

Abstract
Defects in alternative splicing are frequently found in human tumors and result either from mutations in splicing-regulatory elements of specific cancer genes or from changes in the regulatory splicing machinery. RNA splicing regulators have emerged as a new class of oncoproteins and tumor suppressors, and contribute to disease progression by modulating RNA isoforms involved in the hallmark cancer pathways. Thus dysregulation of alternative RNA splicing is fundamental to cancer and provides a potentially rich source of novel therapeutic targets. Here we review the alterations in splicing regulatory factors detected in human tumors, as well as the resulting alternatively spliced isoforms that impact cancer hallmarks, and discuss how they contribute to disease pathogenesis. RNA splicing is a highly regulated process and, as such, the regulators are themselves tightly regulated. Differential transcriptional and post-transcriptional regulation of splicing factors modulates their levels and activities in tumor cells. Furthermore, the composition of the tumor microenvironment can also influence which isoforms are expressed in a given cell type and impact drug responses. Finally, we summarize current efforts in targeting alternative splicing, including global splicing inhibition using small molecules blocking the spliceosome or splicing-factor-modifying enzymes, as well as splice-switching RNA-based therapeutics to modulate cancer-specific splicing isoforms.
Graphical Abstract
INTRODUCTION
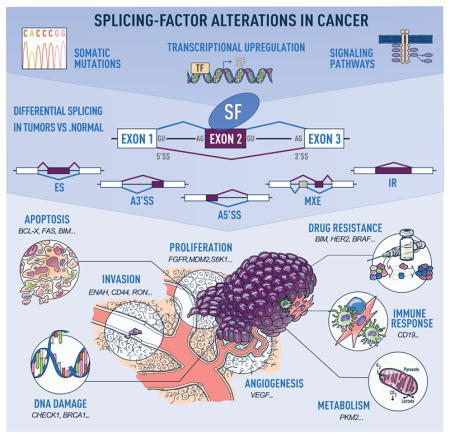
Cancers arise as a consequence of the dysregulation of cellular homeostasis and of its multiple control mechanisms. Alternative RNA splicing is a key step of post-transcriptional gene expression regulation. It contributes to proteomic and functional diversity by enabling the production of distinct RNA isoforms from a single gene. Alternative splicing provides transcriptional plasticity by controlling which RNA isoforms are expressed at a given time point in a given cell type. Cancer cells subvert this process to produce isoforms that benefit cell proliferation or migration, or unable escape from cell death (Figure 1)1.
Figure 1. Alternative-splicing alterations in cancer.

Human tumors exhibit recurrent mutations in, or changes in the levels of, splicing regulatory factors, the latter of which can occur due to copy number changes, or alterations in the transcriptional, post-transcriptional, or post-translational regulation of splicing factors in response to signaling changes (top panel). These changes in splicing-factor levels lead to alterations in the splicing of their downstream targets, promoting events that follow one of the following patterns: exon skipping (ES), alternative 5′ or 3′ splice site (SS) selection (A5′SS or A3′SS), inclusion of mutually exclusive exons (MXE), or intron retention (IR) (middle panel). Misregulated splicing of isoforms involved in key cellular pathways contributes to tumor initiation and progression. Examples of cancer hallmarks and associated tumor isoforms are indicated (bottom panel).
RNA splicing is a highly controlled process that relies on cis-regulatory elements and trans-regulatory factors. The core splicing machinery, the spliceosome, removes introns and joins exons together to generate a mature mRNA molecule. This machinery assembles on the pre-mRNA molecule on specific sequences located at the exon-intron boundaries and that define the 3′ and 5′ splice sites (SSs) and the branch point site (BPS). The core human spliceosome, together with associated regulatory factors, comprise more than 300 proteins and five small nuclear RNAs (snRNAs), and catalyze both constitutive and regulated alternative splicing2–5. The architecture of the spliceosome undergoes dynamic remodeling in preparation for, during, and after the splicing reaction (Figure 2). In addition to the core spliceosome, regulatory proteins are involved in modulating the splicing reaction, and act as splicing activators or repressors by binding to exonic or intronic enhancer or silencer elements.
Figure 2. Components of the core and regulatory splicing machinery that exhibit alterations in human tumors.

(A) Graphical representation of the stepwise assembly of spliceosomal complexes on a pre-mRNA molecule and catalysis of the splicing reaction to generate mature spliced mRNA. First, the ATP-independent binding of U1 snRNP to the 5′ splice site (5′SS) of the intron initiate the assembly of the “Early” or E complex on the pre-mRNA. In addition, SF1 and U2AF2 bind respectively to the branch point site (BPS) and the polypyrimidine tract (Py-tract). In the second step, the ATP-dependent interaction of U2 snRNP with the BPS leads to the formation of the A complex. This interaction is stabilized by the SF3a and SF3b protein complexes, as well as U2AF2 and U2AF1, and leads the displacement of SF1 from the BPS. Recruitment of the pre-assembled U4/U6/U5 tri-snRNP marks the formation of the catalytically inactive B complex. Major conformational changes, including release of U1 and U4, lead to spliceosome activation and formation of the B* complex. The first catalytic step of splicing, generates the C complex and results in the formation of the lariat. Complex C performs the second catalytic step of splicing, which results in the joining of the two exons. Post-splicing the spliceosome disassembles in an orderly manner, releasing the mRNA, as well as the lariat bound by U2/U5/U6. The snRNP are then further dissociated and recycled. (B) Spliceosomal core factors that exhibit recurrent somatic mutations in human tumors are listed next each complex (colored boxes) and are shown in more details for complexes E and A (right panels). In addition to core splicing factors, regulatory splicing factors (SF) that can bind to exonic or intronic splicing enhancer (ESE or ISE) or silencer (ESS or ISS) sequences to fine-tune splicing are also found altered in human tumors (grey boxes).
Defects in alternative splicing are frequently found in human tumors and result either from mutations in splicing-regulatory elements of specific cancer genes or from changes in the regulatory splicing machinery6. Alterations of the splicing machinery are particularly important in cancer because they affect a network of downstream splicing targets, whereas a mutation affecting splicing of a single gene often affects only one isoform. RNA splicing regulators have recently emerged as a new class of oncoproteins or tumor suppressors, and contribute to disease progression by modulating RNA isoforms involved in the hallmarks cancer pathways. Dysregulation of alternative splicing is a fundamental process in cancer and provides a potentially rich source of novel therapeutic targets and biomarkers for disease progression. A better understanding of the regulators of the splicing machinery is a crucial step in understanding the role of RNA splicing in cancer. Here we review the alterations in splicing regulatory factors detected in human tumors, as well as the alternatively spliced isoforms that impact cancer hallmarks, and discuss how they contribute to disease pathogenesis. Finally, we summarize current efforts in targeting alternative splicing as cancer therapeutics.
ALTERATIONS IN SPLICING REGULATORY COMPONENTS
Splicing-factor mutations associated with malignancies
Recurrent somatic mutations in components of the human splicing machinery occur in human tumors, most frequently in hematological malignancies7, suggesting that splicing-factor alterations are a hallmark of cancer. Interestingly, the two most frequently mutated splicing factors are SF3B1, a core component of U2 snRNP involved in BPS selection, and SRSF2, a serine/arginine-rich (SR) protein that acts both in alternative and constitutive splicing and interacts with U1 snRNP (Figure 2). Mutations in other splicing factors have been also been identified, and the list is growing every day as more human tumors are sequenced. However, the functional consequences of most of these mutations and their roles in tumor progression remain to be characterized.
SF3B1 (alias SF3B155)
SF3B1, the key protein component of U2 snRNP, is crucial for formation of the spliceosomal A complex. SF3B1 interacts directly with the RNA-recognition motif (RRM) of U2AF2 as well as with SF3B14a, thus creating a stable complex that directs the recognition of the BPS by U2 snRNA5. SF3B1 also interacts with nucleosomes suggesting that chromatin structure can modulate its splicing functions8. Recurrent somatic SF3B1 mutations occur in myelodysplastic syndromes (MDS), including 83% of refractory anemia with ringed sideroblasts (RARS), an MDS variant with erythroid dysplasia and favorable outcomes, and 76% of refractory cytopenia with multilineage dysplasia and ringed sideroblasts (RCMD-RS) which carries a less favorable survival rate9–12. SF3B1 mutations cluster in exons 12–15, which encode HEAT repeats, a region important for the association of SF3B1 with SF3B14a13. SF3B1 missense mutations alter the recognition of alternative or cryptic 3′SS leading to differential splicing of transcripts, 70% of which are novel isoforms and 50% undergo nonsense-mediated decay (NMD)14,15. In mouse models, this differential exon usage disrupts key pathways in hematopoiesis and iron metabolism and blocks erythroid differentiation, thus providing a basis for the pathogenesis of RARS and RCMD-RS16,17. The K700E missense mutation, which accounts for more than half of SF3B1 mutations in MDS patients and is associated with a better prognosis, promotes splicing of an isoform of the erythroid lineage transcription factor TAL1 that reduces erythroid differentiation in vitro10,13,18. SF3B1 mutations are also detected in other cancers including 15% of chronic lymphocytic leukemia (CLL), in which SF3B1 mutations are associated with an anti-apoptotic role and correlate with poor overall prognosis12,19,20. Additionally, the K700E mutation is detected in 3% of pancreatic and 1.8% of breast cancers, both of which exhibit alterations in RNA splicing patterns21,22. SF3B1 mutations also occur in 1% of cutaneous melanomas and 20% of uveal melanomas in which they are associated with aberrant splicing, chromosome 3 disomy, and intermediate prognosis23–26.
SRSF2 (alias SC35)
The splicing factor SRSF2 belongs to the SR protein family and is involved in regulation of both alternative and constitutive splicing. SRSF2 coordinates recognition of the 5′ and 3′ SS by the U1 and U2 snRNPs, respectively. SR proteins recognize enhancer and silencer sequences in pre-mRNA exons and introns and thereby favor exon inclusion or skipping by recruiting or inhibiting spliceosome assembly5. Mutations in SRSF2 are frequently observed in hematologic malignancies including 10% of MDS, 31–47% of chronic myelomonocytic leukemia (CMML), and 2% of acute myeloid leukemia (AML)13. SRSF2 mutations in MDS are associated with decreased overall survival and increased progression rate from MDS to AML27. Interestingly, SRSF2 missense mutations cluster at proline 95, in a region proximal to the RRM domain, which confers the RNA binding specificity13,28. In mouse models, blood lineage-specific SRSF2 knockout (KO) or heterozygous expression of Srsf2P95H causes defective hematopoiesis29. The SRSF2P95H mutation induces splicing changes in mouse and human myeloid cell models, which likely result from alterations in pre-mRNA sequences recognized by the RRM of SRSF229–32. Indeed, mutant SRSF2 exhibits increased binding specificity for the CCNG consensus sequence, whereas wild-type SRSF2 recognize both CCNG and GGNG sequences29. This alteration in sequence specificity leads to the inclusion of a premature termination codon (PTC)-containing exon in EZH2, a histone methyltransferase implicated in the pathogenesis of MDS29. Finally, deletion of Ezh2 in mouse hematopoietic stem cells causes MDS, providing a causal link between the SRSF2 mutation, EZH2 loss of function, and MDS33.
U2AF1, ZRSR2, RBM10 and other splicing-factor mutations
While SF3B1 and SRSF2 are the most frequently mutated splicing factors in hematologic malignancies, other factors also exhibit recurrent mutations in MDS.
U2AF1 (alias U2AF35) is involved in the formation of the spliceosomal E complex. As a heterodimer with U2AF2 (U2AF65), it is responsible for the recognition of the 3′SS and BPS as well as for stabilizing U2 snRNA binding to the BPS5. In addition to MDS, U2AF1 is also mutated in 3% of lung adenocarcinomas34. Missense mutations in U2AF1 occur almost exclusively at S34 and Q157, thus affecting the C-terminal zinc finger domain. Expression of U2AF1S34F in HeLa cells leads to an increase in PTC-containing transcripts, suggesting global splicing defects13. U2AF1 mutants disrupt proliferation in HeLa cells and exhibit a decreased ability to reconstitute the hematopoietic system when introduced into mouse hematopoietic stem cells, thereby convoluting the link between these mutations and MDS13. However, a recent study described a gain-of-function role for mutant U2AF135. When overexpressed in human hematopoietic progenitor cells, U2AF1S34F promotes lineage-specific splicing changes, most notably in H2AFY and STRAP isoforms, which are not rescued by co-expression of wild-type U2AF1. These splicing isoforms disrupt normal erythroid and granulomyelocytic differentiation in hematopoietic progenitors35. Interestingly, expression of the canonical isoforms is capable of rescuing the differentiation defect35. Taken together, these findings suggest that mutant U2AF1 blocks terminal differentiation of hematopoietic cells, but does not grant a growth or survival advantage, and may therefore require further mutational hits to lead to MDS.
ZRSR2 is involved in the recognition of 3′SS in both major and minor introns, a class of intronic sequences recognized by the minor U12-dependent spliceosome36. In addition, ZRSR2 also promotes the removal of the intron lariat and stitching of the adjacent exons37. ZRSR2 mutations in MDS lead to the retention of minor introns without affecting the major introns38. In contrast to the hotspot mutations in other factors, ZRSR2 mutations are widely distributed and create loss-of-function mutants, thus suggesting that ZRSR2 functions as a tumor suppressor13,38.
The RNA-binding protein RBM10 is a component of the pre-spliceosomal complex A. Mutations in RBM10 are associated with the TARP syndrome, an X-linked recessive disorder with congenital heart malformation and developmental abnormalities, often associated with neonatal lethality39. Somatic mutations in RBM10 are found in lung adenocarcinoma34,40, including 21% of invasive lung adenocarcinoma 41, as well as less frequently in non-anaplastic thyroid cancers42, colorectal carcinoma43, pancreatic adenocarcinoma44 and intraductal papillary mucinous neoplasm45. RBM10 mutations are widely distributed and create loss-of-function mutants, indicating that RBM10 functions as a tumor suppressor46. Furthermore, the presence of RBM10 mutations is associated with a significant reduction in RBM10 expression in lung tumors, and is accompanied by changes in proliferation rates and in alternative splicing of RBM10 target genes47. For example, missense or truncating RBM10 mutations found in lung cancer patients disrupts RBM10-mediated regulation of NUMB splicing, inducing a pro-proliferative isoform46. Conversely, in pancreatic cancer, RBM10 mutations are associated with longer survival in spite of histological features of aggressive disease44.
Mutations in other components of the spliceosome, e.g., PRPF40B, U2AF2, SF3A1, or SF1, occur sporadically in MDS patients. PRPF40B interacts with SF1 and U2AF2 to enhance the inclusion of exons with weak SSs, and regulates splicing of apoptotic isoforms of FAS and BCL-x48. U2AF2 is involved in 3′SS recognition, and in some cases can promote exon skipping49,50. SF3A1 interacts with both the U1 snRNA and U2 snRNP complex to mediate communication between the 5′SS and 3′SS complexes51. Additionally, PRPF40B and U2AF2 are also upregulated or downregulated in several solid tumors52, including melanoma, where U2AF2 promotes metastasis by regulating splicing of CD4453.
Alterations in splicing-factor levels
In solid tumors, splicing factors exhibit frequent changes at the copy number or expression levels but are rarely mutated54. Splicing factors bind directly to pre-mRNA and regulate their downstream targets in a concentration-dependent manner55; thus, changes in splicing-factor levels cause splicing deregulation in tumors even in the absence of mutations. Two major protein families play a critical role in the regulation of alternative splicing through recognition of intronic or exonic enhancer and silencer sequences. The serine/arginine-rich(SR) protein family is composed of 12 members (SRSF1-12) containing SR domains that contribute to protein-protein interactions, and one or two RRMs that allow sequence-directed binding55. Heterogenous nuclear ribonucleoproteins (hnRNPs), which are a large and structurally diverse family of RNA-binding proteins with diverse roles in splicing, mRNA transport, and translation, often function as antagonists to SR-protein-regulated alternative splicing events56. Below, we discuss in more detail several RNA-binding proteins that exhibit expression changes in human tumors, and for which there is compelling in vitro or in vivo evidence that their alterations affect cellular processes involved in transformation (Figure 3).
Figure 3. Recurrent splicing-factor alterations detected in human tumors.

Genomic alterations including expression changes and recurrent somatic mutations in splicing factors detected in more than 2% of tumors in several cohorts of patients, including TCGA data, are indicated per tumor type. Splicing-factor upregulation are depicted in red, downregulation in blue, and somatic mutations in green (See legend for details). Several splicing factors can be found both upregulated and downregulated in tumors of the same tissue, suggesting that distinct splicing-factor genomic alterations are associated with distinct tumor subtypes within the same tissue. AML: acute myeloid leukemia; AML/MDS: acute myeloid leukemia myelodysplastic syndrome; CMML: chronic myelomonocytic leukemia; HN: head and neck; MDS w/o RS: myelodysplastic syndrome without ringed sideroblasts; RARS/RCMD: refractory anemia with ringed sideroblasts and refractory cytopenia with multilineage dysplasia and ringed sideroblasts; MPN: myeloproliferative neoplasm. See references in text.
SRSF1 (alias ASF/SF2)
SRSF1 is a proto-oncogene that controls alternative splicing but also regulates other steps of RNA metabolism57. SRSF1 is frequently upregulated in breast, lung, colon and bladder tumors, in part due to an amplification of Chr.17q2358–60. In breast cancer models, SRFS1 overexpression promotes transformation in vivo and in vitro by enhancing proliferation and decreasing apoptosis58. Additionally, SRSF1 acts synergistically with MYC, and their co-expression correlates with higher tumor grade and decreased survival in breast and lung cancer patients58,60–62. SRSF1 oncogenic activity relies on the regulation of splicing isoforms involved in apoptosis (e.g., BCL2L1, BCL2L11, BIN1), cell growth (e.g., RPS6KB1), cell survival (e.g., MKNK2), or motility (e.g., RON)58–60,63,64. In lung cancer, SRSF1 upregulation is associated cisplatin and topotecan resistance65.
SRSF3 (alias SRp20)
In addition to its role in splicing regulation, SRSF3 is also involved in transcription termination, IRES-dependent viral RNA translation, and homologous recombination-mediated DNA repair66–68. Additionally, SRSF3 together with SRSF1 associates with hypo-phosphorylated chromatin, and controls G0/G1 re-entry69. SRSF3 is overexpressed in lung, breast, ovarian, stomach, bladder, colon, liver, and oral tumors, in part due to copy number changes on Chr.6p2170–72, and shows variable expression in renal cancers52,70. SRSF3 acts as a proto-oncogene as its overexpression is capable of transforming human fibroblasts in vitro, while its depletion causes growth arrest of cancer cell lines70. SRSF3 regulates alternative splicing of genes involved in tumorigenesis, such as isoforms PKM2 that alters cell metabolism or TP53β that induces cellular senescence73,74.
SRSF6 (alias SRp55)
SRSF6 is frequently upregulated in breast, lung, pancreatic and colon cancers, in part due to an amplification of its locus60. SRSF6-overexpression synergizes with MYC to promote transformation of lung epithelial cell lines75, while, its knockdown in lung carcinoma cells decreases proliferation and prevents tumor formation in immunocompromised mice75. SRSF6 promotes pro-oncogenic splice variants of the insulin receptor INSR, the tumor suppressor DLG1, and the downstream effector of the MAPK pathway MKNK275. SRSF6 is also upregulated in multiple subtypes of skin cancer, and its overexpression in murine skin promotes splicing of cassette exons, coordinates wound healing, and induces hyperplasia76. Conversely, PLX4720, a BRAF inhibitor, induces SRSF6 expression in BRAFV600E melanoma cell lines, which in turn promotes splicing of the pro-apoptotic isoform BIM-S leading to increased cell death77. While SRSF6 upregulation also correlates with increased BIM-S expression post-treatment, continued exposure to PLX4720 leads to drug resistance78. Finally, SRSF6 is downregulated in kidney tumors, which could indicate cell type-specific functions52.
Other SR proteins
Other SR proteins are also altered in human tumors but have less well-defined roles in transformation. SRSF5 is upregulated in breast tumors with lymph node metastasis79 and oral tumors80. SRSF5 or SRSF7 are upregulated in lung cancer, and their knockdown impacts cell proliferation81. Interestingly, SRSF5 shows broad downregulation in breast, lung, liver, and kidney tumors52. SRSF4 regulates alternative splicing events leading to cell death in cisplatin treated breast cancer cells82. Renal tumors show a broad differential expression of various SR proteins83. Co-expression of these splicing factors may indicate that the robust network of splicing changes in cancer cells is due to an imbalance among multiple splicing factors rather than differential splicing regulated by a single splicing factor.
TRA2β
TRA2β is an SR-like protein that regulates alternative splicing and is essential for embryonic development84. Overexpression of TRA2β occurs in lung, breast, ovarian, cervical, prostate, colon, and central nervous system tumors, where it correlates with an aggressive phenotype, whereas downregulation is detected in thyroid and renal cancers52,71,85–91. TRA2β-overexpression promotes proliferation in human lung carcinoma cells, while its knockdown induces apoptosis85. TRA2β overexpression in human glioma cells promotes proliferation and migration89, and TRA2β KO leads to defects in murine brain development, highlighting the importance of TRA2β homeostasis in neurogenesis92. TRA2β regulates the inclusion of CD44 exons v4 and v5 in breast tumors90, and inclusion of estrogen receptor alpha ERα exon 7, creating a dominant negative isoform in endometrial tumors93. Interestingly, lung tumors exhibit a rare fusion protein between TRA2β and DNAH5 that preferentially localizes to the cytoplasm, activates ERK1/2 through inhibition of SIRT6, and promotes lung cancer94.
hnRNPA1
hnRNPA1 regulates alternative splicing and translation, and is overexpressed in blood, lung, and colorectal malignancies52,95–98. hnRNPA1 upregulation in lung adenocarcinoma is associated with increased tumor staging; conversely, hnRNPA1 knockdown decreases cell proliferation and induces cell cycle arrest in lung cancer cell lines97. In response to ultraviolet radiation, hnRNPA1 expression is increased in skin cells, consequently modulating splicing of HDM2 and promoting cell survival by activating the mTOR pathway99,100. Furthermore, hnRNPA1 is upregulated in AML, where it functions to prevent myeloid differentiation by binding to the 3′-UTR and thereby preventing translation of C/EBPα mRNA, a critical transcription factor for myelopoiesis98.
hnRNPA2/B1
hnRNPA2/B1, a splicing regulator closely related to hnRNPA1, is frequently overexpressed in lung, breast, colorectal, and brain tumors52,101–103. Up-regulation of hnRNPA2/B1 in bronchial lavage specimens predicts the diagnosis of a lung neoplasm with high sensitivity and specificity101, and its degree of overexpression correlates with microsatellite instability104, increased tumor stage, and decreased overall survival105. hnRNPA2/B1 mediates its tumorigenic effect in glioblastoma through alternative splicing of key oncogenes and tumor suppressors. For example, hnRNPA2/B1 overexpression causes skipping of RON exon 11, creating an oncogenic isoform involved in cell motility; skipping of exon 11 in the insulin receptor INSR leading to an isoform with altered substrate specificity that binds to broader range of mitogens; or inclusion of exon 12a in the tumor suppressor BIN1m creating an isoform that is unable to stimulate apoptosis102.
hnRNPK
hnRNPK is a splicing factor that can act as a tumor suppressor but also exhibits oncogenic functions. Heterozygous deletion of 9q, where hnRNPK is located, is a characteristic of AML and results in hnRNPK decreased expression and haplo-insufficiency106,107. hnRNPK interacts directly with C/EBPα mRNA, and heterozygous hnRNPK KO mice express low levels of the C/EBPα p42 isoform and eventually develop abnormal myelopoiesis98,106. hnRNPK expression is also decreased in renal tumors52. Consistent with its role as a tumor suppressor, hnRNPK is an HDM2-regulated cofactor for p53, and its expression increases upon DNA damage108. Furthermore, hnRNPK knockdown leads to defects in DNA-repair and to increased DNA damage after gamma-irradiation108,109. However, hnRNPK also exhibits oncogenic functions and is upregulated in breast, colorectal, and pancreatic cancer tissues and cell lines 110–113. For example, inhibition of hnRNPK in human cancer cells decreases cell motility, whereas its upregulation increases proliferation and migration110,114. In colorectal and pancreatic tumors and cell lines, oncogenic hnRNPK is translocated from the nucleus to the cytoplasm, thus suggesting a potential explanation for its ability to act as either an oncogene or a tumor suppressor112–114.
