

Abstract
Anti-HIV-1 envelope broadly neutralizing monoclonal antibodies (bNAbs) isolated from memory B-cells may not fully represent HIV-1 neutralizing profiles measured in plasma. Accordingly, we characterized near pan-neutralizing antibodies extracted directly from the plasma of two “elite neutralizers.” Circulating anti-gp120 polyclonal antibodies were deconvoluted using proteomics to guide lineage analysis of bone marrow plasma cells. In both subjects, a single lineage of anti-CD4-binding site (CD4bs) antibodies explained the plasma neutralizing activity. Importantly, members of these lineages potently neutralized 89–100% of a multi-tier 117 pseudovirus panel, closely matching the specificity and breadth of the circulating antibodies. X-ray crystallographic analysis of one monoclonal, N49P7, suggested a unique ability to bypass the CD4bs Phe43 cavity, while reaching deep into highly conserved residues of Layer 3 of the gp120 inner domain, likely explaining its extreme potency and breadth. Further direct analyses of plasma anti-HIV-1 bNAbs should provide new insights for developing antibody-based antiviral agents and vaccines.
eTOC
An HIV-neutralizing antibody achieves near pan-neutralizing activity through recognition of a highly conserved, difficult to access region of the viral gp120 glycoprotein.
Main Text
HIV is an integrating retrovirus that rapidly establishes chronic infection in CD4+ T cells, with their subsequent depletion, leading to collapse of the adaptive immune system. This fundamental characteristic means that prevention of HIV infection by a vaccine will be largely antibody-mediated. Regardless of how antibodies protect, they must recognize epitopes on the HIV envelope glycoprotein, Env, or its subunits gp120 and gp41, which mediate viral attachment and entry.
Anti-Env antibody responses in a minority of HIV-infected persons comprise broad neutralizing activity against diverse viral variants (Sajadi et al., 2016; Sajadi et al., 2011; Scheid et al., 2009; Simek et al., 2009; Walker et al., 2010). It is widely held that these broadly neutralizing responses can be used to guide the development of HIV vaccines and monoclonal antibodies (mAbs) to prevent and treat HIV infection. Implementation of this concept requires three types of information. First, highly conserved epitopes associated with potent neutralization sensitivity must be defined in Env. Significant steps in this direction came with identification of broadly neutralizing anti-Env mAbs from memory B cell pools of so called “elite neutralizers” (Simek et al., 2009; Walker et al., 2009). Broadly neutralizing antibodies identify six epitope clusters (reviewed in (Haynes and Mascola, 2017) including trimer apex gp120 epitopes comprised of V1/V2 sequences and glycan, N332-glycan V3 epitopes, high-mannose epitopes (Trkola et al., 1995), the CD4 binding site (CD4bs) of gp120 (Wu et al., 2010), the membrane proximal region (MPER) (reviewed in (Kwong and Mascola, 2012)), and hybrid epitopes at the gp120-gp41 interface (Huang et al., 2014). Second, the features of broadly neutralizing antibodies that arise in multiple individuals, versus rare subjects, must be fully characterized including both physicochemical properties of the circulating antibodies (Sajadi et al., 2016; Sajadi et al., 2012) and host factors enabling the evolution of broadly neutralizing antibodies (Dugast et al., 2017; Ranasinghe et al., 2015; Rusert et al., 2016; Sajadi et al., 2011). Third, the aggregate nature of the polyclonal environment in which broadly neutralizing activities evolve, persist, and function must be understood. Collectively, this information can be used to delineate whether and how certain epitope specificity groups should be avoided or targeted in order to deliberately achieve potent and broad neutralization through vaccination.
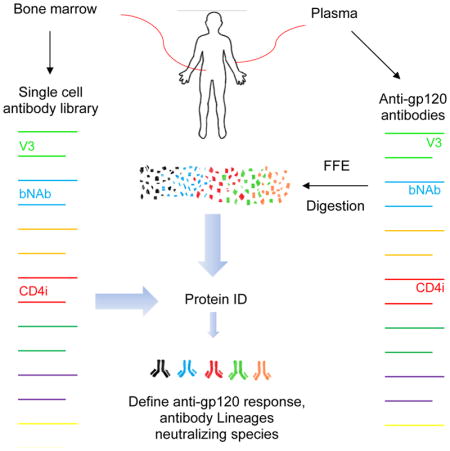
To date, the interrelationships between broadly neutralizing antibodies and the circulating plasma anti-HIV envelope repertoires that harbor them have been examined only indirectly. Typical approaches involved protein fractionation, antigen depletion, and/or infectivity analyses using viral envelopes with targeted mutations (Dhillon et al., 2007; Li et al., 2009; Sather and Stamatatos, 2010). These methods, of necessity, provide an inadequate picture of milieu in which polyclonal anti-Env antibodies evolve to achieve neutralization breadth. Alternatively, intensive efforts have been applied toward the derivation of broadly neutralizing mAbs from memory B cell pools. However, it is known that memory B cell repertoires can differ from bone plasma cell repertoires (Briney et al., 2014; Halliley et al., 2015), and we have shown a limitation of this approach is discordance between anti-Env specificities in the memory B cell pool and circulation (Guan et al., 2009). Perhaps because of these reasons, oftentimes the HIV neutralization profiles of the plasma appear to be different than those of the memory B cell-derived mAbs (Guan et al., 2009; Scheid et al., 2009; Walker et al., 2009). For this reason, we focused on bone-marrow plasma cell repertoires to isolate mAbs from our cohort of elite neutralizers.
