located in the 3′UTR of target mRNAs;57 however miRNAs binding other regions, such as 5′UTR, have also been described.63
After the discovery of miRNAs, polymorphisms affecting miRNA function were identified by several approaches. Modern bioinformatic and statistical analyses, such as GWAS, combined with RNA sequencing and CLIP (cross-linking immunoprecipitation) data, represent key tools in the identification of genetic variants of miRNA binding sites and their impact on gene expression.
Functional variants can be divided into two groups, depending on whether they generate or disrupt miRNA binding sites in target mRNA.64,65 Additionally, Single Nucleotide Polymorphisms (SNPs) and genetic variants in general can modify the secondary structure of the mRNA by affecting the accessibility to binding sites, or by altering the presence of the miRNA-binding site in the mature mRNA.66,17
To study polymorphisms affecting miRNA-binding sites, compared to SNP located in others regions, Lu and colleagues analysed the genotype and the mRNA expression in four populations, as part of the international HapMap Project. They found that compared to introns, 3′UTRs contain higher numbers of SNPs associated with changes in mRNA expression levels.67
Using SNP data, including those from the 1,000 Genomes Project, Richardson and colleagues performed a genome-wide scan of SNPs that disrupt or create new miRNA recognition element site. Specifically, the authors identified 2,723 SNPs disrupting, and 22,295 SNPs creating new miRNA binding sites. Additionally, by analysis of co-expression and eQTL data, they also identified four SNPs with a clear functional role. Among them, rs907091, localised in the IZKF3 gene, a transcription factor important for B-cell activation, created a new binding site for mir-326 with a potential role in autoimmune diseases.68
The correlation between genetic variants at the 3′UTR and miRNA function has been extensively studied and are often associated with diseases. In 2016, Ghanbari and colleagues performed an analysis to identify genetic variants in miRNA genes and in miRNA-binding sites associated with Alzheimer Disease (AD). They found 237 variants in 206 miRNA genes and 42,855 variants in miRNA-binding sites present in AD-GWAS.69 Among the 42,855 variants located in the miRNA-binding sites, they found 10 of them located in the 3′UTR of nine genes, including rs6857, which is predicted to create a target site for miRNA-320e in the 3′UTR of the poliovirus receptor-related 2 (PVRL2) gene.69
As SNPs perturbing miRNA-mRNA regulation can induce aberrant expression of autism-related genes, Vaishnavi and colleagues developed a systematic computational pipeline that integrates data from established databases. Using stringent selection criteria, they were able to identify 9 SNPs modulating and 12 creating new miRNA-mRNA regulation in the 3′UTR of autism-associated genes.70 This paper provides valuable candidate SNPs affecting autism pathogenesis but unfortunately, as for other studies, further functional experiments are needed to validate the predicted data.
Furthermore, in a recent study, Zhang and colleagues identified a genetic variant (rs61764370) localized in the 3′UTR of the Kirsten rat sarcoma viral oncogene homolog (KRAS) that interfered with miRNA/mRNA interaction, and increased risk of developing metastasis in osteosarcoma.71 Using several approaches, the authors demonstrated that the SNP interferes with the interaction between 3′UTR of KRAS mRNA and the miRNA let-7a, thus increasing KRAS protein level and influencing disease outcome.71
Together these studies demonstrate the relevance of dissecting genetic variants in the 3′UTR, particularly those involved in the interaction between miRNA and mRNA, and highlight the importance of genetic variants located in miRNA-binding sites in human diseases.
RNA-binding proteins and 3′UTR regulation
Genetic variants that modify the binding sites of RBPs in the 3′UTR can influence mRNA stability, translation efficiency and localization, by affecting the RNA-binding sequence and domain.
AU-rich elements (AREs) are RNA-binding domains recognized by certain RBPs, such as Human antigen (Hu) R (HuR), and ARE/poly(U)-binding/degradation factor 1 (AUF1), both implicated in controlling mRNA stability.72,73 AREs are present in the 3′UTR of TNF mRNA and modulate TNF production at post-transcriptional level. Di Marco and colleagues showed that two polymorphisms (GAU and CAU trinucleotide insertions), localized in the 3′UTR of TNF mRNA, affect the binding of RBPs, with consequent reduction in the TNF protein expression a mouse model. Importantly, they showed that the polymorphism reduced HuR binding affinity to the ARE, thereby decreasing the production of TNF protein in macrophages.74
Another example is represented by the PPP1R3 gene, encoding the muscle-specific glycogen-targeting regulatory PP1 subunit, which is involved in the regulation of glycogen synthesis in skeletal muscle. Xia and colleagues identified a polymorphism (ARE) in the 3′UTR of the PPP1R3 gene, which reduces the distance between two mRNA-destabilizing sequence ATTTA. The polymorphism is characterized by a 10-nucleotide (allele ARE1) versus a 2-nucleotide interval (allele ARE2). Interestingly, ARE2 was associated with insulin resistance, increased prevalence of type 2 diabetes and reduced expression of this PPP1R3 subunit, causing a reduction in the half-life of the corresponding mRNA. Three proteins of 43, 80, and 139 kDa seem to bind the polymorphic ARE region and the less stable ARE2 allele shows higher protein binding, suggesting the role of the ARE2 in reducing mRNA stability.75
A complex polymorphism, a 6-nucleotide insertion/deletion in the 3′-untranslated region of the thymidylate synthase (TS) gene, affects mRNA stability by modulating the binding of AUF1 to TS mRNA. Pullmann and colleagues demonstrated that AUF1 has higher affinity for the deletion in the 3′UTR TS mRNA, consequently rendering it less stable, compared to the insertion in the same site. Additionally, they demonstrated that AUF1 overexpression preferentially suppressed the deletion allele.76
These studies demonstrate the importance of the identification of genetic variants at the 3′UTR affecting RPBs, as potential predisposing factors for complex diseases, their course, prognosis and complications.
Introns in 3′UTRs
Analogous to what was found in the 5′UTR, also the presence of an intron in the 3′UTRs may influence gene expression. 3′UTRs are generally much longer than 5′UTRs, but relatively few 3′UTRs (<5%) contain introns.46 The reason could be partially explained by nonsense-mediated decay, by which transcript degradation would be typically signalled by an intron downstream of the stop codon.77 In addition, splicing signals within 3′UTRs have been suggested to have reduced maintaining selection, being the 3′UTRs better able to tolerate loss of intron integrity than other gene regions; consequently, 3′UTRs tend to be longer with fewer introns compared to 5′UTRs.78
RiboSNitches, structural variation and RNA regulation
RNA folding to a specific conformation represents an essential step for the function of mRNAs. Structured elements in the UTRs of specific mRNAs can control gene expression and consequently affect physiological processes and disease onset. Today, the extent to which RNA conformational modifications impact the RNA function is still largely unexplored. RNA secondary structure (RSS) differences may have profound implications not only regarding RNA stability, protein binding and translation, but also in disease predisposition and personalized medicine.
RiboSNitch are Single Nucleotide Variant (SNV), found in the UTRs of mRNA transcripts as well as in ncRNAs, that alters the secondary structure of an RNA transcript.79 They are analogous to bacterial riboswitches – RNA elements that adopt a different conformation after binding specific small molecules, leading to gene expression changes.80,81 With riboSNitches, it is the base changes, rather than the binding of a small molecule, that promote RSS rearrangements.82 Experiments have suggested that riboSNitches are not isolated peculiarities: astudy of RSS in a human family trio, identified more than 1900 transcribed variants, corresponding to 15% of all transcribed SNVs that could alter local RNA structure and hence the “RNA folding landscape”.83
To predict the impact of a genetic variant in RNA conformation, several algorithms have been developed.84,85 For example, applying the SNPfold algorithm to all known disease-associated SNPs from the Human Gene Mutation Database, and mapping in the UTRs, Halvorsen and colleagues identified 6 diseases (hyperferritinemia cataract syndrome, β-thalassemia, cartilage-hair hypoplasia, retinoblastoma, chronic obstructive pulmonary disease, and hypertension) where multiple SNPs, in the UTRs of disease-associated genes were predicted to cause RNA conformational change.79
By changing the RNA conformation, a riboSNitch can alter the binding of RBPs and miRNAs that interact with the transcript. The interaction between iron responsive element (IRE) and IRE-binding protein (IREBP) in the ferritin light chain (FTL) RNA, requires both a correct IRE sequence and an exact RNA conformation. Both elements allow the RNA interaction with IREBP, which ultimately lead to translational repression. Mutation in any residue that shifts the structure of the IRE, is able to prevent IREBP binding, leading to increased FTL translation and hyperferritinemia phenotype.