Other hnRNPs
Upregulation of hnRNPM is detected in metastatic breast tumor115. hnRNPM regulates epithelial-mesenchymal transition (EMT) in breast epithelial cells, in part by promoting splicing of the CD44s isoform, and by altering TGF-β signaling115. hnRNPM upregulation is also a poor prognostic factor for Ewing’s sarcoma, where inhibition of the PI3K/AKT/mTOR pathway causes broad transcriptome changes mediated by hnRNPM-regulated splicing events116. Additionally, hnRNPH1 contributes to the aggressiveness of glioblastoma via alternative splicing of IG20/MADD and RON, creating anti-apoptotic and pro-motility protein isoforms117. Moreover, hnRNPC is upregulated in lung and colorectal cancers, and downregulated in kidney cancers 52. hnRNPC acts as a tumor suppressor and alters DNA damage repair by binding to BRCA1, BRCA2, RAD51, and BRIP1 mRNA and modulating the inclusion of intronic Alu transposable elements118. hnRNPE1 upregulation in pancreatic cancer is associated with metastasis and promotes alternative splicing of integrin β1, a transmembrane protein involved in cell adhesion119. Finally, PTBP1, also known as hnRNPI is upregulated in breast, brain, colon, endometrial, and ovarian tumors and cell lines120–124. PTBP1-overexpression increases proliferation, anchorage-independent growth, and invasion in cancer cell lines, but does not transform murine fibroblasts124,125.
Other splicing factors
The epithelial-specific splicing factors ESRP1 and ESRP2 affect splicing of target genes involved in EMT, including CD44, ENAH, FGFR2, and RAC1126–131. They are often upregulated in normal epithelium but downregulated in invasive fronts132. Paradoxically, they have been assigned both a tumor suppressor and an oncogenic function133–135.
Similarly, the splicing factor RBFOX2 has been linked with EMT, and regulates splicing targets in breast, pancreatic and colon tumors128,136–138.
Additionally, splicing factors RBM5 and 10 are found upregulated or downregulated in several solid tumors, and are implicated in the splicing of apoptotic proteins BAX and BCL-x, and the notch pathway regulator NUMB34,139–143.
Finally, QKI downregulation is a common event in several solid tumors and is associated with poor prognosis144–146. Interestingly, MYB-QKI fusions have been identified as a driver event in glioma147.
Defects in pathways regulating splicing factors
Alterations in splicing-factor levels can be explained by gene amplifications or deletions only in a fraction of the tumors that exhibit splicing-factor defects52,54. RNA splicing is a highly regulated process and hence the splicing regulators are themselves tightly regulated. Differential regulation of splicing factors can thus affect their levels and activities in tumors even in the absence of copy-number changes or mutations. Here we discuss examples of transcriptional and post-transcriptional regulation that could explain the defects in splicing-factor levels observed in tumors (Figure 4).
Figure 4. Defects in splicing-factor regulation lead to changes in splicing-factor levels, activity, and cellular localization.

Schematic representation of the transcriptional, post-transcriptional, and post-translational steps that impact the expression of a splicing factor (SF). See text for specific examples and references.
Transcriptional Regulation
The transcription factor MYC is a well-studied oncogene that is overactive in a variety of cancers. However, part of MYC’s oncogenic potential may result from its ability to regulate splicing factors at the transcriptional level. The oncogenic factor SRSF1 is a direct transcriptional target of MYC, and synergizes with MYC to promote tumorigenesis in breast and lung tumors58,62. In gliomas, c-MYC drives the expression of PTBP1, hnRNPA1, and hnRNPA2/B1, all of which favor splicing of the PKM2 isoform used in aerobic glycolysis148. PTBP1 or hnRNPA1 are directly regulated by n-MYC in neuroblastomas, where they controls cell survival and correlate with a worse prognosis149. Conversely, knockdown of hnRNPA1 or hnRNPA2 reduces splicing of PKM2 and alters cell metabolism150. Moreover, MYC-driven tumors exhibit differential expression of spliceosomal components or their regulators, e.g., BUD31 and PRMT5, as well as of their downstream targets151,152. In addition to MYC, other pathways control splicing-factor transcriptional activation. In colorectal tumors, the Wnt signaling pathway, which is frequently dysregulated through APC mutation, directly controls SRSF3 level 153,154. The transcription factors Ets1 and HSF1 mediate basal and oxidant-stress responses by inducing TRA2β expression in colorectal cancer88. Together, these pathways represent key points for potential targeted therapies that could be used to disrupt splicing regulators in tumors.
Alternative splicing and nonsense-mediated mRNA decay
The expression of many RNA-binding proteins is regulated through the splicing of their own pre-mRNA. SR proteins auto-regulate their levels by enhancing the inclusion of PTC-containing cassette exons, termed “poison exons”, within their mRNA. These transcripts are degraded by NMD creating a negative feedback loop when SR-protein levels become elevated155–159. Although auto-regulation has not been experimentally demonstrated for all SR proteins, poison exons are highly conserved throughout evolution, and isoforms containing these ultraconserved regions are detected in human 160,161. While SR proteins auto-regulate through inclusion of poison exons, auto-regulation of hnRNPs involves both inclusion and skipping of PTC-containing regions161–164.
Splicing factors can also cross-regulate the expression of other RNA-binding proteins, through splicing of their respective ultraconserved regions155,162. In murine cells, exogenous SRSF3 enhances inclusion of its own poison exon, while SRSF1 overexpression inhibits SRSF3 exon inclusion155. Similarly, RBFOX2, which coordinates mesenchymal splicing networks in cancer tissues, regulates alternative splicing of a number of different RNA-binding proteins137,165,166. Alternative splicing of murine Quaking, Qk, generates three isoforms Qk5, Qk6, and Qk7, that exhibit both auto- and cross-regulation. Specifically, Qk5 enhances expression of total Qk mRNA while also binding to its own 3′-UTR and downregulates Qk5 protein expression. Qk6 negatively regulates protein expression of Qk5, while also stimulating translation of Qk6 mRNA167. Human lung tumors express high levels of QKI5 vs. QKI6168, suggesting that this extensive network of auto and cross-regulation could exist in humans and that a similar mode of regulation may exist across other splicing factors.
Regulation by lncRNAs
Long non-coding RNA (lncRNAs) are involved in the regulation of alternative splicing, for example by facilitating splicing-factor binding to exonic splicing silencer or intronic splicing silencer elements. lncRNAs PCGEM1 and BC200 regulate alternative splicing of AR and BCL-x, respectively, through interaction with hnRNPA1, hnRNPA2/B1, or U2AF65169,170. Moreover, MALAT1 modulates alternative splicing by influencing SR protein subnuclear localization171. Additionally, LINC01133 sequesters SRSF6, and its knockdown allows SRSF6 to promote EMT and metastasis in colorectal cancer mouse models172.
Regulation by miRNAs
MicroRNAs (miRNA) can act as tumor suppressors or as oncogenes and can play a role in the regulation of splicing-factor expression. Expression of SRSF7 is regulated by miR-30a-5p and miR-181a-5p in renal tumors, and this miRNA-mediated suppression of SRSF7 alters splicing patterns83,173. Conversely, SRSF7 regulates splicing and expression of these miRNAs, thus forming a negative feedback loop173. miR-30a-5p is upregulated in glioma cells by Wnt signaling and acts as an oncogene, perhaps superseding the pro-tumorigenic roles of either SRSF7 and miR-30a-5p in different cancers174. Additionally, SRSF1 is the target of miR-28, miR-505, miR-10a, and miR-10b175,176. The oncogenic lymphoma/leukemia-related factor LRF represses miR-28 and miR-505 expression and potentially leads to increased SRSF1 expression in tumors175. Upregulation of miR-10a and miR-10b in response to retinoic acid causes terminal differentiation of neuroblastoma cells, possibly through repression of SRSF1 levels176. Additionally, miR-10a and miR-10b also target TRA2β, which promotes proliferation in glioblastoma cells89,176. Finally, miR-451 targets hnRNPA1 in human leukemia cells, potentially acting as a tumor suppressor by repressing hnRNPA1 expression98.
Post-translational regulation
SR proteins undergo extensive post-translational modifications which impacts their subcellular localization and thus activity. For example, phosphorylation of the C-terminal RS domain by SR-specific protein kinases (SRPKs) allows nuclear import via interactions with transportin-SR2177–179. Once in the nucleus, Cdc-like kinases (CLK) control the nuclear distribution of SR proteins180–182. Additionally, SRPK and CLK kinases can alter the functionality of SR proteins independently of their effect on splicing-factor localization. For example, CLK2-mediated phosphorylation prevents the auto-regulation of TRA2β157. CLK2 acts as an oncogene in breast cancer where it alters splicing, possibly linking the regulation of splicing-factor phosphorylation and splicing dysregulation in cancer183. Moreover, the oncogenic kinase AKT directly phosphorylates SRSF1, SRSF7, and SRSF5184,185. AKT promotes phosphorylation and subsequent activation of SRPKs, thereby indirectly regulating SR proteins186. Finally, SRPK1 is overexpressed in various cancer types including breast, colon, pancreatic, prostate, and ovarian187–189.
TUMOR-ASSOCIATED ALTERNATIVELY SPLICED ISOFORMS
The hallmarks of cancer described by Hanahan and Weinberg can be used to understand the capabilities acquired by cells during tumor development and progression190. These ten hallmarks include a cancer cell’s ability to sustain proliferation, avoid cell death, invade and metastasize, and even deregulate cellular energetics. Alternative splicing leads to the production of tumor-associated isoforms that function within these hallmarks to promote tumorigenesis. Here we describe several alternative splicing events, providing compelling evidence for their role in tumorigenesis and discuss how these isoforms relate to the cancer hallmarks (Figure 5).
Figure 5. Tumor-associated isoforms representative of the cancer hallmarks.


Splicing event type, isoform structure, tumor expression, and experimental evidence for selected alternative splicing isoforms detected in human tumors. For simplicity, only the alternatively spliced sequences and the flanking exons and are shown (not at scale). The type of splicing event is indicated: ES: exon skipping; MXE: mutually exclusive exons; 5′ASS: 5′ alternative splice site selection; IR: intron retention. The corresponding isoforms are shown in red or blue and their respective functions are indicated when known, to the right of the schematic figure of the isoform. The cancer hallmark associated with the red isoform is indicated in the left-hand column (See legend for details). ‘Tumor types,’ indicates, using dark-colored rectangles, the expression of tumor-associated isoforms in each of the indicated tumor types (‘other tumors’ include: adrenal, gallbladder, ampullary, bone, and brain; ‘gynecological tumors’ include: ovarian, cervical, and uterine; ‘head and neck’ tumors include: oral, head and neck, tongue, esophageal, and thyroid). ‘Experimental evidence’ indicates, using dark gray rectangles, the expression and functional evidence for each isoform based the following experiments: (i) overexpression (OE) or knockdown (KD) in cell lines, (ii) tumor xenografts, (iii) expression in primary tissue, or (iv) expression in TCGA RNA-sequencing data. Known splicing regulatory proteins are listed for each gene. See text for references.
Isoforms sustaining proliferation
RPS6KB1
The gene RPS6KB1 encodes the protein S6K1, a substrate of mTOR, which controls translation and cell growth. The full-length protein is produced from the RPS6KB1-isoform 1 (RPS6KB1-1), whereas inclusion of three cassette exons 6a, 6b, and 6c generates the shorter isoform 2 (RPS6KB1-2)60. A PTC in exon 6c causes the shorter isoform to lack a portion of the kinase domain60,191. Alternative splicing of RPS6KB1-2 is regulated by SRSF1, an oncogenic factor overexpressed in human breast tumors60. High levels of RPS6KB1-2 are detected in breast and lung cancer cell lines and primary tissues191,192. Expression of RPS6KB1-2 in non-transformed cell lines promotes transformation, whereas knockdown in breast, prostate, and lung cancer cells decreases proliferation and tumor growth. Conversely, knockdown of RPS6KB1-1 in cancer cell lines induced transformation191,192. These data suggest that RPS6KB1-1 plays a role as a tumor suppressor whereas RPS6KB1-2 contributes to cell proliferation and tumor growth via mTORC1 and 4E-BP1 phosphorylation.
FGFR
FGFR1, FGFR2 and FGFR3 belong to the fibroblast growth factor receptor (FGFR) family, members of which are involved in cell proliferation and migration during embryologic development, but also during tissue repair and wound healing of adult tissue. These receptors contain a cytoplasmic kinase domain, a transmembrane domain, and an extracellular ligand binding portion that consists of three immunoglobulin domains, Ig-I to III193. The second half of Ig-II is generated by alternative splicing of one of two mutually exclusive exons -exon 8 or 9- to generate isoform FGFR-IIIb or FGFR-IIIc respectively. This differential splicing, regulated by splicing factors hnRNPH1, hnRNPF, ESRP1, and ESRP2, changes the binding specificity of the FGF ligand130,193,194. Increased levels of FGFR-IIIc isoforms are detected in a variety of tumors, and correlate with tumor progression, increased grading, and invasiveness195–197. For example, increased expression of FGFR2-IIIc is detected in renal, endometrial, pancreatic and colorectal carcinoma compared to normal tissues195,198–200. FGFR2-IIIb, but not FGFR2-IIIc, is expressed in non-transformed and non-invasive breast cancer cells, whereas FGFR2-IIIc is detected in invasive cell lines201. Most renal tumor samples have high levels of FGFR2-IIIc, but those tumors with high FGFR2-IIIb expression have a lower grade and stage, and are associated with longer survival198. Furthermore, expression of FGFR-IIIb isoforms is tumor-suppressive in vitro and in vivo in bladder cancer cells, whereas FGFR-IIIc isoforms promote tumor growth, invasion, and metastasis in colorectal, pancreatic, and cervical cancer cells196,197,200,202,203. However, a high FGFR-IIIc/IIIb ratio is not always correlated with poor prognosis199, suggesting that FGFR isoforms may exhibit different functions in specific tissues or tumors.
In addition to their role in cell proliferation, FGFR isoforms also impact EMT, a key step in tumor dissemination and metastasis. The expression of FGFR-IIIb isoforms correlates with epithelial markers and FGFR-IIIc with mesenchymal markers, in prostate, bladder, and renal carcinoma primary tumors and cell lines198,204–206. Finally, expression of FGFR2-IIIc induces human epithelial keratinocyte cells to acquire mesenchymal characteristics207.
MKNK2
Mnk2, a kinase in the MAPK pathway encoded by the MKNK2 gene, has two main spliced isoforms, Mnk2-a and Mnk2–b, which differ in their C terminal domain. Inclusion of exon 13a produces Mnk2-a, the full-length protein isoform, whereas skipping of exon 13a and inclusion of exon 13b encodes Mnk2-b, an isoform lacking the MAPK domain208. Both isoforms are capable of phosphorylating the translation initiation factor eIF4E, which promotes cell growth; however, Mnk2-a is also capable of phosphorylating p38 in response to stress leading to cell death209. SRSF1 regulates MNKN2 alternative splicing, promoting Mnk2-b and decreasing Mnk2-a expression60,62. Normal breast, lung, and colon tissues express higher levels of Mnk2-a than Mnk2-b, whereas the corresponding tumors exhibit a shift towards the Mnk2-b isoform208,209. Since Mnk2-b lacks the MAPK domain, it is unable to activate the p38 stress response, tipping the balance to promote cell growth208,209. Isoform-specific overexpression or knockdown experiments demonstrate that Mnk2-a overexpression inhibits soft-agar colony formation, whereas Mnk2-b expression or Mnk2-a knockdown increases transformation209.
HRAS
H-ras belongs to the Ras GTPase family, a class of proto-oncogenes regulating proliferation, survival, and differentiation. These proteins exert their stimulatory effects when bound to GTP but become inactive when GTP is hydrolyzed to GDP. Alternative splicing of HRAS produces two distinct proteins, p19 and p21. The full-length isoform, p21, contains exons 0 through 4B, whereas isoform p19 includes the alternative exon IDX between exons 3 and 4a. This exon contains a stop codon, and therefore IDX-containing transcripts produce a truncated protein210,211. p19 is unable to bind GTP and therefore cannot function like other Ras proteins; however, the unique C-terminus of p19 allows binding to the scaffolding protein RACK1212, There are limited data regarding the relative expression of p19 and p21 in tumors; however, evidence suggests that p19 may serve as a tumor suppressor, and that its ectopic expression delays G1/S transition213.
CCND1
CCND1 undergoes alternative splicing to generate two isoforms: cyclin D1a, the conventional isoform, and cyclin D1b, which lacks the C-terminal protein domains. Usage of an alternative 5′SS in exon 4 introduces a PTC, and the resulting D1b isoform lacks the GSK-3β phosphorylation site encoded by exon 5. This causes the protein to remain in the nucleus214,215. Increased expression of CCND1b is observed in breast, lung and prostate tumors216–219. However, both CCND1a and CCND1b are expressed in lymphoma, bladder, cervical, esophageal and breast cancer215,220–223. CCND1b expression, but not CCND1a, correlates with tumor grade, metastasis, and patient survival in lung and breast cancer218,222,224. Interestingly, both isoforms enhance tumor formation, although through different mechanisms. Cyclin D1a promotes cell proliferation and G1/S transition, while cyclin D1b impacts invasion and metastasis215,217,220,222,225,226. Cyclin D1b is unable to phosphorylate RB, which is required for cell cycle progression, thus the D1b isoform lacks the proliferative effects of D1a214,216,220,221. However, the role of cyclin D1b in promoting tumor growth remains controversial with several studies claiming it activates proliferation222,224 while others stating it inhibits proliferation221. Therefore, tumors that express both cyclin isoforms may have advantages in proliferation as well as invasion, and the two isoforms are likely to play distinct roles in different cell types.
Isoforms preventing cell death
BCL2L1
BCL2L1, a member of the Bcl-2 family, generates two isoforms, BCL-xL and BCL-xS, which have opposing functions in apoptosis; the first prevents apoptosis while the latter promotes it 227. BCL-xS is generated via an alternative 5′SS in exon 2 (Figure 4), and lacks the exons encoding the Bcl-2 homology domains, BH1 and BH2, but still includes BH3 and BH4228. Sam68 modulated by FBI-1, RBM4, PTBP1, or RBM25 upregulates BCL-xS expression229–233, whereas SRSF1 promotes BCL-xL splicing 234. Increased expression of BCL-xL and decreased expression of BCL-xS are detected in lymphoma, glioma, myeloma, and neuroblastoma cell lines and primary tumors235–238. Expression of the pro-apoptotic BCL-xS isoform in cancer cell lines decreases cell viability and sensitizes cells to chemotherapy and radiation236,239,240. Conversely, expression of BCL-xL promotes cell survival and increases resistance to apoptosis following chemotherapy238,241–243. Therefore BCL-xS can antagonize the protective effects of BCL-xL.
FAS
FAS is a member of the TNF-receptor superfamily known for promoting the extrinsic pathway of apoptosis. An alternatively spliced isoform, soluble Fas (sFAS), is produced by the skipping of exon 6, which encodes the transmembrane domain, and therefore the protein cannot localize to the plasma membrane244. Alternative splicing of FAS is controlled by multiple regulators, including EWS, hnRNPA1, and TIA1, all of which promote exon 6 inclusion245–247, whereas RBM5 and PTBP1 favor exon 6 skipping248. A genome-wide siRNA screen identified close to 200 additional genes that may be implicated in regulating FAS alternative splicing249. sFas is expressed in leukemias and lymphomas250–252, as well as in solid tumors including renal, cervical, endometrial, ovarian, and bladder cancer253–256. Expression of sFas is inversely correlated with patient survival and tumor progression in leukemia, gynecological, and bladder tumors252,254–257.
BIN1
BIN1 is tumor suppressor that functions by interacting with and inhibiting c-MYC. Inclusion of exon 12A of BIN1 generates a protein isoform that no longer binds Myc and therefore eliminates BIN1 tumor-suppressive function258. SRSF1 overexpression promotes inclusion of exon 12A, and this isoform plays a role in escaping cell death in SRSF1-dependent breast tumors58,60. Expression of the BIN1+12A isoform is detected in melanoma and breast cancer cell lines60,259.
CASP2
Caspase 2 is an initiator of apoptosis and functions as a tumor suppressor. Splicing of CASP-2 generates two main isoforms; skipping of exon 9 produces the pro-apoptotic Casp-2L protein isoform, whereas exon 9 inclusion results in a PTC, leading to the anti-apoptotic Casp-2S isoform262,263. Splicing of CASP2 is regulated by RBM5, which promotes exon 9 inclusion260, and SRSF3, which promotes exon 9 skipping261. The major isoform in adult tissue is Casp-2L, whereas Casp-2S is found in brain and muscle tissues262,263. Expression of CASP2 isoforms is detected in various cancers and immortalized cell lines261,264,265.
MCL1
The BH3-containing member of the Bcl-2 family, MCL1, has three major isoforms which differ in their apoptotic potential266,267. Full length MCL1-L includes exons 1 to 3 and displays anti-apoptotic activity. Skipping of exon 2 introduces a PTC, leading to the MCL1-S isoform, which contains the BH3 domain but lacks the BH1, BH2, and transmembrane domains. Finally, truncation of exon 1 produces the MCL1-ES isoform which maintains the BH1-BH3 and transmembrane domains but lacks the PEST domain, a site of cleavage and phosphorylation for caspases268. SRSF1 is one of the regulators of MCL1 splicing in cancer cell lines269. Increased expression of Mcl-1L is observed in oral cancers and basal cell carcinoma compared to normal tissues270,271. High MCL1-L expression correlates with increased tumor size and decreased survival in oral cancers272, as well as with resistance to treatment in oral squamous cell carcinoma273. Interestingly, melanocytes upregulate MCL-1L in response to UVB radiation, which protects them against apoptosis; whereas melanoma cell lines that have elevated MCL1-L expression without UV exposure are resistant to apoptosis274. Finally, MCL1-ES differs from other Bcl-2 family members in that it does not depend on BAX/BAK homodimerization for apoptotic activity. Interestingly, MCL1-ES neutralizes the effects of MCL1-L, and MCL-1ES apoptotic activation is enhanced by MCL1-L expression275. Conversely, MCL1-S apoptotic activity is inhibited by MCL1-L266, suggesting that the different isoforms promote apoptosis via distinct mechanisms.