Circulating polyclonal responses can be deconvoluted by convergent proteomic and genomic analyses (Boutz et al., 2014; Doria-Rose et al., 2014; Wine et al., 2015). Antigen-specific immunoglobulin (Ig) recovered from plasma is subjected to peptidase digestion and mass spectroscopy to reveal the amino acid sequences of Ig fragments. The resolved sequences are then assembled into whole Ig molecules using matched genetic databases as templates. This general strategy has been used recapitulate the CDR3 repertoires in rabbits immunized against Concholepas concholepas hemocyanin (Wine et al., 2013), or in humans vaccinated with tetanus toxoid (Lavinder et al., 2014). Recently, Williams et al. used a similar approach to match clonal lineages of anti-gp41 neutralizing antibodies in plasma with selected cognate memory B cell pools (Williams et al., 2017). However, this study did not address the entire polyclonal anti-envelope response, and it did not examine neutralizing anti-gp120 responses in circulation.
Here we employed a matched genomic and proteomic approach to deconvolute a broadly neutralizing response directed against gp120 from bone marrow derived plasma cells. Our first test subject, N60 (referred to as Subject 1 in a previous publication (Sajadi et al., 2012)) belongs to a previously reported Natural Viral Suppressor (NVS) cohort of HIV subtype B-infected who control HIV replication without medication, some of whom also exhibit persistent titers of very broad and potent neutralizing antibodies (Sajadi et al., 2016; Sajadi et al., 2012).
We have previously found that multiple HIV-infected subjects harbor broad and potent neutralizing activities with highly shared biochemical determinants, such as basic isoelectric points (pI), and specificities for binding epitopes on monomeric gp120 (Sajadi et al., 2016; Sajadi et al., 2011; Sajadi et al., 2012). Serum antibodies from N60 were able to neutralize 90% of 118 multi-clade Tier 2 and 3 panel of viruses (referred to in this paper as the “global panel”) (Figure 1). The duration of such neutralization potency and breadth over a 5-year period was confirmed by sequential testing of N60 plasma against a cross-clade, multi-tier panel of viruses (Table S1).
Figure 1. Neutralization of N60 parent and affinity purified samples.

Pro A/G= Affinity purified antibody from a Protein A/G column; gp120= Affinity purified antibody from a monomeric gp120 column; gp120-IgG1= Affinity purified antibody from a gp120 followed by IgG1 column; IC50= Inhibitory concentration 50 (ug/ml); T/F= transmitted/founder; NC= not characterized. Total IgG from patient N60 was purified by Protein A/G and tested against a 118 multitier and multiclade pseudovirus panel (starting titer 528ug/ml). Parent sample demonstrates considerable breadth, which was also seen in the gp120 and gp120-IgG1 (starting titer 50ug/ml).
The broadly neutralizing plasma antibodies were isolated from N60 plasma by affinity chromatography with monomeric gp120 (Sajadi et al., 2016; Sajadi et al., 2011; Sajadi et al., 2012) (Figure 1). The recovered gp120 affinity fraction from N60 comprises approximately 2% of the starting mass of IgG antibody. Similar recoveries of anti-gp120 antibodies (0.6%–2% of starting mass of IgG antibody) were obtained from the plasma of other HIV infected individuals in our cohort, some of whom were not elite neutralizers (data not shown). In accordance with our previous studies of anti-Env antibody responses (Sajadi et al., 2016; Sajadi et al., 2011; Sajadi et al., 2012), broadly neutralizing activity in N60 plasma was recovered by sequential protein A/G affinity chromatography, gp120 affinity chromatography, and IgG1 affinity chromatography (Figure 1).
Two lines of evidence showed that few if any epitope specificities beyond those on monomeric gp120 were linked with the N60 polyclonal neutralizing response. First, the plasma antibody fraction that flowed through the gp120 affinity column exhibited negligible neutralization activity against a panel of 12 HIV Tier 1–3 pseudoviruses, compared to unfractionated plasma (Table S1). Second, the neutralizing breadth of purified N60 anti-gp120 IgG1 Ig largely matched that of unfractionated plasma (Figure 1).
We have shown a light chain bias in the anti-HIV envelope response (Sajadi et al., 2016). Thus, to further enrich N60 broadly neutralizing antibodies, affinity purified anti-gp120 plasma antibodies were further fractionated by affinity chromatography with antibodies selective for human κ or λ light chains. The bulk anti-gp120 IgG1 κ or λ Ig fractions were then subjected to free flow isoelectric focusing (FFE) to separate individual antibody species according to their pI. In our previous study (Sajadi et al., 2012), the broadly neutralizing antibodies were localized to fractions with more basic pIs. In the current study, neutral and basic IgG1 κ anti-gp120 fractions were tested for neutralization against two Tier 2 pseudoviruses, confirming the localization of broadly neutralizing antibodies to basic pIs (Figure S1 and data not shown). ELISA analysis confirmed that the broadly neutralizing antibody fractions contained IgG reactive with gp120 and gp120 core, and did not bind to D368R gp120, whose single point mutation at position 368 abrogates binding of CD4bs antibodies to HIV-1 gp120 (Figure S1) (Li et al., 2007). These fractions were also less reactive with Full length single-chain (FLSC), a covalent gp120BaL-sCD4 complex, in which the CD4bs is blocked (Figure S1) (Fouts et al., 2000). In comparison, more acidic fractions favored binding to gp120, FLSC, D368R gp120, but not the gp120 core (Figure S1).
