

Abstract
Small molecules containing cyclopropane–heteroatom linkages are commonly needed in medicinal chemistry campaigns, yet are problematic to prepare using existing methods. To address this issue, a scalable Chan–Lam cyclopropylation reaction using potassium cyclopropyl trifluoroborate has been developed. With phenol nucleophiles, the reaction effects O-cyclopropylation, whereas with 2-pyridones, 2-hydroxybenzimidazoles, and 2-aminopyridines the reaction brings about N-cyclopropylation. The transformation is catalyzed by Cu(OAc)2 and 1,10-phenanthroline and employs 1 atm O2 as the terminal oxidant. This method is operationally convenient to perform and provides a simple, strategic disconnection toward the synthesis of cyclopropyl aryl ethers and cyclopropyl amine derivatives bearing an array of functional groups.
Graphical abstract
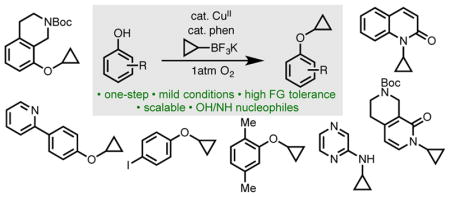
Introduction
Cyclopropyl groups are prevalent in drug molecules and bioactive compounds,[1] with 8 of the top 200 best-selling therapeutics from last year containing a cyclopropyl moiety.[2] As such, methods for introducing these substituents are of significant interest in both academia and the pharmaceutical industry. The goal of the present study was to expand the synthetic toolkit for appending cyclopropyl groups to phenols and aza-heterocycles.
Medicinal Chemistry Considerations
The rationale for incorporating cyclopropyl substituents in the context of medicinal chemistry merits a brief introduction. Sound decision-making in drug discovery campaigns is often guided by disciplined optimization of lipophilic efficiency (LipE).[3] Examination of the cyclopropyl substituent reveals that in addition to its unique electronic properties, the C–H bond dissociation energies of this group (106 Kcal/mol) are significantly stronger than those of the ring-opened i-propyl (98 Kcal/mol) and n-propyl (98 Kcal/mol) analogs.[4] This data suggests that one might expect improved metabolic stability toward oxidative clearance, amongst other desirable properties, when employing this functionality. To further interrogate this concept, a pairwise analysis of matched molecular pairs (MMPs) for a series of alkyl group transforms was examined using the Pfizer database and tools developed by our computational ADME group.[5]
For this analysis, we focused on mining MMPs for transforms of interest which contained clearance endpoints from Pfizer’s high-throughput human liver microsome (HLM) assay. The transforms examined and the corresponding statistics are summarized below (Figure 1). The data shows that, on average, cyclopropyl ring-opening transforms (i-Pr, 1a and n-Pr, 1b) along with ring expansion transforms (cyclobutyl, 1c and cyclopentyl, 1d) result in an increase in HLM clearance. It is important to point out the intrinsic variability in the HLM assay, as illustrated by the relatively wide confidence intervals observed. However, general trends are useful and these increases in microsomal clearance can be rationalized as a result of increasing lipophilicity across the first four aforementioned transformations.[6] To demonstrate this point, we evaluated the transform of ethyl 1e (clogP ~1.7) to cyclopropyl 2 (clogP ~1.6), having approximately equal lipophilic contributions, and found no meaningful change in clearance time (82%±20%) (entry 5). Therefore, it is not surprising that methyl-bearing MMPs 1f (clogP Me~1.1) show a 30% increase in microsomal stability, and thus represent the only transform examined with a clear metabolic advantage to cyclopropyl substitution. Improving metabolic stability in a series is a common medicinal chemistry objective in order to improve pharmacokinetic properties.[7] In light of this analysis, new methodologies to introduce the cyclopropyl motif are warranted and believed to be of broad interest due to the privileged nature of this functionality.
Figure 1.

Transformations examined to probe trends in microsomal stability.
While methodology to access cyclopropyl amines has been vigorously pursued in recent years,[8] cyclopropyl ethers have received less attention. Nevertheless, small molecules containing cyclopropyl aryl ethers have a range of biological activities across several different medical indications (Figure 2).[9] Despite their utility, current methods for their synthesis remain limited and sometimes incompatible with structurally complex, heteroatom-rich substrates. This gap in synthetic methodology motivated the present study.
Figure 2.

Representative bioactive compounds bearing cyclopropylaryl ethers.[9]
In terms of synthetic strategy, the most prudent retrosynthetic disconnection of a cyclopropyl aryl ether is of the O–c-Pr bond to give the corresponding phenol and a cyclopropyl source. The alternative disconnection of the Ar–O bond is complicated by the fact that cyclopropanol readily rearranges to form propanal.[10] Indeed, this retrosynthetic logic underlies all existing methods (Scheme 1).[11–13] In one approach, a multi-step sequence consisting of O-alkylation with a 1,2-dihaloethane electrophile, base-mediated E2 elimination, and Simmons–Smith cyclopropanation[11] is used. While this route provides access to the desired compounds, it requires at least three steps, uses reagents with poor functional group compatibility, and can be low-yielding (Scheme 1a).[11b] A second approach is the reaction between a cyclopropyl halide electrophile and a phenoxide nucleophile in a Williamson ether synthesis (Scheme 1b).[12] Unfortunately, cyclopropyl halides are poor electrophiles in SN2 reactions and can undergo competitive E2 elimination. Thus, high temperatures (typically ≥150 °C) are required to enable addition of the aryloxide into the corresponding in situ formed cyclopropene. Under these forcing conditions, functional group compatibility can be limited and formation of undesired allyl-containing byproducts is sometimes observed. A third strategy was reported in 1999 by Hollingworth and coworkers. This method exploits a sulfur-stabilized carbocation in a modified SN1 reaction to form the key O–C(cyclopropyl) bond. Subsequent oxidation/reduction then excises the sulfide (Scheme 1c).[13] While inventive, this route nonetheless suffers from limited phenol substrate scope due to the difficulty of the SN1 step with electron-poor phenols and requires subsequent oxidation/reduction steps that are incompatible with many functional groups.
Scheme 1.

Comparisons of different methods for O-cyclopropylation of phenols.[11–13]
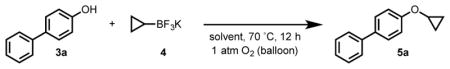
The limitations of these existing methods prompted us to consider an alternative, whereby a phenol could react with a nucleophilic cyclopropyl source in an oxidative coupling reaction. We envisioned that a reaction of this type would avoid the aforementioned complications with cyclopropyl halide electrophiles and would not require any additional concession steps. Herein, we describe the realization of this idea and report a new method for generating cyclopropyl aryl ethers using Chan–Lam-type coupling. While the initial focus was on phenols, it was later found that this protocol is also efficient for N-cyclopropylation with select aza-heterocycles (vide infra).