Isoforms rewiring cell metabolism
PKM
The two isoforms of pyruvate kinase, a key glycolytic enzyme, are formed by the splicing of one of two mutually exclusive exons that share 56 amino acids but differ at 22 residues276. Inclusion of PKM exon 9 produces the constitutively active PKM1, while inclusion of exon 10 encodes PKM2. Both PKM isoforms perform the same catalytic function, but PKM2 can switch between the active and inactive state276. PKM2 expression is regulated either by repressing inclusion of exon 9 via binding of PTBP1, hnRNPA1, or hnRNPA2, or by promoting exon 10 inclusion via binding of SRSF3. Both splicing events increase expression of PKM2 relative to PKM173,148,150. PKM2 is detected in most embryonic as well as proliferating adult tissues, with the exception of muscle, brain and bladder, which express only PKM1276. Increased PKM2 levels are reported in many human solid tumors and correlate with decreased patient survival, advanced stage and poor prognosis285. PKM2 knockdown inhibits tumor progression and metastasis in vivo and in vitro in ovarian, gastric, colon, liver, and esophageal cancer models277,278,282,285. Conversely, cancer cells engineered to express PKM1 in place of PKM2 convert from aerobic glycolysis to mitochondrial respiration and are unable to form tumors after xenotransplantation286,287. However, other studies suggest that PKM2 is not necessary for tumor growth in colon cancer cell lines or in a breast cancer mouse model288–290. PKM2 plays a role in cancer metabolism and activates the PI3K/Akt pathway291. The ability to inhibit PKM2 activity is important for cell proliferation in vivo and in vitro292, and allows cells to respond to signaling and environmental cues276.
Isoforms promoting angiogenesis
VEGFA
The growth factor VEGFA stimulates blood vessel formation by promoting proliferation and migration of endothelial cells. The VEGFA transcript undergoes alternative splicing in two distinct regions to produce protein isoforms of variable length. Inclusion of variable exons 6a, 6b, 7a, or 7b encodes the VEGFAxxx isoforms, where ‘xxx’ refers to the final number of amino acids293. In addition, inclusion of variable exon 8b, instead of exon 8a, at the 3′ end of the transcript produces the anti-angiogenic VEGFAxxxb isoforms294. VEGFAxxxb splicing is promoted by SRSF6 overexpression, whereas SRSF1 and SRSF5 overexpression promote VEGFAxxx295. Adult human tissues express predominantly anti-angiogenic VEGFxxxb isoforms, such as the common VEGF165b isoform, but their expression often decreases as tumors progress296. Decreased expression of VEGF165b is found in human metastatic melanoma and prostate tumors297,298. A shift in expression from VEGF165b to VEGF165 occurs in colon and squamous cell carcinoma tumors80,299. However neither the expression of VEGFAxxx nor of VEGFAxxxb correlates with patient survival in head and neck tumors300. Finally, overexpression of VEGF165b reduces tumor growth in mouse xenograft models of colon, renal, prostate, or soft tissue tumors299,301,302. In vitro VEGFA165b binds to the same receptor as do VEGFA165 and with the same affinity; however, VEGFA165b is unable to stimulate the VEGF signaling pathway. Thus, anti-angiogenic VEGFAxxxb isoforms can inhibit VEGFAxxx-mediated angiogenesis298.
Isoforms enabling cell invasion and metastatic dissemination
CD44
CD44 is a transmembrane glycoprotein that binds hyaluronic acid and functions in cell division, survival, and adhesion. The CD44s isoform contains exons 1–5 and 16–20, whereas inclusion of any of the variable exons 6–10 generates one of the CD44v isoforms303. Regulators of CD44 alternative splicing include ESRP1, hnRNPA1, and SRSF2, all of which promote CD44v splicing126,304–306, whereas hnRNPL inhibits CD44v expression307. CD44v isoforms are expressed in both normal and tumor tissues, but their expression frequently increases in gastric, ovarian, bladder, colon, and prostate tumors304,308–314. CD44v expression is often associated with tumor progression, is frequently found in recurrent tumors, and correlates with increased grading312,313,315. The role of CD44v isoforms in tumor progression remains a topic of discussion. Expression of exogenous CD44v8-10 increases tumor initiation frequency in gastric cancer models309, and, similarly, CD44v9 facilitates invasion in prostate cancer312,313. However, some ovarian tumors express higher levels of CD44s than CD44v, and patients expressing CD44v8-10 have longer survival rates311,316. Finally, EMT not only affects, but is also affected by CD44 isoform expression. Epithelial cells express predominantly CD44v, but switch to CD44s after undergoing EMT in breast and ovarian tumor models304,311. CD44s is required for EMT in breast and ovarian cancer models, and its expression enhances migration304,316. CD44s expression also induces a mesenchymal phenotype, increases cell invasion, and results in poor differentiation and distant metastasis in gallbladder cancer models317. However, CD44v expressing gallbladder cancer cells are still highly tumorigenic even though exhibit decreased invasive potential317.
ENAH
ENAH, also known as Mena, regulates actin nucleation and polymerization and modulates cell morphology and motility. Splicing of the ENAH transcript generates three main isoforms, which play different roles in tumor progression. Inclusion of exon INV produces MENA-INV, inclusion of exon 11a produces MENA11a, and skipping of exon 6 produces MENAΔv6318. Alternative splicing of MENA is regulated by ESPR1 and ESPR2319. The ratio of Mena isoforms varies between normal and tumor tissues. For example, breast tumors express high levels of Mena11a or MenaINV, while limited to no expression of these isoforms is detected in normal tissue. In addition, MenaINV expression increases with tumor grade, metastasis and tumor progression, and is accompanied by a decrease in Mena11a320–322. Expression of both pan-Mena and Mena11a increases in lung tumors compared to normal tissue; however, low Mena11a expression correlates with decreased survival rates in lung cancer patients, and patients expressing high levels of Mena11a do significantly better323. Expression of Mena11a also correlates with epithelial markers and decreased invasion, whereas MenaINV and MenaΔv6 expression correlate with mesenchymal markers, and increased invasion and metastasis318,321,323–325. Knockdown of Mena11a in breast cancer cell lines decreases cell migration, and ectopic Mena11a expression reduces lamellipodia protrusion319.
MSTR1 (alias RON)
The receptor tyrosine kinase RON (MSTR1) is a member of the MET proto-oncogene family, which is implicated in tumor progression. Exons 5, 6, 11, and 19 undergo alternative splicing to produce four isoforms: RONΔex11 (RONΔ165), RONΔex5-6 (RONΔ160), RONΔex5-6-11 (RONΔ155), and RONΔex19 (RONΔ170)326,327. Splicing of RONΔ165 is regulated by SRSF159,328. RONΔ165, RONΔ160 and RONΔ155 are constitutively active, whereas RONΔ170 is a kinase-defective isoform that inhibits tumorigenesis by other active RON isoforms326,327,329. RONΔ160 likely exerts its tumorigenic potential by increasing β-catenin expression330. RONΔ165, RONΔ160, or RONΔ155 are expressed in human primary colon, ovarian, breast, and brain tumors, as well as in gastric and lung cancer cell lines59,328,329,331–333. Ectopic expression of RONΔ160 or RONΔ155 promotes tumor formation and lung metastasis in NIH3T3 xenograft mouse models331. Additionally, expression of RONΔ160 or RONΔ155, but not RONΔ165, induces anchorage-independent growth in colon cancer cells lines330. However, RONΔ165 expression increases motility and invasiveness in cancer cell lines59,329.
RAC1
Rac1 is a member of the Rho GTPase family, which is involved in signaling for cell motility and proliferation. Inclusion of RAC1 exon 3b produces the constitutively active RAC1b isoform, which contains 19 additional amino acids behind the switch II domain, a region important for Rac1 interaction with regulators and effectors334,335. SRSF1 is one of the regulators of RAC1 alternative splicing336. Rac1b has accelerated guanosine diphosphate (GDP)/guanosine triphosphate (GTP) exchange and impaired GTP hydrolysis, thus leading to prolonged signaling activity335,337,338. Furthermore, RAC1b is unable to interact with RHO-GDI, to signal downstream PAK1 and JNK kinases, or to activate the RelB pathway339,340, but can negatively regulate RAC1 activity341. RAC1b is expressed in breast, thyroid, colorectal, and lung tumors336,342–344. Increased expression of RAC1b in thyroid tumors correlates with metastasis and poor clinical outcome343. Rac1b expression in colon and thyroid cancer cell lines sustains cell survival by stimulating G1/S progression and protecting cells from apoptosis345–347.
KLF6
KLF6 belongs to the Kruppel-like family of transcription factors which regulate cell proliferation, differentiation, and survival. KLF6-SV1 uses an alternative 5′SS that causes a frame-shift and produces a protein isoform that contains 21 novel amino acids but lacks all three of the zinc finger domains348. Alternative splicing of KLF6 is regulated by SRSF1, TGF-β1, and Ras signaling349,350. Increased KLF6-SV1 expression is observed in prostate, lung, ovarian, brain, breast, pancreatic, and liver tumors, and correlates with poor patient survival349,351–356. Full-length KLF6-FL can act as a tumor suppressor, whereas KLF6-SV1 is oncogenic. KLF6-SV1 knockdown in lung, ovarian, colon, and brain cancer cells increases apoptosis, whereas its overexpression promotes proliferation and survival349,352,353. Expression of KLF6-SV1 increases cell survival, migration and invasion in breast cancer cell lines, but has no effect on proliferation354. Ectopic KLF6-SV1 expression does not alter tumor size, but increases metastasis incidence, in mice xenograft experiments354. Furthermore, KLF6-SV1 knockdown prevents tumor formation, while knockdown of the full-length isoform increases tumor growth, in ovarian cancer xenograft models. Finally, expression of KLF6-FL is associated with epithelial markers, whereas KLF6-SV1 is associated with a mesenchymal phenotype354.
Isoforms enabling drug resistance
BCL2L11 (alias BIM)
The BH3-only protein, BIM, is a pro-apoptotic protein encoded by BCL2L11. The three major isoforms, BIM-EL, BIM-L, and BIM-S, are pro-apoptotic but differ in their activity, BIM-S being most active357. In addition, two isoforms, BIMγ1 and BIMγ2, are generated by alternative splicing of exon 3, which contains a stop codon and results in a truncated protein that lacks the BH3 domain and thus lacks the pro-apoptotic activity58. SRSF1-overexpression induces alternative splicing of BIM to promote BIMγ1 and BIMγ2 splicing58. PTBP1 and hnRNPC promote exon 3 skipping and expression of the pro-apoptotic BIM isoforms358. The BIMγ isoforms are expressed in leukemia, lung, and breast cancer cells58,359,360. Ectopic expression of BIMγ1 reduces apoptosis levels in mammary epithelial cells58. Finally, expression of BIM isoforms has been linked to drug response in tumors. High levels of BIM-EL correlate with a better induction of apoptosis in response to tyrosine kinase inhibitors in EGFR-mutant lung and HER2-amplified breast tumor models and predict responses in treatment-naïve patients361. In addition, expression of BIMγ isoforms in lung cancer patients with a BIM polymorphism increases resistance to tyrosine kinase inhibitors360.
HER2 (alias ErbB2)
HER2 is a tyrosine kinase from the EGFR family, frequently amplified or overexpressed in breast tumors. Skipping of exon 20 encodes d16HER2, a constitutively active protein that lacks 16 amino acids in the extracellular domain, and is primarily detected in breast tumors362–364. Alternative splicing of HER2 exon 20 is regulated by SRSF3 and hnRNPH1365. Expression of d16HER2 increases proliferation, induces EMT and invasion, and decreases sensitivity to the HER2-targeting antibody trastuzumab363,366–370. d16HER2 expression allows breast cancer cells to evade trastuzumab-induced apoptosis by upregulating Bcl-2 and activating SRC, a kinase involved in proliferation and migration370,371.
SPLICING ALTERATIONS AND THE TUMOR MICROENVIRONMENT
Our current understanding of RNA splicing alterations relies on the expression of splicing isoforms and their regulators in tumor cells. However, solid tumors are composed of a mixture of cell types in addition to cancer cells, including fibroblasts, various immune cell types, and endothelial cells, all of which influence tumor progression and drug responses372. Although cell-type specific splicing has been described, we know very little about splicing alterations in these cell types in the tumor context.
Matrix stiffness and composition affects RNA splicing
The local microenvironment, or niche, plays important roles in cell fate, cancer onset, and malignant evolution373. A major component of the niche is the extracellular matrix (ECM), a complex network of macromolecules with distinctive physical, biochemical, and biomechanical properties that undergoes remodeling during metastasis. Yet, it remains unclear how the ECM composition impacts splicing isoforms and their regulators during tumor progression and metastasis. Interestingly, cells grown in 3D cultures on an ECM exhibit different splicing profiles compared to the same cells grown on plastic, suggesting that ECM stiffness and composition can influence splicing choices64,374. Matrix stiffness can alter splicing, for example, through differential phosphorylation and activation of splicing regulators from the SR protein family375. Additionally, signaling through ECM proteins and integrin engagement can impact tumor initiation and metastasis, and can also selectively alter splicing. For example, laminin 511 promotes self-renewal and tumor initiation by engaging the α6Bβ1 integrin splice variant376. The expression of the α6Bβ1 isoform is repressed by the splicing factor ESRP1 and depends on VEGF autocrine signalling257. Furthermore, ECM proteins are themselves regulated by splicing, and often undergo splicing switches during tumor progression. For example, the oncofetal ED-A and pro-angiogenic ED-B fibronectin isoforms differ in their integrin binding domain and show differential assembly into fibrils377,378. Malignant cells express high levels of ED-A fibronectin and its receptor, α5β1 integrin, both of which have been linked to radiation resistance379. Interestingly, aberrant ECM can also alter fibronectin splicing in non-malignant cells379. Similarly, tenascin-c expresses unique alternative splice forms in breast tumors380. In both patient samples and cell culture models, these ECM splicing isoforms have been linked to invasiveness379. Osteopontin SPP1 is another ECM protein that is overexpressed in various cancers and promotes oncogenic features. Splicing of SPP1 generates multiple isoforms that play a role in cancer development and progression through their surface receptors CD44 and integrins381. Finally, metastatic lesions exhibit alterations in splicing isoforms that impact cell polarity, cell-cell interactions, and EMT. Examples of metastasis-specific splicing events include isoforms of fibronectin FN1, Tenascin C TNC, CD44, ENAH, and RAC1321,382,383. Alterations in upstream splicing regulators that control metastasis-associated splicing isoforms are found in human tumors115,128.
Splicing isoforms and immune cell functions
Other key components of the tumor microenvironment are immune cells, which can either promote or inhibit tumor growth384. RNA splicing controls multiple regulatory steps in immune cell development and function385. Transcriptome-wide studies identified a repertoire of splicing isoforms expressed in specific immune-cell types, and linked many of these events with lineage differentiation386; however, it is not known how changes in the immune cell repertoire impact splicing patterns in human tumors.
Alternative splicing plays a role in the control of innate immunity. For example, SF3A1 regulates the splicing of genes involved in Toll-like receptor (TLR) signaling in macrophages, and controls the production of positive regulators of TLR signaling, IRAK1, CD14, and IKKβ, as well as negative regulators sTLR4 and Rab7b387. Another example is the splicing of the MyD88s isoform, which limits innate immune activation downstream of TLR signaling. MyD88s splicing is controlled by Eftdu2, SF3A1, and SF3B1388,389. Moreover, inclusion of an alternative TLR4 exon generates a soluble isoform that inhibits TNF-α and NF-κB signaling in macrophages, thereby acting as a negative feedback mechanism. Similarly, soluble isoforms of membrane receptors, such as IL-4R, -5R, and -6R, are frequently generated by splicing in immune cells385. Furthermore, alternative splicing plays a role in class switch from IgM to IgD during B-cell differentiation and also impacts the generation of a secreted form of IgM385. In addition, splicing can increase the transcript diversity of IgE by generating isoforms that are either secreted or membrane-bound. Finally, loss of the RNA-binding protein HuR results in defective class-switching and leads to B-cell death390,391.
The best-studied examples of functional splicing events in T-cell differentiation are the cell-surface glycoproteins CD44 and CD45 (PTPRC). Splicing of CD44, which is regulated by Sam68, produces an alternative CD44v isoform that is involved in both lymphocyte activation and metastasis, as described above385. CD45 isoforms, named RA/RB/RC/RO, are expressed in different patterns in functionally distinct T-cell populations, and CD45 splicing serves as a feedback mechanism to maintain T-cell homeostasis385. Briefly, naïve T-cells express a CD45 isoform that includes at least one of the variable exons 4, 5, and 6, each which encode an extracellular domain that is heavily glycosylated and thus prevents CD45 homodimerization392. Upon T-cell activation, skipping of CD45 variable exons allows homodimerization at the cell surface, which leads to an inactive form and decreased signaling through the T-cell receptor. HNRNPLL, HNRNPL, SRSF2, PTBP1, HNRNPE2, and HNRNPA1 have been all implicated in the regulation of CD45 splicing393–397. Moreover, stimulation of the T-cell receptor induces splicing changes in immune-related targets including CD45, Fyn, TRAF3, BRD8, and TRIM397–399.
SPLICING MODULATION AS CANCER THERAPEUTICS
Modulation of RNA splicing can provide novel therapeutic targets for oncology. Splicing modulation can be achieved either by fine-tuning the level or activity of splicing regulators, thus affecting the network of their downstream splicing targets, or by precisely targeting a single spliced isoform expressed in cancer cells (Figure 6).
Figure 6. Therapeutic strategies to target splicing alterations in tumors rely either on broad-spectrum splicing inhibition or on isoform-specific modulation.

(A) Small molecules targeting components of the spliceosome (e.g., SF3B1, or tri-sRNP) block their activity by preventing assembly of a functional spliceosome into the pre-mRNA, and thus globally inhibit splicing. Alternatively, broad splicing inhibition can be achieved by targeting the enzymes that modulating the activity of splicing regulatory factors (SF), using for example small molecules inhibitors of CLKs or SRPKs, two kinase families that regulate the phosphorylation and thus the activity of SR proteins. Compounds that affect splicing factor poly-ubiquitination and proteasomal degradation (e.g. sulfonamides) can also induce broad changes in splicing profiles. On the other hand, isoform specific inhibition can be achieved by using splice-switching antisense oligonucleotides (ASOs) that bind in a sequence-specific manner and modulate the outcome of a specific splicing isoform. (B) ASOs can promote exon skipping or inclusion by blocking the 5′SS, an exonic silencer (ESS), or enhancer element (ESE) or by preventing the usage of a mutant (MUT)/cryptic splice site. See text for details. (C) Properties of ASO chemistries are currently used for splicing-modulation. See text for details. 2′-MOE/PS: 2′-O-(2-methoxyethyl)/phosphorothioate; 2′-OMe/PS: 2′ O-methyl/phosphorothioate; PMO: phosphorodiamidate morpholino oligomer; LNA: locked nucleic acid.
Small molecules modulating the activity or levels of splicing regulators
Compounds that affect global splicing efficiency or SS selection have been identified over the years and their number is steadily increasing400. The molecular mechanisms of action of these agents are progressively being elucidated. The first group of compounds that impact splicing includes spliceostatins, sudemycins, and FD-895 and its parent pladienolides molecules or their derivatives (e.g. E7107 or FR901464), which all act directly on the core spliceosomal component SF3B1401,402,403,404 (Figure 6A). Interestingly, only a fraction of the splicing events (~10%) are affected by SF3B1 inhibition, suggesting that some SS, likely weak SS, are more sensitive than others to spliceosomal inhibitors17,29. Isoginkgetin, another splicing inhibitor, acts by preventing recruitment of the U4/U5/U6 tri-snRNPs, which leads to accumulation of the spliceosomal complex A405. Additionally, several small molecules alter the activity of splicing-factors, for example by targeting their regulatory kinases. Molecules such as NB-506, SRPIN34, diospyrin D1, and TG003 reduce SR-protein phosphorylation through the inhibition of proteins from the SRPK, CLK, DYRK, or topoisomerase families and thus modulate splicing of SR-protein targets406,407. Finallysulfonamides, a class of cancer drugs that achieve efficacy in a subset of cancer patients, have been recently shown to act by reducing the expression of splicing factor RBM39 (alias CAPERα) through a novel mode of targeted proteasomal degradation408,409. Treatment of cancer cell lines with sulfonamides triggers the association of RBM39 with the CUL4- DCAF15 ubiquitin ligase, leading to RBM39 poly-ubiquitination and proteasomal degradation408,409. Degradation of RBM39 leas to aberrant pre-mRNA splicing in a set of target genes408,409. Interestingly, DCAF15 expression and copy number correlated with sulfonamides sensitivity, suggesting that regulators of splicing-factor degradation could constitute promising drug targets.
While effective, these small molecules often lack specificity, and their exact mechanisms of actions are not always well understood, which could potentially lead to off-target effects and limit their clinical application. Interestingly, in vitro and in vivo data suggest that cancer cells are more sensitive than normal cells to global splicing inhibition, thus providing a therapeutic window that could be exploited even when using broad-spectrum splicing inhibitors151,152,410,411. Even though initial trials of an SF3B1 inhibitor, E7107, in solid tumors were suspended due to unexpected toxicity, newer inhibitors, such as H3B-8800, are currently being tested in phase I trials for hematological malignancies412,413.
Splice-switching RNA-based therapeutics
RNA-based therapeutics offer the potential to target virtually any molecule, especially those lacking a catalytic activity that could be inhibited, or those not amenable to targeted antibody approaches414. FDA approval of Spinraza™, which is the first splicing-correcting therapy and uses antisense oligonucleotides (ASO) to treat spinal muscular atrophy (SMA), has opened the field for RNA-based approaches to target splicing defects415. Splice-switching ASOs are 15- to 30-mer long chemically modified RNA molecules that can redirect a specific splicing event in order to prevent the production of a truncated or mutated protein, or to generate a specific protein isoform. Their specificity comes from their complimentary binding to a unique sequence on the mRNA, thus affecting only the targeted spliced isoform. Splice-switching ASOs can be designed to specifically target (i) a 5′ or 3′SS, thus blocking its usage, (ii) a splicing enhancer sequence, thus preventing binding of a splicing activator and promoting exon skipping, or (iii) a splicing silencer sequence, thus preventing binding of a repressor and promoting exon inclusion416 (Figure 6B). Another splice-switching strategy is the use of bifunctional oligonucleotides made of an antisense portion that determines target specificity, and a non-hybridizing tail that recruits proteins or RNA/protein complexes that modulate SS selection417–419.