Based on the above observations, we used plasma derived antibodies and bone marrow plasma cells from N60 to establish a basic strategy for deconvoluting the plasma anti-gp120 polyclonal neutralizing response. First, the broadly neutralizing IgG fraction of serum from N60 was enriched as described above (Sajadi et al., 2011; Sajadi et al., 2012). The enriched broadly neutralizing fraction was subjected to enzymatic digestion with trypsin, chymotrypsin, and/or Glu-C as described in Methods and peptide fragments were subjected to LC-MS/MS to calculate mass assignments, from which their amino acid sequences could be deduced. Next, the peptide sequences were used to identify the Ig H and L chain genes from which they originated.
Peptide sequences were aligned and assembled using as templates 1,022 authentically paired Ig H and L chain amino acid sequences translated from the patient-specific bone marrow plasma cells and circulating plasmablasts database derived by single-cell, paired chain sequencing (described in Methods). Antibodies were identified from three complementary fractionation approaches, using a search algorithm relying on the presence of unique peptides (peptides found in individual antibodies in the database rather than those shared among many) (see Methods).
The Fab sequences of the 15 identified H and L gene pairs were combined with a generic IgG1 backbone order to construct synthetic mAb expression plasmids protein generation (see Methods) (Guan et al., 2009). This construction strategy was appropriate, as the plasma broadly neutralizing antibodies were exclusively of the IgG1 isotype (Figure 1).
FFE was then used to compare the electrophoretic behaviors of the reconstructed mAbs versus bulk polyclonal anti-gp120 plasma antibodies. As shown in Figure 2, the plasma antibodies tended to focus at highest concentrations within two series of fractions peaking at lower and higher pH ranges in the gradient (which ranged pH 6.5 to pH 9.5). Fractions containing plasma antibody peptide/sequences used for mAb reconstruction (see above and Methods) also ran in these regions. In the case of IgG1 κ, fractions near the center of the gradient (~fractions 45–55) yielded no unique antibodies (the peptides that derived from this region did match some of the neighboring mAbs on either side, but at a lower frequency). Similarly, for IgG1 λ, fractions above pH 8 contained too little protein to perform downstream analysis. Importantly, the pI values of the reconstructed mAbs derived from the FFE or gel isoelectric focusing approaches corresponded to those of the plasma fractions that yielded their identifying peptides (compare bars and arrows in Figure 2). This consistency indicated that the identified and reconstructed mAbs contained assemblages of amino acid sequences authentic to the plasma antibodies.
Figure 2. Free flow electrophoretic fractionation of N60 plasma anti-gp120 polyclonal antibodies and reconstructed anti-gp120 mAbs.

The gray line indicates the pH (right Y axis) gradient across the fractions created by the FFE procedure. Anti-gp120 κ light chain (top) or anti-gp120 λ light chain (bottom) polyclonal plasma antibody preparations were processed separately (see main text and Methods). The plasma antibody protein concentrations (left Y axis) detected across fractions are shown by the black trace. FFE analyses of identified and reconstructed mAbs (see main text) are depicted by horizontal bars. Bar width spans 75–85% of the total amount of antibodies in the FFE fraction, as determined by ELISA. Eight mAbs (checkered bars) were identified by evaluating peptides from individual FFE fractions of anti-gp120 plasma antibodies. The FFE fraction reflecting the most coverage and unique peptide pairings is indicted for each mAb by a matched-color arrow. Four additional mAbs (hatched colored bars) were identified by evaluating peptides from IEF gel fractionation. Searches using peptides digested from bulk plasma anti-gp120 antibodies identified 1 additional mAbs (solid colored bars). One other mAb (criss-cross colored bars) was identified by homology search of the Ig gene database. The pH gradient shown is for the polyclonal N60IgG1 anto-gp120 κ fraction; pH gradients from each monoclonal run overlapped the trace shown, with a variance of up to 0–5 fractions in either direction.
The reconstructed mAbs were characterized for source-cell subset and lineage relationships (Table S2, Figure S2). All of the anti-gp120 mAb sequences were found in bone marrow CD138− and CD138+ populations by single cell PCR (summarized in Table S2). Only one mAb (N60P22) could also be detected in the circulating plasmablast population (Table S2). This mAb had the second highest frequency of any in the bone marrow, so it could be that the frequency of the mAbs in the plasmablast population is less than the bone marrow, or they can only be detected for a short time, as occurs with vaccination or acute infections. A homology search (multiple sequence alignment) of the bone marrow database did not reveal any of the ancestral forms of any of the mAbs identified, implying that even though the mAbs were found in all bone marrow compartments, including the long-lived plasma cells (CD138+, CD19−), their longevity may be limited for unknown reasons.