Several molecular biological techniques have been developed to interrogate RNA structure at single-nucleotide resolution, including SHAPE.86 This technique was applied to show that the correct FTL conformational structure can be restored by a group of SNPs in LD, namely a structure-stabilizing haplotype (SSH). This may explain some cases of strong LD between SNPs and also indicate a set of “causal SNPs”, rather than a single “causal mutation” for some phenotypes. Moreover, a comprehensive analysis of human genetic variation highlighted that SSHs are common in mRNA and they generally stabilize the RBP target sites.87
Similarly, using SHAPE, Kutchko and colleagues identified 3 different functional conformations of the 5′UTR of retinoblastoma 1 (RB1), also finding that private SNVs in two patients with retinoblastoma caused the collapse of the RNA structural ensemble, leading to a specific RNA abnormal conformation.88
Beyond retinoblastoma, riboSNitches have also been found in the H19 gene, a long non-coding RNA involved in several cancers. Li and colleagues observed that rs2839698 GA/AA genotypes increase the risk of colorectal cancer in the Chinese populations compared with the GG genotype. Interestingly, the A allele generates an important conformational change in the folding structure of H19 that may cause the loss of the target binding site for some miRNAs, while creating a binding site for other miRNAs.89
A further example of a pathogenic RiboSNitch and its potential use in personalized medicine is represented by the SNP rs12455792, localized in the 5′UTR of the SMAD4, a gene involved in blood vessel remodeling and matrix maintenance. Wang et al. demonstrated that the CT or TT genotypes were associated with reduced transcriptional activity, altered RNA folding structure, and decreased SMAD4 expression, as well as significantly elevated risk of thoracic aortic aneurysm and dissection (TAAD). Moreover, using computational analysis and other approaches, they showed that the lower SMAD4 expression might be due to a reduced function of a RNA hairpin structure. Additionally, SMAD4 mRNA abundance, assessed in freshly frozen aorta tissues from TAAD patients, was significantly higher in CC genotype than in CT or TT genotypes, suggesting rs12455792 as a predictor of TAAD progression.90
Overall, these findings indicate that riboSNitches are an exciting and active research area and likely represent an important set of genetic variants, the characterization of which should ultimately be very useful in identifying causal variants, both in the UTRs and beyond.
Nonsense-mediated decay
Another way that polymorphisms in UTRs can influence gene expression is through nonsense-mediated decay (NMD) of mRNA. NMD is a safeguard mechanism that prevents cells from generating deleterious truncated proteins. It degrades abnormal mRNAs that contain a premature termination codon (PTC). NMD can also target normal, non-mutant, transcripts thus regulating gene expression and impacting several physiological processes such as cell differentiation, response to stress, neuronal development, and the onset of various diseases.91 Aberrant splicing, long 3′UTR and uORF are some of the mechanisms implicated in NMD activation.92
An illustrative example with important ramifications is AUF1, which targets mRNAs containing AU-rich elements (AREs) for rapid cytoplasmic turnover. Alternative splicing generates five variants of AUF1 mRNA, which have different 3′UTRs. The generation of alternative 3′UTR can affect AUF1 expression by two mechanisms: AUF1 protein directly binding AUF1 3′UTR splice variants that retain intron 9 (affected by the alternative splicing), and activation of the mRNA NMD pathway. Two of the AUF1 3′UTR variants position the translational termination codon more than 50 nucleotides upstream of an exon-exon junction, creating a potential triggering signal for NMD. Disruption of cellular NMD pathways by gene specific knockdown enhanced the mRNA expression of these two AUF1 isoforms, with stabilization of each transcript. Additionally, quantification of AUF1 mRNA 3′UTR splice variants during murine embryonic development showed that the expression of NMD-sensitive AUF1 mRNAs is specifically enhanced as development proceeds, contributing to dynamic changes in AUF1 3′UTR structures during embryogenesis.93
Using microarray analysis, Kim and colleagues revealed that the level of cyclin-dependent kinase inhibitor 1A (CDKN1A; also known as Waf1/p21) mRNAs increases in cells depleted of cellular NMD factors. Interestingly, p21 mRNA contains an uORF, which is a NMD-inducing feature. Using several approaches, they identified the uORF in CDKN1A mRNA as a negative modulator of translation of the main downstream ORF, thus providing additional biological evidence of the possible role of NMD in diverse biological pathways.94
NMD has been implicated in the onset of several diseases; for example, NMD-induced loss-of-function was shown to contribute to the onset of certain cancers. Hu and colleagues developed an algorithm to predict NMD and applied it to somatic mutations, finding 73,000 mutations that are predicted to elicit NMD and are associated with significant reduction of gene expression in tumour suppressor genes.95 Interestingly, half of the hypermutated stomach adenocarcinomas are characterized by NMD-eliciting mutations in two genes implicated in translation initiation (LARP4B and EIF5B). Together these results underline the key role of NMD in human pathophysiology.
WHAT IS KNOWN FROM GWAS STUDIES
GWAS data are increasing rapidly, and thus the scientific community needs to develop appropriate tools to manage systematically the large amount of information available. With this goal in mind, several databases collecting GWAS data related to diseases and quantitative parameters have been developed.
For example, the NHGRI GWAS Catalog,12 is a quality-controlled, manually curated collection of all those published genome-wide association studies which assay at least 100,000 SNPs. The NHGRI GWAS Catalog contains all SNP-trait associations with p-values < 1.0 × 10−5.11
With the aim to evaluate the distribution and impact of variants at UTRs, we extracted data from a recent version of this catalog (www.ebi.ac.uk/gwas; accessed 2017-09-19, version v1.0).
As all the reported variants in the GWAS Catalog are annotated based on their genetic position, we estimated that the 3.7% of the 57,671 variants included in this dataset are localised in the UTRs. In particular, 1,652 of them map in the 3′UTR, representing 2.9% of the total variants, while 442 are in the 5′UTR, reaching the 0.8% (table 1).
Table 1. Summary of GWAS associations in reported in the UTRs.
The table reports, from left to right: the number of variants included in the GWAS Catalog (accession date: 2017-09-19, version 1.0), the significance threshold (p value) considered, the number of variants in the UTRs, and the number of variants in the 5′UTR and 3′UTR. In parentheses are the percentages with respect to variants mapping in the UTRs and percentages with respect to variants present in the entire genome (indicated as “total”) are specified.
| Total N | Significance threshold | N in UTR | N in 5′UTR (% to the UTR; % to the total) |
N in 3′UTR (% to the UTR; % to the total) |
|---|---|---|---|---|
| 57,671 | p value<9×10−6 | 2,094 | 442 (21.1; 0.8) | 1,652 (78.9; 2.9) |
| p value<5×10−8 | 1,362 | 321 (23.6; 0.6) | 1,041 (76.4; 1.8) |
Thus, almost 80% of the UTR associated variants reported in the literature are localised in the 3′UTR region. However, as the 3′UTRs are, on average, much longer than 5′UTRs, we adjusted the number of identified variants per the UTRs length, observing a comparable number of associated variants in both UTRs (table 2). These estimations were obtained by exploiting genomic data from the Ensembl database (http://grch37.ensembl.org/biomart/, database: Ensembl Genes 91, dataset: Human Genes - GRCh37.p13).
Table 2. Genomic distribution of UTRs associations.
The table reports, from left to right: the UTR type, the number of variants included in the GWAS Catalog and localised in the UTRs, the average length of the 5′UTR and 3′UTR expressed in bp and, in parentheses, the number of genes used in the calculation, the genomic length calculated as the product of the average length for the number of genes considered, and the number of associated variants per each kb of UTRs’ length. The average length has been calculated using all genes associated with an Ensembl Identifier and having an UTR.
| Region type | N GWAS variants in the region | Average length (N genes) | Genomic length | N variants per kb |
|---|---|---|---|---|
| 5′UTR | 442 | 151.5 bp (18,714) | 2,835 kb | 0.16 |
| 3′UTR | 1,652 | 872.2 bp (18,770) | 16,371 kb | 0.10 |
Moreover, taking advantage of this catalog, we searched for variants mapping the UTRs that affect disease risk and/or quantitative parameters. For instance, among diseases, genetic variants at UTRs are more frequently associated with immunological, neoplastic and neurological pathologies (figure 1, panel A). Similarly, among the quantitative traits, we observed that the large part of known UTR associations were involved in immune-related, haematological, anatomical parameters and metabolic traits, while a small number of variants affect inflammatory and cardiological phenotypes (figure 1, panel B).
Figure 1. Distribution of UTR variants among associated diseases and quantitative traits.

Representation of UTR variants distributed among associated diseases and quantitative traits, as reported in GWAS Catalog. Variants are considered at 5′UTR and 3′UTR jointly (indicated as “total”), and separately. Diseases are categorized in seven non-overlapping classes (panel A), while quantitative traits in nine categories (panel B). In each panel, the percentage of variants associated with phenotypes in the defined categories is reported.
Furthermore, we selected the strongest and most common associations mapping in the untranslated regions; in particular, we extracted 15 disease-related and 35 trait-related SNPs, whose p-values represent the top 5% percentile for each category, considering a p-value threshold lower than 5×10−8 and a minor allelic frequency - MAF > 5% (table 3 and table 4).
Table 3. Examples of genetic variants in UTRs reported in the GWAS Catalog and associated with diseases.