Natural unmodified DNA or RNA oligonucleotides are vulnerable to nuclease degradation and are unstable in vivo. Chemical modification of the phosphate backbone and/or the ribose ring can produce stable molecules with high substrate specificity, low toxicity, low immunogenicity, and that limit RNAse H degradation420. ASO designed to activate RNAse H cleavage will not be discussed here as they do not modulate alternative splicing but trigger degradation of their mRNA target. Several distinct ASO chemistries are currently used for splicing-modulation (Figure 6C). A common backbone modification uses phosphorothioates (PS) at the nucleotide link420. PS-ASOs are more hydrophobic, more nuclease resistant, and bind with higher affinity than ASOs with unmodified phosphodiester linkages421. PS-ASOs are often combined with ribose modifications such as 2′-O-(2-methoxyethyl) (2′-MOE) or 2′ O-methyl (2′-OMe)420. Uniformly modified 2′-MOE/PS ASOs are effective when administered in saline by nearly all routes of administration and their tissue half-lives ranges from 2 to 4 weeks, but can even achieve 6 months in the central nervous system421. Another type of modification uses locked nucleic acid (LNA), which increases binding affinity and reduces off-target effects by allowing the usage of shorter sequences that are less likely to partially hybridize to non-target sequences416. A distinct class of backbone chemistry uses phosphorodiamidate linkages in morpholino oligomers (PMO or morpholino)416. PMOs are neutrally charged and provide better specificity and display lower toxicity than PS-ASOs. However, PMOs often need to be conjugated to a delivery moiety for in vivo delivery. Finally, peptide nucleic acid (PNA) offer specificity similar to PMO, but their low water solubility limits their use416.
However, efficient delivery to the target organ still remains one of the major challenges in the field of RNA-based therapeutics. The two challenging steps involve getting the ASO to the tissue of therapeutic interest and then delivering it to the correct intracellular compartment422. In addition to naked formulations, ASO modifications, carriers and other approaches are currently being tested to increase splicing efficiency, lower the dosage, enable tissue-specific delivery, and limit toxicity and off-target effects416. In vivo, ASOs can be injected either systemically, or directly into the specific organ where the correction needs to be achieved416. For example, the FDA-approved 2′-MOE/PS ASO, SpinrazaTM, is delivered intrathecal in saline, and achieves a 4–6 months half-life in the cerebrospinal fluid after initial clearance423. Eteplirsen™, the first splice-switching PMO to received FDA-approval for Duchenne Muscular atrophy is delivered by intravenous infusion. Renal clearance plays a major role in ASOs pharmacokinetics and biodistribution422. PS-ASOs bind to plasma proteins and slow their renal clearance, thus allowing broader tissue distribution, whereas uncharged PMO are cleared much faster and accumulate at lower levels422. Finally, efforts to deliver ASOs to specific tissues are ongoing422. The most promising targeted approach utilizes ASOs conjugated with an N-acetylgalactosamine (GalNac) that allows effective uptake by hepatocytes via an asialoglycoprotein receptor dependent mechanism422. Novel ASO delivery strategies are rapidly emerging. Yet, ASOs delivery to tumors will certainly face similar challenges as the delivery of other cancer drugs and will require further optimization to efficiently delivery therapeutics to cancer patients.
ASO-mediated correction of cancer-associated splicing isoforms can be achieved in vitro in human cell lines and in vivo in xenograft tumor models (Table 1). For example, ASO targeting of a splicing enhancer that regulates inclusion of exon 23 of the transcription factor STAT3 can shift expression from the STAT3α to the STAT3β isoform424. Induction of STAT3β, an isoform that lacks the C-terminal transactivation domain, leads to apoptosis and cell-cycle arrest in breast cancer cells, as well as to tumor regression in xenograft breast cancer models424. Another example is the ASO-mediated skipping of MDM4 exon 6 to decreases MDM4 protein abundance, an oncoprotein that inhibits p53-mediated tumor suppression425. Tumors express high levels of MDM4 as a result of a splicing switch between the NMD-degraded MDM4-S isoform expressed in normal cells, and the full-length exon 6-containing MDM4-L isoform produced in cancer cells. Skipping of MDM4 exon 6 decreases tumor growth in patient-derived xenograft models of melanoma and lymphoma425. RNA-based therapeutics are currently being tested in the clinic in lymphoma and lung cancer patients to downregulate STAT3 expression426.
Table 1.
Cancer-associated human isoforms targeted by splice-switching ASO
| Gene name | ASO chemistry | Type of splicing correction | Tumor type | Tested in cell lines | Tested in vivo | References |
|---|---|---|---|---|---|---|
| STAT3 | PMO | exon 23 skipping | breast | √ | √ | 424 |
| MDM4 | PMO | exon 6 skipping | skin, lymphoma | √ | √ | 425 |
| ERBB4 | LNA | exon 26 skipping | breast | √ | √ | 444 |
| BCL2L1 | 2′-MOE/PS | exon 2 skipping | skin | √ | √ | 239,240,445 |
| GLDC | 2′-MOE/PS | exon 7 skipping | lung | √ | √ | 446 |
| PKM2 | 2′-MOE/PS | exon 9 inclusion | brain | √ | n.d. | 447 |
| MCL1 | PMO | exon 2 skipping | skin | √ | n.d. | 271 |
| MDM2 | PNA | exon 4 skipping | uterine | √ | n.d. | 448 |
| BRCA2 | 2′-OMe/PS | cryptic exon skipping | breast | √ | n.d. | 449 |
| IL5R | 2′-MOE/PS | exon 5 skipping | lymphoma | √ | n.d. | 450 |
| FGFR1 | PMO | exon α inclusion | brain | √ | n.d. | 451 |
| MSTR1 | PMO | exon 11 skipping | breast and stomach | √ | n.d. | 452 |
| USP5 | PMO | alternative 5′SS | brain | √ | n.d. | 453 |
PMO: phosphorodiamidate morpholino oligomer; LNA: locked nucleic acid; PNA: peptide nucleic acid; 2′-MOE/PS: 2′-O-(2-methoxyethyl)/phosphorothioate; 2′-OMe/PS: 2′ O-methyl/phosphorothioate; n.d.: not determined.
Cancer drugs affecting RNA splicing
Alternative splicing is modulated by a variety of cellular responses, including body temperature changes, circadian rhythm, exposure to radiations, as well chemotherapies427–430. Transcriptome-wide studies identified a repertoire of splicing isoforms expressed after treatment with the cancer drugs camptothecin, doxorubicin, or cisplatin82,431–433. A large fraction of these transcripts function in pathways frequently disrupted in cancer, i.e., cell cycle, DNA repair, genetic instability, and replicative immortality429. Additionally, treatment with gemcitabine, a first line chemotherapy for pancreatic cancer, leads to drug-resistance and is associated with a splicing switch to the oncogenic isoforms MKNK2-b and PKM2, as well as with the upregulation of SRSF1 and PTBP1434. Finally, cancer drugs can be combined with splicing-modulating compounds; for example amiloride potentiates the effect of imatinib in CML, and sudemycin enhances the effects of ibrutinib in CLL435.
Interestingly, changes that affect SS selection can affect resistance to targeted cancer therapies. For example, treatment with vemurafenib, a BRAFV600 inhibitor, selects resistant cells expressing an alternatively spliced BRAF isoform that lacks the RAS-binding domain that normally regulates BRAF dimerization and activation436. Similarly, the BRCA1Δ11q isoform, a variant lacking the majority of exon 11, promotes resistance to PARP inhibition and cisplatin437. Moreover, expression of the oncogenic BARD1β splicing isoform impairs homologous recombination and sensitize colon cancer cells to PARP inhibition even in BRCA1 wild-type cells438. Finally, the selection for pre-existing alternatively spliced CD19 isoforms bearing a compromised epitope explains resistance to CART-19 immunotherapy in B-ALL patients439.
CONCLUSION
Since RNA splicing was discovered forty years ago, our understanding of its role in human diseases has been expanding, but many questions remain unanswered. The recent years have undoubtedly shown that alterations in RNA splicing are frequent in tumors and contribute to disease pathogenicity. Cancer screening panels currently include splicing-factor mutations among the mutated genes in hematological malignancies. Alterations in splicing-factor levels and dysregulation of downstream splicing targets are tumor characteristics shared by many cancers. Interestingly, these factors can act either as tumor suppressors or as oncogenes, depending on the tumor type, suggesting cell-type-specific functions and targets. Splicing alterations represent a novel and rich source of potential therapeutic targets, and several clinical trials are currently testing them in cancer patients, such as SF3B1 inhibitors in MDS patients as described above412,413. The advances in RNA-based therapeutics will likely accelerate the development of splicing-modulating compounds as cancer therapeutics. Additionally, advances in other fields may be applied to address current challenges in delivery and efficacy of RNA-based therapeutics. For example, specific delivery to leukocytes can been achieved by loading siRNA onto lipid-based nanoparticles coated with anti-CD38 monoclonal antibodies440. This approach was proven effective at inhibiting cyclin D1 in vivo, suppressing tumor growth and prolonging survival of mice xenografted with human lymphoma cells440, thus opening a new avenue for the treatment of hematological malignancies. Similar targeted strategies could be utilized to deliver splice-switching ASOs to the cells of therapeutic interest and increase their efficacy in tumors.
Alterations in splicing-factor levels are often detected in human tumors, yet only a fraction of these tumors exhibit copy number changes. Thus understanding the transcriptional and post-transcriptional regulation of splicing factors is critically needed to open new direction for drug targets. The pathways that control splicing-factor homeostasis in relevant normal or tumor tissues are not well understood, and it remains unclear how they become dysregulated in tumors. Another underexplored area is how the coupling of alternative splicing with NMD impacts splicing-factor regulation in tumors. Given that cancer cells exhibit differences in the regulation of NMD, the link between these two regulatory pathways in tumorigenesis warrants further attention and may provide novel therapeutic opportunities441,442. In addition, tumors often exhibit alterations in multiple splicing factors, and thus understanding the regulatory networks of RNA-binding proteins and their targets will be crucial for the development of effective splicing-factor inhibitors.
Importantly, all previous studies exploring the role of splicing in human cancer are based on bulk tumor material, which contains a majority of tumor cells together with other cell types that have been shown to impact tumor development and drug response. Yet, whether oncogenic splicing isoforms are present in each individual cell type remains unknown. Variations in splicing patterns have rarely been studied at the single-cell level, and these differences have the potential to contribute to the heterogeneity in drug response. Interestingly, a bimodal variation in splicing patterns was observed among single dendritic cells, suggesting that single cells can exhibit distinct splicing isoforms443. Dissecting splicing heterogeneity in tumors at the single-cell level will likely be required for to ensure the success of future splicing-modulating cancer therapies.
Acknowledgments
We thank Stephen Sampson and Juergen Scharner for reading and editing the manuscript, and Matt Wimsatt for assistance with graphical art in Figure 1. This work was supported by the National Cancer Institute (R00CA178206 to OA) and The Jackson Laboratory. The authors have no conflict of interest to declare.
ABBREVIATIONS
- ASO
antisense oligonucleotides
- BH
Bcl-2 homology domain
- BPS
branch point site
- CLL
chronic lymphocytic leukemia
- CMML
chronic myelomonocytic leukemia
- ECM
extracellular matrix
- EMT
epithelial-mesenchymal transition
- HEAT
Huntington, Elongation Factor 3, PR65/A, TOR domain
- lncRNA
long non-coding RNA
- MDS
myelodysplastic syndromes
- miRNA
microRNA
- NMD
nonsense-mediated decay
- PMO
phosphorodiamidate morpholino oligomer
- PTC
premature termination codon
- RARS
refractory anemia with ringed sideroblasts
- RCMD-RS
refractory cytopenia with multilineage dysplasia and ringed sideroblasts
- RNA
ribonucleic acid
- RRM
RNA-recognition Motif
- SMA
spinal muscular atrophy
- SR
serine/arginine-rich
- sRNA
small nuclear RNA
- snRNP
small nuclear ribonucleoproteins
- SS
splice site
- TLR
Toll-like receptor
References
- 1.Biamonti G, Catillo M, Pignataro D, Montecucco A, Ghigna C. The alternative splicing side of cancer. Semin Cell Dev Biol. 2014;32:30–36. doi: 10.1016/j.semcdb.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 2.Bertram K, et al. Cryo-EM Structure of a Pre-catalytic Human Spliceosome Primed for Activation. Cell. 2017;170(4):701–713 e711. doi: 10.1016/j.cell.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 3.Hegele A, et al. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell. 2012;45(4):567–580. doi: 10.1016/j.molcel.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X, Yan C, Hang J, Finci LI, Lei J, Shi Y. An Atomic Structure of the Human Spliceosome. Cell. 2017;169(5):918–929 e914. doi: 10.1016/j.cell.2017.04.033. [DOI] [PubMed] [Google Scholar]
- 5.Wahl MC, Luhrmann R. SnapShot: Spliceosome Dynamics I. Cell. 2015;161(6):1474–e1471. doi: 10.1016/j.cell.2015.05.050. [DOI] [PubMed] [Google Scholar]
- 6.Climente-Gonzalez H, Porta-Pardo E, Godzik A, Eyras E. The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 2017;20(9):2215–2226. doi: 10.1016/j.celrep.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida K, Ogawa S. Splicing factor mutations and cancer. Wiley Interdiscip Rev RNA. 2014;5(4):445–459. doi: 10.1002/wrna.1222. [DOI] [PubMed] [Google Scholar]
- 8.Kfir N, Lev-Maor G, Glaich O, Alajem A, Datta A, Sze SK, Meshorer E, Ast G. SF3B1 association with chromatin determines splicing outcomes. Cell Rep. 2015;11(4):618–629. doi: 10.1016/j.celrep.2015.03.048. [DOI] [PubMed] [Google Scholar]
- 9.Malcovati L, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011;118(24):6239–6246. doi: 10.1182/blood-2011-09-377275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papaemmanuil E, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi D, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118(26):6904–6908. doi: 10.1182/blood-2011-08-373159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 14.Alsafadi S, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7:10615. doi: 10.1038/ncomms10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Darman RB, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015;13(5):1033–1045. doi: 10.1016/j.celrep.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 16.Dolatshad H, et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia. 2015;29(8):1798. doi: 10.1038/leu.2015.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obeng EA, et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell. 2016;30(3):404–417. doi: 10.1016/j.ccell.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin S, et al. Splicing factor SF3B1K700E mutant dysregulates erythroid differentiation via aberrant alternative splicing of transcription factor TAL1. PLoS One. 2017;12(5):e0175523. doi: 10.1371/journal.pone.0175523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quesada V, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44(1):47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 20.Te Raa GD, et al. The impact of SF3B1 mutations in CLL on the DNA-damage response. Leukemia. 2015;29(5):1133–1142. doi: 10.1038/leu.2014.318. [DOI] [PubMed] [Google Scholar]
- 21.Biankin AV, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maguire SL, et al. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol. 2015;235(4):571–580. doi: 10.1002/path.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45(2):133–135. doi: 10.1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong Y, Krauthammer M, Halaban R. Rare SF3B1 R625 mutations in cutaneous melanoma. Melanoma Res. 2014;24(4):332–334. doi: 10.1097/CMR.0000000000000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin M, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45(8):933–936. doi: 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robertson AG, et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell. 2017;32(2):204–220 e215. doi: 10.1016/j.ccell.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng X, Zhan Z, Naren D, Li J, Yan T, Gong Y. Prognostic value of SRSF2 mutations in patients with de novo myelodysplastic syndromes: A meta-analysis. PLoS One. 2017;131(6):621–635. doi: 10.1371/journal.pone.0185053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meggendorfer M, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML) Blood. 2012;120(15):3080–3088. doi: 10.1182/blood-2012-01-404863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim E, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell. 2015;27(5):617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komeno Y, et al. SRSF2 Is Essential for Hematopoiesis, and Its Myelodysplastic Syndrome-Related Mutations Dysregulate Alternative Pre-mRNA Splicing. Mol Cell Biol. 2015;35(17):3071–3082. doi: 10.1128/MCB.00202-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, Manley JL. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci U S A. 2015;112(34):E4726–4734. doi: 10.1073/pnas.1514105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kon A, et al. Physiological Srsf2 P95H expression causes impaired hematopoietic stem cell functions and aberrant RNA splicing in mice. Blood. 2017 doi: 10.1182/blood-2017-01-762393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sashida G, et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun. 2014;5:4177. doi: 10.1038/ncomms5177. [DOI] [PubMed] [Google Scholar]
- 34.Imielinski M, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yip BH, et al. The U2AF1S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J Clin Invest. 2017;127(6):2206–2221. doi: 10.1172/JCI91363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turunen JJ, Niemela EH, Verma B, Frilander MJ. The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA. 2013;4(1):61–76. doi: 10.1002/wrna.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen H, Zheng X, Luecke S, Green MR. The U2AF35-related protein Urp contacts the 3′ splice site to promote U12-type intron splicing and the second step of U2-type intron splicing. Genes Dev. 2010;24(21):2389–2394. doi: 10.1101/gad.1974810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madan V, et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun. 2015;6:6042. doi: 10.1038/ncomms7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnston JJ, Teer JK, Cherukuri PF, Hansen NF, Loftus SK, Center NIHIS, Chong K, Mullikin JC, Biesecker LG. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet. 2010;86(5):743–748. doi: 10.1016/j.ajhg.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vinayanuwattikun C, et al. Elucidating Genomic Characteristics of Lung Cancer Progression from In Situ to Invasive Adenocarcinoma. Sci Rep. 2016;6:31628. doi: 10.1038/srep31628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ibrahimpasic T, et al. Genomic Alterations in Fatal Forms of Non-Anaplastic Thyroid Cancer: Identification of MED12 and RBM10 as Novel Thyroid Cancer Genes Associated with Tumor Virulence. Clin Cancer Res. 2017;23(19):5970–5980. doi: 10.1158/1078-0432.CCR-17-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giannakis M, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016 doi: 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Witkiewicz AK, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Furukawa T, et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep. 2011;1:161. doi: 10.1038/srep00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hernandez J, Bechara E, Schlesinger D, Delgado J, Serrano L, Valcarcel J. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol. 2016;13(4):466–472. doi: 10.1080/15476286.2016.1144004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao J, et al. Functional analysis reveals that RBM10 mutations contribute to lung adenocarcinoma pathogenesis by deregulating splicing. Sci Rep. 2017;7:40488. doi: 10.1038/srep40488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becerra S, Montes M, Hernandez-Munain C, Sune C. Prp40 pre-mRNA processing factor 40 homolog B (PRPF40B) associates with SF1 and U2AF65 and modulates alternative pre-mRNA splicing in vivo. RNA. 2015;21(3):438–457. doi: 10.1261/rna.047258.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agrawal AA, Salsi E, Chatrikhi R, Henderson S, Jenkins JL, Green MR, Ermolenko DN, Kielkopf CL. An extended U2AF(65)-RNA-binding domain recognizes the 3′ splice site signal. Nat Commun. 2016;7:10950. doi: 10.1038/ncomms10950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho S, Moon H, Loh TJ, Jang HN, Liu Y, Zhou J, Ohn T, Zheng X, Shen H. Splicing inhibition of U2AF65 leads to alternative exon skipping. Proc Natl Acad Sci U S A. 2015;112(32):9926–9931. doi: 10.1073/pnas.1500639112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharma S, Wongpalee SP, Vashisht A, Wohlschlegel JA, Black DL. Stem-loop 4 of U1 snRNA is essential for splicing and interacts with the U2 snRNP-specific SF3A1 protein during spliceosome assembly. Genes Dev. 2014;28(22):2518–2531. doi: 10.1101/gad.248625.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sebestyen E, Singh B, Minana B, Pages A, Mateo F, Pujana MA, Valcarcel J, Eyras E. Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome Res. 2016;26(6):732–744. doi: 10.1101/gr.199935.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang P, et al. CD82 suppresses CD44 alternative splicing-dependent melanoma metastasis by mediating U2AF2 ubiquitination and degradation. Oncogene. 2016;35(38):5056–5069. doi: 10.1038/onc.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anczuków O, Krainer AR. Splicing-factor alterations in cancers. RNA. 2016;22(9):1285–1301. doi: 10.1261/rna.057919.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417(1):15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 56.Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet. 2016;135(8):851–867. doi: 10.1007/s00439-016-1683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Das S, Krainer AR. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol Cancer Res. 2014;12(9):1195–1204. doi: 10.1158/1541-7786.MCR-14-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anczukow O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, Krainer AR. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19(2):220–228. doi: 10.1038/nsmb.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, Green MR, Riva S, Biamonti G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell. 2005;20(6):881–890. doi: 10.1016/j.molcel.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 60.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14(3):185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ezponda T, Pajares MJ, Agorreta J, Echeveste JI, Lopez-Picazo JM, Torre W, Pio R, Montuenga LM. The oncoprotein SF2/ASF promotes non-small cell lung cancer survival by enhancing survivin expression. Clin Cancer Res. 2010;16(16):4113–4125. doi: 10.1158/1078-0432.CCR-10-0076. [DOI] [PubMed] [Google Scholar]