Overall, the dominant N60 plasma anti-gp120 response arose from 15 mAbs from 7 distinct lineages (Table S2), as shown in the neighbor-joining phylogeny tree (see Methods) of the entire BM antibodies (Figure S2). Lineage 1 was distinguished by 1–2 heavy chain and 1–5 κ light chain gene usage as well as a relatively high degree of somatic hypermutation (33–42% amino acid variation in the heavy chain). Lineage 1 mAbs resemble previously reported broadly neutralizing anti-CD4bs antibodies, which are also assigned to VH1–2 and Vκ1–5 gene families that exhibit the signature deletion in the CDRL3 (Table S2, Figure S3A) (West et al., 2012; Zhou et al., 2010). These antibodies had basic pIs (Figure 2).
In a previous study of N60 (Sajadi et al., 2016), we determined that roughly 50% of the total anti-gp120 plasma response involved antibodies with λ light chains. On fractionation based on isoelectric point, the bulk of the λ antibodies focused to just a handful of fractions (Figure 2, Figure S1), indicating one distinct family targeting a single epitope. As only Lineage 7 expressed λ light chains, it is evident that cross-reactive anti-V3 antibodies account for the bulk of the circulating anti-gp120 response in N60 (potentially up to 1% of the total circulating IgG). Such representation in the polyclonal anti-gp120 response is in accordance with the immunodominant nature of the V3 loop as evinced in studies of anti-gp120 responses in other cohorts (Javaherian et al., 1989; LaRosa et al., 1990; Vogel et al., 1994).
Neutralization testing of the mAbs against 15 pseudoviruses from Tiers 1–3 showed that Lineage 1 comprised the broadest and most potent activity, followed by Lineages 2, 7, and 5 (Table S3). Expanded testing of Lineage 1 mAbs revealed neutralization breadth approaching the coverage observed with the polyclonal plasma anti-gp120 Ig (Figure 3). Two mAbs, N60P1.1 and N60P25.1, demonstrated particularly strong neutralizing activity (Figure 3), matching 70% and 73% of the affinity purified plasma Ig breadth, respectively. Thus, broad plasma neutralizing activity in N60 appears to arise from Lineage 1, representing a monospecific fraction of the anti-gp120 repertoire at the time point evaluated. Anti-CD4bs mAbs from Lineage 2, which appeared in high frequency but did not have strong breadth and potency, contributed minimally to the overall plasma profile. In any case, the concurrence of other types of poorly neutralizing anti-gp120 antibodies does not appear to abrogate the presence and activity of broadly neutralizing antibodies in circulation.
Figure 3. Neutralization activity of N60 plasma derived anti-Env antibodies (alone and in combination).

A panel of pseudoviruses representing the indicated HIV-1 strains (listed in the left column) that were sensitive to the bulk plasma N60 gp120-Ig were tested against neutralizing anti-CD4bs antibodies from Lineage 1 and 2. For Lineage 2, only one mAb N60P22 was tested, as the other was a closely related clone (98% sequence homology). IC50 values are color-coded according to the color key on the left: the greater the neutralization, the darker red the color; grey represents no neutralization (IC50>25ug/ml). Taken together, the anti-CD4bs mAbs neutralized 89% of the viruses that were sensitive to bulk plasma anti-gp120 Ig. An equimolar mix of the mAbs called N60mAb Mix1 (all CD4bs, CD4i, and variable loop antibodies with > 5% sequence divergence) was tested at equimolar concentrations neutralized 79% of the pseudoviruses, and N60mAb Mix2 (all CD4bs antibodies with > 5% sequence divergence at equimolar concentrations) neutralized 89% of the pseudoviruses. IC50=Inhibitory Concentration 50 in ug/ml.
Despite their breadth and potency, none of the anti-CD4bs mAbs from Lineage 1 matched the full breadth of the polyclonal plasma anti-gp120 Ig recovered from N60. Considered collectively, the anti-CD4bs mAbs neutralized 89% of the viruses that were sensitive to bulk anti-gp120 plasma Ig. Resistance to the mAbs was independent of virus clade or Tier (Figure 3). Notably, one resistant pseudovirus was neutralized by the Lineage 5 anti-CD4i mAb, N60P39. Thus, the combined profiles of six mAbs including N60P39 could cover 90% of the viruses neutralized by bulk anti-gp120 plasma Ig. To determine if greater breadth could be established by mAb mixtures, the panel of viruses sensitive to bulk anti-gp120 plasma Ig were tested against an equimolar pool of all anti-CD4 BS mAbs, or an equimolar combination of represented mAbs from all lineages (related clones with <5% amino acid sequence diversity were not included). These mAb pools covered, respectively, 89 and 79% of the neutralizing activity breadth mediated by the plasma Ig (Figure 3). These results suggest that the plasma neutralizing response in N60 could involve a cryptic anti-gp120 specificity (active against a small subset of test viruses resistant to the identified mAbs) and/or a molar ratio of specificities that we could not readily duplicate in vitro. Nevertheless, we were able to recapitulate 90% of the serum activity in this test case.