The table reports, from left to right: the UTR region where each variant is located; the affected disease; the gene where the variant is predicted to map; the disease-associated polymorphism (SNP); the risk allele frequency; its statistical significance, expressed as p value; the impact of the associated variant on disease, expressed as odds ratio; and the Pubmed ID of the paper where the result has been reported, as indicated in GWAS Catalog.
| UTR region | Disease | Mapped gene | SNP | Risk allele frequency | p value | Odds ratio | Pubmed ID |
|---|---|---|---|---|---|---|---|
| 5′UTR | Duodenal ulcer | PSCA | rs2294008 | 0.37 | 2.E-33 | 1.84 | 22387998 |
| Primary biliary cholangitis | IL21R | rs2189521 | 0.70 | 4.E-16 | 1.41 | 28425483 | |
| Chronic hepatitis B infection | CD40 | rs1883832 | 0.37 | 3.E-15 | 1.19 | 25802187 | |
|
| |||||||
| 3′UTR | Testicular germ cell tumor | KITLG | rs995030 | 0.80 | 2.E-50 | 2.26 | 23666240 |
| Venous thromboembolism | F5 | rs6427196 | 0.93 | 4.E-51 | 2.07 | 23650146 | |
| Dementia and core Alzheimer’s disease neuropathologic changes | PVRL2 | rs6857 | 0.29 | 2.E-62 | 1.61 | 25188341 | |
| Chronic hepatitis B infection | HLA-DPB1 | rs9277535 | 0.58 | 1.E-70 | 1.52 | 25802187 | |
| Chronic lymphocytic leukemia | IRF4 | rs9391997 | 0.49 | 9.E-22 | 1.35 | 26956414 | |
| Atrial fibrillation | TBX5 | rs883079 | 0.42 | 5.E-15 | 1.18 | 28416822 | |
| Type 2 diabetes | LOC105375716, SLC30A8 | rs3802177 | 0.70 | 2.E-18 | 1.16 | 24509480 | |
| Coronary heart disease | ZPR1 | rs964184 | 0.13 | 1.E-17 | 1.13 | 21378990 | |
| Epithelial ovarian cancer | HOXD3 | rs711830 | 0.32 | 3.E-15 | 1.12 | 28346442 | |
| Myocardial infarction | CELSR2 | rs7528419 | 0.80 | 1.E-15 | 1.11 | 26343387 | |
| Breast cancer | SLC4A7 | rs4973768 | 0.47 | 2.E-30 | 1.10 | 23535729 | |
| Inflammatory bowel disease | FEN1 | rs4246215 | 0.34 | 2.E-15 | 1.08 | 23128233 | |
Table 4. Examples of genetic variants in UTRs reported in GWAS Catalog and associated with quantitative parameters.
The table reports, from left to right: the UTR region where each variant is located, the associated quantitative trait, the gene where the variant is predicted to map, the associated polymorphism (SNP), the frequency of the tested allele, its statistical significance (p value), the impact of the associated variant on the trait, and the Pubmed ID of the paper where the result has been reported, as indicated in GWAS Catalog.
| UTR region | Quantitative trait | Mapped gene | SNP | Tested allele frequency | P-value | Effect of the tested allele | PubmedID |
|---|---|---|---|---|---|---|---|
| 5′UTR | HDL cholesterol levels | APOA5 | rs651821 | 0.72 | 9.E-47 | 0.19 | 28334899 |
| Complement C3 and C4 levels | GTF2H4 | rs1052693 | 0.36 | 3.E-48 | 0.10 | 23028341 | |
| Mean platelet volume | ODF3 | rs11604127 | 0.24 | 7.E-163 | 0.12 | 27863252 | |
| Acylcarnitine levels | SLC16A9 | rs1171614 | 0.20 | 2.E-81 | 0.10 | 26068415 | |
| Platelet count | ODF3 | rs11604127 | 0.24 | 4.E-103 | 0.09 | 27863252 | |
| Height | CNPY2 | rs3809128 | 0.21 | 7.E-35 | 0.08 | 25429064 | |
| Monocyte percentage of white cells | LTBR | rs10849448 | 0.76 | 4.E-46 | 0.06 | 27863252 | |
| Hip circumference adjusted for BMI | GDF5 | rs143384 | 0.57 | 1.E-31 | 0.04 | 25673412 | |
| Mean corpuscular hemoglobin | TBX6 | rs3809627 | 0.40 | 5.E-33 | 0.04 | 27863252 | |
| Serum ferritin levels | PMS1 | rs5742933 | 0.22 | 2.E-10 | 0.11 | 25162662 | |
|
| |||||||
| 3′UTR | Triglycerides | ZPR1 | rs964184 | 0.13 | 7.E-240 | 16.95 | 20686565 |
| Soluble ICAM-1 | ICAM1 | rs281437 | 0.30 | 3.E-10 | 10.10 | 18604267 | |
| Platelet count | SH2B3 | rs739496 | 0.11 | 7.E-12 | 8.25 | 25705162 | |
| Caffeine metabolism (plasma 1,7-dimethylxanthine (paraxanthine) to 1,3,7-trimethylxanthine (caffeine) ratio) | AHR | rs11400459 | 0.64 | 5.E-10 | 6.23 | 27702941 | |
| LDL cholesterol | CELSR2 | rs629301 | 0.22 | 1.E-170 | 5.65 | 20686565 | |
| Cholesterol, total | CELSR2 | rs629301 | 0.22 | 6.E-131 | 5.41 | 20686565 | |
| Age-related macular degeneration | NELFE | rs522162 | 0.93 | 2.E-10 | 2.33 | 23577725 | |
| Menarche (age at onset) | TRIM66 | rs4929923 | 0.36 | 1.E-08 | 2.30 | 21102462 | |
| Hypertriglyceridemia | ZPR1 | rs964184 | 0.30 | 5.E-35 | 1.77 | 23505323 | |
| Alcohol consumption (drinkers vs non-drinkers) | OAS3 | rs2072134 | 0.16 | 3.E-16 | 1.58 | 28485404 | |
| Blood protein levels | LEPR | rs17415296 | 0.19 | 4.E-229 | 1.40 | 28240269 | |
| Allergic sensitization | STAT6 | rs1059513 | 0.90 | 1.E-14 | 1.30 | 23817571 | |
| Obesity | KCNMA1 | rs2116830 | 0.80 | 3.E-10 | 1.26 | 21708048 | |
| Allergic sensitization | IL1RL1 | rs3771175 | 0.86 | 5.E-11 | 1.20 | 23817571 | |
| QT interval | LIG3 | rs1052536 | 0.53 | 6.E-25 | 0.98 | 24952745 | |
| End-stage coagulation | MCF2L | rs10665 | 0.88 | 2.E-47 | 0.85 | 23381943 | |
| Cholesterol, total | HMGCR | rs12916 | 0.40 | 5.E-74 | 0.68 | 24097068 | |
| Resting heart rate | KIAA1755 | rs6123471 | 0.46 | 7.E-72 | 0.60 | 27798624 | |
| Plasma omega-6 polyunsaturated fatty acid levels (gamma-linolenic acid) | FADS1 | rs174546 | 0.66 | 5.E-171 | 0.52 | 26584805 | |
| Urinary metabolites (H-NMR features) | ACADS | rs3916 | 0.28 | 2.E-22 | 0.40 | 24586186 | |
| HDL Cholesterol - Triglycerides (HDLC-TG) | APOA5 | rs2266788 | 0.09 | 5.E-13 | 0.39 | 21386085 | |
| Red blood cell fatty acid levels | FADS1 | rs174545 | 0.33 | 8.E-90 | 0.37 | 25500335 | |
| Metabolic syndrome | APOA5 | rs2266788 | 0.09 | 2.E-09 | 0.26 | 21386085 | |
| Metabolite levels (lipoprotein measures) | ZPR1 | rs964184 | 0.86 | 8.E-66 | 0.24 | 27005778 | |
| IgE levels | STAT6 | rs1059513 | 0.11 | 2.E-12 | 0.12 | 22075330 | |
| Cognitive function | TOMM40 | rs10119 | 0.29 | 6.E-09 | 0.04 | 25644384 | |
Among the SNPs associated with diseases (table 3), we cite some examples mapping in 5′UTRs, and showing a moderate impact (expressed as odds ratio) in cancer or autoimmune pathologies. For example, Tanikawa and colleagues found the SNP rs2294008 to be associated with duodenal ulcer in the PSCA gene, encoding prostate stem cell antigen. This gene is a good candidate, being highly expressed in several tissues, such as bladder, placenta, colon, kidney, and stomach, and also being detected in pancreatic and bladder cancers. The SNP risk allele for the disease encodes a translation initiation codon upstream of the reported site, thus changing protein localization from the cytoplasm to the cell surface.96
Another interesting variant that recently emerged from GWAS is rs2189521, associated with primary biliary cholangitis (PBC) in the IL21 receptor gene (IL21R).97 Qiu and colleagues reported that the risk allele for PBC regulates differential IL21R expression; this variant is also highly correlated with multiple SNPs in the IL21R region, suggesting that variation in IL21R expression may explain this signal. By applying several histochemical experiments, they showed that the enhanced expression in PBC livers (in the hepatic portal tracks) of IL21R and of its ligand, IL21, support an involvement of IL21 signalling pathway deregulation in the disease mechanism.
Likewise, among SNPs mapping in the 3′UTR, one of the strongest associations is the SNP rs6427196, localized in the coagulation factor V gene (F5) and found associated, in a large meta-analysis, with venous thromboembolism (odds ratio = 2.07, p-value = 4×10−51).98
Another variant with a strong effect (odd ratio = 2.26, p-value = 2×10−50) is rs995030, associated with testicular germ cell tumour in KITLG, encoding the ligand for the receptor tyrosine kinase KIT.99 The KIT–KITLG system regulates the survival, proliferation and migration of germ cells, and mutations in this gene confer an increased tumour risk in a mouse model of the disease. Although the gene may explain the association, no correlation has been found so far between rs995030 and variation in KITLG expression.100
Among the strongest association of genetic variants in UTRs with quantitative traits (table 4), the following two examples are particularly informative.