- 62.Das S, Anczukow O, Akerman M, Krainer AR. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 2012;1(2):110–117. doi: 10.1016/j.celrep.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leu S, Lin YM, Wu CH, Ouyang P. Loss of Pnn expression results in mouse early embryonic lethality and cellular apoptosis through SRSF1-mediated alternative expression of Bcl-xS and ICAD. J Cell Sci. 2012;125(Pt 13):3164–3172. doi: 10.1242/jcs.100859. [DOI] [PubMed] [Google Scholar]
- 64.Anczukow O, et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol Cell. 2015;60(1):105–117. doi: 10.1016/j.molcel.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang L, et al. Genomic Landscape Survey Identifies SRSF1 as a Key Oncodriver in Small Cell Lung Cancer. PLoS Genet. 2016;12(4):e1005895. doi: 10.1371/journal.pgen.1005895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cui M, Allen MA, Larsen A, Macmorris M, Han M, Blumenthal T. Genes involved in pre-mRNA 3′-end formation and transcription termination revealed by a lin-15 operon Muv suppressor screen. Proc Natl Acad Sci U S A. 2008;105(43):16665–16670. doi: 10.1073/pnas.0807104105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bedard KM, Daijogo S, Semler BL. A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 2007;26(2):459–467. doi: 10.1038/sj.emboj.7601494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He X, Zhang P. Serine/arginine-rich splicing factor 3 (SRSF3) regulates homologous recombination-mediated DNA repair. Mol Cancer. 2015;14:158. doi: 10.1186/s12943-015-0422-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loomis RJ, Naoe Y, Parker JB, Savic V, Bozovsky MR, Macfarlan T, Manley JL, Chakravarti D. Chromatin binding of SRp20 and ASF/SF2 and dissociation from mitotic chromosomes is modulated by histone H3 serine 10 phosphorylation. Mol Cell. 2009;33(4):450–461. doi: 10.1016/j.molcel.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jia R, Li C, McCoy JP, Deng CX, Zheng ZM. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int J Biol Sci. 2010;6(7):806–826. doi: 10.7150/ijbs.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iborra S, Hirschfeld M, Jaeger M, Zur Hausen A, Braicu I, Sehouli J, Gitsch G, Stickeler E. Alterations in expression pattern of splicing factors in epithelial ovarian cancer and its clinical impact. Int J Gynecol Cancer. 2013;23(6):990–996. doi: 10.1097/IGC.0b013e31829783e3. [DOI] [PubMed] [Google Scholar]
- 72.Peiqi L, Zhaozhong G, Yaotian Y, Jun J, Jihua G, Rong J. Expression of SRSF3 is Correlated with Carcinogenesis and Progression of Oral Squamous Cell Carcinoma. Int J Med Sci. 2016;13(7):533–539. doi: 10.7150/ijms.14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Z, Chatterjee D, Jeon HY, Akerman M, Vander Heiden MG, Cantley LC, Krainer AR. Exon-centric regulation of pyruvate kinase M alternative splicing via mutually exclusive exons. J Mol Cell Biol. 2012;4(2):79–87. doi: 10.1093/jmcb/mjr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang Y, Horikawa I, Ajiro M, Robles AI, Fujita K, Mondal AM, Stauffer JK, Zheng ZM, Harris CC. Downregulation of splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene. 2013;32(22):2792–2798. doi: 10.1038/onc.2012.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cohen-Eliav M, Golan-Gerstl R, Siegfried Z, Andersen CL, Thorsen K, Orntoft TF, Mu D, Karni R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol. 2013;229(4):630–639. doi: 10.1002/path.4129. [DOI] [PubMed] [Google Scholar]
- 76.Jensen MA, Wilkinson JE, Krainer AR. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol. 2014;21(2):189–197. doi: 10.1038/nsmb.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang CC, et al. Apoptosis of human melanoma cells induced by inhibition of B-RAFV600E involves preferential splicing of bimS. Cell Death Dis. 2010;1:e69. doi: 10.1038/cddis.2010.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lai F, Jiang CC, Farrelly ML, Zhang XD, Hersey P. Evidence for upregulation of Bim and the splicing factor SRp55 in melanoma cells from patients treated with selective BRAF inhibitors. Melanoma Res. 2012;22(3):244–251. doi: 10.1097/CMR.0b013e328353eff2. [DOI] [PubMed] [Google Scholar]
- 79.Huang CS, Shen CY, Wang HW, Wu PE, Cheng CW. Increased expression of SRp40 affecting CD44 splicing is associated with the clinical outcome of lymph node metastasis in human breast cancer. Clin Chim Acta. 2007;384(1–2):69–74. doi: 10.1016/j.cca.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 80.Biselli-Chicote PM, et al. Overexpression of Antiangiogenic Vascular Endothelial Growth Factor Isoform and Splicing Regulatory Factors in Oral, Laryngeal and Pharyngeal Squamous Cell Carcinomas. Asian Pac J Cancer Prev. 2017;18(8):2171–2177. doi: 10.22034/APJCP.2017.18.8.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim HR, et al. SRSF5: a novel marker for small-cell lung cancer and pleural metastatic cancer. Lung Cancer. 2016;99:57–65. doi: 10.1016/j.lungcan.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 82.Gabriel M, et al. Role of the splicing factor SRSF4 in cisplatin-induced modifications of pre-mRNA splicing and apoptosis. BMC Cancer. 2015;15:227. doi: 10.1186/s12885-015-1259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Piekielko-Witkowska A, Wiszomirska H, Wojcicka A, Poplawski P, Boguslawska J, Tanski Z, Nauman A. Disturbed expression of splicing factors in renal cancer affects alternative splicing of apoptosis regulators, oncogenes, and tumor suppressors. PLoS One. 2010;5(10):e13690. doi: 10.1371/journal.pone.0013690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mende Y, Jakubik M, Riessland M, Schoenen F, Rossbach K, Kleinridders A, Kohler C, Buch T, Wirth B. Deficiency of the splicing factor Sfrs10 results in early embryonic lethality in mice and has no impact on full-length SMN/Smn splicing. Hum Mol Genet. 2010;19(11):2154–2167. doi: 10.1093/hmg/ddq094. [DOI] [PubMed] [Google Scholar]
- 85.Ji L, et al. Transformer 2beta (Tra2beta/SFRS10) positively regulates the progression of NSCLC via promoting cell proliferation. J Mol Histol. 2014;45(5):573–582. doi: 10.1007/s10735-014-9582-3. [DOI] [PubMed] [Google Scholar]
- 86.Gabriel B, Zur Hausen A, Bouda J, Boudova L, Koprivova M, Hirschfeld M, Jager M, Stickeler E. Significance of nuclear hTra2-beta1 expression in cervical cancer. Acta Obstet Gynecol Scand. 2009;88(2):216–221. doi: 10.1080/00016340802503021. [DOI] [PubMed] [Google Scholar]
- 87.Diao Y, Wu D, Dai Z, Kang H, Wang Z, Wang X. Prognostic value of transformer 2beta expression in prostate cancer. Int J Clin Exp Pathol. 2015;8(6):6967–6973. [PMC free article] [PubMed] [Google Scholar]
- 88.Kajita K, et al. Ets1 and heat shock factor 1 regulate transcription of the Transformer 2beta gene in human colon cancer cells. J Gastroenterol. 2013;48(11):1222–1233. doi: 10.1007/s00535-012-0745-2. [DOI] [PubMed] [Google Scholar]
- 89.Yang L, Tao T, Wang Y, Bao Z, He X, Cui G. Knocking down the expression of TRA2beta inhibits the proliferation and migration of human glioma cells. Pathol Res Pract. 2015;211(10):731–739. doi: 10.1016/j.prp.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 90.Watermann DO, Tang Y, Zur Hausen A, Jager M, Stamm S, Stickeler E. Splicing factor Tra2-beta1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene. Cancer Res. 2006;66(9):4774–4780. doi: 10.1158/0008-5472.CAN-04-3294. [DOI] [PubMed] [Google Scholar]
- 91.Fischer DC, Noack K, Runnebaum IB, Watermann DO, Kieback DG, Stamm S, Stickeler E. Expression of splicing factors in human ovarian cancer. Oncol Rep. 2004;11(5):1085–1090. [PubMed] [Google Scholar]
- 92.Storbeck M, Hupperich K, Gaspar JA, Meganathan K, Martinez Carrera L, Wirth R, Sachinidis A, Wirth B. Neuronal-specific deficiency of the splicing factor Tra2b causes apoptosis in neurogenic areas of the developing mouse brain. PLoS One. 2014;9(2):e89020. doi: 10.1371/journal.pone.0089020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hirschfeld M, Ouyang YQ, Jaeger M, Erbes T, Orlowska-Volk M, Zur Hausen A, Stickeler E. HNRNP G and HTRA2-BETA1 regulate estrogen receptor alpha expression with potential impact on endometrial cancer. BMC Cancer. 2015;15:86. doi: 10.1186/s12885-015-1088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li F, et al. Identification of TRA2B-DNAH5 fusion as a novel oncogenic driver in human lung squamous cell carcinoma. Cell Res. 2016;26(10):1149–1164. doi: 10.1038/cr.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mayeda A, Krainer AR. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell. 1992;68(2):365–375. doi: 10.1016/0092-8674(92)90477-t. [DOI] [PubMed] [Google Scholar]
- 96.Park SJ, et al. Heterogeneous nuclear ribonucleoprotein A1 post-transcriptionally regulates Drp1 expression in neuroblastoma cells. Biochim Biophys Acta. 2015;1849(12):1423–1431. doi: 10.1016/j.bbagrm.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu X, Zhou Y, Lou Y, Zhong H. Knockdown of HNRNPA1 inhibits lung adenocarcinoma cell proliferation through cell cycle arrest at G0/G1 phase. Gene. 2016;576(2 Pt 2):791–797. doi: 10.1016/j.gene.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 98.Song L, et al. microRNA-451-modulated hnRNP A1 takes a part in granulocytic differentiation regulation and acute myeloid leukemia. Oncotarget. 2017;8(33):55453–55466. doi: 10.18632/oncotarget.19325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Feng J, Li L, Tong L, Tang L, Wu S. The Involvement of Splicing Factor hnRNP A1 in UVB-induced Alternative Splicing of hdm2. Photochem Photobiol. 2016;92(2):318–324. doi: 10.1111/php.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Feng J, Liao Y, Xu X, Yi Q, He L, Tang L. hnRNP A1 promotes keratinocyte cell survival post UVB radiation through PI3K/Akt/mTOR pathway. Exp Cell Res. 2017 doi: 10.1016/j.yexcr.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 101.Fielding P, Turnbull L, Prime W, Walshaw M, Field JK. Heterogeneous nuclear ribonucleoprotein A2/B1 up-regulation in bronchial lavage specimens: a clinical marker of early lung cancer detection. Clin Cancer Res. 1999;5(12):4048–4052. [PubMed] [Google Scholar]
- 102.Golan-Gerstl R, Cohen M, Shilo A, Suh SS, Bakacs A, Coppola L, Karni R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71(13):4464–4472. doi: 10.1158/0008-5472.CAN-10-4410. [DOI] [PubMed] [Google Scholar]
- 103.Zhou J, Allred DC, Avis I, Martinez A, Vos MD, Smith L, Treston AM, Mulshine JL. Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2/B1 (hnRNP-A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res Treat. 2001;66(3):217–224. doi: 10.1023/a:1010631915831. [DOI] [PubMed] [Google Scholar]
- 104.Zhou J, Nong L, Wloch M, Cantor A, Mulshine JL, Tockman MS. Expression of early lung cancer detection marker: hnRNP-A2/B1 and its relation to microsatellite alteration in non-small cell lung cancer. Lung Cancer. 2001;34(3):341–350. doi: 10.1016/s0169-5002(01)00254-9. [DOI] [PubMed] [Google Scholar]
- 105.Qu XH, Liu JL, Zhong XW, Li XI, Zhang QG. Insights into the roles of hnRNP A2/B1 and AXL in non-small cell lung cancer. Oncol Lett. 2015;10(3):1677–1685. doi: 10.3892/ol.2015.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gallardo M, et al. hnRNP K Is a Haploinsufficient Tumor Suppressor that Regulates Proliferation and Differentiation Programs in Hematologic Malignancies. Cancer Cell. 2015;28(4):486–499. doi: 10.1016/j.ccell.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sweetser DA, et al. Delineation of the minimal commonly deleted segment and identification of candidate tumor-suppressor genes in del(9q) acute myeloid leukemia. Genes Chromosomes Cancer. 2005;44(3):279–291. doi: 10.1002/gcc.20236. [DOI] [PubMed] [Google Scholar]
- 108.Moumen A, Masterson P, O’Connor MJ, Jackson SP. hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell. 2005;123(6):1065–1078. doi: 10.1016/j.cell.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 109.Wiesmann N, Strozynski J, Beck C, Zimmermann N, Mendler S, Gieringer R, Schmidtmann I, Brieger J. Knockdown of hnRNPK leads to increased DNA damage after irradiation and reduces survival of tumor cells. Carcinogenesis. 2017;38(3):321–328. doi: 10.1093/carcin/bgx006. [DOI] [PubMed] [Google Scholar]
- 110.Gao R, Yu Y, Inoue A, Widodo N, Kaul SC, Wadhwa R. Heterogeneous nuclear ribonucleoprotein K (hnRNP-K) promotes tumor metastasis by induction of genes involved in extracellular matrix, cell movement, and angiogenesis. J Biol Chem. 2013;288(21):15046–15056. doi: 10.1074/jbc.M113.466136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mandal M, Vadlamudi R, Nguyen D, Wang RA, Costa L, Bagheri-Yarmand R, Mendelsohn J, Kumar R. Growth factors regulate heterogeneous nuclear ribonucleoprotein K expression and function. J Biol Chem. 2001;276(13):9699–9704. doi: 10.1074/jbc.M008514200. [DOI] [PubMed] [Google Scholar]
- 112.Carpenter B, McKay M, Dundas SR, Lawrie LC, Telfer C, Murray GI. Heterogeneous nuclear ribonucleoprotein K is over expressed, aberrantly localised and is associated with poor prognosis in colorectal cancer. Br J Cancer. 2006;95(7):921–927. doi: 10.1038/sj.bjc.6603349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhou R, Shanas R, Nelson MA, Bhattacharyya A, Shi J. Increased expression of the heterogeneous nuclear ribonucleoprotein K in pancreatic cancer and its association with the mutant p53. Int J Cancer. 2010;126(2):395–404. doi: 10.1002/ijc.24744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Inoue A, Sawata SY, Taira K, Wadhwa R. Loss-of-function screening by randomized intracellular antibodies: identification of hnRNP-K as a potential target for metastasis. Proc Natl Acad Sci U S A. 2007;104(21):8983–8988. doi: 10.1073/pnas.0607595104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu Y, et al. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev. 2014;28(11):1191–1203. doi: 10.1101/gad.241968.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Passacantilli I, Frisone P, De Paola E, Fidaleo M, Paronetto MP. hnRNPM guides an alternative splicing program in response to inhibition of the PI3K/AKT/mTOR pathway in Ewing sarcoma cells. Nucleic Acids Res. 2017;45(21):12270–12284. doi: 10.1093/nar/gkx831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lefave CV, Squatrito M, Vorlova S, Rocco GL, Brennan CW, Holland EC, Pan YX, Cartegni L. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011;30(19):4084–4097. doi: 10.1038/emboj.2011.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Anantha RW, Alcivar AL, Ma J, Cai H, Simhadri S, Ule J, Konig J, Xia B. Requirement of heterogeneous nuclear ribonucleoprotein C for BRCA gene expression and homologous recombination. PLoS One. 2013;8(4):e61368. doi: 10.1371/journal.pone.0061368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jiang P, Li Z, Tian F, Li X, Yang J. Fyn/heterogeneous nuclear ribonucleoprotein E1 signaling regulates pancreatic cancer metastasis by affecting the alternative splicing of integrin beta1. Int J Oncol. 2017;51(1):169–183. doi: 10.3892/ijo.2017.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.He X, Ee PL, Coon JS, Beck WT. Alternative splicing of the multidrug resistance protein 1/ATP binding cassette transporter subfamily gene in ovarian cancer creates functional splice variants and is associated with increased expression of the splicing factors PTB and SRp20. Clin Cancer Res. 2004;10(14):4652–4660. doi: 10.1158/1078-0432.CCR-03-0439. [DOI] [PubMed] [Google Scholar]
- 121.He X, Pool M, Darcy KM, Lim SB, Auersperg N, Coon JS, Beck WT. Knockdown of polypyrimidine tract-binding protein suppresses ovarian tumor cell growth and invasiveness in vitro. Oncogene. 2007;26(34):4961–4968. doi: 10.1038/sj.onc.1210307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jin W, McCutcheon IE, Fuller GN, Huang ES, Cote GJ. Fibroblast growth factor receptor-1 alpha-exon exclusion and polypyrimidine tract-binding protein in glioblastoma multiforme tumors. Cancer Res. 2000;60(5):1221–1224. [PubMed] [Google Scholar]
- 123.Takahashi H, et al. Significance of Polypyrimidine Tract-Binding Protein 1 Expression in Colorectal Cancer. Mol Cancer Ther. 2015;14(7):1705–1716. doi: 10.1158/1535-7163.MCT-14-0142. [DOI] [PubMed] [Google Scholar]
- 124.Wang C, Norton JT, Ghosh S, Kim J, Fushimi K, Wu JY, Stack MS, Huang S. Polypyrimidine tract-binding protein (PTB) differentially affects malignancy in a cell line-dependent manner. J Biol Chem. 2008;283(29):20277–20287. doi: 10.1074/jbc.M803682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.He X, Arslan AD, Ho TT, Yuan C, Stampfer MR, Beck WT. Involvement of polypyrimidine tract-binding protein (PTBP1) in maintaining breast cancer cell growth and malignant properties. Oncogenesis. 2014;3:e84. doi: 10.1038/oncsis.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen L, et al. Snail Driving Alternative Splicing of CD44 by ESRP1 Enhances Invasion and Migration in Epithelial Ovarian Cancer. Cell Physiol Biochem. 2017;43(6):2489–2504. doi: 10.1159/000484458. [DOI] [PubMed] [Google Scholar]
- 127.Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, Xing Y, Carstens RP. Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol Cell Biol. 2012;32(8):1468–1482. doi: 10.1128/MCB.06536-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7(8):e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, Guo W, Xing Y, Carstens RP. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J. 2010;29(19):3286–3300. doi: 10.1038/emboj.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33(5):591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Warzecha CC, Shen S, Xing Y, Carstens RP. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol. 2009;6(5):546–562. doi: 10.4161/rna.6.5.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ishii H, Saitoh M, Sakamoto K, Kondo T, Katoh R, Tanaka S, Motizuki M, Masuyama K, Miyazawa K. Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J Biol Chem. 2014;289(40):27386–27399. doi: 10.1074/jbc.M114.589432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Horiguchi K, et al. TGF-beta drives epithelial-mesenchymal transition through deltaEF1-mediated downregulation of ESRP. Oncogene. 2012;31(26):3190–3201. doi: 10.1038/onc.2011.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Leontieva OV, Ionov Y. RNA-binding motif protein 35A is a novel tumor suppressor for colorectal cancer. Cell Cycle. 2009;8(3):490–497. doi: 10.4161/cc.8.3.7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yae T, et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun. 2012;3:883. doi: 10.1038/ncomms1892. [DOI] [PubMed] [Google Scholar]
- 136.Lapuk A, et al. Exon-level microarray analyses identify alternative splicing programs in breast cancer. Mol Cancer Res. 2010;8(7):961–974. doi: 10.1158/1541-7786.MCR-09-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Venables JP, et al. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol Cell Biol. 2013;33(2):396–405. doi: 10.1128/MCB.01174-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Danan-Gotthold M, Golan-Gerstl R, Eisenberg E, Meir K, Karni R, Levanon EY. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res. 2015;43(10):5130–5144. doi: 10.1093/nar/gkv210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Oh JJ, Razfar A, Delgado I, Reed RA, Malkina A, Boctor B, Slamon DJ. 3p21.3 tumor suppressor gene H37/Luca15/RBM5 inhibits growth of human lung cancer cells through cell cycle arrest and apoptosis. Cancer Res. 2006;66(7):3419–3427. doi: 10.1158/0008-5472.CAN-05-1667. [DOI] [PubMed] [Google Scholar]
- 140.Rintala-Maki ND, Goard CA, Langdon CE, Wall VE, Traulsen KE, Morin CD, Bonin M, Sutherland LC. Expression of RBM5-related factors in primary breast tissue. J Cell Biochem. 2007;100(6):1440–1458. doi: 10.1002/jcb.21134. [DOI] [PubMed] [Google Scholar]
- 141.Bechara EG, Sebestyen E, Bernardis I, Eyras E, Valcarcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell. 2013;52(5):720–733. doi: 10.1016/j.molcel.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 142.Mourtada-Maarabouni M, Williams GT. RBM5/LUCA-15--tumour suppression by control of apoptosis and the cell cycle? ScientificWorldJournal. 2002;2:1885–1890. doi: 10.1100/tsw.2002.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sutherland LC, Wang K, Robinson AG. RBM5 as a putative tumor suppressor gene for lung cancer. J Thorac Oncol. 2010;5(3):294–298. doi: 10.1097/JTO.0b013e3181c6e330. [DOI] [PubMed] [Google Scholar]
- 144.Lu W, et al. QKI impairs self-renewal and tumorigenicity of oral cancer cells via repression of SOX2. Cancer Biol Ther. 2014;15(9):1174–1184. doi: 10.4161/cbt.29502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhao Y, et al. The tumor suppressing effects of QKI-5 in prostate cancer: a novel diagnostic and prognostic protein. Cancer Biol Ther. 2014;15(1):108–118. doi: 10.4161/cbt.26722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zong FY, et al. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet. 2014;10(4):e1004289. doi: 10.1371/journal.pgen.1004289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bandopadhayay P, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet. 2016;48(3):273–282. doi: 10.1038/ng.3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463(7279):364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang S, et al. MYCN controls an alternative RNA splicing program in high-risk metastatic neuroblastoma. Cancer Lett. 2016;371(2):214–224. doi: 10.1016/j.canlet.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci U S A. 2010;107(5):1894–1899. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Koh CM, et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015;523(7558):96–100. doi: 10.1038/nature14351. [DOI] [PubMed] [Google Scholar]
- 152.Hsu TY, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525(7569):384–388. doi: 10.1038/nature14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Goncalves V, Matos P, Jordan P. The beta-catenin/TCF4 pathway modifies alternative splicing through modulation of SRp20 expression. RNA. 2008;14(12):2538–2549. doi: 10.1261/rna.1253408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Corbo C, Orru S, Gemei M, Noto RD, Mirabelli P, Imperlini E, Ruoppolo M, Vecchio LD, Salvatore F. Protein cross-talk in CD133+ colon cancer cells indicates activation of the Wnt pathway and upregulation of SRp20 that is potentially involved in tumorigenicity. Proteomics. 2012;12(12):2045–2059. doi: 10.1002/pmic.201100370. [DOI] [PubMed] [Google Scholar]