Previously we reported that multiple Clade B HIV infected patients expressed broadly neutralizing plasma antibody responses with similar biochemical characteristics (Sajadi et al., 2016; Sajadi et al., 2011; Sajadi et al., 2012), such as basic pIs. We posited that this trend reflected shared inter-subject specificity for neutralizing gp120 epitopes. To test this hypothesis, we applied the same deconvolution procedures and selection algorithms described above to another NVS cohort subject, N49, who exhibited a near pan-neutralization response, able to neutralize 99% of a 117 pseudovirus “global panel” of Tier 1–3 isolates (Figure 4). Similar to the N60 responses, the neutralizing antibodies from N49 plasma could be recovered from plasma by gp120 affinity chromatography (recovered gp120 affinity fraction known to be approximately 1% of the starting mass of IgG antibody for this patient); however, in this case the IgG1 λ fraction contained the broad neutralizing antibodies. As for N60, 1,313 natively paired heavy and light chain gene sequences were generated from single bone marrow plasma cells and peripheral plasmablasts, and matched up to the peptide sequences from mass spectrometry.
Figure 4. Neutralization activity of N49 plasma and P series mAbs.

A “global panel” of 117 HIV-1 psuedovirus Tier 1-3 isolates (individual viruses listed on the left column) were tested against all N49 plasma and CD4bs antibodies. IC50 values are color-coded according to the color key on the left: the greater the neutralization, the darker red the color; white represents no neutralization (IC50>50ug/ml). The individual mAbs showed extreme breadth with N49P6, N49P7, and N49P11 exhibiting 100% breadth, N49P7.1 exhibiting 99% breadth, and N49P9 exhibiting 89% breadth. IC50=Inhibitory Concentration 50 in ug/ml. NC=not classified
The broadly neutralizing antibodies in N49 plasma fell into two lineages, distinguished by different light chain gene usage (Table S4). Similar to the N60 Lineage 1 broadly neutralizing antibodies, the N49 mAbs all exhibited basic pIs and VH1-2 gene usage. However, all of the N49 mAbs used λ light chain genes, while also containing deletions in CDRL1 and CDRL3. The binding characteristics of this N49 lineage also matched the N60 neutralizing antibodies, reflecting anti-CD4bs specificity. These antibodies bind to monomeric gp120, have little to no binding to D368R and FLSC in both ELISA and Biacore (Figures S3B and S4).
A distinguishing feature of the N49 anti-CD4bs mAbs was that they exhibited remarkably broad neutralizing activity when tested against a multi-clade, 117 pseudoviruses “global panel.” As shown in Figure 4, N49 mAbs N49P6, N49P7, and N49P11 each neutralized 100% of the viruses. The mAbs from N49 were fundamentally distinguished from N60 in the sense that three identified plasma mAbs (N49P6, N49P7, and N49P11) completely recapitulated the antiviral breadth of affinity purified anti-gp120 plasma IgG. The N49 P series mAbs also had high potency, with ability to neutralize 42–86% of all viruses at under 1ug/ml. mAb N49 P7 had complete breadth (ability to neutralize all 117 pseudovirus “global panel”) and the highest potency (86.4%). The breadth and potency of the N49 group of mAbs was compared other mAbs reported to have substantial breadth (3BNC117, 8ANC195, 10-1074, 10E8, N6, PG9, PG16, PGDM1400, PGT121, PGT128, PGT145, PGT151, NIH45–46, VRC01, and VRC07), using the same neutralization assay and panel. The breadth of the N49 mAbs surpassed those of all mAbs tested (Table 1). The potency of the broadest and most potent N49 P series mAb, N49P7, was compared to N6. N49P7 has a mean and median IC50 (0.44ug/ml and 0.10 ug/ml, respectively) compared to N6 (1.15ug/ml and 0.09 ug/ml, respectively) in this assay. We next compared the performance of N49P7 to the top 2 mAbs we tested (10E8 and N6), and to the only bNAb we did not test (DH511-2) (Williams et al., 2017). On comparing neutralizing activity to viruses that were resistant to at least one of those 3 mAbs, N49P7 could neutralize all at the best overall breadth and potency (Figure 5). N49P7 was further tested in an additional panel of an additional 53 pseudoviruses, not present in the “global panel” but which has been used to characterize other pan-neutralizing mAbs (Huang et al., 2016). As shown in Figure S5, mAb N49P7 was effective against 94% of this panel. Considering these data in aggregate with what was measured in the “global panel,” mAb N49P7 was able to neutralize 98.2% of all 170 test viruses.
Table 1. Comparison of neutralization breadth between those derived from N49 and other bNAbs.
Comparison of N49P7 to other bNAbs tested in same 117 pseudovirus “global panel.” Percentages are given as values of viruses neutralized (IC50 <50ug/ml)/viruses tested. All testing was done in the same lab. N49 P series tested against the full 117 pseudovirus panel of Tier 1–3 isolates. All other mAbs tested against 114–117 pseudoviruses, except for VRC01, which was tested against 96. Neutralization at titers >50ug/ml were considered non-neutralizing B) Comparison of N49P7 to other bNAbs. Viruses resistant to N6, DH511-2, or 10E8 are shown (IC50 values for DH511-2 obtained from (Williams et al., 2017)). N49P7 shows the greatest breadth and overall potency. NT = Not tested
| mAb | % neutralization |
|---|---|
| N49P6 | 100 |
| N49P7 | 100 |
| N49P7.1 | 99 |
| N49P11 | 100 |
| N49P9 | 89 |
| N6 | 98 |
| 10E8 | 97 |
| VRC07 | 92 |
| 3BNC117 | 90 |
| VRC01 | 88 |
| NIH 45–46 | 86 |
| PG9 | 85 |
| PG16 | 82 |
| PGDM1400 | 80 |
| PGT145 | 74 |
| PGT121 | 66 |
| PGT151 | 66 |
| 8ANC195 | 65 |
| PGT128 | 60 |
| 10–1074 | 57 |
Figure 5. Comparison of potency and breadth of resistant viruses between N49P7, N6, DH511-2, and 10E8.