Reaction Optimization
At the outset, we took inspiration from recent reports on Chan–Lam N-cyclopropylation for the preparation of cyclopropylamine derivatives.[8] Of particular importance for our investigation, Mudryk and coworkers reported that N-cyclopropyl 4-nitrobenzenesulfonamide (pKa(DMSO) 13.9 for NsNH2)[14] underwent Chan–Lam coupling with cyclopropyl boronic acid,[8d] lending credence to the notion that phenols (pKa(DMSO) = 10–19) would be of an appropriate acidity and would be sufficiently nucleophilic to be competent reaction partners. This approach was further motivated by other recent reports on Chan–Lam O- and N-alkylation using alkyl boronates/boronic acids.[15–17] To the best of our knowledge, there is only one example in which a phenol has been reported to react with a cyclopropylboronic acid in the presence of a copper promoter. In a 2010 patent, Cacatian and coworkers report a single reaction proceeding in 13% yield using 1 equivalent of Cu(OAc)2 and 2 equivalents of bipyridine.[18]
Based on these earlier precedents, we began our reaction optimization efforts using cyclopropyl boronic acid as the coupling partner in the presence of catalytic quantities of copper(II) salts. While we did observe product formation under these conditions, we soon turned to the more stable potassium trifluoroborate salt due to inconsistent results with cyclopropyl boronic acid, which we believe stems from its decomposition during reaction setup (evidenced by a visible yellowing of the typically white solid).[19] To our delight, the use of potassium cyclopropyl trifluoroborate nearly doubled the yield and improved reproducibility. Using 1 atm O2 as the terminal oxidant, we were pleased to find that the reaction led to consistent product formation with only 10% Cu(OAc)2 (Table 1). Interestingly, the use of bidentate ligands dramatically improved the efficiency of the reaction, with 1,10-phenanthroline ultimately providing the highest yield (entries 10 and 11). The optimal solvent mixture was found to be 3:1 toluene:H2O. We believe that water is necessary for solubilizing the reaction components and promoting transmetalation by facilitating formation of the mixed boronate species via hydrolysis.[20] Various bases were screened (entries 5–6), and K2CO3 proved to be optimal. It was found that the reaction vessel must be purged with an O2 balloon and left under an atmosphere of O2 for the duration of the reaction to achieve optimal yields. Simply running the reaction open to air or purging the reaction vessel with O2 and sealing it resulted in reduced yields (entries 11 and 12). The reaction proceeded with good to high yields over a large variety of copper(II) species. Notably, copper(II) formate (Cu(HCO2)2), a copper(II) species that is seldom used in Chan–Lam coupling was found to be nearly as effective as the optimal catalyst, Cu(OAc)2. Ultimately, Cu(OAc)2 was used for subsequent studies because of its efficiency in the reaction and its widespread availability.
Table 1.
Optimization of reaction conditions for O-cyclopropylation of phenols.a
| |||||
|---|---|---|---|---|---|
| Entry | CuII (10 mol%) | Ligand | Base (2 equiv) | Solvent | Yield (%)b |
| 1 | Cu(OAc)2 | 20% pyridine | --- | DCE/H2O (2:1) | 24 |
| 2 | Cu(OAc)2 | 20% pyridine | --- | toluene/H2O (2:1) | 35 |
| 3 | Cu(OAc)2 | 20% pyridine | --- | MeCN/H2O (2:1) | 21 |
| 4 | --- | 20% pyridine | --- | toluene/H2O (2:1) | 0 |
| 5 | Cu(OAc)2 | 20% pyridine | KOAc | toluene/H2O (2:1) | 41 |
| 6 | Cu(OAc)2 | 20% pyridine | K2CO3 | toluene/H2O (2:1) | 47 |
| 7 | Cu(OAc)2 | --- | K2CO3 | toluene/H2O (2:1) | 0 |
| 8 | Cu(OAc)2 | 20% pyridine | K2CO3 | toluene/H2O (1:1) | 43 |
| 9 | Cu(OAc)2 | 20% pyridine | K2CO3 | toluene/H2O (3:1) | 36 |
| 10 | Cu(OAc)2 | 10% bipy | K2CO3 | toluene/H2O (2:1) | 71 |
| 11c | Cu(OAc)2 | 10% bipy | K2CO3 | toluene/H2O (2:1) | 49 |
| 12 | Cu(OAc)2 | 10% phen | K2CO3 | toluene/H2O (2:1) | 84 |
| 13 | Cu(HCO2)2 | 10% phen | K2CO3 | toluene/H2O (2:1) | 82 |
| 14 | Cu(OTf)2 | 10% phen | K2CO3 | toluene/H2O (2:1) | 63 |
phen = 1,10-phenanthroline, bipy = 2,2′-bipyridine.
Reaction conditions (unless otherwise specified): 3a (0.3 mmol), 4 (0.9 mmol), solvent, 1 atm O2 (balloon), 12 h, 70 °C.
1 H NMR yield using dibromomethane as internal standard.
Reaction run under air instead of an O2 balloon.
Substrate Scope
Having optimized the reaction conditions, the scope of phenol nucleophiles was next examined (Table 2). Phenols with diverse functional groups, differing electronic properties, and varied substitution patterns were tested. In most cases, the desired product was obtained in moderate to high yield (5a–r). Phenols containing one or more electron-donating substituents at the ortho-, meta-, or para- position on the aromatic ring were generally high-yielding, with the reaction tolerating ethers, thioethers, and alkyl groups (5a–5g). Substitution at the ortho-position led to diminished yields, likely due to steric hindrance (5d). Furthermore, the reaction proceeds smoothly in the presence of halide substituents (5h–k), which presents the opportunity to diversify the products via subsequent cross-coupling. The introduction of strong electron-withdrawing groups at either the meta- or para-positions led to lower yields, requiring higher catalyst loading (5l–o). Moreover, in these cases, competitive formation of the undesired O-allylated byproduct was observed. We postulate that this is due to the attenuated nucleophilicity of the corresponding electron-poor phenoxides, which could potentially reduce the affinity of the substrate for the Cu(II) center and/or raise the activation energy for the C–O reductive elimination step. Although the yields are generally lower for electron-poor phenols, it is important to note that the reaction is nevertheless compatible with a wide variety of functional groups that one might encounter in medicinal chemistry, such as esters, nitriles, halogens, and nitro groups, providing preparatively useful quantities of the product in all of these cases. Notably, methyl ester does not hydrolyze under the reaction conditions and gives the desired cyclopropylated product (5l). We were pleased to observe that the reaction proceeded to a modest extent in the presence of various heterocycles (5p–r), including those that could potentially bind to copper.
Table 2.
Substrate scope of phenol O-cyclopropylation.a

|
All reactions were run on 0.3 mmol scale unless noted otherwise.
Percentages correspond to isolated yields.
25 mol% Cu(OAc)2, 12.5 mol% phen.
25 mol% Cu(OAc)2, 25 mol% phen.
We also established that the reaction could be performed on larger scale. In particular, 6 mmol of our standard substrate, p-phenyl phenol (3a), yielded 81% (>1 gram) of the desired cyclopropylated product 5a.
Though these reaction conditions are not generally effective in promoting N-cyclopropylation of nitrogen nucleophiles, extensive screening of potential reaction partners revealed three classes of aza-heterocycles, 2-pyridones,[21] 2-aminopyridines,[22] and 2-hydroxybenzimidazoles, are reactive substrates using this method (Table 3). Interestingly, subjecting simple 2-hydroxybenzimidazole to the reaction conditions resulted in bis-N,N′-cyclopropylation in high yield (8a). Although 2-hydroxpyridine itself reacted smoothly to form product 9b, other derivatives were cyclopropylated in lower yields (9a–g). 2-Aminopyridine derivatives were also reactive; in this case, the exo-cyclic nitrogen proved to be the more reactive position (10a–c). In compound 10c the free phenol is left unfunctionalized, likely due to the two electron-withdrawing carbonyl substituents, which are expected to attenuate reactivity.
Table 3.
Substrate scope of azaheterocycle N-cyclopropylation.a

|
All reactions were run on a 0.3–0.6 mmol scale. Percentages correspond to isolated yields.
13% of the mono-cyclopropylated product (8a′) was also isolated.
With 10% Cu(OAc)2 and 10% phen under otherwise identical conditions: 49% 8a (bis), 15% 8a′ (mono). The connectivity of 8a′ was confirmed by single-crystal X-ray diffraction (see SI).
25 mol% Cu(OAc)2, 25 mol% phen.
Conclusion
In summary, we have developed a Chan–Lam protocol for O-cyclopropylation of phenols and N-cyclopropylation of select azaheterocycles. The reaction uses a simple copper(II) precatalyst with 1 atm O2 as the terminal oxidant and allows for a range of structurally and functionally diverse nucleophiles to be smoothly coupled with potassium cyclopropyl trifluoroborates in good to high yields. A broad range of functional groups, including halides, ethers, esters, heterocycles, and nitro groups were tolerated. Compared to existing synthetic methods, this approach has several advantages. It is operationally convenient to perform, uses commercially available reagents, employs an inexpensive copper(II) catalyst, has high levels of functional group tolerance, and gives minimal byproduct formation in most cases. We envision that this reaction will find immediate use in medicinal chemistry laboratories as a tool for late-stage installation of cyclopropyl groups. Additionally, in the long term, it could potentially be adapted for use on larger scale in early- or late-stage development.