- 155.Jumaa H, Nielsen PJ. The splicing factor SRp20 modifies splicing of its own mRNA and ASF/SF2 antagonizes this regulation. EMBO J. 1997;16(16):5077–5085. doi: 10.1093/emboj/16.16.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sureau A, Gattoni R, Dooghe Y, Stevenin J, Soret J. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 2001;20(7):1785–1796. doi: 10.1093/emboj/20.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Stoilov P, Daoud R, Nayler O, Stamm S. Human tra2-beta1 autoregulates its protein concentration by influencing alternative splicing of its pre-mRNA. Hum Mol Genet. 2004;13(5):509–524. doi: 10.1093/hmg/ddh051. [DOI] [PubMed] [Google Scholar]
- 158.Saltzman AL, Kim YK, Pan Q, Fagnani MM, Maquat LE, Blencowe BJ. Regulation of multiple core spliceosomal proteins by alternative splicing-coupled nonsense-mediated mRNA decay. Mol Cell Biol. 2008;28(13):4320–4330. doi: 10.1128/MCB.00361-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sun S, Zhang Z, Sinha R, Karni R, Krainer AR. SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat Struct Mol Biol. 2010;17(3):306–312. doi: 10.1038/nsmb.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature. 2007;446(7138):926–929. doi: 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- 161.Ni JZ, et al. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 2007;21(6):708–718. doi: 10.1101/gad.1525507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rossbach O, Hung LH, Schreiner S, Grishina I, Heiner M, Hui J, Bindereif A. Auto- and cross-regulation of the hnRNP L proteins by alternative splicing. Mol Cell Biol. 2009;29(6):1442–1451. doi: 10.1128/MCB.01689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.McGlincy NJ, Tan LY, Paul N, Zavolan M, Lilley KS, Smith CW. Expression proteomics of UPF1 knockdown in HeLa cells reveals autoregulation of hnRNP A2/B1 mediated by alternative splicing resulting in nonsense-mediated mRNA decay. BMC Genomics. 2010;11:565. doi: 10.1186/1471-2164-11-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hase ME, Yalamanchili P, Visa N. The Drosophila heterogeneous nuclear ribonucleoprotein M protein, HRP59, regulates alternative splicing and controls the production of its own mRNA. J Biol Chem. 2006;281(51):39135–39141. doi: 10.1074/jbc.M604235200. [DOI] [PubMed] [Google Scholar]
- 165.Jangi M, Boutz PL, Paul P, Sharp PA. Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev. 2014;28(6):637–651. doi: 10.1101/gad.235770.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jangi M, Sharp PA. Building robust transcriptomes with master splicing factors. Cell. 2014;159(3):487–498. doi: 10.1016/j.cell.2014.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fagg WS, Liu N, Fair JH, Shiue L, Katzman S, Donohue JP, Ares M., Jr Autogenous cross-regulation of Quaking mRNA processing and translation balances Quaking functions in splicing and translation. Genes Dev. 2017;31(18):1894–1909. doi: 10.1101/gad.302059.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.de Miguel FJ, et al. A large-scale analysis of alternative splicing reveals a key role of QKI in lung cancer. Mol Oncol. 2016;10(9):1437–1449. doi: 10.1016/j.molonc.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhang Z, et al. Regulation of androgen receptor splice variant AR3 by PCGEM1. Oncotarget. 2016;7(13):15481–15491. doi: 10.18632/oncotarget.7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Singh R, Gupta SC, Peng WX, Zhou N, Pochampally R, Atfi A, Watabe K, Lu Z, Mo YY. Regulation of alternative splicing of Bcl-x by BC200 contributes to breast cancer pathogenesis. Cell Death Dis. 2016;7(6):e2262. doi: 10.1038/cddis.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tripathi V, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39(6):925–938. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kong J, Sun W, Li C, Wan L, Wang S, Wu Y, Xu E, Zhang H, Lai M. Long non-coding RNA LINC01133 inhibits epithelial-mesenchymal transition and metastasis in colorectal cancer by interacting with SRSF6. Cancer Lett. 2016;380(2):476–484. doi: 10.1016/j.canlet.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 173.Boguslawska J, Sokol E, Rybicka B, Czubaty A, Rodzik K, Piekielko-Witkowska A. microRNAs target SRSF7 splicing factor to modulate the expression of osteopontin splice variants in renal cancer cells. Gene. 2016;595(2):142–149. doi: 10.1016/j.gene.2016.09.031. [DOI] [PubMed] [Google Scholar]
- 174.Wang Z, Dai X, Chen Y, Sun C, Zhu Q, Zhao H, Liu G, Huang Q, Lan Q. MiR-30a-5p is induced by Wnt/beta-catenin pathway and promotes glioma cell invasion by repressing NCAM. Biochem Biophys Res Commun. 2015;465(3):374–380. doi: 10.1016/j.bbrc.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 175.Verduci L, Simili M, Rizzo M, Mercatanti A, Evangelista M, Mariani L, Rainaldi G, Pitto L. MicroRNA (miRNA)-mediated interaction between leukemia/lymphoma-related factor (LRF) and alternative splicing factor/splicing factor 2 (ASF/SF2) affects mouse embryonic fibroblast senescence and apoptosis. J Biol Chem. 2010;285(50):39551–39563. doi: 10.1074/jbc.M110.114736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Meseguer S, Mudduluru G, Escamilla JM, Allgayer H, Barettino D. MicroRNAs-10a and -10b contribute to retinoic acid-induced differentiation of neuroblastoma cells and target the alternative splicing regulatory factor SFRS1 (SF2/ASF) J Biol Chem. 2011;286(6):4150–4164. doi: 10.1074/jbc.M110.167817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kataoka N, Bachorik JL, Dreyfuss G. Transportin-SR, a nuclear import receptor for SR proteins. J Cell Biol. 1999;145(6):1145–1152. doi: 10.1083/jcb.145.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lai MC, Lin RI, Huang SY, Tsai CW, Tarn WY. A human importin-beta family protein, transportin-SR2, interacts with the phosphorylated RS domain of SR proteins. J Biol Chem. 2000;275(11):7950–7957. doi: 10.1074/jbc.275.11.7950. [DOI] [PubMed] [Google Scholar]
- 179.Koizumi J, Okamoto Y, Onogi H, Mayeda A, Krainer AR, Hagiwara M. The subcellular localization of SF2/ASF is regulated by direct interaction with SR protein kinases (SRPKs) J Biol Chem. 1999;274(16):11125–11131. doi: 10.1074/jbc.274.16.11125. [DOI] [PubMed] [Google Scholar]
- 180.Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996;15(2):265–275. [PMC free article] [PubMed] [Google Scholar]
- 181.Duncan PI, Stojdl DF, Marius RM, Scheit KH, Bell JC. The Clk2 and Clk3 dual-specificity protein kinases regulate the intranuclear distribution of SR proteins and influence pre-mRNA splicing. Exp Cell Res. 1998;241(2):300–308. doi: 10.1006/excr.1998.4083. [DOI] [PubMed] [Google Scholar]
- 182.Ngo JC, Chakrabarti S, Ding JH, Velazquez-Dones A, Nolen B, Aubol BE, Adams JA, Fu XD, Ghosh G. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol Cell. 2005;20(1):77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 183.Yoshida T, et al. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Res. 2015;75(7):1516–1526. doi: 10.1158/0008-5472.CAN-14-2443. [DOI] [PubMed] [Google Scholar]
- 184.Blaustein M, et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol. 2005;12(12):1037–1044. doi: 10.1038/nsmb1020. [DOI] [PubMed] [Google Scholar]
- 185.Patel NA, Chalfant CE, Watson JE, Wyatt JR, Dean NM, Eichler DC, Cooper DR. Insulin regulates alternative splicing of protein kinase C beta II through a phosphatidylinositol 3-kinase-dependent pathway involving the nuclear serine/arginine-rich splicing factor, SRp40, in skeletal muscle cells. J Biol Chem. 2001;276(25):22648–22654. doi: 10.1074/jbc.M101260200. [DOI] [PubMed] [Google Scholar]
- 186.Zhou Z, et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell. 2012;47(3):422–433. doi: 10.1016/j.molcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hayes GM, Carrigan PE, Miller LJ. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res. 2007;67(5):2072–2080. doi: 10.1158/0008-5472.CAN-06-2969. [DOI] [PubMed] [Google Scholar]
- 188.Mavrou A, et al. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene. 2015;34(33):4311–4319. doi: 10.1038/onc.2014.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Odunsi K, Mhawech-Fauceglia P, Andrews C, Beck A, Amuwo O, Lele S, Black JD, Huang RY. Elevated expression of the serine-arginine protein kinase 1 gene in ovarian cancer and its role in Cisplatin cytotoxicity in vitro. PLoS One. 2012;7(12):e51030. doi: 10.1371/journal.pone.0051030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 191.Ben-Hur V, Denichenko P, Siegfried Z, Maimon A, Krainer A, Davidson B, Karni R. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013;3(1):103–115. doi: 10.1016/j.celrep.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mei H, Wang Y, Fan J, Lin Z. Alternative splicing of S6K1 promotes non-small cell lung cancer survival. Tumour Biol. 2016;37(10):13369–13376. doi: 10.1007/s13277-016-5253-1. [DOI] [PubMed] [Google Scholar]
- 193.Gong SG. Isoforms of receptors of fibroblast growth factors. J Cell Physiol. 2014;229(12):1887–1895. doi: 10.1002/jcp.24649. [DOI] [PubMed] [Google Scholar]
- 194.Mauger DM, Lin C, Garcia-Blanco MA. hnRNP H and hnRNP F complex with Fox2 to silence fibroblast growth factor receptor 2 exon IIIc. Mol Cell Biol. 2008;28(17):5403–5419. doi: 10.1128/MCB.00739-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Matsuda Y, Hagio M, Seya T, Ishiwata T. Fibroblast growth factor receptor 2 IIIc as a therapeutic target for colorectal cancer cells. Mol Cancer Ther. 2012;11(9):2010–2020. doi: 10.1158/1535-7163.MCT-12-0243. [DOI] [PubMed] [Google Scholar]
- 196.Kawase R, Ishiwata T, Matsuda Y, Onda M, Kudo M, Takeshita T, Naito Z. Expression of fibroblast growth factor receptor 2 IIIc in human uterine cervical intraepithelial neoplasia and cervical cancer. Int J Oncol. 2010;36(2):331–340. [PubMed] [Google Scholar]
- 197.Sonvilla G, et al. Fibroblast growth factor receptor 3-IIIc mediates colorectal cancer growth and migration. Br J Cancer. 2010;102(7):1145–1156. doi: 10.1038/sj.bjc.6605596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhao Q, et al. Tumor-specific isoform switch of the fibroblast growth factor receptor 2 underlies the mesenchymal and malignant phenotypes of clear cell renal cell carcinomas. Clin Cancer Res. 2013;19(9):2460–2472. doi: 10.1158/1078-0432.CCR-12-3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Peng WX, Kudo M, Fujii T, Teduka K, Naito Z. Altered expression of fibroblast growth factor receptor 2 isoform IIIc: relevance to endometrioid adenocarcinoma carcinogenesis and histological differentiation. Int J Clin Exp Pathol. 2014;7(3):1069–1076. [PMC free article] [PubMed] [Google Scholar]
- 200.Ishiwata T, Matsuda Y, Yamamoto T, Uchida E, Korc M, Naito Z. Enhanced expression of fibroblast growth factor receptor 2 IIIc promotes human pancreatic cancer cell proliferation. Am J Pathol. 2012;180(5):1928–1941. doi: 10.1016/j.ajpath.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Cha JY, Lambert QT, Reuther GW, Der CJ. Involvement of fibroblast growth factor receptor 2 isoform switching in mammary oncogenesis. Mol Cancer Res. 2008;6(3):435–445. doi: 10.1158/1541-7786.MCR-07-0187. [DOI] [PubMed] [Google Scholar]
- 202.Ricol D, et al. Tumour suppressive properties of fibroblast growth factor receptor 2-IIIb in human bladder cancer. Oncogene. 1999;18(51):7234–7243. doi: 10.1038/sj.onc.1203186. [DOI] [PubMed] [Google Scholar]
- 203.Kornmann M, Ishiwata T, Matsuda K, Lopez ME, Fukahi K, Asano G, Beger HG, Korc M. IIIc isoform of fibroblast growth factor receptor 1 is overexpressed in human pancreatic cancer and enhances tumorigenicity of hamster ductal cells. Gastroenterology. 2002;123(1):301–313. doi: 10.1053/gast.2002.34174. [DOI] [PubMed] [Google Scholar]
- 204.Oltean S, Sorg BS, Albrecht T, Bonano VI, Brazas RM, Dewhirst MW, Garcia-Blanco MA. Alternative inclusion of fibroblast growth factor receptor 2 exon IIIc in Dunning prostate tumors reveals unexpected epithelial mesenchymal plasticity. Proc Natl Acad Sci U S A. 2006;103(38):14116–14121. doi: 10.1073/pnas.0603090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yan G, Fukabori Y, McBride G, Nikolaropolous S, McKeehan WL. Exon switching and activation of stromal and embryonic fibroblast growth factor (FGF)-FGF receptor genes in prostate epithelial cells accompany stromal independence and malignancy. Mol Cell Biol. 1993;13(8):4513–4522. doi: 10.1128/mcb.13.8.4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66(23):11271–11278. doi: 10.1158/0008-5472.CAN-06-2044. [DOI] [PubMed] [Google Scholar]
- 207.Ranieri D, Rosato B, Nanni M, Magenta A, Belleudi F, Torrisi MR. Expression of the FGFR2 mesenchymal splicing variant in epithelial cells drives epithelial-mesenchymal transition. Oncotarget. 2016;7(5):5440–5460. doi: 10.18632/oncotarget.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Scheper GC, Parra JL, Wilson M, Van Kollenburg B, Vertegaal AC, Han ZG, Proud CG. The N and C termini of the splice variants of the human mitogen-activated protein kinase-interacting kinase Mnk2 determine activity and localization. Mol Cell Biol. 2003;23(16):5692–5705. doi: 10.1128/MCB.23.16.5692-5705.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Maimon A, et al. Mnk2 alternative splicing modulates the p38-MAPK pathway and impacts Ras-induced transformation. Cell Rep. 2014;7(2):501–513. doi: 10.1016/j.celrep.2014.03.041. [DOI] [PubMed] [Google Scholar]
- 210.Cohen JB, Broz SD, Levinson AD. Expression of the H-ras proto-oncogene is controlled by alternative splicing. Cell. 1989;58(3):461–472. doi: 10.1016/0092-8674(89)90427-3. [DOI] [PubMed] [Google Scholar]
- 211.Guil S, Darzynkiewicz E, Bach-Elias M. Study of the 2719 mutant of the c-H-ras oncogene in a bi-intronic alternative splicing system. Oncogene. 2002;21(36):5649–5653. doi: 10.1038/sj.onc.1205722. [DOI] [PubMed] [Google Scholar]
- 212.Guil S, de La Iglesia N, Fernandez-Larrea J, Cifuentes D, Ferrer JC, Guinovart JJ, Bach-Elias M. Alternative splicing of the human proto-oncogene c-H-ras renders a new Ras family protein that trafficks to cytoplasm and nucleus. Cancer Res. 2003;63(17):5178–5187. [PubMed] [Google Scholar]
- 213.Camats M, Kokolo M, Heesom KJ, Ladomery M, Bach-Elias M. P19 H-ras induces G1/S phase delay maintaining cells in a reversible quiescence state. PLoS One. 2009;4(12):e8513. doi: 10.1371/journal.pone.0008513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Solomon DA, Wang Y, Fox SR, Lambeck TC, Giesting S, Lan Z, Senderowicz AM, Conti CJ, Knudsen ES. Cyclin D1 splice variants, Differential effects on localization, RB phosphorylation, and cellular transformation. J Biol Chem. 2003;278(32):30339–30347. doi: 10.1074/jbc.M303969200. [DOI] [PubMed] [Google Scholar]
- 215.Lu F, Gladden AB, Diehl JA. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res. 2003;63(21):7056–7061. [PubMed] [Google Scholar]
- 216.Wang Y, et al. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res. 2008;68(14):5628–5638. doi: 10.1158/0008-5472.CAN-07-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wu FH, Luo LQ, Liu Y, Zhan QX, Luo C, Luo J, Zhang GM, Feng ZH. Cyclin D1b splice variant promotes alphavbeta3-mediated adhesion and invasive migration of breast cancer cells. Cancer Lett. 2014;355(1):159–167. doi: 10.1016/j.canlet.2014.08.044. [DOI] [PubMed] [Google Scholar]
- 218.Li R, et al. Expression of cyclin D1 splice variants is differentially associated with outcome in non-small cell lung cancer patients. Hum Pathol. 2008;39(12):1792–1801. doi: 10.1016/j.humpath.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 219.Burd CJ, et al. Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc Natl Acad Sci U S A. 2006;103(7):2190–2195. doi: 10.1073/pnas.0506281103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kim CJ, Nishi K, Isono T, Okuyama Y, Tambe Y, Okada Y, Inoue H. Cyclin D1b variant promotes cell invasiveness independent of binding to CDK4 in human bladder cancer cells. Mol Carcinog. 2009;48(10):953–964. doi: 10.1002/mc.20547. [DOI] [PubMed] [Google Scholar]
- 221.Wang N, Wei H, Yin D, Lu Y, Zhang Y, Jiang D, Jiang Y, Zhang S. Cyclin D1b overexpression inhibits cell proliferation and induces cell apoptosis in cervical cancer cells in vitro and in vivo. Int J Clin Exp Pathol. 2014;7(7):4016–4023. [PMC free article] [PubMed] [Google Scholar]
- 222.Millar EK, et al. Cyclin D1b protein expression in breast cancer is independent of cyclin D1a and associated with poor disease outcome. Oncogene. 2009;28(15):1812–1820. doi: 10.1038/onc.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Krieger S, Gauduchon J, Roussel M, Troussard X, Sola B. Relevance of cyclin D1b expression and CCND1 polymorphism in the pathogenesis of multiple myeloma and mantle cell lymphoma. BMC Cancer. 2006;6:238. doi: 10.1186/1471-2407-6-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wei M, et al. Knocking down cyclin D1b inhibits breast cancer cell growth and suppresses tumor development in a breast cancer model. Cancer Sci. 2011;102(8):1537–1544. doi: 10.1111/j.1349-7006.2011.01969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Augello MA, et al. Convergence of oncogenic and hormone receptor pathways promotes metastatic phenotypes. J Clin Invest. 2013;123(1):493–508. doi: 10.1172/JCI64750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kim CJ, Tambe Y, Mukaisho KI, Sugihara H, Kawauchi A, Inoue H. Akt-dependent activation of Erk by cyclin D1b contributes to cell invasiveness and tumorigenicity. Oncol Lett. 2016;12(6):4850–4856. doi: 10.3892/ol.2016.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74(4):597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 228.Akgul C, Moulding DA, Edwards SW. Alternative splicing of Bcl-2-related genes: functional consequences and potential therapeutic applications. Cell Mol Life Sci. 2004;61(17):2189–2199. doi: 10.1007/s00018-004-4001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol. 2007;176(7):929–939. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhou A, Ou AC, Cho A, Benz EJ, Jr, Huang SC. Novel splicing factor RBM25 modulates Bcl-x pre-mRNA 5′ splice site selection. Mol Cell Biol. 2008;28(19):5924–5936. doi: 10.1128/MCB.00560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Bielli P, Bordi M, Di Biasio V, Sette C. Regulation of BCL-X splicing reveals a role for the polypyrimidine tract binding protein (PTBP1/hnRNP I) in alternative 5′ splice site selection. Nucleic Acids Res. 2014;42(19):12070–12081. doi: 10.1093/nar/gku922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wang Y, Chen D, Qian H, Tsai YS, Shao S, Liu Q, Dominguez D, Wang Z. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell. 2014;26(3):374–389. doi: 10.1016/j.ccr.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Bielli P, Busa R, Di Stasi SM, Munoz MJ, Botti F, Kornblihtt AR, Sette C. The transcription factor FBI-1 inhibits SAM68-mediated BCL-X alternative splicing and apoptosis. EMBO Rep. 2014;15(4):419–427. doi: 10.1002/embr.201338241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Moore MJ, Wang Q, Kennedy CJ, Silver PA. An alternative splicing network links cell-cycle control to apoptosis. Cell. 2010;142(4):625–636. doi: 10.1016/j.cell.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Xerri L, Parc P, Brousset P, Schlaifer D, Hassoun J, Reed JC, Krajewski S, Birnbaum D. Predominant expression of the long isoform of Bcl-x (Bcl-xL) in human lymphomas. Br J Haematol. 1996;92(4):900–906. doi: 10.1046/j.1365-2141.1996.423958.x. [DOI] [PubMed] [Google Scholar]
- 236.Li Z, Li Q, Han L, Tian N, Liang Q, Li Y, Zhao X, Du C, Tian Y. Pro-apoptotic effects of splice-switching oligonucleotides targeting Bcl-x pre-mRNA in human glioma cell lines. Oncol Rep. 2016;35(2):1013–1019. doi: 10.3892/or.2015.4465. [DOI] [PubMed] [Google Scholar]
- 237.Tu Y, et al. BCL-X expression in multiple myeloma: possible indicator of chemoresistance. Cancer Res. 1998;58(2):256–262. [PubMed] [Google Scholar]
- 238.Dole MG, Jasty R, Cooper MJ, Thompson CB, Nunez G, Castle VP. Bcl-xL is expressed in neuroblastoma cells and modulates chemotherapy-induced apoptosis. Cancer Res. 1995;55(12):2576–2582. [PubMed] [Google Scholar]
- 239.Mercatante DR, Bortner CD, Cidlowski JA, Kole R. Modification of alternative splicing of Bcl-x pre-mRNA in prostate, breast cancer cells, analysis of apoptosis and cell death. J Biol Chem. 2001;276(19):16411–16417. doi: 10.1074/jbc.M009256200. [DOI] [PubMed] [Google Scholar]
- 240.Taylor JK, Zhang QQ, Wyatt JR, Dean NM. Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nat Biotechnol. 1999;17(11):1097–1100. doi: 10.1038/15079. [DOI] [PubMed] [Google Scholar]
- 241.Coluccia AM, Perego S, Cleris L, Gunby RH, Passoni L, Marchesi E, Formelli F, Gambacorti-Passerini C. Bcl-XL down-regulation suppresses the tumorigenic potential of NPM/ALK in vitro and in vivo. Blood. 2004;103(7):2787–2794. doi: 10.1182/blood-2003-09-3144. [DOI] [PubMed] [Google Scholar]
- 242.Hayward RL, Macpherson JS, Cummings J, Monia BP, Smyth JF, Jodrell DI. Antisense Bcl-xl down-regulation switches the response to topoisomerase I inhibition from senescence to apoptosis in colorectal cancer cells, enhancing global cytotoxicity. Clin Cancer Res. 2003;9(7):2856–2865. [PubMed] [Google Scholar]
- 243.Lebedeva I, Rando R, Ojwang J, Cossum P, Stein CA. Bcl-xL in prostate cancer cells: effects of overexpression and down-regulation on chemosensitivity. Cancer Res. 2000;60(21):6052–6060. [PubMed] [Google Scholar]
- 244.Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263(5154):1759–1762. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- 245.Paronetto MP, Bernardis I, Volpe E, Bechara E, Sebestyen E, Eyras E, Valcarcel J. Regulation of FAS exon definition and apoptosis by the Ewing sarcoma protein. Cell Rep. 2014;7(4):1211–1226. doi: 10.1016/j.celrep.2014.03.077. [DOI] [PubMed] [Google Scholar]
- 246.Oh H, et al. hnRNP A1 contacts exon 5 to promote exon 6 inclusion of apoptotic Fas gene. Apoptosis. 2013;18(7):825–835. doi: 10.1007/s10495-013-0824-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Izquierdo JM, Majos N, Bonnal S, Martinez C, Castelo R, Guigo R, Bilbao D, Valcarcel J. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell. 2005;19(4):475–484. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 248.Bonnal S, Martinez C, Forch P, Bachi A, Wilm M, Valcarcel J. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Mol Cell. 2008;32(1):81–95. doi: 10.1016/j.molcel.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 249.Tejedor JR, Papasaikas P, Valcarcel J. Genome-wide identification of Fas/CD95 alternative splicing regulators reveals links with iron homeostasis. Mol Cell. 2015;57(1):23–38. doi: 10.1016/j.molcel.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 250.Liu JH, Wei S, Lamy T, Li Y, Epling-Burnette PK, Djeu JY, Loughran TP., Jr Blockade of Fas-dependent apoptosis by soluble Fas in LGL leukemia. Blood. 2002;100(4):1449–1453. [PubMed] [Google Scholar]
- 251.Inaba H, Komada Y, Li QS, Zhang XL, Tanaka S, Azuma E, Yamamoto H, Sakurai M. mRNA expression of variant Fas molecules in acute leukemia cells. Am J Hematol. 1999;62(3):150–158. doi: 10.1002/(sici)1096-8652(199911)62:3<150::aid-ajh4>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 252.Kamihira S, Yamada Y, Tomonaga M, Sugahara K, Tsuruda K. Discrepant expression of membrane and soluble isoforms of Fas (CD95/APO-1) in adult T-cell leukaemia: soluble Fas isoform is an independent risk factor for prognosis. Br J Haematol. 1999;107(4):851–860. doi: 10.1046/j.1365-2141.1999.01792.x. [DOI] [PubMed] [Google Scholar]