Variants in the apolipoprotein A5 (APOA5) gene are associated with lipids levels, mainly HDL cholesterol, and with related dysfunctions including the metabolic syndrome. Several of these variants map in UTRs, such as the SNP rs651821 localized in the 5′UTR region.101 By searching this SNP in a recently published catalog of gene expression data (https://www.gtexportal.org/home/),102 we observed that it is an eQTL (in the adipose – subcutaneous specific tissue) for a long non coding RNA gene (RP11-109L13.1) located 400 kb downstream the APOA5 gene. The effective implication of this gene in lipid modulation is suggested by a second variant mapping in the 3′UTR of the gene and showing pleiotropic effects on triglycerides and HDL-C levels: the SNP rs2266788.103 Interestingly, in a more recent work, Caussy and colleagues showed that the less frequent allele of rs2266788, belonging to APOA5 haplotype 2 (APOA5*2), reduces APOA5 expression at the post-transcriptional level by creating a functional target site for miRNA485-5p, mainly expressed in the liver. Therefore, the increased level of triglycerides in the presence of APOA5*2 could be caused by the APOA5 downregulation mediated by miRNA485-5p.104
After genetic variants associated with diseases or parameters have been identified, it is important to establish their specific gene localization and, consequently, their functional effects. To this end, several tools, such as the Ensembl Variant Effect Predictor (VEP),105 are now available. However, since the localization of a variant may be different in different isoforms of the same gene, the variant can be predicted to map in UTRs (in one isoform) as well as in introns, in non-sense mediated decay (NMD) or non coding transcripts, in regulatory regions, in transcription factors (TF) binding sites or even outside the gene (in alternative isoforms) (figure 2). This introduces an extra layer of complexity in genetic data interpretation.
Figure 2. Alternative annotations of 5′UTR variants.

The pie graph summarizes the 5′UTR variants reported in the GWAS Catalog and shows their alternative predicted localization due to the presence of gene isoforms.
An important tool to estimate the detrimental effect of a variant is represented by the Combined Annotation Dependent Depletion score (C-score).106 This score combines different information, such as the variant consequence on DNA gene sequence, its impact on expression, acetylation, and methylation, and the conservation score of the region, in a single metric. The higher the C-score, the more deleterious is the variant; to identify potentially pathogenic variants, Kircher and colleagues suggested using a cut-off value between 10 and 20. In this way, the C-scores give important information about the different allelic impact of a variant, its functional role and pathogenicity; also allowing to rank causal variants in a genome sequence. We calculated the C-score for UTR variants in the GWAS Catalog data and obtained values ranging from 0.001 to 22.1, with a relatively low mean value (mean=5.89), indicating that variants in UTRs are mainly benign. We then prioritized only those variants unequivocally mapping in UTRs from VEP annotation (table 5). Among the most deleterious variant, rs1128334 (C-score = 16.2) was notable in that it was associated with systemic lupus erythematosus risk and was located in the 3′UTR of the ETS1 gene. ETS1 encodes the transcription factor C-ets-1, involved in a wide range of immune functions, including Th17 cell development and terminal differentiation of B lymphocytes. When evaluating allelic expression in peripheral blood mononuclear cells, the risk allele showed lower ETS1 expression levels.107
Table 5. C-score values for UTR-associated variants with diseases and related traits.
The table reports, from left to right: the UTR region where each selected variant is located; the rsID of the polymorphism (SNP); the associated quantitative trait or disease; the gene where the variant is predicted to map; the statistical significance of the variant, expressed as p value; its impact on the trait/disease; the C-score; and the Pubmed ID of the paper where the result has been reported, as indicated in GWAS Catalog.
| UTR region | SNP | Trait/disease | Mapped gene | p-value | Effect of the tested allele | C-score | PubmedID |
|---|---|---|---|---|---|---|---|
| 5′UTR | rs2236293 | Blood protein levels | TMEM8B | 5.0E-14 | 0.33 | 17.96 | 28240269 |
| rs149698681 | Granulocyte percentage of myeloid white cells | CAPN3 | 2.0E-10 | 0.08 | 13.83 | 27863252 | |
|
| |||||||
| 3′UTR | rs78378222 | Mean corpuscular hemoglobin | TP53 | 6.0E-09 | 0.10 | 17.97 | 27863252 |
| rs45474992 | Monocyte count | BBC3 | 3.0E-09 | 0.06 | 17.19 | 27863252 | |
| rs1128334 | Systemic lupus erythematosus | ETS1 | 7.0E-12 | 1.39 | 16.20 | 26663301 | |
| rs2297991 | Cholesterol, total | GPAM | 8.0E-10 | 0.04 | 15.46 | 25961943 | |
| rs11553699 | Reticulocyte fraction of red cells | TMEM120B, RHOF | 3.0E-09 | 0.03 | 14.74 | 27863252 | |
| rs6796 | Mean platelet volume | KDELR2 | 5.0E-15 | 0.03 | 14.21 | 27863252 | |
| rs16850073 | Monocyte percentage of white cells | CXCL6 | 5.0E-13 | 0.03 | 13.39 | 27863252 | |
| rs4233366 | Asthma | PPOX - ADAMTS4 | 5.0E-15 | 1.09 | 12.63 | 27182965 | |
An example of 3′UTR genetic regulation predisposing to autoimmunity
Since the early 1900s, it was postulated that a qualitative condition such as the presence/absence of a given pathology could be caused by multiple quantitative traits, each of which is influenced by a number of genetic variants.108 This model fits particularly well into the context of complex traits and common diseases in which many variants with small effect are involved in disease predisposition.109 In this context, the study of quantitative trait variation is a valuable approach to dissect the predisposition to complex diseases through the analysis of the biomedical parameters in population cohort individuals without the use of case-control strategies that rely on differences between patients vs healthy individuals. The dissection of quantitative trait variation in the general population shows several advantages including the large sample size and the collection of raw data unaffected by the pathology itself or by the drug treatment. This will increase the accuracy and robustness of data and thus will harness the power to detect associated variants in GWAS studies.
The resulting genetic association data of quantitative traits will then be compared with case-control studies for pathologies in order to identify those genetic variants associated with both a quantitative trait and a disease. This approach can reveal disease-related endophenotypes, thus helping on one hand to identify causal variant(s) at a locus and, on the other hand, to elucidate disease etio-pathogenesis.
By applying this approach to autoimmune diseases and immune-related quantitative parameters, we recently identified a genetic variant localized in the 3′UTR of the TNFSF13B gene that increased the stability and translation of the corresponding mRNA, and having a pivotal role in autoimmunity.17
TNFSF13B encodes the protein BAFF (B-cell activating factor), a cytokine primarily produced by monocytes and neutrophils, and involved in the development, survival and differentiation of B cells.110,111 The variant predisposing to autoimmunity results from the combination of a deletion with a polymorphism (GCTGT>A), referred to as BAFF-var. BAFF-var generates an alternative polyadenylation signal, leading to a mixed population of mRNAs characterized by long and short 3′ UTRs, in contrast to the wild-type allele, which produces only a long 3′UTR transcript. We observed that individuals carrying one copy of BAFF-var had a 35% reduction of the long transcript. Notably, the production of the shorter 3′UTR transcript was responsible for an increase of the RNA stability and translation due to the absence of a miRNA binding site for miR-15a, resulting in a rise of soluble BAFF. However, the increase in the mRNA expression explained only about 24 to 27% of the higher amount of soluble BAFF, indicating that an increase in translation level was also probably involved. The strong increase in the serum concentration of the soluble BAFF protein (about 19% per BAFF-var allele) led to other important downstream events closely linked to BAFF function, such as a rise of the number of circulating B cells and immunoglobulins, mainly IgG, IgA and IgM. Additionally, we observed a reduction of circulating monocytes, a phenomenon probably due to a negative feedback mechanism to compensate the augmented production of soluble BAFF in these cells. Overall, the immune system dysregulation caused by BAFF-var led to increased risk of multiple sclerosis and systemic lupus erythematosus (figure 3).
Figure 3. BAFF-var effects at the transcription, protein and cellular level.

Representation of the localization of BAFF-var within TNFSF13B gene and its effects on the generation of mRNAs with different 3′UTR lengths. The number and location of microRNA sites are reported. BAFF-var creates an alternative polyadenylation signal that generates a shorter 3′UTR transcript lacking a miRNA binding site. In contrast to the wild type allele which produces only with long 3′UTR, BAFF-var leads to a mixed population of mRNAs with long and short 3′UTRs, resulting in higher production of sBAFF. In turn, the increased sBAFF levels lead to higher numbers of B cells and immunoglobulins, reduced levels of monocytes, and increased risk for autoimmunity.
Interestingly, BAFF-var is particularly frequent in Sardinia (26.5%), where the main study was conducted, with decreasing frequency when assaying from Southern (5.7%) to Northern European populations (1.8%). This high allele frequency in Sardinia allowed us to identify the association with multiple sclerosis at a genome-wide significance level using a relatively small sample set of about 3,000 patients and a similar number of controls.