Experimental Section
General Information
Unless otherwise noted, all materials were used as received from commercial sources without further purification. Consistently high yields were obtained with potassium cyclopropyltrifluoroborate purchased from Boron Molecular. Batches from other suppliers sometimes afforded lower yields; we therefore recommend calibrating the reaction using the 4-phenylphenol substrate and purifying the trifluoroborate salt via soxhlet extraction if yields are lower than expected. 1H and 13C spectra were recorded on Bruker DRX-500 and AV-600. Spectra were internally referenced to SiMe4 or solvent signals. The following abbreviations (or combinations thereof) were used to explain multiplicities: b = broad, s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. High-resolution mass spectra (HRMS) were recorded on an Agilent ESI-TOF (LC/MSD TOF); compounds that could not be ionized under these conditions were measured on an Agilent 5973 Inert GC-MS mass spectrometer. Compounds with no mass spec data reported in this manuscript could not be ionized on either of these machines. Melting points were measured on a MEL-TEMP II (Laboratory Devices, USA) apparatus and are uncorrected
General Procedure for O- and N-Cyclopropylation of Phenols and Azaheterocycles
To a 5-mL scintillation vial equipped with a Teflon-coated magnetic stir bar were added the phenol substrate (0.3 mmol), cyclopropyl potassium trifluoroborate (133.2 mg, 0.9 mmol), Cu(OAc)2, 1,10-phenanthroline, and K2CO3 (83.0 mg, 0.6 mmol). The vial was charged with toluene (0.5 mL) and water (0.15 mL) and sealed with a septum cap. The septum was pierced with a 21G needle connected to an O2 balloon. The septum cap was partially unscrewed to purge the vial for 5 seconds and then resealed by tightening the cap. The reaction was stirred under O2 (balloon) at 70 °C for 12 h, after which it was quenched with sat. NH4Cl (aq.) and extracted with EtOAc. The combined organics were dried over MgSO4, filtered and concentrated in vacuo. 1H NMR yields were determined by integration of the cyclopropyl product peak relative to 1.0 equiv dibromomethane as internal standard. Crude products were then purified by silica flash column chromatography. The optimal catalyst/ligand loading was found to vary depending on the substrate employed:
-
General Procedure A (typical phenol substrates):
Cu(OAc)2 (5.4 mg, 10 mol %), 1,10-phenanthroline (5.4 mg, 10 mol %)
-
General Procedure B (electron-poor phenols and azaheterocycles):
Cu(OAc)2 (13.6 mg, 25 mol%), 1,10-phenanthroline (6.8 mg, 12.5 mol%)
-
General Procedure C (Pfizer examples, more valuable substrates)
Cu(OAc)2 (13.6 mg, 25 mol%), 1,10-phenanthroline (13.5 mg, 25 mol%)
Characterization of New Compounds
Data for O-cyclopropyl aryl ether products 5a–5r and N-cyclopropylated azaheretocycle products 8a–8d, 9a–9g, 10a–10c as well as starting material S4 are included below. Original NMR spectra can be found in the supporting information. Analytical data for compounds S1–S3 have been reported elsewhere.
4-Cyclopropoxy-1,1′-biphenyl (5a)
The title compound was prepared from 4-phenylphenol (54 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as a white solid (52.9 mg, 84% yield). mp = 109–110 °C; 1H NMR (500 MHz, CDCl3) δ 7.65–7.49 (m, 4H), 7.42 (t, J = 7.7 Hz, 2H), 7.33–7.28 (m, 1H), 7.12 (d, J = 8.7 Hz, 1H), 3.81–3.75 (m, 1H), 0.81 (d, J = 4.5 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 128.9, 128.2, 126.9, 115.4, 51.0, 6.4.
1-(tert-Butyl)-4-cyclopropoxybenzene (5b)
The title compound was prepared from 4-(tert-butyl) phenol (45 mg, 0.3 mmol) according to general procedure A. Purification by silica gel 13 chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as a yellow oil (41.0 mg, 72% yield). 1H NMR (500 MHz, CDCl3) δ 7.30 (d, J = 8.8 Hz, 2H), 6.98 (d, J = 8.8 Hz, 2H), 3.71 (dq, J = 5.9, 4.4 Hz, 1H), 1.30 (s, 9H), 0.76 (d, J = 4.1 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 157.0, 144.0, 126.5, 114.8, 100.0, 51.1, 34.5, 32.0, 6.6; GC/MS (EI) m/z calcd for C13H18O [M]+ 190.1358, found 190.
1,3-Di-tert-butyl-5-cyclopropoxybenzene (5c)
The title compound was prepared from 3,5-di-tert-butylphenol (61.8 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as a colorless oil (30.9 mg, 42% yield). 1H NMR (500 MHz, CDCl3) δ 7.04 (t, J = 1.7 Hz, 1H), 6.92 (d, J = 1.6 Hz, 2H), 3.79–3.60 (m, 1H), 1.32 (s, 18H), 0.77 (d, J = 6.4, 3.6, 1.6 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 158.6, 152.2, 115.3, 109.4, 50.6, 35.1, 31.6, 6.3; GC/MS (EI) m/z calcd for C17H26O [M]+ 246.1984, found 246.
2-Cyclopropoxy-1,4-dimethylbenzene (5d)
The title compound was prepared from 2,5-dimethylphenol (36.6 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as a yellow oil (22.3 mg, 46% yield). 1H NMR (500 MHz, CDCl3) δ 7.03–6.93 (m, 2H), 6.69 (dt, J = 7.4, 1.2 Hz, 1H), 3.79–3.64 (m, 1H), 2.35 (s, 3H), 2.13 (s, 3H), 0.88–0.64 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 157.3, 136.8, 130.7, 123.7, 121.4, 113.4, 50.9, 21.9, 16.1, 6.7.
(4-Cyclopropoxyphenyl)(methyl)sulfane (5e)
The title compound was prepared from 4- (methylthio)phenol (42 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as a yellow solid (32.4 mg, 60% yield). mp = 52–55 °C; 1H NMR (500 MHz, CDCl3) δ 7.28 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 3.75–3.67 (m, 1H), 2.44 (s, 3H), 0.76 (d, J = 3.0 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 157.9, 130.4, 129.5, 116.1, 51.3, 18.4, 6.6.
1-(Benzyloxy)-4-cyclopropoxybenzene (5f)
The title compound was prepared from 4- (benzyloxy)phenol (60 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as an off-white solid (45.0 mg, 63% yield). mp = 77–79 °C; 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 7.6 Hz, 2H), 7.36 (t, J = 7.5 Hz, 2H), 7.31 (d, J = 7.2 Hz, 1H), 6.96 (d, J = 9.2 Hz, 2H), 6.89 (d, J = 8.2 Hz, 2H), 5.00 (s, 2H), 3.66 (dt, J = 5.5, 2.4 Hz, 1H), 0.73 (d, J = 3.2 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 153.6, 153.6, 137.7, 129.0, 128.3, 127.9, 116.1, 116.1, 116.1, 100.0, 71.1, 51.5, 6.6.
1-Cyclopropoxy-3-methoxybenzene (5g)
The title compound was prepared from 3-methoxyphenol (37.2 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as a colorless oil (27.5 mg, 56% yield). 1H NMR (500 MHz, CDCl3) δ 7.18 (t, J = 8.2 Hz, 1H), 6.67 (ddd, J = 8.1, 2.3, 0.8 Hz, 1H), 6.62 (t, J = 2.4 Hz, 1H), 6.53 (ddd, J = 8.2, 2.4, 0.8 Hz, 1H), 3.79 (d, J = 0.5 Hz, 3H), 3.75–3.69 (m, 1H), 0.77 (d, J = 4.5 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 130.2, 107.8, 106.8, 101.8, 55.7, 51.2, 6.6; GC/MS (EI) m/z calcd for C10H12O2 [M]+ 164.0837, found 164.