- 253.Nonomura N, Nishimura K, Ono Y, Fukui T, Harada Y, Takaha N, Takahara S, Okuyama A. Soluble Fas in serum from patients with renal cell carcinoma. Urology. 2000;55(1):151–155. doi: 10.1016/s0090-4295(99)00379-9. [DOI] [PubMed] [Google Scholar]
- 254.Konno R, Takano T, Sato S, Yajima A. Serum soluble fas level as a prognostic factor in patients with gynecological malignancies. Clin Cancer Res. 2000;6(9):3576–3580. [PubMed] [Google Scholar]
- 255.Midis GP, Shen Y, Owen-Schaub LB. Elevated soluble Fas (sFas) levels in nonhematopoietic human malignancy. Cancer Res. 1996;56(17):3870–3874. [PubMed] [Google Scholar]
- 256.Mizutani Y, Yoshida O, Bonavida B. Prognostic significance of soluble Fas in the serum of patients with bladder cancer. J Urol. 1998;160(2):571–576. [PubMed] [Google Scholar]
- 257.Goel HL, et al. Regulated splicing of the alpha6 integrin cytoplasmic domain determines the fate of breast cancer stem cells. Cell Rep. 2014;7(3):747–761. doi: 10.1016/j.celrep.2014.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Sakamuro D, Elliott KJ, Wechsler-Reya R, Prendergast GC. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat Genet. 1996;14(1):69–77. doi: 10.1038/ng0996-69. [DOI] [PubMed] [Google Scholar]
- 259.Ge K, DuHadaway J, Du W, Herlyn M, Rodeck U, Prendergast GC. Mechanism for elimination of a tumor suppressor: aberrant splicing of a brain-specific exon causes loss of function of Bin1 in melanoma. Proc Natl Acad Sci U S A. 1999;96(17):9689–9694. doi: 10.1073/pnas.96.17.9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Fushimi K, Ray P, Kar A, Wang L, Sutherland LC, Wu JY. Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proc Natl Acad Sci U S A. 2008;105(41):15708–15713. doi: 10.1073/pnas.0805569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Jang HN, et al. Exon 9 skipping of apoptotic caspase-2 pre-mRNA is promoted by SRSF3 through interaction with exon 8. Biochim Biophys Acta. 2014;1839(1):25–32. doi: 10.1016/j.bbagrm.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Wang L, Miura M, Bergeron L, Zhu H, Yuan J. Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78(5):739–750. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- 263.Bergeron L, et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12(9):1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Solier S, et al. Topoisomerase I and II inhibitors control caspase-2 pre-messenger RNA splicing in human cells. Mol Cancer Res. 2004;2(1):53–61. [PubMed] [Google Scholar]
- 265.Han C, Zhao R, Kroger J, Qu M, Wani AA, Wang QE. Caspase-2 short isoform interacts with membrane-associated cytoskeleton proteins to inhibit apoptosis. PLoS One. 2013;8(7):e67033. doi: 10.1371/journal.pone.0067033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Bae J, Leo CP, Hsu SY, Hsueh AJ. MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J Biol Chem. 2000;275(33):25255–25261. doi: 10.1074/jbc.M909826199. [DOI] [PubMed] [Google Scholar]
- 267.Bingle CD, Craig RW, Swales BM, Singleton V, Zhou P, Whyte MK. Exon skipping in Mcl-1 results in a bcl-2 homology domain 3 only gene product that promotes cell death. J Biol Chem. 2000;275(29):22136–22146. doi: 10.1074/jbc.M909572199. [DOI] [PubMed] [Google Scholar]
- 268.Le Gouill S, Podar K, Harousseau JL, Anderson KC. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle. 2004;3(10):1259–1262. doi: 10.4161/cc.3.10.1196. [DOI] [PubMed] [Google Scholar]
- 269.Gautrey HL, Tyson-Capper AJ. Regulation of Mcl-1 by SRSF1 and SRSF5 in cancer cells. PLoS One. 2012;7(12):e51497. doi: 10.1371/journal.pone.0051497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Mallick S, Patil R, Gyanchandani R, Pawar S, Palve V, Kannan S, Pathak KA, Choudhary M, Teni TR. Human oral cancers have altered expression of Bcl-2 family members and increased expression of the anti-apoptotic splice variant of Mcl-1. J Pathol. 2009;217(3):398–407. doi: 10.1002/path.2459. [DOI] [PubMed] [Google Scholar]
- 271.Shieh JJ, Liu KT, Huang SW, Chen YJ, Hsieh TY. Modification of alternative splicing of Mcl-1 pre-mRNA using antisense morpholino oligonucleotides induces apoptosis in basal cell carcinoma cells. J Invest Dermatol. 2009;129(10):2497–2506. doi: 10.1038/jid.2009.83. [DOI] [PubMed] [Google Scholar]
- 272.Palve V, Mallick S, Ghaisas G, Kannan S, Teni T. Overexpression of Mcl-1L splice variant is associated with poor prognosis and chemoresistance in oral cancers. PLoS One. 2014;9(11):e111927. doi: 10.1371/journal.pone.0111927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Palve VC, Teni TR. Association of anti-apoptotic Mcl-1L isoform expression with radioresistance of oral squamous carcinoma cells. Radiat Oncol. 2012;7:135. doi: 10.1186/1748-717X-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Fukumoto T, Iwasaki T, Okada T, Hashimoto T, Moon Y, Sakaguchi M, Fukami Y, Nishigori C, Oka M. High expression of Mcl-1L via the MEK-ERK-phospho-STAT3 (Ser727) pathway protects melanocytes and melanoma from UVB-induced apoptosis. Genes Cells. 2016;21(2):185–199. doi: 10.1111/gtc.12330. [DOI] [PubMed] [Google Scholar]
- 275.Kim JH, Bae J. MCL-1ES induces MCL-1L-dependent BAX- and BAK-independent mitochondrial apoptosis. PLoS One. 2013;8(11):e79626. doi: 10.1371/journal.pone.0079626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Dayton TL, Jacks T, Vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016;17(12):1721–1730. doi: 10.15252/embr.201643300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Chao TK, Huang TS, Liao YP, Huang RL, Su PH, Shen HY, Lai HC, Wang YC. Pyruvate kinase M2 is a poor prognostic marker of and a therapeutic target in ovarian cancer. PLoS One. 2017;12(7):e0182166. doi: 10.1371/journal.pone.0182166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Shiroki T, et al. Enhanced expression of the M2 isoform of pyruvate kinase is involved in gastric cancer development by regulating cancer-specific metabolism. Cancer Sci. 2017;108(5):931–940. doi: 10.1111/cas.13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Liu ZX, Hong L, Fang SQ, Tan GH, Huang PG, Zeng Z, Xia X, Wang XX. Overexpression of pyruvate kinase M2 predicts a poor prognosis for patients with osteosarcoma. Tumour Biol. 2016;37(11):14923–14928. doi: 10.1007/s13277-016-5401-7. [DOI] [PubMed] [Google Scholar]
- 280.Mohammad GH, Olde Damink SW, Malago M, Dhar DK, Pereira SP. Pyruvate Kinase M2 and Lactate Dehydrogenase A Are Overexpressed in Pancreatic Cancer and Correlate with Poor Outcome. PLoS One. 2016;11(3):e0151635. doi: 10.1371/journal.pone.0151635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Lu W, et al. Up-regulation of PKM2 promote malignancy and related to adverse prognostic risk factor in human gallbladder cancer. Sci Rep. 2016;6:26351. doi: 10.1038/srep26351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Liu WR, et al. PKM2 promotes metastasis by recruiting myeloid-derived suppressor cells and indicates poor prognosis for hepatocellular carcinoma. Oncotarget. 2015;6(2):846–861. doi: 10.18632/oncotarget.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Zhu H, Luo H, Zhu X, Hu X, Zheng L, Zhu X. Pyruvate kinase M2 (PKM2) expression correlates with prognosis in solid cancers: a meta-analysis. Oncotarget. 2017;8(1):1628–1640. doi: 10.18632/oncotarget.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Azoitei N, Becher A, Steinestel K, Rouhi A, Diepold K, Genze F, Simmet T, Seufferlein T. PKM2 promotes tumor angiogenesis by regulating HIF-1alpha through NF-kappaB activation. Mol Cancer. 2016;15:3. doi: 10.1186/s12943-015-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Wang W, He Q, Sun J, Liu Z, Zhao L, Lu Z, Zhou X, Wang A. Pyruvate kinase M2 deregulation enhances the metastatic potential of tongue squamous cell carcinoma. Oncotarget. 2017;8(40):68252–68262. doi: 10.18632/oncotarget.19291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 287.Anastasiou D, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8(10):839–847. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Cortes-Cros M, et al. M2 isoform of pyruvate kinase is dispensable for tumor maintenance and growth. Proc Natl Acad Sci U S A. 2013;110(2):489–494. doi: 10.1073/pnas.1212780110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Israelsen WJ, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155(2):397–409. doi: 10.1016/j.cell.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Lau AN, et al. PKM2 is not required for colon cancer initiated by APC loss. Cancer Metab. 2017;5:10. doi: 10.1186/s40170-017-0172-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.He Y, et al. Pyruvate kinase isoform M2 (PKM2) participates in multiple myeloma cell proliferation, adhesion and chemoresistance. Leuk Res. 2015;39(12):1428–1436. doi: 10.1016/j.leukres.2015.09.019. [DOI] [PubMed] [Google Scholar]
- 292.Israelsen WJ, Vander Heiden MG. Pyruvate kinase: Function, regulation and role in cancer. Semin Cell Dev Biol. 2015;43:43–51. doi: 10.1016/j.semcdb.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol. 1991;5(12):1806–1814. doi: 10.1210/mend-5-12-1806. [DOI] [PubMed] [Google Scholar]
- 294.Bates DO, Cui TG, Doughty JM, Winkler M, Sugiono M, Shields JD, Peat D, Gillatt D, Harper SJ. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002;62(14):4123–4131. [PubMed] [Google Scholar]
- 295.Nowak DG, et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J Biol Chem. 2010;285(8):5532–5540. doi: 10.1074/jbc.M109.074930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer. 2008;8(11):880–887. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Pritchard-Jones RO, Dunn DB, Qiu Y, Varey AH, Orlando A, Rigby H, Harper SJ, Bates DO. Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br J Cancer. 2007;97(2):223–230. doi: 10.1038/sj.bjc.6603839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Woolard J, et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004;64(21):7822–7835. doi: 10.1158/0008-5472.CAN-04-0934. [DOI] [PubMed] [Google Scholar]
- 299.Varey AH, et al. VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br J Cancer. 2008;98(8):1366–1379. doi: 10.1038/sj.bjc.6604308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Wilkie MD, et al. Relative expression of vascular endothelial growth factor isoforms in squamous cell carcinoma of the head and neck. Head Neck. 2016;38(5):775–781. doi: 10.1002/hed.23959. [DOI] [PubMed] [Google Scholar]
- 301.Rennel E, et al. The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumour growth in mice. Br J Cancer. 2008;98(7):1250–1257. doi: 10.1038/sj.bjc.6604309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Rennel ES, et al. Recombinant human VEGF165b protein is an effective anti-cancer agent in mice. Eur J Cancer. 2008;44(13):1883–1894. doi: 10.1016/j.ejca.2008.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Screaton GR, Bell MV, Bell JI, Jackson DG. The identification of a new alternative exon with highly restricted tissue expression in transcripts encoding the mouse Pgp-1 (CD44) homing receptor, Comparison of all 10 variable exons between mouse, human, and rat. J Biol Chem. 1993;268(17):12235–12238. [PubMed] [Google Scholar]
- 304.Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, Cheng C. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121(3):1064–1074. doi: 10.1172/JCI44540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Zhou ZJ, Dai Z, Zhou SL, Fu XT, Zhao YM, Shi YH, Zhou J, Fan J. Overexpression of HnRNP A1 promotes tumor invasion through regulating CD44v6 and indicates poor prognosis for hepatocellular carcinoma. Int J Cancer. 2013;132(5):1080–1089. doi: 10.1002/ijc.27742. [DOI] [PubMed] [Google Scholar]
- 306.Loh TJ, et al. SC35 promotes splicing of the C5-V6-C6 isoform of CD44 pre-mRNA. Oncol Rep. 2014;31(1):273–279. doi: 10.3892/or.2013.2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Loh TJ, et al. hnRNP L inhibits CD44 V10 exon splicing through interacting with its upstream intron. Biochim Biophys Acta. 2015;1849(6):743–750. doi: 10.1016/j.bbagrm.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 308.Fox SB, Fawcett J, Jackson DG, Collins I, Gatter KC, Harris AL, Gearing A, Simmons DL. Normal human tissues, in addition to some tumors, express multiple different CD44 isoforms. Cancer Res. 1994;54(16):4539–4546. [PubMed] [Google Scholar]
- 309.Lau WM, Teng E, Chong HS, Lopez KA, Tay AY, Salto-Tellez M, Shabbir A, So JB, Chan SL. CD44v8-10 is a cancer-specific marker for gastric cancer stem cells. Cancer Res. 2014;74(9):2630–2641. doi: 10.1158/0008-5472.CAN-13-2309. [DOI] [PubMed] [Google Scholar]
- 310.Choi ES, Kim H, Kim HP, Choi Y, Goh SH. CD44v8-10 as a potential theranostic biomarker for targeting disseminated cancer cells in advanced gastric cancer. Sci Rep. 2017;7(1):4930. doi: 10.1038/s41598-017-05247-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Sosulski A, et al. CD44 Splice Variant v8-10 as a Marker of Serous Ovarian Cancer Prognosis. PLoS One. 2016;11(6):e0156595. doi: 10.1371/journal.pone.0156595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Omara-Opyene AL, Qiu J, Shah GV, Iczkowski KA. Prostate cancer invasion is influenced more by expression of a CD44 isoform including variant 9 than by Muc18. Lab Invest. 2004;84(7):894–907. doi: 10.1038/labinvest.3700112. [DOI] [PubMed] [Google Scholar]
- 313.Terpe HJ, Storkel S, Zimmer U, Anquez V, Fischer C, Pantel K, Gunthert U. Expression of CD44 isoforms in renal cell tumors, Positive correlation to tumor differentiation. Am J Pathol. 1996;148(2):453–463. [PMC free article] [PubMed] [Google Scholar]
- 314.Woodman AC, Sugiyama M, Yoshida K, Sugino T, Borgya A, Goodison S, Matsumura Y, Tarin D. Analysis of anomalous CD44 gene expression in human breast, bladder, and colon cancer and correlation of observed mRNA and protein isoforms. Am J Pathol. 1996;149(5):1519–1530. [PMC free article] [PubMed] [Google Scholar]
- 315.Wu CL, Chao YJ, Yang TM, Chen YL, Chang KC, Hsu HP, Shan YS, Lai MD. Dual role of CD44 isoforms in ampullary adenocarcinoma: CD44s predicts poor prognosis in early cancer and CD44nu is an indicator for recurrence in advanced cancer. BMC Cancer. 2015;15:903. doi: 10.1186/s12885-015-1924-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Bhattacharya R, Mitra T, Chaudhuri SR, Roy SS. Mesenchymal splice isoform of CD44 (CD44s) promotes EMT/invasion and imparts stem-like properties to ovarian cancer cells. J Cell Biochem. 2017 doi: 10.1002/jcb.26504. [DOI] [PubMed] [Google Scholar]
- 317.Miwa T, Nagata T, Kojima H, Sekine S, Okumura T. Isoform switch of CD44 induces different chemotactic and tumorigenic ability in gallbladder cancer. Int J Oncol. 2017;51(3):771–780. doi: 10.3892/ijo.2017.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Di Modugno F, et al. Splicing program of human MENA produces a previously undescribed isoform associated with invasive, mesenchymal-like breast tumors. Proc Natl Acad Sci U S A. 2012;109(47):19280–19285. doi: 10.1073/pnas.1214394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Balsamo M, et al. The alternatively-included 11a sequence modifies the effects of Mena on actin cytoskeletal organization and cell behavior. Sci Rep. 2016;6:35298. doi: 10.1038/srep35298. of the scientific advisory board for MetaStat, a company that is developing cancer biomarkers that involve use of Mena and Mena-isoform specific antibodies. MetaStat did not provide any funding to support this work, and this study is not directly relevant to MetaStat, however, the disclosed relationship could be perceived as a potential conflict of interest. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Tanaka N, Yoshida H, Suzuki Y, Harigaya K. Relative expression of hMena11a and hMenaINV splice isoforms is a useful biomarker in development and progression of human breast carcinoma. Int J Oncol. 2014;45(5):1921–1928. doi: 10.3892/ijo.2014.2591. [DOI] [PubMed] [Google Scholar]
- 321.Oudin MJ, Hughes SK, Rohani N, Moufarrej MN, Jones JG, Condeelis JS, Lauffenburger DA, Gertler FB. Characterization of the expression of the pro-metastatic Mena(INV) isoform during breast tumor progression. Clin Exp Metastasis. 2016;33(3):249–261. doi: 10.1007/s10585-015-9775-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Goswami S, et al. Identification of invasion specific splice variants of the cytoskeletal protein Mena present in mammary tumor cells during invasion in vivo. Clin Exp Metastasis. 2009;26(2):153–159. doi: 10.1007/s10585-008-9225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Bria E, et al. Prognostic impact of alternative splicing-derived hMENA isoforms in resected, node-negative, non-small-cell lung cancer. Oncotarget. 2014;5(22):11054–11063. doi: 10.18632/oncotarget.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Di Modugno F, et al. Molecular cloning of hMena (ENAH) and its splice variant hMena+11a: epidermal growth factor increases their expression and stimulates hMena+11a phosphorylation in breast cancer cell lines. Cancer Res. 2007;67(6):2657–2665. doi: 10.1158/0008-5472.CAN-06-1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Philippar U, et al. A Mena invasion isoform potentiates EGF-induced carcinoma cell invasion and metastasis. Dev Cell. 2008;15(6):813–828. doi: 10.1016/j.devcel.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Lu Y, Yao HP, Wang MH. Multiple variants of the RON receptor tyrosine kinase: biochemical properties, tumorigenic activities, and potential drug targets. Cancer Lett. 2007;257(2):157–164. doi: 10.1016/j.canlet.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 327.Krishnaswamy S, Mohammed AK, Amer OE, Tripathi G, Alokail MS, Al-Daghri NM. Recepteur d’Origine nantais (RON) tyrosine kinase splicing variants lacking exons 18 and 19 occur ubiquitously in lung cancer. Int J Clin Exp Med. 2015;8(11):20778–20786. [PMC free article] [PubMed] [Google Scholar]
- 328.Mayer S, Hirschfeld M, Jaeger M, Pies S, Iborra S, Erbes T, Stickeler E. RON alternative splicing regulation in primary ovarian cancer. Oncol Rep. 2015;34(1):423–430. doi: 10.3892/or.2015.3995. [DOI] [PubMed] [Google Scholar]
- 329.Collesi C, Santoro MM, Gaudino G, Comoglio PM. A splicing variant of the RON transcript induces constitutive tyrosine kinase activity and an invasive phenotype. Mol Cell Biol. 1996;16(10):5518–5526. doi: 10.1128/mcb.16.10.5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Xu XM, Wang D, Shen Q, Chen YQ, Wang MH. RNA-mediated gene silencing of the RON receptor tyrosine kinase alters oncogenic phenotypes of human colorectal carcinoma cells. Oncogene. 2004;23(52):8464–8474. doi: 10.1038/sj.onc.1207907. [DOI] [PubMed] [Google Scholar]
- 331.Zhou YQ, He C, Chen YQ, Wang D, Wang MH. Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: generation of different splicing RON variants and their oncogenic potential. Oncogene. 2003;22(2):186–197. doi: 10.1038/sj.onc.1206075. [DOI] [PubMed] [Google Scholar]
- 332.Eckerich C, Schulte A, Martens T, Zapf S, Westphal M, Lamszus K. RON receptor tyrosine kinase in human gliomas: expression, function, and identification of a novel soluble splice variant. J Neurochem. 2009;109(4):969–980. doi: 10.1111/j.1471-4159.2009.06027.x. [DOI] [PubMed] [Google Scholar]
- 333.Krishnaswamy S, Mohammed AK, Tripathi G, Alokail MS, Al-Daghri NM. Splice variants of the extracellular region of RON receptor tyrosine kinase in lung cancer cell lines identified by PCR and sequencing. BMC Cancer. 2017;17(1):738. doi: 10.1186/s12885-017-3747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Singh A, Karnoub AE, Palmby TR, Lengyel E, Sondek J, Der CJ. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene. 2004;23(58):9369–9380. doi: 10.1038/sj.onc.1208182. [DOI] [PubMed] [Google Scholar]
- 335.Fiegen D, Haeusler LC, Blumenstein L, Herbrand U, Dvorsky R, Vetter IR, Ahmadian MR. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J Biol Chem. 2004;279(6):4743–4749. doi: 10.1074/jbc.M310281200. [DOI] [PubMed] [Google Scholar]
- 336.Goncalves V, Henriques AF, Pereira JF, Neves Costa A, Moyer MP, Moita LF, Gama-Carvalho M, Matos P, Jordan P. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. RNA. 2014;20(4):474–482. doi: 10.1261/rna.041376.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Matos P, Collard JG, Jordan P. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J Biol Chem. 2003;278(50):50442–50448. doi: 10.1074/jbc.M308215200. [DOI] [PubMed] [Google Scholar]
- 338.Beausoleil E, et al. Structure-activity relationship of isoform selective inhibitors of Rac1/1b GTPase nucleotide binding. Bioorg Med Chem Lett. 2009;19(19):5594–5598. doi: 10.1016/j.bmcl.2009.08.037. [DOI] [PubMed] [Google Scholar]
- 339.Matos P, Jordan P. Rac1, but not Rac1B, stimulates RelB-mediated gene transcription in colorectal cancer cells. J Biol Chem. 2006;281(19):13724–13732. doi: 10.1074/jbc.M513243200. [DOI] [PubMed] [Google Scholar]
- 340.Melzer C, Hass R, von der Ohe J, Lehnert H, Ungefroren H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun Signal. 2017;15(1):19. doi: 10.1186/s12964-017-0175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Nimnual AS, Taylor LJ, Nyako M, Jeng HH, Bar-Sagi D. Perturbation of cytoskeleton dynamics by the opposing effects of Rac1 and Rac1b. Small GTPases. 2010;1(2):89–97. doi: 10.4161/sgtp.1.2.14427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, Harbeck N, Schmitt M, Lengyel E. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000;19(26):3013–3020. doi: 10.1038/sj.onc.1203621. [DOI] [PubMed] [Google Scholar]
- 343.Faria M, Capinha L, Simoes-Pereira J, Bugalho MJ, Silva AL. Extending the Impact of RAC1b Overexpression to Follicular Thyroid Carcinomas. Int J Endocrinol. 2016;2016:1972367. doi: 10.1155/2016/1972367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Zhou C, et al. The Rac1 splice form Rac1b promotes K-ras-induced lung tumorigenesis. Oncogene. 2013;32(7):903–909. doi: 10.1038/onc.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Matos P, Jordan P. Increased Rac1b expression sustains colorectal tumor cell survival. Mol Cancer Res. 2008;6(7):1178–1184. doi: 10.1158/1541-7786.MCR-08-0008. [DOI] [PubMed] [Google Scholar]
- 346.Faria M, Matos P, Pereira T, Cabrera R, Cardoso BA, Bugalho MJ, Silva AL. RAC1b overexpression stimulates proliferation and NF-kB-mediated anti-apoptotic signaling in thyroid cancer cells. PLoS One. 2017;12(2):e0172689. doi: 10.1371/journal.pone.0172689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Li G, et al. Rac1b enhances cell survival through activation of the JNK2/c-JUN/Cyclin-D1 and AKT2/MCL1 pathways. Oncotarget. 2016;7(14):17970–17985. doi: 10.18632/oncotarget.7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Narla G, et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005;65(13):5761–5768. doi: 10.1158/0008-5472.CAN-05-0217. [DOI] [PubMed] [Google Scholar]