Viruses resistant to N6, DH511-2, or 10E8 are shown (IC50 values for DH511-2 obtained from (Williams et al., 2017)). N49P7 shows the greatest breadth and overall potency. NT = Not tested
To define the molecular basis for the broad potencies of N60 and N49 P mAbs series we solved the crystal structures of N60P23 and N49P7 Fabs in complex with HIV-1 93TH057 gp120 (Table S5). N60P23, a clone of N60 P1.1 that has a 1 amino acid (aa) difference in the light chain, exhibited an epitope footprint with intermolecular contacts similar to those of VRC01 and other previously described CD4bs antibodies (Figure S6 and Table S6). In contrast, N49P7 bound to gp120 in a unique manner (Figure 6). The deletion in the CDRL1 (not found in N6) combined with the rotation/tilting of the light chain ‘opens’ the variable light (VL) side of the N49P7 antigen binding site to accommodate different lengths of the highly variable loops D, E and V5 (Figure S7). Changes in the length of gp120 loop V5 and the length (and glycosylation status) of loop E that cause steric clashes with an antibody light chain were described previously as mechanisms of HIV-1 resistance to VRC01-class antibodies (Lynch et al., 2015). These signatures, along with a normal length CDRH3 (19 aa) and unique sequence signatures within CDRH2 (not seen in VRC01 and N6, Figure S7), allow N49P7 to bypass the Phe43 cavity affording it an unusual capacity to contact the gp120 inner domain at residues 97, 102 and 124 of Layer 2, and 472–480 of Layer 3 (Figure 6 and Figure S7). Overall N49P7 contributes 207 Å2 of its buried surface area (BSA) to the gp120 inner domain which is much higher than N6 and the VRC01 Abs class (Table S6). The gp120 inner domain harbors some of the most conserved amino acid sequences in the HIV envelope, located within structural Layers 1, 2 and 3 and a consolidation of 8 β-strands (Finzi et al., 2010). Other CD4bs antibodies such as N6 and VRC01 also display contacts with the inner domain; however, these are less prominent. Although NIH 45–46 is not as broad and potent and N49P7 (Table 1), it has previously shown to have more contacts with the inner domain (mostly Layer 2) than VRC01 (Diskin et al., 2011), and it was postulated that this may the reason for the breadth. N49P7 is unique in the number of inner domain contacts (especially Layer 3). N49P7 exhibits a lower outer domain to inner domain buried surface area ratio (3.74) compared to N6 (7.58) and VRC01 (9.68), which shows it has substantially more contacts with the inner domain (Table S6). Thus, the N49P7 paratope recognizes a mixed inner domain/CD4-binding site (termed here the iCD4bs) containing some of the most conserved sequences in gp120. These features help explain the extreme neutralization breadth of this antibody.
Figure 6. Crystal structure of N49P7 Fab-gp12093TH057 coree complex.

(A) Ribbon diagram of complex with the complementarity-determining regions (CDRs) of N49P7 Fab contributing to the gp120 binding (from light chain CDR L1 and CDR L3 and from heavy chain CDR H1, CDR H2 and CDR H3, see also Table S6) colored as shown. The gp120 outer and inner domains are colored in black and gray, respectively. The D (S274 – T283), E5 (F353 – T358), V5 (T455 - N465) and the CD4 binding (Q362 - G372) loops are colored in cyan, orange, violet and magenta, respectively. G54 of N49P7 Fab is highlighted in red and sugars at positions 276 (Loop D) and 355 (Loop E) are shown as sticks. (B) A blow up view into the network of interactions of N49P7 Fab with residues of the inner domain of gp120. Inner domain Layers 2 and 3 are colored pale green and beige, respectively. Two hydrogen bonds (CDR H2 N52 and Layer 3 N474; CDR H3 E100E and Layer 2 K97) and a salt bridge (CDR H3 R100C and Layer 3 D477) are formed at the N49P7 Fab – gp120 inner domain interface. (C) Gp12093TH057 coree and N49P7 CDR contact residues mapped onto the primary sequence. Contact residues are defined by a 5 Å cutoff and marked above the sequence. Side chain (+) and main chain (−) contacts are colored based on contact type; hydrophobic in green, hydrophilic in blue, or both in black. Buried surface residues as determined by PISA are shaded. (D) Blow up views into the Fab - gp12093TH057 coree interfaces of N49P7 Fab, N60P23 Fab and N6 Fab (PDB code: 5te6). The molecular surface is displayed over gp120 molecules with outer domain loops: D, X5, V5 and CD4 binding and inner domain Layers 2 and 3 and the 7-stranded β-sandwich with CDRs colored as in panels A and B. The Fab residue at the position equivalent to F43 of CD4 is shown as sticks (Gly, His and Tyr in N49P7, N60P23 and N6, respectively). (E) The buried surface area (BSA) of gp120 residues involved in binding to N49P7, N60P23 and N6 are shown as bars (bottom panel) and their conservation among HIV-1 isolates (top panel). The conservation of the residue at particular position is shown as % difference from the HXBc2 sequence. Only unique sequences in the database having an equivalent residue at each position were included in the calculated percentage representing approximately 32,000 sequences on average. Conserved inner domain residues uniquely targeted by N49P7 are highlighted in red.