1-Cyclopropoxy-4-fluorobenzene (5h)
The title compound was prepared from 4-fluorophenol (33.6 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as a yellow oil (22.6 mg, 50% yield). 1H NMR (500 MHz, CDCl3) δ 7.08–6.87 (m, 4H), 3.80–3.56 (m, 1H), 0.84–0.67 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 116.2, 116.1, 116.0, 51.6, 30.1, 6.6.
1-Chloro-4-cyclopropoxybenzene (5i)
The title compound was prepared from 4-chlorophenol (38.4 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as an orange oil (26.7 mg, 53% yield). 1H NMR (500 MHz, CDCl3) δ 7.32 (dd, J = 10.7, 5.9 Hz, 2H), 7.06 (t, J = 8.3 Hz, 2H), 3.79 (tt, J = 5.9, 2.9 Hz, 1H), 0.85 (d, J = 5.3 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 129.6, 127.3, 126.2, 116.6, 51.5, 6.6.
1-Bromo-4-cyclopropoxybenzene (5j)
The title compound was prepared from 4-bromophenol (51.9 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as a yellow oil (34.5 mg, 54% yield). 1H NMR (500 MHz, CDCl3) δ 7.37 (d, J = 7.1 Hz, 2H), 6.92 (d, J = 7.1 Hz, 2H), 3.70 (dd, J = 6.0, 3.4 Hz, 1H), 0.77 (ddt, J = 5.4, 3.4, 1.9 Hz, 4H), 13C NMR (150 MHz, CDCl3) δ 132.6, 117.2, 113.6, 51.4, 30.1, 6.6; GC/MS (EI) m/z calcd for C9H9OBr [M]+ 211.9837, found 212.
1-Iodo-4-cyclopropoxybenzene (5k)
The title compound was prepared from 4-iodophenol (66.0 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as an orange solid (46.6 mg, 60% yield). mp = 49–50 °C; 1H NMR (500 MHz, CDCl3) δ 7.69–7.56 (d, 2H), 6.99–6.81 (d, 2H), 3.78 (m, J = 6.1, 2.8 Hz, 1H), 0.85 (m, J = 7.5, 2.8 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 159.0, 138.3, 117.6, 83.2, 51.1, 6.3; GC/MS (EI) m/z calcd for C9H9OI [M]+ 259.9698, found 260.
Methyl 4-cyclopropoxybenzoate (5l)
The title compound was prepared from methyl 4-hydroxybenzoate (45.6 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as a yellow oil (19.6 mg, 34% yield). 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.9 Hz, 2H), 7.06 (d, J = 8.8 Hz, 2H), 3.88 (s, 2H), 3.78 (m, J = 6.1, 3.2 Hz, 1H), 0.97–0.65 (d, 4H); 13C NMR (150 MHz, CDCl3) δ 167.3, 163.2, 131.9, 123.3, 115.1, 52.3, 51.6, 6.7.
4-(Cyclopropyloxy)benzonitrile (5m)
The title compound was prepared from 4-cyanophenol (35.6 mg, 0.3 mmol) according to General Procedure B. The standard reaction conditions afforded an inseparable mixture of the desired O-cyclopropylated product, as well as O-allylated product S1. After aqueous workup, the mixture was redissolved in H2O (0.8 mL) and acetone (6.2 mL), and treated with OsO4 (2.5 wt% in i-PrOH, 0.15 mL, 0.015 mmol, 5 mol%) and NMO (53 mg, 0.45 mmol, 1.5 equiv). The reaction was stirred at rt, after which it was quenched with aqueous sodium bisulfite, extracted with EtOAc, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica flash column chromatography (eluent: 2% EtOAc/Hex to 5% EtOAc/Hex) afforded the title compound as a colorless oil (13.0 mg, 27% yield). 1H NMR (500 MHz, CDCl3) δ 7.60–7.56 (m, 2H), 7.12–7.07 (m, 2H), 3.78 (tt, J = 6.0, 3.0 Hz, 1H), 0.85–0.81 (m, 2H), 0.82–0.78 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 162.6, 134.0, 119.4, 116.0, 104.4, 51.5, 6.4.
4-(Allyloxy)benzonitrile (S1):[23]
1H NMR (500 MHz, CDCl3) δ 7.63–7.55 (m, 2H), 6.99–6.94 (m, 2H), 6.03 (ddd, J = 22.5, 10.6, 5.3 Hz, 1H), 5.48–5.38 (m, 1H), 5.36–5.28 (m, 1H), 4.59 (dt, J = 5.3, 1.6 Hz, 2H).
3-(Cyclopropyloxy)benzonitrile (5n)
The title compound was prepared from 3-cyanophenol (35.6 mg, 0.3 mmol) according to General Procedure B. The standard reaction conditions afforded an inseparable mixture of the desired O-cyclopropylated product, as well as O-allylated product S2. After aqueous workup, the mixture was redissolved in H2O (0.8 mL) and acetone (6.2 mL), and treated with OsO4 (2.5 wt% in i-PrOH, 0.15 mL, 0.015 mmol, 5 mol%) and NMO (53 mg, 0.45 mmol, 1.5 equiv). The reaction was stirred at rt, after which it was quenched with aqueous sodium bisulfite, extracted with EtOAc, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica flash column chromatography (eluent: 2% EtOAc/Hex to 5% EtOAc/Hex) afforded the title compound as a colorless oil (14.1 mg, 30% yield). 1H NMR (500 MHz, CDCl3) δ 7.37 (t, J = 8.0 Hz, 1H), 7.34–7.32 (m, 1H), 7.27–7.23 (m, 2H), 3.75 (tt, J = 6.0, 3.0 Hz, 1H), 0.85–0.81 (m, 2H), 0.81–0.78 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 159.3, 130.4, 124.9, 120.5, 118.9, 118.1, 113.2, 51.4, 6.4.
2-(Allyloxy)benzonitrile (S2):[24]
1H NMR (500 MHz, CDCl3) 7.19–7.14 (m, 3H), 6.88 (d, J = 8.8 Hz, 1H), 6.81 (d, J = 8.3 Hz, 2H), 5.99–6.06 (m, 1H), 5.42 (d, J = 17.7, 1H), 5.33 (d, J = 10.7 Hz, 1H), 4.56 (d, J = 5.3 Hz, 2H).
1-(Cyclopropyloxy)-3-nitrobenzene (5o)
The title compound was prepared from 3-nitrophenol (41.7 mg, 0.3 mmol) according to General Procedure B. The standard reaction conditions afforded an inseparable mixture of the desired O-cyclopropylated product, as well as O-allylated product S3. After aqueous workup, the mixture was redissolved in H2O (0.8 mL) and acetone (6.2 mL), and treated with OsO4 (2.5 wt% in i-PrOH, 0.15 mL, 0.015 mmol, 5 mol%) and NMO (53 mg, 0.45 mmol, 1.5 equiv). The reaction was stirred at rt, after which it was quenched with aqueous sodium bisulfite, extracted with EtOAc, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica flash column chromatography (eluent: 2% EtOAc/Hex to 10% EtOAc/Hex) afforded the title compound as a yellow oil (14.8 mg, 28% yield). 1H NMR (500 MHz, CDCl3) δ 7.92 (t, J = 2.3 Hz, 1H), 7.86–7.81 (m, 1H), 7.42 (t, J = 8.2 Hz, 1H), 7.34–7.30 (m, 1H), 3.82 (tt, J = 6.2, 2.9 Hz, 1H), 0.89–0.84 (m, 2H), 0.84–0.79 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 160.0, 149.5, 130.2, 122.3, 116.4, 109.9, 51.9, 6.7.