A possible explanation of the high frequency of BAFF-var is its positive selection due to a selective pressure acting in Sardinia and, in general, in Southern Europe. The most plausible candidate is malaria infection, as suggested by the correlation between the frequency of this variant vs malaria prevalence across Europe before its eradication (~1950).112 Additionally, BAFF transgenic mice survived lethal Plasmodium yoelii malaria, and there is evidence that the malaria parasite can prevent long-term immunity dysregulating the dendritic cells producing BAFF.113
This is a classic example of hygiene hypothesis, in which a genetic variant for a long time positively selected because protective for an infectious disease, predisposes for autoimmunity once the incidence of infection is strongly reduced.114 This is because, while the environmental factors can change very fast, on the contrary the selective pressure needs many generations to be nullified.
Conclusions and perspectives
Genome-wide association studies (GWAS) have uncovered new areas of investigation into the association between different diseases/traits and a large number of genetic loci. Since 2005,115 GWAS have revolutionized the study of complex traits, yielding to-date more than 24,218 unique SNP-trait associations from 2,518 publications. This somehow implies that, quite frequently, genotype and phenotype are linked, more generally, the mechanisms by which each gene affects the disorders remain largely unknown, and often are not discussed within the reports. It requires the support of functional indications and tools for downstream statistical and bioinformatic analyses.
Toward this aim, the first test to perform after finding a genetic variant associated with a phenotype is to assess the variant activity predictions, using the already mentioned C-score and VEP analysis tool, and others such as PredictSNP2 (https://loschmidt.chemi.muni.cz/predictsnp2/)116. They are able to visualize, annotate and prioritize such data to guide the analyst toward a more focussed work hypothesis. However, if they do not make concrete predictions about the role of the polymorphisms, there are many additional databases to consult. For example, if a polymorphism changes the DNA sequence necessary for the binding of a transcription factor (TF), this can be tested by searching in TF databases,117 but it gives a probabilistic answer that will likely require further tests.
Moreover, a polymorphism does not necessarily regulate the nearest gene. For instance, a systematic study of complex phenotypes and associations in the GWAS Catalog found evidence that affected genes are often up to 2 MB from the associated SNP.118 To address this issue, a number of techniques have been developed, usually variations on the chromosome capture technique,119 which allows the isolation and identification of chromosome sequences that interact with one another. While the data are dependent on the tissue in which the experiment was performed, the state of the art is quite advanced now, and a browser has been constructed to allow visualization of this interacting regions information (http://promoter.bx.psu.edu/hi-c/).120
Another issue to consider is that the spectrum of RNA products of RNA polymerase II has expanded considerably over the last years. It now includes numerous non-coding RNA products, including some that function as enhancers (enhancer RNAs) and thus influence transcription.121 Another important milestone that can aid researchers to correlate genomic data with disease and other phenotypic traits, for example by identifying elements responsible for tissue-specific expression, was the ENCODE project (https://www.encodeproject.org/). Because of the great importance of these expression elements, EnhancerAtlas has been assembled.122 This atlas provides expression information from 105 Human cell lines or tissues, using eight different measures of enhancer activity and DNA accessibility to RNA polymerase II. Consulting these diverse sources may allow more specific hypotheses to be formulated, and ultimately tested. When these hypotheses are validated, they may offer more specific information: cell of interest, enhancer or TF of interest, even revealing non-proximal genes that are actually mediating the phenotype. So, in the end, the route may be longer, but the destination more satisfying, when reached.
Other useful bioinformatic tools, although still in an early phase of verification, are represented by several algorithms developed to test for riboSNitch activity in transcripts and have had some success.79,84,86
The lack of an immediate answer when a trait modifying genetic association is detected may first be disheartening, but it is likely just a consequence of the under-appreciated role of non-protein based mechanisms as phenotypic mediators. The spectrum of RNA species and especially RNA polymerase II products is widening.123 However, it should be kept in mind that as exploration of these different elements and mechanisms, and their cellular roles proceeds, explaining genetic associations with complex traits could provide even more precise indications of disease mechanisms and points of possible intervention than mere amino acid changes. Ultimately, these discoveries will set the stage for more precise and effective interventions.
Table 6. Summary of results described in this overview.
The table reports, from left to right: the affected gene and its trans regulator (if any); the UTR region where the studied genetic variant is localized; the associated disease; the reference where the mechanism reported has been described.
| Affected gene_trans regulator | UTR region | Associated disease | Reference |
|---|---|---|---|
| uORF | |||
| SPINK1 | 5′UTR | Hereditary pancreatitis | Calvo, 2009 |
| HR | 5′UTR | Marie Unna hereditary hypotrichosis | Wen, 2009 |
| HT3A | 5′UTR | Bipolar disorder | Niesler, 2001 |
| IRES | |||
| c-MYC_ITAFs and YB-1 | 5′UTR | Multiple myeloma | Cobbold, 2010 |
| connexin-32 | 5′UTR | Charcot-Marie-Tooth disease | Hudder, 2000 |
| RNA-binding protein | |||
| TPH2_unknown | 5′UTR | Behavioural traits and psychiatric disorders | Chen GL, 2008 |
| FMR1_TDP-43 | 5′UTR | Fragile X-associated tremor/ataxia syndrome | He, 2014 |
| 5′UTR deletion | |||
| ATP7B | 5′UTR | Wilson disease | Cullen 2003 |
| CCG repeat | |||
| FMR1 | 5′UTR | Fragile X syndrome | Yifan Zhou, 2016 |
| Kozak sequence | |||
| β-globin | 5′UTR | β-thalassaemia | Angioletti, 2004 |
| 3′UTR length and polyadenylation signal | |||
| HTT | 3′UTR | Huntington disease | Romo, 2017 |
| FOXP3 | 3′UTR | Polyendocrinopathy, enteropathy, X-linked | Bennet 2001 |
| α and β globin | 3′UTR | Thalassemia | Orkin 1985; Rund 1992; Higgs 1983 |
| TNFSF13B | 3′UTR | Multiple sclerosis, Systemic lupus erythematous | Steri, 2017 |
| MicroRNA | |||
| IZKF3_miR 326 | 3′UTR | Autoimmune diseases | Richardson, 2011 |
| PVRL_miR 320 | 3′UTR | Alzheimer Disease | Ghanbari, 2016 |
| KRAS_miR let-7a | 3′UTR | Metastasis in osteosarcoma | Zhang, 2017 |
| APOA5_miR-485-5p | 3′UTR | Metabolic syndrome | Caussy et al, 2014 |
| TNFSF13B_miR 15a | 3′UTR | Multiple sclerosis | Steri, 2017 |
| RNA-binding protein | |||
| TNFα_HuR | 3′UTR | Host defence | Di Marco, 2001 |
| PPP1R3_unknown | 3′UTR | Type 2 diabetes | Xia, 1999 |
| TS_AUF1 | 3′UTR | Rheumatoid arthritis and several malignancies | Pullmann, 2006 |
| Alternative translation initiation codon | |||
| PSCA | 5′UTR | Duodenal ulcer | Tanikawa, 2012 |
| Unknown mechanism | |||
| APOA5 | 5′UTR | Metabolic syndrome | Zhou, 2013 |
| ETS1 | 3′UTR | Systemic lupus erythematosus | Yang, 2010 |
| F5 | 3′UTR | Venous thromboembolism | Tang et al, 2013 |
| IL21R | 5′UTR | Primary biliary cholangitis | Qiu, 2017 |
| KITLG | 3′UTR | Testicular germ cell tumor | Ruark, 2013 |
Sidebar title: Key concepts.
Allele: each form of a gene present in a specific chromosomal position. A genetic variant is characterised by at least two alleles, thus two alternative forms of it.
Allelic frequency: the number of times a specific allele is observed in a population divided by the total number of copies of all the alleles at that particular genetic locus in the same population.
Quantitative trait: is any measurable phenotype, such as height, weight and the level of cells in the blood, which have a continuous distribution. Quantitative traits are generally regulated by several genetic variants and by environmental factors, thus they can be considered as complex traits.
Endophenotype: any quantitative trait with a clear genetic connection with a disease condition.
GWAS: is a genetic study that assesses the association of genetic variants localized in the entire genome with a phenotype. For GWAS, the significance threshold, expressed as the p value of the statistical test used, is generally 5×10−8, which corresponds to the nominal threshold of 0.05 corrected for the number of independent genetic variants assessed (estimated to be at least 106), thus the higher the number of genetic variants interrogated, the lower the p value threshold.
Pleiotropy: phenomenon by which a gene can influence two or more phenotypes.
Expression quantitative trait locus (eQTL): is a genetic variant that contributes to the variation of the mRNA level of one or more genes.
Long non coding RNA (lncRNA): transcript longer than 200 nucleotides that is not translated into protein and can regulate gene transcription and translation.
Linkage Disequilibrium (LD): non random association between alleles at different loci. If two or more alleles at each locus are frequently present together, it can render the task of finding the allele responsible of the association, the “causal” allele, difficult, or impossible.
Haplotype block: a set of closely linked DNA alleles on one chromosome that are often inherited together.
RNA folding landscape: the set of folding conformations that a single RNA transcript may assume.
Acknowledgments
We thank Francesco Cucca for supporting the entire work, Edoardo Fiorillo and Valentina Serra for helpful comments and suggestions on the manuscript. We acknowledge funding from the Intramural Research Program of the NIH, National Institute on Aging, contracts number N01-AG-1-2109 and HHSN271201100005C, and from the Horizon 2020 Research and Innovation Program of the European Union (grant 633964).