Discussion
Most studies of broadly neutralizing antibody responses employ a reductionist approach that starts with the random isolation of a monoclonal antibodies from memory B cell pools using preselected antigen “baits.” Such neutralizing antibodies are then used as heuristic guides to construct a theoretical picture of the plasma response landscape that comprised their persistence and function. One of several caveats to this approach is that circulating and memory B cell responses can be discordant (Guan et al., 2009). In this study antibody sequences were directly extracted from plasma, thus enabling us to reconstruct the neutralizing antibody lineages extant within circulating polyclonal anti-gp120 responses. This approach clearly defines commonalities in broadly neutralizing polyclonal response profiles among multiple individuals, which informs HIV vaccine development; it elucidates whether and how related antibody specificities cooperate to generate the gross plasma neutralizing profiles measured in vitro; it allows unambiguous pairings with any contemporaneous viral genomes, thus facilitating a direct understanding of how HIV infection persists in the presence of pan-neutralizing activity.
Given the concept that successful adaptive immunity to HIV infection provides a blueprint for vaccine design, we focused on deconvoluting polyclonal anti-Env plasma responses with broadly neutralizing features that are shared among different HIV-infected subjects (Rusert et al., 2016; Sajadi et al., 2012). Accordingly, the two subjects investigated here, N60 and N49, exhibited pauciclonal, anti-gp120 plasma species with similar biochemical properties (Sajadi et al., 2012). Six insights emerged from our deconvolution exercise.
First, our results agree with earlier predictions (Sajadi et al., 2012) that plasma neutralizing responses with shared biochemical characteristics target similar epitopes. The plasma neutralizing antibodies in subjects N49 and N60 were determined to be pauciclonal, with basic isoelectric points, indicating similar paratope composition (Sajadi et al., 2012). We show here that the key neutralizing antibodies in both subjects indeed share overlapping epitope specificities involving the CD4bs.
Second, in agreement with previous studies (Rusert et al., 2016; Walker et al., 2010) we find that the overall polyclonal anti-gp120 plasma response is restricted to a limited number of specificities and antibody species. We could trace over 50% of the anti-gp120 antibodies in N60 plasma, thus up to 1% of the total circulating IgG1 to one anti-V3 antibody family comprising only two members.
Third, the dominant anti-gp120 response in N60 derives from seven separate events producing 7 clonal lineages (Figure S2 and Table S2). This range differs from what is typically seen in the memory B cell compartment, where there is a comparatively larger number of epitopes and antibodies (Scheid et al., 2009), which are often discordant with the plasma response (Guan et al., 2009). In the latter study, the plasma anti-gp120 antibodies were found in the N60 memory B cell pool alongside antibody species targeting epitopes not found in circulation. We cannot eliminate the possibility that other species found the memory B cell pool are entirely absent in the polyclonal anti-gp120 IgG recovered by affinity fractionation; however, they would have to exist at minor levels beneath our threshold of detection.
Fourth, the broad and potent neutralizing antibodies were clearly persistent and functional in plasma despite being only a minor fraction of the total circulating IgG response. The frequency of N60 anti-gp120 polyclonal antibody found in circulation (2%) is similar to the frequencies found by us in the bone marrow (3.2%), which agrees with previous reports of a direct correlation between bone marrow frequencies and serum titers (Montezuma-Rusca et al., 2015). It has been argued that “non-neutralizing” anti-gp120 responses prevent broadly neutralizing antibody responses from evolving, persisting and/or or acting in circulation (de Taeye et al., 2015; Georgiev et al., 2015; Sanders et al., 2013). Whether pre-existing, non-neutralizing anti-envelope responses impede the appearance of broadly neutralizing antibodies in circulation is a matter of continuing investigation. In any case, deconvolution of the anti-gp120 response in N60 plasma shows that once broadly neutralizing antibodies arise in circulation they are not necessarily erased by contemporaneous non-neutralizing antibody responses over the course of 4 years (Table S1).
Fifth, anti-CD4bs antibodies are responsible for nearly all of the broad and potent neutralization breadth seen in N60 plasma, and can explain all of the extreme breadth in N49 plasma (Figure 3, Figure 4). However, it was noteworthy that we were unable to totally recapitulate the neutralization breadth of the N60 polyclonal response with mixtures of N60 mAbs. This discrepancy suggests that the plasma neutralizing response in N60 could involve the collective action of other anti-gp120 specificities (active against the small subset of test viruses resistant to the identified mAbs) acting at molar ratios that we could not readily be duplicated in vitro. In comparison, N49 was distinguished from N60 in the sense that three identified plasma mAbs (N49P6, N49P7, and N49P11) recapitulated the antiviral breadth of affinity purified anti-gp120 plasma IgG, as each neutralized all viruses tested. Members of the N49 P series bNAbs are the broadest and most potent naturally occurring anti-gp120 antibodies described to date, capable of neutralizing viruses that are missed by other broadly neutralizing mAbs (including N6, DH411-2, and 10E8).