1-(Allyloxy)-3-nitrobenzene (S3):[25]
1H NMR (600 MHz, CDCl3) δ 7.25–7.80 (m, 4H), 6.11–5.99 (m, 1H), 5.45 (dd, J = 17.2, 1.5 Hz, 1H), 5.37–5.32 (m, 1H), 4.63 (dt, J = 5.3, 1.5 Hz, 2H).
tert-butyl-8-cyclopropoxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (5p)
The title compound was prepared from tert-butyl-8-hydroxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (74.8 mg, 0.300 mmol) according to General Procedure C. Purification was performed using flash column chromatography (12g SiO2, Isco, 100% Hept. to 10% EtOAc) to afford the title compound as a colorless oil (27.0 mg, 31% yield). 1H NMR (600 MHz, CDCl3) δ 7.19–7.12 (m, 1 H) 7.08 (d, J = 8.07 Hz, 1 H) 6.77 (br d, J = 7.34 Hz, 1 H) 4.45 (br s, 2 H) 3.80–3.72 (m, 1H) 3.64 (br t, J = 5.69 Hz, 2 H) 2.82 (br t, J = 5.50 Hz, 2 H) 1.52 (s, 9 H) 0.78 (br d, J = 3.48 Hz, 4 H); 13C NMR (150 MHz, CDCl3) δ 155.48 (br s, 1C), 154.99 (br s, 1C), 135.85 (br s, 1C), 126.57, 122.49 (br s, 1C), 121.08 (br s, 1C), 109.62, 79.55, 50.76, 41.53 (br s, 1C), 40.97–40.39 (m, 1C), 29.00 (br s, 1C), 28.49 (s, 3C), 6.20 (br s, 2C); HRMS (ESI-TOF) m/z calcd for C17H23NO3 + [M+Na]+ = 312.1570, found 312.1551.
2-(4-cyclopropoxyphenyl)pyridine (5q)
The title compound was prepared from 4-(pyridin-2-yl)phenol (51.3 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as an off-white solid (26.6 mg, 42% yield). mp = 82–84 °C; 1H NMR (500 MHz, CDCl3) δ 8.66 (d, J = 4.8 Hz, 1H), 7.97–7.93 (m, 2H), 7.71 (dd, J = 23.5, 7.8 Hz, 2H), 7.22–7.11 (m, 3H), 3.80 (t, J = 4.6 Hz, 1H), 0.87–0.74 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 160.1, 157.1, 149.3, 137.1, 132.1, 128.3, 121.6, 120.1, 115.4, 51.1, 6.4; HRMS (ESI-TOF) m/z calcd for C14H14NO [M+H]+ 212.1070, found 212.1064.
2-(4-Cyclopropoxyphenyl)thiophene (5r)
The title compound was prepared from 4-(thiophen-2-yl)phenol (52 mg, 0.3 mmol) according to General Procedure A. Purification by silica gel chromatography with 10:1 hexanes:EtOAc as the eluent gave the product as an off-white solid (39.5 mg, 61% yield). mp = 88–89 °C; 1H NMR (500 MHz, CDCl3) δ 7.71–7.53 (m, 2H), 7.32–7.24 (m, 2H), 7.27–7.03 (m, 3H), 3.94–3.76 (m, 1H), 0.88 (d, J = 5.1 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 159.0, 144.8, 128.3, 128.0, 127.5, 124.3, 122.5, 115.8, 51.3, 6.7; GC/MS (EI) m/z calcd for C13H12OS [M]+ 216.0609, found 216.
1,3-Dicyclopropyl-1,3-dihydro-2H-benzimidazol-2-one (8a)
The title compound was prepared from 2-hydroxybenzimidazole (40.2 mg, 0.3 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: 5% EtOAc/Hexane to 20% EtOAc/Hexane) afforded the title compound as a white solid (31.8 mg, 49% yield), along with separable mono-cyclopropylated product 8a′. mp = 92–94 °C; 1H NMR (500 MHz, CDCl3) δ 7.21–7.13 (m, 2H), 7.12–7.04 (m, 2H), 2.83 (tt, J = 7.0, 3.7 Hz, 2H), 1.11–1.05 (m, 4H), 0.99 (ddd, J = 8.0, 5.4, 3.8 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 155.0, 130.1, 121.2, 108.4, 22.5, 6.0; HRMS (ESI-TOF) m/z calcd for C13H15N2O [M+H]+ 215.1184, found 215.1180.
1-Cyclopropyl-1H-benzimidazol-2-ol (8a′)
The title compound was prepared according to the procedure described above for 8a and was isolated as a white solid (7.7 mg, 15% yield). mp = 184–188 °C; 1H NMR (CDCl3, 500 MHz) δ 10.03 (brs, 1H, OH), 7.23–7.19 (m, 1H), 7.14–7.04 (m, 3H), 2.91 (tt, J = 7.0, 3.6 Hz, 1H), 1.18–1.09 (m, 2H), 1.08–1.02 (m, 2H); 13C NMR (CDCl3, 150 MHz) δ 156.1, 131.5, 127.6, 121.7, 121.4, 109.5, 108.9, 22.4, 6.2; HRMS (ESI-TOF) m/z calcd for C10H11N2O [M+H]+ 175.0871, found 175.0876; X-ray (single-crystal): Colorless block crystals of X-ray diffraction quality were obtained by slow evaporation of a solution of 8a′ in CDCl3 (CCDC 1584673).[26]
1-Cyclopropyl-3-methyl-1,3-dihydro-2H-benzimidazol-2-one (8b)
The title compound was prepared from 1-methyl-1,3-dihydro-2H-benzimidazol-2-one (44.5 mg, 0.3 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: 10% EtOAc/Hex) afforded the title compound as a white solid (43.0 mg, 76% yield). mp = 84–86 °C; 1H NMR (500 MHz, CDCl3) δ 7.20–7.15 (m, 1H), 7.10–7.05 (m, 2H), 6.95–6.90 (m, 1H), 3.36 (s, 3H), 2.86 (tt, J = 7.1, 3.7 Hz, 1H), 1.11–1.06 (m, 2H), 1.02–0.97 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 154.8, 130.3, 129.8, 121.3, 121.1, 108.5, 107.2, 27.0, 22.6, 6.1; HRMS (ESI-TOF) m/z calcd for C11H13N2O [M+H]+ 189.1028, found 189.1022.
3-Cyclopropylbenzo[d]oxazol-2(3H)-one (8c)
The title compound was prepared from 2-hydroxybenzoxazolinone (40.5 mg, 0.3 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: 5 % EtOAc/Hex to 10% EtOAc/Hex) afforded the title compound as a white solid (45.3 mg, 86% yield). mp = 62–63 °C; 1H NMR (500 MHz, CDCl3) δ 7.21–7.13 (m, 3H), 7.12–7.08 (m, 1H), 2.92 (tt, J = 7.0, 3.6 Hz, 1H), 1.15–1.08 (m, 2H), 1.08–1.02 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 154.4, 142.3, 132.0, 123.8, 122.5, 109.9, 109.2, 23.3, 6.0; HRMS (ESI-TOF) m/z calcd for C9H9N2O2 [M+H]+ 177.0659, found 177.0659.
6-Chloro-3-cyclopropyl-1,3-benzoxazol-2(3H)-one (8d)
The title compound was prepared from 6-chlorobenzoxazol-2(3H)-one (50.9 mg, 0.3 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: 10% EtOAc/Hex to 20% EtOAc/Hex) afforded the title compound as an orange solid (44.3 mg, 70% yield). mp 110–112 °C; 1H NMR (500 MHz, CDCl3) δ 7.18–7.12 (m, 2H), 7.08–7.03 (m, 1H), 2.90 (tt, J = 7.0, 3.6 Hz, 1H), 1.15–1.07 (m, 2H), 1.05–0.98 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 154.0, 142.6, 130.8, 128.1, 123.9, 110.8, 109.7, 23.4, 6.0; GC/MS (EI) m/z calcd for C10H8ClNO2 [M]+ 209.0244, found 209.