Footnotes
Authors declare no conflict of interest.
Contributor Information
Maristella Steri, Istituto di Ricerca Genetica e Biomedica, Consiglio Nazionale delle Ricerche (CNR), Monserrato, Cagliari, Italy.
M. Laura Idda, Laboratory of Genetics and Genomics, National Institute on Aging, National Institute of Health, Baltimore, MD.
Michael B. Whalen, Istituto di Biofisica, Consiglio Nazionale delle Ricerche (CNR), Povo, Trento, Italy
Valeria Orrù, Istituto di Ricerca Genetica e Biomedica, Consiglio Nazionale delle Ricerche (CNR), Monserrato, Cagliari, Italy.
References
- 1.Sampson JN, Jacobs K, Wang Z, Yeager M, Chanock S, Chatterjee N. A two-platform design for next generation genome-wide association studies. Genet Epidemiol. 2012 May;36(4):400–8. doi: 10.1002/gepi.21634. [DOI] [PubMed] [Google Scholar]
- 2.Voight BF, Kang HM, Ding J, Palmer CD, Sidore C, Chines PS, Burtt NP, Fuchsberger C, Li Y, Erdmann J, et al. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8(8):e1002793. doi: 10.1371/journal.pgen.1002793. doi: 10.1371/journal.pgen.1002793. Epub 2012 Aug 2. Erratum in: PLoS Genet. 2013 Apr;9(4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis Res Ther. 2011 Feb 1;13(1):101. doi: 10.1186/ar3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Telenti A, Pierce LC, Biggs WH, di Iulio J, Wong EH, Fabani MM, Kirkness EF, Moustafa A, Shah N, Xie C, et al. Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci U S A. 2016 Oct 18;113(42):11901–11906. doi: 10.1073/pnas.1613365113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016 Oct;48(10):1279–83. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sidore C, Busonero F, Maschio A, Porcu E, Naitza S, Zoledziewska M, Mulas A, Pistis G, Steri M, Danjou F, et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet. 2015 Nov;47(11):1272–1281. doi: 10.1038/ng.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.1000 Genomes Project Consortium. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015 Oct 1;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slatkin M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 2008 Jun;9(6):477–85. doi: 10.1038/nrg2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wellcome Trust Case Control Consortium; Australo-Anglo-American Spondylitis Consortium (TASC) Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007 Nov;39(11):1329–37. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids Research. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Condit CM, Achter PJ, Lauer I, Sefcovic E. The changing meanings of “mutation:” A contextualized study of public discourse. Hum Mutat. 2002 Jan;19(1):69–75. doi: 10.1002/humu.10023. [DOI] [PubMed] [Google Scholar]
- 14.Brookes AJ. The essence of SNPs. Gene. 1999 Jul 8;234(2):177–86. doi: 10.1016/s0378-1119(99)00219-x. [DOI] [PubMed] [Google Scholar]
- 15.Jansen RP. mRNA localization: message on the move. Nat Rev Mol Cell Biol. 2001 Apr;2(4):247–56. doi: 10.1038/35067016. [DOI] [PubMed] [Google Scholar]
- 16.Mignone F, Gissi C, Liuni S, Pesole G. Untranslated regions of mRNAs. Genome Biol. 2002;3(3):REVIEWS0004. doi: 10.1186/gb-2002-3-3-reviews0004. Epub 2002 Feb 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steri M, Orrù V, Idda ML, Pitzalis M, Pala M, Zara I, Sidore C, Faà V, Floris M, Deiana M, et al. Overexpression of the Cytokine BAFF and Autoimmunity Risk. N Engl J Med. 2017 Apr 27;376(17):1615–1626. doi: 10.1056/NEJMoa1610528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cenik C, Derti A, Mellor JC, Berriz GF, Roth FP. Genome-wide functional analysis of human 5′ untranslated region introns. Genome Biol. 2010;11(3):R29. doi: 10.1186/gb-2010-11-3-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Angioletti M, Lacerra G, Sabato V, Carestia C. Beta+45 G --> C: a novel silent beta-thalassaemia mutation, the first in the Kozak sequence. Br J Haematol. 2004 Jan;124(2):224–31. doi: 10.1046/j.1365-2141.2003.04754.x. [DOI] [PubMed] [Google Scholar]
- 20.Pichon X, Wilson LA, Stoneley M, Bastide A, King HA, Somers J, Willis AE. RNA binding protein/RNA element interactions and the control of translation. Curr Protein Pept Sci. 2012 Jun;13(4):294–304. doi: 10.2174/138920312801619475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araujo PR, Yoon K, Ko D, Smith AD, Qiao M, Suresh U, Burns SC, Penalva LO. Before It Gets Started: Regulating Translation at the 5′ UTR. Comp Funct Genomics. 2012;2012:475731. doi: 10.1155/2012/475731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005 Sep 2;309(5740):1559–63. doi: 10.1126/science.1112014. Erratum in: Science. 2006 Mar 24;311(5768):1713. [DOI] [PubMed] [Google Scholar]
- 23.Cullen LM, Prat L, Cox DW. Genetic variation in the promoter and 5′ UTR of the copper transporter, ATP7B, in patients with Wilson disease. Clin Genet. 2003 Nov;64(5):429–32. doi: 10.1034/j.1399-0004.2003.00160.x. [DOI] [PubMed] [Google Scholar]
- 24.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013 Jan;38(1):23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Y, Kumari D, Sciascia N, Usdin K. CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol Autism. 2016 Oct 6;7:42. doi: 10.1186/s13229-016-0105-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol. 1987 Aug 20;196(4):947–50. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- 27.Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci U S A. 2009 May 5;106(18):7507–12. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barbosa C, Peixeiro I, Romão L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013;9(8):e1003529. doi: 10.1371/journal.pgen.1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wethmar K, Smink JJ, Leutz A. Upstream open reading frames: molecular switches in (patho)physiology. Bioessays. 2010 Oct;32(10):885–93. doi: 10.1002/bies.201000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U, Landt O, Becker M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000 Jun;25(2):213–6. doi: 10.1038/76088. [DOI] [PubMed] [Google Scholar]
- 31.Wen Y, Liu Y, Xu Y, Zhao Y, Hua R, Wang K, Sun M, Li Y, Yang S, Zhang XJ, et al. Loss-of-function mutations of an inhibitory upstream ORF in the human hairless transcript cause Marie Unna hereditary hypotrichosis. Nat Genet. 2009 Feb;41(2):228–33. doi: 10.1038/ng.276. [DOI] [PubMed] [Google Scholar]
- 32.Niesler B, Flohr T, Nöthen MM, Fischer C, Rietschel M, Franzek E, Albus M, Propping P, Rappold GA. Association between the 5′ UTR variant C178T of the serotonin receptor gene HTR3A and bipolar affective disorder. Pharmacogenetics. 2001 Aug;11(6):471–5. doi: 10.1097/00008571-200108000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell SF, Walker SE, Algire MA, Park EH, Hinnebusch AG, Lorsch JR. The 5′-7-methylguanosine cap on eukaryotic mRNAs serves both to stimulate canonical translation initiation and to block an alternative pathway. Mol Cell. 2010 Sep 24;39(6):950–62. doi: 10.1016/j.molcel.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988 Jul 28;334(6180):320–5. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- 35.Martínez-Salas E, Piñeiro D, Fernández N. Alternative Mechanisms to Initiate Translation in Eukaryotic RNAs. Comp Funct Genomics. 2012;2012:391546. doi: 10.1155/2012/391546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cobbold LC, Wilson LA, Sawicka K, King HA, Kondrashov AV, Spriggs KA, Bushell M, Willis AE. Upregulated c-myc expression in multiple myeloma by internal ribosome entry results from increased interactions with and expression of PTB-1 and YB-1. Oncogene. 2010 May 13;29(19):2884–91. doi: 10.1038/onc.2010.31. [DOI] [PubMed] [Google Scholar]
- 37.Hudder A, Werner R. Analysis of a Charcot-Marie-Tooth disease mutation reveals an essential internal ribosome entry site element in the connexin-32 gene. J Biol Chem. 2000 Nov 3;275(44):34586–91. doi: 10.1074/jbc.M005199200. [DOI] [PubMed] [Google Scholar]
- 38.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008 Jun 18;582(14):1977–86. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007 Jun;8(6):479–90. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Auweter SD, Oberstrass FC, Allain FH. Sequence-specific binding of single-stranded RNA: is there a code for recognition? Nucleic Acids Res. 2006;34(17):4943–59. doi: 10.1093/nar/gkl620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Serretti A, Liappas I, Mandelli L, Albani D, Forloni G, Malitas P, Piperi C, Politis A, Tzavellas EO, Papadopoulou-Daifoti Z, et al. TPH2 gene variants and anxiety during alcohol detoxification outcome. Psychiatry Res. 2009 May 15;167(1–2):106–14. doi: 10.1016/j.psychres.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 42.Chi S, Teng L, Song JH, Zhou C, Pan WH, Zhao RL, Zhang C. Tryptophan hydroxylase 2 gene polymorphisms and poststroke anxiety disorders. J Affect Disord. 2013 Jan 10;144(1–2):179–82. doi: 10.1016/j.jad.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 43.Chen GL, Vallender EJ, Miller GM. Functional characterization of the human TPH2 5′ regulatory region: untranslated region and polymorphisms modulate gene expression in vitro. Hum Genet. 2008 Jan;122(6):645–57. doi: 10.1007/s00439-007-0443-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He F, Krans A, Freibaum BD, Taylor JP, Todd PK. TDP-43 suppresses CGG repeat-induced neurotoxicity through interactions with HnRNP A2/B1. Hum Mol Genet. 2014 Oct 1;23(19):5036–51. doi: 10.1093/hmg/ddu216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pesole G, Mignone F, Gissi C, Grillo G, Licciulli F, Liuni S. Structural and functional features of eukaryotic mRNA untranslated regions. Gene. 2001 Oct 3;276(1–2):73–81. doi: 10.1016/s0378-1119(01)00674-6. [DOI] [PubMed] [Google Scholar]