- 349.Yea S, et al. Ras promotes growth by alternative splicing-mediated inactivation of the KLF6 tumor suppressor in hepatocellular carcinoma. Gastroenterology. 2008;134(5):1521–1531. doi: 10.1053/j.gastro.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Botella LM, Sanz-Rodriguez F, Komi Y, Fernandez LA, Varela E, Garrido-Martin EM, Narla G, Friedman SL, Kojima S. TGF-beta regulates the expression of transcription factor KLF6 and its splice variants and promotes co-operative transactivation of common target genes through a Smad3-Sp1-KLF6 interaction. Biochem J. 2009;419(2):485–495. doi: 10.1042/BJ20081434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Narla G, et al. A germline DNA polymorphism enhances alternative splicing of the KLF6 tumor suppressor gene and is associated with increased prostate cancer risk. Cancer Res. 2005;65(4):1213–1222. doi: 10.1158/0008-5472.CAN-04-4249. [DOI] [PubMed] [Google Scholar]
- 352.DiFeo A, Feld L, Rodriguez E, Wang C, Beer DG, Martignetti JA, Narla G. A functional role for KLF6-SV1 in lung adenocarcinoma prognosis and chemotherapy response. Cancer Res. 2008;68(4):965–970. doi: 10.1158/0008-5472.CAN-07-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Camacho-Vanegas O, et al. Functional inactivation of the KLF6 tumor suppressor gene by loss of heterozygosity and increased alternative splicing in glioblastoma. Int J Cancer. 2007;121(6):1390–1395. doi: 10.1002/ijc.22809. [DOI] [PubMed] [Google Scholar]
- 354.Hatami R, et al. KLF6-SV1 drives breast cancer metastasis and is associated with poor survival. Sci Transl Med. 2013;5(169):169ra112. doi: 10.1126/scitranslmed.3004688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Hartel M, Narla G, Wente MN, Giese NA, Martignoni ME, Martignetti JA, Friess H, Friedman SL. Increased alternative splicing of the KLF6 tumour suppressor gene correlates with prognosis and tumour grade in patients with pancreatic cancer. Eur J Cancer. 2008;44(13):1895–1903. doi: 10.1016/j.ejca.2008.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Liu X, et al. KLF6 loss of function in human prostate cancer progression is implicated in resistance to androgen deprivation. Am J Pathol. 2012;181(3):1007–1016. doi: 10.1016/j.ajpath.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. Embo J. 1998;17(2):384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Juan WC, Roca X, Ong ST. Identification of cis-acting elements and splicing factors involved in the regulation of BIM Pre-mRNA splicing. PLoS One. 2014;9(4):e95210. doi: 10.1371/journal.pone.0095210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Isobe K, et al. Association of BIM Deletion Polymorphism and BIM-gamma RNA Expression in NSCLC with EGFR Mutation. Cancer Genomics Proteomics. 2016;13(6):475–482. doi: 10.21873/cgp.20010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Ng KP, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18(4):521–528. doi: 10.1038/nm.2713. [DOI] [PubMed] [Google Scholar]
- 361.Faber AC, et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1(4):352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Kwong KY, Hung MC. A novel splice variant of HER2 with increased transformation activity. Mol Carcinog. 1998;23(2):62–68. doi: 10.1002/(sici)1098-2744(199810)23:2<62::aid-mc2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 363.Castiglioni F, Tagliabue E, Campiglio M, Pupa SM, Balsari A, Menard S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer. 2006;13(1):221–232. doi: 10.1677/erc.1.01047. [DOI] [PubMed] [Google Scholar]
- 364.Wada R, Yagihashi S, Naito Z. mRNA expression of delta-HER2 and its clinicopathological correlation in HER2-overexpressing breast cancer. Mol Med Rep. 2016;14(6):5104–5110. doi: 10.3892/mmr.2016.5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Gautrey H, Jackson C, Dittrich AL, Browell D, Lennard T, Tyson-Capper A. SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol. 2015;12(10):1139–1151. doi: 10.1080/15476286.2015.1076610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Alajati A, Sausgruber N, Aceto N, Duss S, Sarret S, Voshol H, Bonenfant D, Bentires-Alj M. Mammary tumor formation and metastasis evoked by a HER2 splice variant. Cancer Res. 2013;73(17):5320–5327. doi: 10.1158/0008-5472.CAN-12-3186. [DOI] [PubMed] [Google Scholar]
- 367.Huynh FC, Jones FE. MicroRNA-7 inhibits multiple oncogenic pathways to suppress HER2Delta16 mediated breast tumorigenesis and reverse trastuzumab resistance. PLoS One. 2014;9(12):e114419. doi: 10.1371/journal.pone.0114419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Mitra D, Brumlik MJ, Okamgba SU, Zhu Y, Duplessis TT, Parvani JG, Lesko SM, Brogi E, Jones FE. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8(8):2152–2162. doi: 10.1158/1535-7163.MCT-09-0295. [DOI] [PubMed] [Google Scholar]
- 369.Tilio M, et al. Irreversible inhibition of Delta16HER2 is necessary to suppress Delta16HER2-positive breast carcinomas resistant to Lapatinib. Cancer Lett. 2016;381(1):76–84. doi: 10.1016/j.canlet.2016.07.028. [DOI] [PubMed] [Google Scholar]
- 370.Cittelly DM, Das PM, Salvo VA, Fonseca JP, Burow ME, Jones FE. Oncogenic HER2{Delta}16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors. Carcinogenesis. 2010;31(12):2049–2057. doi: 10.1093/carcin/bgq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Castagnoli L, et al. Activated d16HER2 homodimers and SRC kinase mediate optimal efficacy for trastuzumab. Cancer Res. 2014;74(21):6248–6259. doi: 10.1158/0008-5472.CAN-14-0983. [DOI] [PubMed] [Google Scholar]
- 372.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Srebrow A, Blaustein M, Kornblihtt AR. Regulation of fibronectin alternative splicing by a basement membrane-like extracellular matrix. FEBS Lett. 2002;514(2–3):285–289. doi: 10.1016/s0014-5793(02)02382-7. [DOI] [PubMed] [Google Scholar]
- 375.Bordeleau F, Califano JP, Negron Abril YL, Mason BN, LaValley DJ, Shin SJ, Weiss RS, Reinhart-King CA. Tissue stiffness regulates serine/arginine-rich protein-mediated splicing of the extra domain B-fibronectin isoform in tumors. Proc Natl Acad Sci U S A. 2015;112(27):8314–8319. doi: 10.1073/pnas.1505421112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Chang C, et al. A laminin 511 matrix is regulated by TAZ and functions as the ligand for the alpha6Bbeta1 integrin to sustain breast cancer stem cells. Genes Dev. 2015;29(1):1–6. doi: 10.1101/gad.253682.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Schiefner A, Gebauer M, Skerra A. Extra-domain B in oncofetal fibronectin structurally promotes fibrillar head-to-tail dimerization of extracellular matrix protein. J Biol Chem. 2012;287(21):17578–17588. doi: 10.1074/jbc.M111.303131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Fukuda T, Yoshida N, Kataoka Y, Manabe R, Mizuno-Horikawa Y, Sato M, Kuriyama K, Yasui N, Sekiguchi K. Mice lacking the EDB segment of fibronectin develop normally but exhibit reduced cell growth and fibronectin matrix assembly in vitro. Cancer Res. 2002;62(19):5603–5610. [PubMed] [Google Scholar]
- 379.Nam JM, Onodera Y, Bissell MJ, Park CC. Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin alpha5beta1 and fibronectin. Cancer Res. 2010;70(13):5238–5248. doi: 10.1158/0008-5472.CAN-09-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Borsi L, Carnemolla B, Nicolo G, Spina B, Tanara G, Zardi L. Expression of different tenascin isoforms in normal, hyperplastic and neoplastic human breast tissues. Int J Cancer. 1992;52(5):688–692. doi: 10.1002/ijc.2910520504. [DOI] [PubMed] [Google Scholar]
- 381.Briones-Orta MA, Avendano-Vazquez SE, Aparicio-Bautista DI, Coombes JD, Weber GF, Syn WK. Osteopontin splice variants and polymorphisms in cancer progression and prognosis. Biochim Biophys Acta. 2017;1868(1):93–108 A. doi: 10.1016/j.bbcan.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 382.Lowy CM, Oskarsson T. Tenascin C in metastasis: A view from the invasive front. Cell Adh Migr. 2015;9(1–2):112–124. doi: 10.1080/19336918.2015.1008331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Pelisch F, Khauv D, Risso G, Stallings-Mann M, Blaustein M, Quadrana L, Radisky DC, Srebrow A. Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J Cell Biochem. 2012;113(7):2319–2329. doi: 10.1002/jcb.24103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell. 2016;164(6):1233–1247. doi: 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Martinez NM, Lynch KW. Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol Rev. 2013;253(1):216–236. doi: 10.1111/imr.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Ergun A, Doran G, Costello JC, Paik HH, Collins JJ, Mathis D, Benoist C, ImmGen C. Differential splicing across immune system lineages. Proc Natl Acad Sci U S A. 2013;110(35):14324–14329. doi: 10.1073/pnas.1311839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.O’Connor BP, Danhorn T, De Arras L, Flatley BR, Marcus RA, Farias-Hesson E, Leach SM, Alper S. Regulation of toll-like receptor signaling by the SF3a mRNA splicing complex. PLoS Genet. 2015;11(2):e1004932. doi: 10.1371/journal.pgen.1004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Adib-Conquy M, Adrie C, Fitting C, Gattolliat O, Beyaert R, Cavaillon JM. Up-regulation of MyD88s and SIGIRR, molecules inhibiting Toll-like receptor signaling, in monocytes from septic patients. Crit Care Med. 2006;34(9):2377–2385. doi: 10.1097/01.CCM.0000233875.93866.88. [DOI] [PubMed] [Google Scholar]
- 389.De Arras L, Laws R, Leach SM, Pontis K, Freedman JH, Schwartz DA, Alper S. Comparative genomics RNAi screen identifies Eftud2 as a novel regulator of innate immunity. Genetics. 2014;197(2):485–496. doi: 10.1534/genetics.113.160499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.DeMicco A, Naradikian MS, Sindhava VJ, Yoon JH, Gorospe M, Wertheim GB, Cancro MP, Bassing CH. B Cell-Intrinsic Expression of the HuR RNA-Binding Protein Is Required for the T Cell-Dependent Immune Response In Vivo. J Immunol. 2015;195(7):3449–3462. doi: 10.4049/jimmunol.1500512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Diaz-Munoz MD, et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nat Immunol. 2015;16(4):415–425. doi: 10.1038/ni.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Tong A, Nguyen J, Lynch KW. Differential expression of CD45 isoforms is controlled by the combined activity of basal and inducible splicing-regulatory elements in each of the variable exons. J Biol Chem. 2005;280(46):38297–38304. doi: 10.1074/jbc.M508123200. [DOI] [PubMed] [Google Scholar]
- 393.Preussner M, Schreiner S, Hung LH, Porstner M, Jack HM, Benes V, Ratsch G, Bindereif A. HnRNP L and L-like cooperate in multiple-exon regulation of CD45 alternative splicing. Nucleic Acids Res. 2012;40(12):5666–5678. doi: 10.1093/nar/gks221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Wang HY, Xu X, Ding JH, Bermingham JR, Jr, Fu XD. SC35 plays a role in T cell development and alternative splicing of CD45. Mol Cell. 2001;7(2):331–342. doi: 10.1016/s1097-2765(01)00181-2. [DOI] [PubMed] [Google Scholar]
- 395.Oberdoerffer S, Moita LF, Neems D, Freitas RP, Hacohen N, Rao A. Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL. Science. 2008;321(5889):686–691. doi: 10.1126/science.1157610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Topp JD, Jackson J, Melton AA, Lynch KW. A cell-based screen for splicing regulators identifies hnRNP LL as a distinct signal-induced repressor of CD45 variable exon 4. RNA. 2008;14(10):2038–2049. doi: 10.1261/rna.1212008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Butte MJ, Lee SJ, Jesneck J, Keir ME, Haining WN, Sharpe AH. CD28 costimulation regulates genome-wide effects on alternative splicing. PLoS One. 2012;7(6):e40032. doi: 10.1371/journal.pone.0040032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Martinez NM, et al. Alternative splicing networks regulated by signaling in human T cells. RNA. 2012;18(5):1029–1040. doi: 10.1261/rna.032243.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Ip JY, Tong A, Pan Q, Topp JD, Blencowe BJ, Lynch KW. Global analysis of alternative splicing during T-cell activation. RNA. 2007;13(4):563–572. doi: 10.1261/rna.457207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Salton M, Misteli T. Small Molecule Modulators of Pre-mRNA Splicing in Cancer Therapy. Trends Mol Med. 2016;22(1):28–37. doi: 10.1016/j.molmed.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Convertini P, et al. Sudemycin E influences alternative splicing and changes chromatin modifications. Nucleic Acids Res. 2014;42(8):4947–4961. doi: 10.1093/nar/gku151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Corrionero A, Minana B, Valcarcel J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 2011;25(5):445–459. doi: 10.1101/gad.2014311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem Biol. 2011;6(6):582–589. doi: 10.1021/cb100356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Kashyap MK, et al. Targeting the spliceosome in chronic lymphocytic leukemia with the macrolides FD-895 and pladienolide-B. Haematologica. 2015;100(7):945–954. doi: 10.3324/haematol.2014.122069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.O’Brien K, Matlin AJ, Lowell AM, Moore MJ. The biflavonoid isoginkgetin is a general inhibitor of Pre-mRNA splicing. J Biol Chem. 2008;283(48):33147–33154. doi: 10.1074/jbc.M805556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Pilch B, Allemand E, Facompre M, Bailly C, Riou JF, Soret J, Tazi J. Specific inhibition of serine- and arginine-rich splicing factors phosphorylation, spliceosome assembly, and splicing by the antitumor drug NB-506. Cancer Res. 2001;61(18):6876–6884. [PubMed] [Google Scholar]
- 407.Tazi J, Bakkour N, Soret J, Zekri L, Hazra B, Laine W, Baldeyrou B, Lansiaux A, Bailly C. Selective inhibition of topoisomerase I and various steps of spliceosome assembly by diospyrin derivatives. Mol Pharmacol. 2005;67(4):1186–1194. doi: 10.1124/mol.104.007633. [DOI] [PubMed] [Google Scholar]
- 408.Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS, Nijhawan D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;356(6336) doi: 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- 409.Uehara T, et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat Chem Biol. 2017;13(6):675–680. doi: 10.1038/nchembio.2363. [DOI] [PubMed] [Google Scholar]
- 410.Paolella BR, et al. Copy-number and gene dependency analysis reveals partial copy loss of wild-type SF3B1 as a novel cancer vulnerability. Elife. 2017:6. doi: 10.7554/eLife.23268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11(11):847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 412.Agrawal AA, Yu L, Smith PG, Buonamici S. Targeting splicing abnormalities in cancer. Curr Opin Genet Dev. 2017;48:67–74. doi: 10.1016/j.gde.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 413.Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22(9):976–986. doi: 10.1038/nm.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11(2):125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Wan L, Dreyfuss G. Splicing-Correcting Therapy for SMA. Cell. 2017;170(1):5. doi: 10.1016/j.cell.2017.06.028. [DOI] [PubMed] [Google Scholar]
- 416.Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016;44(14):6549–6563. doi: 10.1093/nar/gkw533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Brosseau JP, et al. Redirecting splicing with bifunctional oligonucleotides. Nucleic Acids Res. 2014;42(6):e40. doi: 10.1093/nar/gkt1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Rigo F, Hua Y, Chun SJ, Prakash TP, Krainer AR, Bennett CF. Synthetic oligonucleotides recruit ILF2/3 to RNA transcripts to modulate splicing. Nat Chem Biol. 2012;8(6):555–561. doi: 10.1038/nchembio.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003;10(2):120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 420.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 421.Crooke ST, Wang S, Vickers TA, Shen W, Liang XH. Cellular uptake and trafficking of antisense oligonucleotides. Nat Biotechnol. 2017;35(3):230–237. doi: 10.1038/nbt.3779. [DOI] [PubMed] [Google Scholar]
- 422.Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44(14):6518–6548. doi: 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, Norris DA, Bennett CF, Bishop KM. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86(10):890–897. doi: 10.1212/WNL.0000000000002445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Zammarchi F, de Stanchina E, Bournazou E, Supakorndej T, Martires K, Riedel E, Corben AD, Bromberg JF, Cartegni L. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc Natl Acad Sci U S A. 2011;108(43):17779–17784. doi: 10.1073/pnas.1108482108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Dewaele M, et al. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J Clin Invest. 2016;126(1):68–84. doi: 10.1172/JCI82534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Hong D, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7(314):314ra185. doi: 10.1126/scitranslmed.aac5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Preussner M, Goldammer G, Neumann A, Haltenhof T, Rautenstrauch P, Muller-McNicoll M, Heyd F. Body Temperature Cycles Control Rhythmic Alternative Splicing in Mammals. Mol Cell. 2017;67(3):433–446 e434. doi: 10.1016/j.molcel.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 428.Munoz MJ, et al. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell. 2009;137(4):708–720. doi: 10.1016/j.cell.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 429.Lambert CA, Garbacki N, Colige AC. Chemotherapy induces alternative transcription and splicing: Facts and hopes for cancer treatment. Int J Biochem Cell Biol. 2017;91(Pt B):84–97. doi: 10.1016/j.biocel.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 430.Shkreta L, Froehlich U, Paquet ER, Toutant J, Elela SA, Chabot B. Anticancer drugs affect the alternative splicing of Bcl-x and other human apoptotic genes. Mol Cancer Ther. 2008;7(6):1398–1409. doi: 10.1158/1535-7163.MCT-08-0192. [DOI] [PubMed] [Google Scholar]
- 431.Solier S, Barb J, Zeeberg BR, Varma S, Ryan MC, Kohn KW, Weinstein JN, Munson PJ, Pommier Y. Genome-wide analysis of novel splice variants induced by topoisomerase I poisoning shows preferential occurrence in genes encoding splicing factors. Cancer Res. 2010;70(20):8055–8065. doi: 10.1158/0008-5472.CAN-10-2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Dutertre M, et al. Cotranscriptional exon skipping in the genotoxic stress response. Nat Struct Mol Biol. 2010;17(11):1358–1366. doi: 10.1038/nsmb.1912. [DOI] [PubMed] [Google Scholar]
- 433.Boutz PL, Bhutkar A, Sharp PA. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015;29(1):63–80. doi: 10.1101/gad.247361.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Calabretta S, Bielli P, Passacantilli I, Pilozzi E, Fendrich V, Capurso G, Fave GD, Sette C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene. 2016;35(16):2031–2039. doi: 10.1038/onc.2015.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Xargay-Torrent S, et al. The splicing modulator sudemycin induces a specific antitumor response and cooperates with ibrutinib in chronic lymphocytic leukemia. Oncotarget. 2015;6(26):22734–22749. doi: 10.18632/oncotarget.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Poulikakos PI, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480(7377):387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Wang Y, et al. The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016;76(9):2778–2790. doi: 10.1158/0008-5472.CAN-16-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Ozden O, et al. Expression of an Oncogenic BARD1 Splice Variant Impairs Homologous Recombination and Predicts Response to PARP-1 Inhibitor Therapy in Colon Cancer. Sci Rep. 2016;6:26273. doi: 10.1038/srep26273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Sotillo E, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5(12):1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Weinstein S, et al. Harnessing RNAi-based nanomedicines for therapeutic gene silencing in B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113(1):E16–22. doi: 10.1073/pnas.1519273113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci U S A. 2003;100(1):189–192. doi: 10.1073/pnas.0136770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Popp MW, Maquat LE. Nonsense-mediated mRNA Decay and Cancer. Curr Opin Genet Dev. 2017;48:44–50. doi: 10.1016/j.gde.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Shalek AK, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498(7453):236–240. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Nielsen TO, Sorensen S, Dagnaes-Hansen F, Kjems J, Sorensen BS. Directing HER4 mRNA expression towards the CYT2 isoform by antisense oligonucleotide decreases growth of breast cancer cells in vitro and in vivo. Br J Cancer. 2013;108(11):2291–2298. doi: 10.1038/bjc.2013.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Bauman JA, Li SD, Yang A, Huang L, Kole R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010;38(22):8348–8356. doi: 10.1093/nar/gkq731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Lin J, Lee JHJ, Paramasivam K, Pathak E, Wang Z, Pramono ZAD, Lim B, Wee KB, Surana U. Induced-Decay of Glycine Decarboxylase Transcripts as an Anticancer Therapeutic Strategy for Non-Small-Cell Lung Carcinoma. Mol Ther Nucleic Acids. 2017;9:263–273. doi: 10.1016/j.omtn.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Wang Z, Jeon HY, Rigo F, Bennett CF, Krainer AR. Manipulation of PK-M mutually exclusive alternative splicing by antisense oligonucleotides. Open Biol. 2012;2(10):120133. doi: 10.1098/rsob.120133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Shiraishi T, Eysturskarth J, Nielsen PE. Modulation of mdm2 pre-mRNA splicing by 9-aminoacridine-PNA (peptide nucleic acid) conjugates targeting intron-exon junctions. BMC Cancer. 2010;10:342. doi: 10.1186/1471-2407-10-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Anczuków O, Buisson M, Leone M, Coutanson C, Lasset C, Calender A, Sinilnikova OM, Mazoyer S. BRCA2 Deep Intronic Mutation Causing Activation of a Cryptic Exon: Opening toward a New Preventive Therapeutic Strategy. Clin Cancer Res. 2012;18(18):4903–4909. doi: 10.1158/1078-0432.CCR-12-1100. [DOI] [PubMed] [Google Scholar]
- 450.Karras JG, McKay RA, Dean NM, Monia BP. Deletion of individual exons and induction of soluble murine interleukin-5 receptor-alpha chain expression through antisense oligonucleotide-mediated redirection of pre-mRNA splicing. Mol Pharmacol. 2000;58(2):380–387. doi: 10.1124/mol.58.2.380. [DOI] [PubMed] [Google Scholar]
- 451.Bruno IG, Jin W, Cote GJ. Correction of aberrant FGFR1 alternative RNA splicing through targeting of intronic regulatory elements. Hum Mol Genet. 2004;13(20):2409–2420. doi: 10.1093/hmg/ddh272. [DOI] [PubMed] [Google Scholar]
- 452.Ghigna C, De Toledo M, Bonomi S, Valacca C, Gallo S, Apicella M, Eperon I, Tazi J, Biamonti G. Pro-metastatic splicing of Ron proto-oncogene mRNA can be reversed: therapeutic potential of bifunctional oligonucleotides and indole derivatives. RNA Biol. 2010;7(4):495–503. doi: 10.4161/rna.7.4.12744. [DOI] [PubMed] [Google Scholar]
- 453.Izaguirre DI, Zhu W, Hai T, Cheung HC, Krahe R, Cote GJ. PTBP1-dependent regulation of USP5 alternative RNA splicing plays a role in glioblastoma tumorigenesis. Mol Carcinog. 2012;51(11):895–906. doi: 10.1002/mc.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]