1-Cyclopropylquinolin-2(1H)-one (9a)
The title compound was prepared from 2-hydroxyquinoline (43.5 mg, 0.3 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: 30% EtOAc/Hex to 50% EtOAc/Hex) afforded the title compound as a yellow oil (41.0 mg, 74% yield). 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.5 Hz, 1H), 7.59 (d, J = 9.5 Hz, 1H), 7.55–7.45 (m, 2H), 7.18 (t, J = 7.5 Hz, 1H), 6.59 (d, J = 9.4 Hz, 1H), 2.93 (tt, J = 7.7, 4.1 Hz, 1H), 1.39–1.30 (m, 2H), 0.93–0.85 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 163.8, 141.1, 139.1, 130.0, 128.6, 122.5, 122.2, 121.0, 115.6, 26.2, 10.5; HRMS (ESI-TOF) m/z calcd for C12H12NO [M+H]+ 186.0913, found 186.0914. 21
1-Cyclopropylpyridin-2-one (9b)
The title compound was prepared from 2-hydroxypyridine (47.6 mg, 0.5 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: 2% MeOH/DCM to 5% MeOH/DCM), followed by preparative TLC (eluent: 5% MeOH/DCM) afforded the title compound as a yellow oil (24.8 mg, 37% yield). The connectivity (i.e., N-versus O-cyclopropylation) was confirmed by NOESY analysis and by the diagnostic carbonyl signal at 164.3 ppm in the 13C NMR spectrum. 1H NMR (500 MHz, CDCl3) δ 8.63–8.57 (m, 2H), 7.87–7.83 (m, 1H), 7.44 (tt, J = 6.8, 1.2 Hz, 1H), 4.64 (tt, J = 7.7, 3.6 Hz, 1H), 2.47–2.42 (m, 2H), 2.19–2.15 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 164.3, 139.0, 136.9, 120.9, 105.7, 32.3, 6.8; HRMS (ESI-TOF) m/z calcd for C8H10NO [M+H]+ 136.0757, found 136.0757.
1-Cyclopropyl-4-methylpyridin-2-one (9c)
The title compound was prepared from 2-hydroxy-4-methylpyridine (32.7 mg, 0.3 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: DCM to 2% MeOH/DCM) afforded the title compound as a pale-yellow oil (31.9 mg, 71% yield). 1H NMR (500 MHz, CDCl3) δ 7.11 (d, J = 7.1 Hz, 1H), 6.29 (s, 1H), 5.92 (dd, J = 7.1, 1.9 Hz, 1H), 3.24 (tt, J = 7.6, 4.2 Hz, 1H), 2.10 (s, 3H), 1.09–1.01 (m, 2H), 0.82–0.75 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 164.2, 150.6, 135.8, 119.2, 108.2, 31.8, 21.2, 6.7; HRMS (ESI-TOF) m/z calcd for C9H12NO [M+H]+ 150.0913, found 150.0912.
1-Cyclopropyl-5-fluoropyridin-2-one (9d)
The title compound was prepared from 2-hydroxy-5-fluoropyridine (33.9 mg, 0.3 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: DCM to 2% MeOH/DCM) afforded the title compound as a pale-yellow oil (29.0 mg, 63% yield). 1H NMR (500 MHz, CDCl3) δ 7.25–7.19 (m, 1H), 7.17 (t, J = 3.9 Hz, 1H), 6.48 (dd, J = 10.0, 5.4 Hz, 1H), 3.30 (tt, J = 7.7, 4.3 Hz, 1H), 1.14–1.09 (m, 2H), 0.86–0.81 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 162.3, 147.1 (d, J = 231 Hz), 131.0 (d, J = 24 Hz), 122.3 (d, J = 37 Hz), 121.5 (d, J = 7 Hz), 32.7, 7.1; 19F NMR (CDCl3, 376 MHz, CDCl3) δ –149.35; HRMS (ESI-TOF) m/z calcd for C8H9FNO [M+H]+ 154.0663, found 154.0663.
1-Cyclopropyl-5-chloropyridin-2-one (9e)
The title compound was prepared from 2-hydroxy-5-chloropyridine (38.9 mg, 0.3 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: DCM to 2% MeOH/DCM), followed by preparative TLC (eluent: 2% MeOH/DCM) afforded the title compound as a yellow oil (33.8 mg, 66% yield). 1H NMR (500 MHz, CDCl3) δ 7.30 (d, J = 2.8 Hz, 1H), 7.21 (dd, J = 9.7, 2.9 Hz, 1H), 6.48 (d, J = 9.7 Hz, 1H), 3.28 (tt, J = 7.6, 4.2 Hz, 1H), 1.15–1.09 (m, 2H), 0.86–0.82 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 162.7, 140.1, 134.7, 121.8, 112.0, 32.9, 31.1, 7.1; HRMS (ESI-TOF) m/z calcd for C8H9ClNO [M+H] 170.0367, found 170.0367.
1-Cyclopropyl-6-methylpyridin-2(1H)-one (9f)
The title compound was prepared from 6-methylpyridin-2(1H)-one (32.7 mg, 0.300 mmol) according to General Procedure C. Purification was performed using flash column chromatography (12 g SiO2, Isco, 100% Hept. to 10% MeOH/EtOAc) to afford the title compound as a pale-yellow gum (27.5 mg, 61% yield). 1H NMR (400MHz, CDCl3) δ 7.15 (dd, J = 9.2, 6.7 Hz, 1H), 6.39 (d, J = 9.2 Hz, 1H), 5.97 (d, J = 6.7 Hz, 1H), 2.83 (tt, J = 7.1, 4.2 Hz, 1H), 2.46 (s, 3H), 1.31–1.23 (m, 2H), 0.92–0.85 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 165.24, 148.59, 138.60, 118.44, 106.83, 28.08, 20.53, 10.33 (s, 2C); HRMS (ESI-TOF) m/z calcd for C9H11NO+ [M+H]+ 150.0913, found 150.0915.
tert-Butyl-7-cyclopropyl-8-oxo-3,4,7,8-tetrahydro-2,7-naphthyridine-2(1H)-carboxylate (9g)
The title compound was prepared from tert-butyl-8-oxo-3,4,7,8-tetrahydro-2,7-naphthyridine-2(1H)-carboxylate (75.1 mg, 0.300 mmol) according to General Procedure C. Purification was performed using flash column chromatography (12g SiO2, Isco, 100% Hept. to 10% MeOH/EtOAc) to afford the title compound as a pale-yellow gum (55.4 mg, 63% yield). 1H NMR (600 MHz, CDCl3) δ 7.12 (br d, J=6.97 Hz, 1 H) 5.95 (br d, J = 6.97 Hz, 1 H) 4.39 (s, 2 H) 3.61 (br t, J = 5.59 Hz, 2 H) 3.34 (dt, J = 7.24, 3.53 Hz, 1 H) 2.64–2.56 (m, 2 H) 1.50 (d, J = 1.10 Hz, 9 H) 1.12 (br d, J = 7.15 Hz, 2 H) 0.83–0.88 (m, 2 H); 13C NMR (150MHz, CDCl3) δ = 162.04, 154.95 (br s, 1C), 144.63 (br s, 1C), 133.18, 123.98 (br s, 1C), 106.95 (br s, 1C), 79.95 (br s, 1C), 42.28 (br s, 1C), 39.47 (br s, 1C), 31.77, 28.52, 28.47 (s, 3C), 6.56 (s, 2C); HRMS (ESI-TOF) m/z calcd for C16H22N2O3 + [M+H]+ = 291.1703, found 291.1707.
N-Cyclopropyl-N-(pyridin-2-yl)acetamide (10a)
The title compound was prepared from 2-acetamidopyridine (40.8 mg, 0.3 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: 1% MeOH/DCM) afforded the title compound as a brown oil (47.0 mg, 89% yield). 1H NMR (500 MHz, CDCl3) δ 8.45 (d, J = 4.7 Hz, 1H), 7.68 (td, J = 7.7, 2.0 Hz, 1H), 7.21–7.10 (m, 2H), 3.11 (tt, J = 7.2, 3.8 Hz, 1H), 2.16 (s, 3H), 0.95–0.84 (m, 2H), 0.60–0.46 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 172.8, 155.3, 148.8, 137.8, 122.2, 121.8, 30.5, 23.6, 9.2; HRMS (ESI-TOF) m/z calcd for C10H13N2O [M+H]+ 177.1022, found 177.1021.