- 46.Hong X, Scofield DG, Lynch M. Intron size, abundance, and distribution within untranslated regions of genes. Mol Biol Evol. 2006 Dec;23(12):2392–404. doi: 10.1093/molbev/msl111. Epub 2006 Sep 15. [DOI] [PubMed] [Google Scholar]
- 47.Di Giammartino DC, Nishida K, Manley JL. Mechanisms and consequences of alternative polyadenylation. Mol Cell. 2011 Sep 16;43(6):853–66. doi: 10.1016/j.molcel.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lianoglou S, Garg V, Yang JL, Leslie CS, Mayr C. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 2013 Nov 1;27(21):2380–96. doi: 10.1101/gad.229328.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smibert P, Miura P, Westholm JO, Shenker S, May G, Duff MO, Zhang D, Eads BD, Carlson J, Brown JB, et al. Global patterns of tissue-specific alternative polyadenylation in Drosophila. 2012 Mar 29;1(3):277–89. doi: 10.1016/j.celrep.2012.01.001. Erratum in: Cell Rep, 2013, Mar 28, 3(3), 969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009 Aug 21;138(4):673–84. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rhinn H, Qiang L, Yamashita T, Rhee D, Zolin A, Vanti W, Abeliovich A. Alternative α-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat Commun. 2012;3:1084. doi: 10.1038/ncomms2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Romo L, Ashar-Patel A, Pfister E, Aronin N. Alterations in mRNA 3′ UTR Isoform Abundance Accompany Gene Expression Changes in Human Huntington’s Disease Brains. Cell Rep. 2017 Sep 26;20(13):3057–3070. doi: 10.1016/j.celrep.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett CL, Brunkow ME, Ramsdell F, O’Briant KC, Zhu Q, Fuleihan RL, Shigeoka AO, Ochs HD, Chance PF. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA-->AAUGAA) leads to the IPEX syndrome. Immunogenetics. 2001 Aug;53(6):435–9. doi: 10.1007/s002510100358. [DOI] [PubMed] [Google Scholar]
- 54.Orkin SH, Cheng TC, Antonarakis SE, Kazazian HH., Jr Thalassemia due to a mutation in the cleavage-polyadenylation signal of the human beta-globin gene. EMBO J. 1985 Feb;4(2):453–6. doi: 10.1002/j.1460-2075.1985.tb03650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rund D, Dowling C, Najjar K, Rachmilewitz EA, Kazazian HH, Jr, Oppenheim A. Two mutations in the beta-globin polyadenylylation signal reveal extended transcripts and new RNA polyadenylylation sites. Proc Natl Acad Sci U S A. 1992 May 15;89(10):4324–8. doi: 10.1073/pnas.89.10.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Higgs DR, Goodbourn SE, Lamb J, Clegg JB, Weatherall DJ, Proudfoot NJ. Alpha-thalassaemia caused by a polyadenylation signal mutation. Nature. 1983 Nov 24–30;306(5941):398–400. doi: 10.1038/306398a0. [DOI] [PubMed] [Google Scholar]
- 57.Felekkis K, Touvana E, Stefanou Ch, Deltas C. microRNAs: a newly described class of encoded molecules that play a role in health and disease. Hippokratia. 2010 Oct;14(4):236–40. [PMC free article] [PubMed] [Google Scholar]
- 58.Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004 Dec 15;18(24):3016–27. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009 Feb;10(2):126–39. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 60.Kim YK, Kim B, Kim VN. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc Natl Acad Sci U S A. 2016 Mar 29;113(13):E1881–9. doi: 10.1073/pnas.1602532113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004 Feb;10(2):185–91. doi: 10.1261/rna.5167604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang G. siRNA and miRNA: an insight into RISCs. Trends Biochem Sci. 2005 Feb;30(2):106–14. doi: 10.1016/j.tibs.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 63.Wongfieng W, Jumnainsong A, Chamgramol Y, Sripa B, Leelayuwat C. 5′-UTR and 3′-UTR Regulation of MICB Expression in Human Cancer Cells by Novel microRNAs. Genes (Basel) 2017 Aug 29;8(9) doi: 10.3390/genes8090213. pii: E213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet. 2006 Dec;38(12):1452–6. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- 65.Jin Y, Lee CG. Single Nucleotide Polymorphisms Associated with MicroRNA Regulation. Biomolecules. 2013 Apr 9;3(2):287–302. doi: 10.3390/biom3020287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hariharan M, Scaria V, Brahmachari SK. dbSMR: a novel resource of genome-wide SNPs affecting microRNA mediated regulation. BMC Bioinformatics. 2009 Apr 16;10:108. doi: 10.1186/1471-2105-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu J, Clark AG. Impact of microRNA regulation on variation in human gene expression. Genome Res. 2012 Jul;22(7):1243–54. doi: 10.1101/gr.132514.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Richardson K, Lai CQ, Parnell LD, Lee YC, Ordovas JM. A genome-wide survey for SNPs altering microRNA seed sites identifies functional candidates in GWAS. BMC Genomics. 2011 Oct 13;12:504. doi: 10.1186/1471-2164-12-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghanbari M, Ikram MA, de Looper HW, Hofman A, Erkeland SJ, Franco OH, Dehghan A. Genome-wide identification of microRNA-related variants associated with risk of Alzheimer’s disease. Sci Rep. 2016 Jun 22;6:28387. doi: 10.1038/srep28387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vaishnavi V, Manikandan M, Munirajan AK. Mining the 3′UTR of autism-implicated genes for SNPs perturbing microRNA regulation. Genomics Proteomics Bioinformatics. 2014 Apr;12(2):92–104. doi: 10.1016/j.gpb.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang S, Hou C, Li G, Zhong Y, Zhang J, Guo X, Li B, Bi Z, Shao M. A single nucleotide polymorphism in the 3′-untranslated region of the KRAS gene disrupts the interaction with let-7a and enhances the metastatic potential of osteosarcoma cells. Int J Mol Med. 2016 Sep;38(3):919–26. doi: 10.3892/ijmm.2016.2661. [DOI] [PubMed] [Google Scholar]
- 72.Peng SS, Chen CY, Xu N, Shyu AB. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 1998 Jun 15;17(12):3461–70. doi: 10.1093/emboj/17.12.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gratacós FM, Brewer G. The role of AUF1 in regulated mRNA decay. Wiley Interdiscip Rev RNA. 2010 Nov-Dec;1(3):457–73. doi: 10.1002/wrna.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Di Marco S, Hel Z, Lachance C, Furneaux H, Radzioch D. Polymorphism in the 3′-untranslated region of TNFalpha mRNA impairs binding of the post-transcriptional regulatory protein HuR to TNFalpha mRNA. Nucleic Acids Res. 2001 Feb 15;29(4):863–71. doi: 10.1093/nar/29.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xia J, Bogardus C, Prochazka M. A type 2 diabetes-associated polymorphic ARE motif affecting expression of PPP1R3 is involved in RNA-protein interactions. Mol Genet Metab. 1999 Sep;68(1):48–55. doi: 10.1006/mgme.1999.2884. [DOI] [PubMed] [Google Scholar]
- 76.Pullmann R, Jr, Abdelmohsen K, Lal A, Martindale JL, Ladner RD, Gorospe M. Differential stability of thymidylate synthase 3′-untranslated region polymorphic variants regulated by AUF1. J Biol Chem. 2006 Aug 18;281(33):23456–63. doi: 10.1074/jbc.M600282200. [DOI] [PubMed] [Google Scholar]
- 77.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 78.Scofield DG, Hong X, Lynch M. Position of the final intron in full-length transcripts: determined by NMD? Mol Biol Evol. 2007 Apr;24(4):896–9. doi: 10.1093/molbev/msm010. Epub 2007 Jan 22. [DOI] [PubMed] [Google Scholar]
- 79.Halvorsen M, Martin JS, Broadaway S, Laederach A. Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet. 2010 Aug 19;6(8):e1001074. doi: 10.1371/journal.pgen.1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mironov AS, Gusarov I, Rafikov R, Lopez LE, Shatalin K, Kreneva RA, Perumov DA, Nudler E. Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell. 2002 Nov 27;111(5):747–56. doi: 10.1016/s0092-8674(02)01134-0. [DOI] [PubMed] [Google Scholar]
- 81.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Genetic control by a metabolite binding mRNA. Chem Biol. 2002 Sep;9(9):1043. doi: 10.1016/s1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 82.Ritz J, Martin JS, Laederach A. Evaluating our ability to predict the structural disruption of RNA by SNPs. BMC Genomics. 2012 Jun 18;13( Suppl 4):S6. doi: 10.1186/1471-2164-13-S4-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, Chang HY. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014 Jan 30;505(7485):706–9. doi: 10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bindewald E, Shapiro BA. RNA secondary structure prediction from sequence alignments using a network of k-nearest neighbor classifiers. RNA. 2006 Mar;12(3):342–52. doi: 10.1261/rna.2164906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hofacker IL, Stadler PF. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics. 2006 May 15;22(10):1172–6. doi: 10.1093/bioinformatics/btl023. [DOI] [PubMed] [Google Scholar]