N-Cyclopropylpyrazin-2-amine (10b)
The title compound was prepared from 2-aminopyrimidine (28.5 mg, 0.3 mmol) according to General Procedure B. Purification by silica flash column chromatography (eluent: 1–2% MeOH/DCM) afforded the title compound as a pale-yellow solid (13.2 mg, 33% yield). mp = 82–85 °C; 1H NMR (600 MHz, CDCl3) δ 8.34 (s, 2H), 6.58 (t, J = 4.8 Hz, 1H), 5.67 (brs, 1H, –NH), 2.77 (dddd, J = 9.2, 7.1, 3.6, 2.1 Hz, 1H), 0.89–0.80 (m, 2H), 0.59–0.52 (m, 2H); 13C NMR(150 MHz, CDCl3) δ 163.4, 158.2, 111.2, 111.2, 23.9, 7.5, 7.5; HRMS (ESI-TOF) m/z calcd for C7H10N3 [M+H]+ 136.0869, found 136.0871.
5-Hydroxy-N1,N1-dimethyl-N3-(5-methylpyridin-2-yl)isophthalamide (S4)
The title compounds was prepared in a similar method as a previously published Pfizer patent,[27] 1H NMR (400 MHz, DMSO-d6) δ 10.64 (s, 1H), 9.97 (s, 1H), 8.21 (s, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.65 (dd, J = 2.0, 8.5 Hz, 1H), 7.47 (s, 1H), 7.43–7.36 (m, 1H), 6.93 (dd, J = 1.3, 2.1 Hz, 1H), 2.98 (br s, 3H), 2.93 (br s, 3H), 2.28 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.87, 165.65, 157.78, 150.36, 148.21, 138.87, 138.40, 136.09, 129.29, 117.45, 117.20, 116.15, 114.92, 35.18, 17.80 (carbon signal suspected under solvent peak); HRMS (ESI-TOF) m/z calcd for C16H17N3O3 + [M+H]+ 300.1343, found 300.1345.
N1-Cyclopropyl-5-hydroxy-N3,N3-dimethyl-N1-(5-methylpyridin-2-yl)isophthalamide (10c)
The title compound was prepared from 5-hydroxy-N1,N1-dimethyl-N3-(5-methylpyridin-2-yl)isophthalamide (S4) (180 mg, 0.6 mmol) according to General Procedure C. Purification by ISCO (0–100% EtOAc/Heptanes) afforded the title compound as a white foam (130 mg, 64%). Structure elucidated by HMBC. 1H NMR (400 MHz, DMSO-d6) δ 9.80 (s, 1 H), 8.26–8.08 (m, 1 H), 7.57 (ddd, J = 8.07, 2.45, 0.61 Hz, 1 H), 7.16 (d, J = 8.07 Hz, 1 H), 6.81 (dd, J = 2.32, 1.47 Hz, 1 H), 6.65 (dd, J = 2.38, 1.41 Hz, 1 H), 6.60 (t, J = 1.41 Hz, 1 H), 3.10 (tt, J = 7.21, 3.73 Hz, 1 H), 2.90 (br s, 3 H), 2.62 (br s, 3 H), 2.23 (s, 3 H), 0.81–0.69 (m, 2 H), 0.63–0.52 (m, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 171.07, 169.65, 157.34, 153.52, 149.13, 139.02, 138.88, 137.71, 131.98, 122.54, 116.94, 115.95, 115.30, 38.98, 34.97, 31.45, 17.74, 8.30; HRMS (ESI-TOF) m/z calcd for C19H21N3O3 + [M+H]+ 340.1656, found 340.1663
Supplementary Material
Acknowledgments
This work was financially supported by the National Institutes of Health (1R35GM125052), The Scripps Research Institute (TSRI), and Pfizer, Inc. We gratefully acknowledge the NSF for a Graduate Research Fellowship (NSF/DGE-1346837, J.D.) and the DAAD for a postdoctoral fellowship (M.L.O.). We further thank Dr. Dee-Hua Huang for assistance with NMR spectroscopy; Dr. Milan Gembicky and Dr. Arnold L. Rheingold for X-ray structure determination; and Dr. Gregory Kauffman, Dr. Eugene Rui, and Dr. Klaus Dress for helpful discussion regarding the MMP analysis.
Footnotes
Supporting Information Available. Photographic depiction of reaction setup, matched molecular pair (MMP) data, X-ray crystallographic data, and 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a recent review, see: Talele TT. J Med Chem. 2016;59:8712–8756. doi: 10.1021/acs.jmedchem.6b00472.
- 2.McGrath NA, Brichacek M, Njardarson JT. J Chem Educ. 2010;87:1348–1349. [Google Scholar]
- 3.For selected references, see: Leeson PD, Springthorpe B. Nat Rev Drug Discov. 2007;6:881–890. doi: 10.1038/nrd2445.Edwards MP, Price DA. Ann Reports Med Chem. 2010;45:381–391.Freeman-Cook KD, Hoffman RL, Johnson TW. Future Med Chem. 2013;5:113–115. doi: 10.4155/fmc.12.208.Meanwell NA. Chem Res Toxicol. 2016;29:564–616. doi: 10.1021/acs.chemrestox.6b00043.
- 4.Tian Z, Fattahi A, Lis L, Kass SR. J Am Chem Soc. 2006;128:17087–17092. doi: 10.1021/ja065348u. [DOI] [PubMed] [Google Scholar]
- 5.Keefer CE, Chang G, Kauffman GW. Bioorg Med Chem. 2011;19:3739–3749. doi: 10.1016/j.bmc.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Stepan AF, Kauffman GW, Keefer CE, Verhoest PR, Edwards MP. J Med Chem. 2013;56:6985–6990. doi: 10.1021/jm4008642. [DOI] [PubMed] [Google Scholar]
- 7.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Chem Res Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 8.For representative examples, see: Buchwald–Hartwig amination: Hofmans S, Berghe TV, Devisscher L, Hassannia B, Lyssens S, Joossens J, Van Der Veken P, Vandenabeele P, Augustyns K. J Med Chem. 2016;59:2041–2053. doi: 10.1021/acs.jmedchem.5b01641.. Cyclization of 2-bromoprop-2-enylamines: Bayliffe FH, Steven A, Ando K, Shipman M. Synlett. 2015;16:1371–1374. Reductive amination: Shaw MH, Croft RA, Whittingham WG, Bower JF. J Am Chem Soc. 2015;137:8054–8057. doi: 10.1021/jacs.5b05215.. For selected examples of copper-catalyzed Chan–Lam N-cyclopropylation, see: Mudryk B, Zheng B, Chen K, Eastgate MD. Org Process Res Dev. 2014;18:520–527.Rossi SA, Shimkin KW, Xu Q, Mori-Quiroz LM, Watson DA. Org Lett. 2013;15:2314–2317. doi: 10.1021/ol401004r.Bénard S, Neuville L, Zhu J. Chem Commun. 2010;46:3393–3395. doi: 10.1039/b925499d.Tsuritani T, Strotman NA, Yamamoto Y, Kawasaki M, Yasuda N, Mase T. Org Lett. 2008;10:1653–1655. doi: 10.1021/ol800376f.. For an example using a cyclopropylbismuth reagent, see: Gagnon A, St-Onge M, Little K, Duplessis M, Barabé F. J Am Chem Soc. 2007;129:44–45. doi: 10.1021/ja0676758.
- 9.(a) Cherian J, Choi I, Nayyar A, Manjunatha UH, Mukherjee T, Lee YS, Boshoff HI, Singh R, Ha YH, Goodwin M, Lakshminarayana SB, Niyomrattanakit P, Jiricek J, Ravindran S, Dick T, Keller TH, Dartois V, Barry CE., III J Med Chem. 2011;54:5639–5659. doi: 10.1021/jm1010644. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Toledo-Sherman LM, Prime ME, Mrzljak L, Beconi MG, Beresford A, Brookfield FA, Brown CJ, Cardaun I, Courtney SM, Dijkman U, Hamelin-Flegg E, Johnson PD, Kempf V, Lyons K, Matthews K, Mitchell WL, O’Connell C, Pena P, Powell K, Rassoulpour A, Reed L, Reindl W, Selvaratnam S, Friley WW, Weddel DA, Went NE, Wheelan P, Winkler C, Winkler D, Wityak J, Yarnold CJ, Yates D, Munoz-Sanjuan I, Dominguez C. J Med Chem. 2015;58:1159–1183. doi: 10.1021/jm501350y. [DOI] [PubMed] [Google Scholar]; (c) Chen S, Bohnert G, Jiang J, Xia Z. Synta Pharmaceuticals Corporation), Compounds for Inflammation and Immune-Related Uses. WO2012151355 A1. World Patent. 2012 Aug 11;; (d) Hop CECA, Wang Y, Kumar S, Elipe MVS, Raab CE, Dean DC, Poon GK, Keohane CA, Strauss J, Chiu SHL, Curtis N, Elliott J, Gerhard U, Locker K, Morrison D, Mortishire-Smith R, Thomas S, Watt AP, Evans DC. Drug Metab Dispos. 2002;30:937–943. doi: 10.1124/dmd.30.8.937. [DOI] [PubMed] [Google Scholar]; (e) Pelcman B, Olofsson K, Katkevics M, Ozola V, Suna E, Kalvins I, Trapencieris P. (Biolipox AB), Indoles Useful in the Treatment of Inflammation. WO2006077364 A1. World Patent. 2006 Jul 27;; (f) Chirasani VR, Sankar R, Senapati S. J Phys Chem B. 2016;120:8254. doi: 10.1021/acs.jpcb.6b01928. [DOI] [PubMed] [Google Scholar]
- 10.(a) Stahl GW, Cottle DL. J Am Chem Soc. 1943;65:1782–1783. [Google Scholar]; (b) Roberts JD, Chambers VC. J Am Chem Soc. 1951;73:3176–3179. [Google Scholar]
- 11.(a) Charette AB, Beauchemin A. Simmons-Smith Cyclopropanation Reaction. In: Overman LE, editor. Organic Reactions. Vol. 58. John Wiley & Sons; New York: 2001. pp. 5–40. [Google Scholar]; (b) Lemoine RC, Petersen AC, Setti L, Chen L, Wanner J, Jekle A, Heilek G, deRosier A, Ji C, Rotstein DM. Bioorg Med Chem Lett. 2010;20:1830–1833. doi: 10.1016/j.bmcl.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Takanobu K, Hiroki S, Hideyuki I, Minoru S, Kouhei A, Tsuyoshi M Takeda Pharmaceutical Company Limited. Heteromonocyclic Compound and Use Thereof. WO2008062905 A2. World Patent. 2008 May 29; For a high-yielding example with cyclopropyl bromide, see: Barr KJ, Beinstock C, Maclean J, Zhang H, Beresis RT, Zhang D Merck, Sharp, and Dohme. 4-Heteroaryl Substituted Benzoic Acid Compounds as Rorgammat Inhibitors and Uses Therof. WO2014026327 A1. World Patent. 2014 Feb 20;
- 13.Hollingworth GJ, Dinnell K, Dickinson LC, Elliott JM, Kulagowski JJ, Swain CJ, Thomson CG. Tetrahedron Lett. 1999;40:2633–2636. [Google Scholar]
- 14.Ludwig M, Pytela O, Vecera M. Collect Czech Chem Commun. 1984;49:2593–2601. [Google Scholar]
- 15.For an example of O-arylation and -alkenylation of alcohols, see: Quach TD, Batey RA. Org Lett. 2003;5:1381–1384. doi: 10.1021/ol034454n.. For examples of O-alkylation using alkyl boronic acids, see: Sueki S, Kuninobu Y. Org Lett. 2013;15:1544–1547. doi: 10.1021/ol400323z.Jacobson CE, Martinez-Muñoz N, Gorin DJ. J Org Chem. 2015;80:7305–7310. doi: 10.1021/acs.joc.5b01077.
- 16.For examples of N-alkylation using alkyl boronic acids/boronates, see: González I, Mosquera J, Guerrero C, Rodríguez R, Cruces J. Org Lett. 2009;11:1677–1680. doi: 10.1021/ol802882k.Larrosa M, Guerrero C, Rodríguez R, Cruces J. Synlett. 2010;14:2101–2105.Naya L, Larrosa M, Rodríguez R, Cruces J. Tetrahedron Lett. 2011;53:769–772.
- 17.For leading references on the mechanism of Chan–Lam coupling see: King AE, Brunold TC, Stahl SS. J Am Chem Soc. 2009;131:5044–5045. doi: 10.1021/ja9006657.King AE, Ryland BL, Brunold TC, Stahl SS. Organometallics. 2009;31:7948–7957. doi: 10.1021/om300586p.Vantourout JC, Miras HN, Isidro-Llobet A, Sproules S, Watson AJB. J Am Chem Soc. 2017;139:4769–4779. doi: 10.1021/jacs.6b12800.
- 18.Cacatian S, Claremon DA, Dillad LW, Fuchs K, Heine N, Jia L, Leftheris K, McKeever B, Morales-Ramos A, Singh S, Venkatamaran S, Wu G, Xu Z, Yuan J, Zhang Y Vitae Pharmaceuticals, Boehringer Ingelheim. Inhibitors of Beta-Secretase. WO2010105179 A3. World Patent. 2010 Nov 11;
- 19.Molander GA, Sandrock DL. Curr Opin Drug Discov Devel. 2009;12:811–823. [PMC free article] [PubMed] [Google Scholar]
- 20.Lennox AJJ, Llyod-Jones GC. Chem Soc Rev. 2014;43:412–443. doi: 10.1039/c3cs60197h. [DOI] [PubMed] [Google Scholar]
- 21.(a) Tambe YB, Sharma S, Pathak A, Reddy LK. Synth Comm. 2012;42:1341–1348. [Google Scholar]; (b) Racine E, Monnier F, Vors JP, Taillefer M. Chem Commun. 2013;49:7412–7414. doi: 10.1039/c3cc42575d. [DOI] [PubMed] [Google Scholar]; (c) Liu C, Lin J, Moslin RM, Weinstein DS, Tokarski JS Bristol-Myers Squibb Company. Imidazopyridazine compounds useful as modulators of IL-12, IL-23 and/or IFN alpha responses. World Patent WO2015089143 A1. 2015 Jun 18;; (d) Martin L, Steurer S, Cockcroft X-L. Boehringer Ingleheim International) New pyridinones and isoquinolinones as inhibitors of the bromodomain brd9. WO2016139361 A1. World Patent. 2016 Sep 9;
- 22.Menet CJM, Schmitt BA, Geney RJJ, Doyle KJ, Peach J, Palmer NJ, Jones GP, Hardy D, Duffy JES Galapagos NV. Imidazo[4,5-C]pyridine Derivatives Useful for the Treatment of Degenerative and Inflammatory Diseases. WO 2013117645 A1. World Patent. 2013 Aug 15;
- 23.Denmark SE, Carson N. Org Lett. 2015;17:5728–5731. doi: 10.1021/acs.orglett.5b02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pincock AL, Pincock JA, Stefanova R. J Am Chem Soc. 2002;124:9768–9778. doi: 10.1021/ja011981y. [DOI] [PubMed] [Google Scholar]
- 25.Park JK, Lackey HH, Ondrusek BA, McQuade DT. J Am Chem Soc. 2011;133:2410–2413. doi: 10.1021/ja1112518. [DOI] [PubMed] [Google Scholar]
- 26.CCDC 1584673 (8a′) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 27.Bai H, Bailey S, Bhumralkar DR, Bi F, Guo F, He M, Humphries PS, Ling AL, Luo J, Nukui S, Zhou R Pfizer, Inc. Fused Phenyl Amido Heterocyclic Compounds for the Prevention and Treatment of Glucokinase-Mediated Diseases. WO2007122482 A1. World Patent. 2007 Nov 1;
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.