- 86.Smola MJ, Rice GM, Busan S, Siegfried NA, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat Protoc. 2015 Nov;10(11):1643–69. doi: 10.1038/nprot.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martin JS, Halvorsen M, Davis-Neulander L, Ritz J, Gopinath C, Beauregard A, Laederach A. Structural effects of linkage disequilibrium on the transcriptome. RNA. 2012 Jan;18(1):77–87. doi: 10.1261/rna.029900.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kutchko KM, Sanders W, Ziehr B, Phillips G, Solem A, Halvorsen M, Weeks KM, Moorman N, Laederach A. Multiple conformations are a conserved and regulatory feature of the RB1 5′ UTR. RNA. 2015 Jul;21(7):1274–85. doi: 10.1261/rna.049221.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li S, Hua Y, Jin J, Wang H, Du M, Zhu L, Chu H, Zhang Z, Wang M. Association of genetic variants in lncRNA H19 with risk of colorectal cancer in a Chinese population. Oncotarget. 2016 May 3;7(18):25470–7. doi: 10.18632/oncotarget.8330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang Y, Huang HY, Bian GL, Yu YS, Ye WX, Hua F, Chen YH, Shen ZY. A Functional Variant of SMAD4 Enhances Thoracic Aortic Aneurysm and Dissection Risk through Promoting Smooth Muscle Cell Apoptosis and Proteoglycan Degradation. EBioMedicine. 2017 Jul;21:197–205. doi: 10.1016/j.ebiom.2017.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nasif S, Contu L, Mühlemann O. Beyond quality control: The role of nonsense-mediated mRNA decay (NMD) in regulating gene expression. Semin Cell Dev Biol. 2017 Sep 1; doi: 10.1016/j.semcdb.2017.08.053. pii: S1084-9521(17)30342-7. [DOI] [PubMed] [Google Scholar]
- 92.Nickless A, Bailis JM, You Z. Control of gene expression through the nonsense-mediated RNA decay pathway. Cell Biosci. 2017 May 19;7:26. doi: 10.1186/s13578-017-0153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Banihashemi L, Wilson GM, Das N, Brewer G. Upf1/Upf2 regulation of 3′ untranslated region splice variants of AUF1 links nonsense-mediated and A+U-rich element-mediated mRNA decay. Mol Cell Biol. 2006 Dec;26(23):8743–54. doi: 10.1128/MCB.02251-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim KM, Cho H, Kim YK. The upstream open reading frame of cyclin-dependent kinase inhibitor 1A mRNA negatively regulates translation of the downstream main open reading frame. Biochem Biophys Res Commun. 2012 Aug 3;424(3):469–75. doi: 10.1016/j.bbrc.2012.06.135. [DOI] [PubMed] [Google Scholar]
- 95.Hu Z, Yau C, Ahmed AA. A pan-cancer genome-wide analysis reveals tumour dependencies by induction of nonsense-mediated decay. Nat Commun. 2017 Jun 26;8:15943. doi: 10.1038/ncomms15943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tanikawa C, Urabe Y, Matsuo K, Kubo M, Takahashi A, Ito H, Tajima K, Kamatani N, Nakamura Y, Matsuda K. A genome-wide association study identifies two susceptibility loci for duodenal ulcer in the Japanese population. Nat Genet. 2012 Mar 4;44(4):430–4. S1–2. doi: 10.1038/ng.1109. [DOI] [PubMed] [Google Scholar]
- 97.Qiu F, Tang R, Zuo X, Shi X, Wei Y, Zheng X, Dai Y, Gong Y, Wang L, Xu P, et al. A genome-wide association study identifies six novel risk loci for primary biliary cholangitis. Nat Commun. 2017 Apr 20;8:14828. doi: 10.1038/ncomms14828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tang W, Teichert M, Chasman DI, Heit JA, Morange PE, Li G, Pankratz N, Leebeek FW, Paré G, de Andrade M, et al. A genome-wide association study for venous thromboembolism: the extended cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Genet Epidemiol. 2013 Jul;37(5):512–521. doi: 10.1002/gepi.21731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ruark E, Seal S, McDonald H, Zhang F, Elliot A, Lau K, Perdeaux E, Rapley E, et al. Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat Genet. 2013 Jun;45(6):686–9. doi: 10.1038/ng.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rapley EA, Turnbull C, Al Olama AA, Dermitzakis ET, Linger R, Huddart RA, Renwick A, Hughes D, Hines S, Seal S, et al. A genome-wide association study of testicular germ cell tumor. Nat Genet. 2009 Jul;41(7):807–10. doi: 10.1038/ng.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhou L, He M, Mo Z, Wu C, Yang H, Yu D, Yang X, Zhang X, Wang Y, Sun J, et al. A genome wide association study identifies common variants associated with lipid levels in the Chinese population. PLoS One. 2013 Dec 30;8(12):e82420. doi: 10.1371/journal.pone.0082420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; et al. Genetic effects on gene expression across human tissues. Nature. 2017 Oct 11;550(7675):204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kraja AT, Vaidya D, Pankow JS, Goodarzi MO, Assimes TL, Kullo IJ, Sovio U, Mathias RA, Sun YV, Franceschini N, et al. A bivariate genome-wide approach to metabolic syndrome: STAMPEED consortium. Diabetes. 2011 Apr;60(4):1329–39. doi: 10.2337/db10-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Caussy C, Charrière S, Marçais C, Di Filippo M, Sassolas A, Delay M, Euthine V, Jalabert A, Lefai E, Rome S, Moulin P. An APOA5 3′UTR variant associated with plasma triglycerides triggers APOA5 downregulation by creating a functional miR-485-5p binding site. Am J Hum Genet. 2014 Jan 2;94(1):129–34. doi: 10.1016/j.ajhg.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biology. 2016 Jun 6;17(1):122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014 Feb 2; doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yang W, Shen N, Ye DQ, Liu Q, Zhang Y, Qian XX, Hirankarn N, Ying D, Pan HF, Mok CC, et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010 Feb 12;6(2):e1000841. doi: 10.1371/journal.pgen.1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fisher R. XV.—The Correlation between Relatives on the Supposition of Mendelian Inheritance. Transactions of the Royal Society of Edinburgh. 1918;52(2):399–433. doi: 10.1017/S0080456800012163. [DOI] [Google Scholar]
- 109.Plomin R, Haworth CM, Davis OS. Common disorders are quantitative traits. Nat Rev Genet. 2009 Dec;10(12):872–8. doi: 10.1038/nrg2670. [DOI] [PubMed] [Google Scholar]
- 110.Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. 2009 Jul;9(7):491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 111.Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK, Thomas M, Kim HY, León MG, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011 Feb 26;377(9767):721–31. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- 112.Tognotti E. Program to eradicate malaria in Sardinia, 1946–1950. Emerg Infect Dis. 2009 Sep;15(9):1460–6. doi: 10.3201/eid1509.081317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu XQ, Stacey KJ, Horne-Debets JM, Cridland JA, Fischer K, Narum D, Mackay F, Pierce SK, Wykes MN. Malaria infection alters the expression of B-cell activating factor resulting in diminished memory antibody responses and survival. Eur J Immunol. 2012 Dec;42(12):3291–301. doi: 10.1002/eji.201242689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002 Sep 19;347(12):911–20. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 115.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005 Apr 15;308(5720):385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bendl J, Musil M, Štourač J, Zendulka J, Damborský J, Brezovský J. PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions. PLoS Comput Biol. 2016 May 25;12(5):e1004962. doi: 10.1371/journal.pcbi.1004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kumar S, Ambrosini G, Bucher P. SNP2TFBS - a database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 2017 Jan 4;45(D1):D139–D144. doi: 10.1093/nar/gkw1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brodie A, Azaria JR, Ofran Y. How far from the SNP may the causative genes be? Nucleic Acids Res. 2016 Jul 27;44(13):6046–54. doi: 10.1093/nar/gkw500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002 Feb 15;295(5558):1306–11. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 120.Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, Cairns J, Wingett SW, Várnai C, Thiecke MJ, et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell. 2016 Nov 17;167(5):1369–1384. e19. doi: 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016 Apr;17(4):207–23. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- 122.Gao T, He B, Liu S, Zhu H, Tan K, Qian J. EnhancerAtlas: a resource for enhancer annotation and analysis in 105 human cell/tissue types. Bioinformatics. 2016 Dec 1;32(23):3543–3551. doi: 10.1093/bioinformatics/btw495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.St Laurent G, Vyatkin Y, Kapranov P. Dark matter RNA illuminates the puzzle of genome-wide association studies. BMC Med. 2014 Jun 12;12:97. doi: 10.1186/1741-7015-12-97. [DOI] [PMC free article] [PubMed] [Google Scholar]