Sixth, the pan-neutralizing properties of N49P7 is likely explained by its access to the iCD4bs. Similar to another pan-neutralizing CD4bs antibody, N6, N49P7 engages conserved residues of the CD4-binding loop. However, N49P7 bypasses the Phe43 cavity and relies more heavily on binding to highly conserved residues of Layer 2 and 3 of inner domain in the direct interior of gp120 (Figure 6). This is notable in that the Phe43 cavity was assumed to be the optimal target for CD4bs antibodies and novel small drugs (Dey and Berger, 2013). We conclude that the novel epitope-paratope relationships reported here provide key insights for understanding how anti-CD4bs antibodies can be used to enable very broad antiviral effects. We are confident that our data can be applied toward the design of antiviral agents capable of suppressing HIV strains that are resistant to other mAbs of this class.
The finding of anti-CD4bs in these two individuals further highlights the importance of the CD4bs epitope as a target of HIV vaccination. Anti-CD4bs naïve B cell precursors have been found in all 15 healthy donors studied with the eOD-GT8 immunogen that was designed to target the germline VRC01-like mAbs (Jardine et al., 2016). Furthermore, the CD4bs is the most common epitope targeted by bNabs, and the detection of related, very broadly neutralizing anti-CD4bs antibodies in multiple patients (Scheid et al., 2009; Wu et al., 2010) supports the concept that such responses might be frequently raised by vaccination. At the same time, these responses arise during the unusual circumstances of chronic HIV infection in those who maintain a large degree of control over viral replication, and the circumstances that drive the anti-gp120 response towards the evolution of such antibodies remain to be defined.
It is encouraging that extremely broad and potent neutralizing antibodies persist and function within a larger plasma milieu of non-neutralizing or poorly neutralizing antibodies. Thus, very broad plasma neutralizing activity may not demand pristine responses against a selected target. Further, the neutralizing antibodies recognize a relatively straightforward monomeric gp120 structure. Collectively, such features support practical efforts to generate broadly neutralizing responses via vaccination with gp120-based immunogens. Regardless of approach, success in neutralization-based vaccine design depends on a clear understanding of the virological and immunological conditions that generate super-broadly neutralizing plasma responses. Longitudinal deconvolution of plasma responses from the time of acute infection will help facilitate efforts in this direction.
Star Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mohammad M. Sajadi (msajadi@ihv.umaryland.edu).
Experimental Model and Subject Details
Human Subjects
The patients identified for this study were selected from the NVS study (Sajadi et al., 2009; Sajadi et al., 2011; Sajadi et al., 2007). NVS patients are defined as having HIV-1 by Western Blot while having an HIV-1 RNA <400 copies/ml for at least 4 measurements and 2 years. N60, an African-American male, met the above definition and was 53 years old (5 years after HIV diagnosis) at the time of bone marrow aspirate. N49, an African American transgender female (MTF) had a higher viral load setpoint, averaging 7,854 HIV-1 copies/ml over 9 years. N49 was 40 years old (21 years after HIV diagnosis) at the time of bone marrow aspirate. Both of the patients’ serum were identified as having the most broad and potent neutralizing activity based on Tier 2 activity in and a cross-clade neutralization panel among 120 patients (Sajadi et al., 2016; Sajadi et al., 2011; Sajadi et al., 2012). All studies were IRB approved and all individuals provided written informed consent.
Cell lines
Human embryonic kidney HEK293T cells (sex: female) were obtained from ATCC (Manassas, VA) and cultured in 5% C02 at 37°C in DMEM medium supplemented with 10% heat inactivated fetal bovine serum (FBS). TZM-bl cells (sex: female) were obtained from the NIH AIDS Reagent Program (Bethesda, MD) and cultured in 5% C02 at 37°C in RPMI medium from Gibco (Gaithersburg, MD) supplemented with 10% heat inactivated FBS with 100 units/ml of penicillin and 100 μg of streptomycin/ml. FreeStyle™ 293-F Cells (sex: female) were obtained from Thermo Fisher Scientific (Waltham, MA) and were cultured in 5% C02 at 37°C in FreeStyle™ 293 Expression Medium from Gibco (Gaithersburg, MD).
Method Details
Proteins and Antigens
Recombinant HIV-1 antigens were generated as described previously (31). Test antigens included the YU2 gp120 core, from which V1, V2, and V3 have been deleted (Wu et al., 1996); the YU2 gp120 core containing the V3 loop (YU2 gp120 core +V3) (Wu et al., 1996); monomeric HIV-1 Ba-Lgp120 (Fouts et al., 2000); D368R Ba-L gp120, which is monomeric HIV-1 Ba-Lgp120 that has a single point mutation at position 387 (Li et al., 2007); and a single chain gp120-CD4 complex (FLSC) presenting a full length CD4-induced Ba-L gp120 structure in which the CD4 binding site is occupied (Fouts et al., 2000). Two monoclonals specific for the C-terminal peptide of HIV-1 gp120 were used: an affinity-purified goat Ab, D7324, purchased from Cliniqa (San Marcos, CA) and JR52, a mouse monoclonal (courtesy of James Robinson, MD) (Robinson et al., 2016). All proteins were expressed by transient transfection of 293T cells and purified by lectin affinity chromatography, as previously described, and dialyzed against PBS prior to use (Fouts et al., 2000). The following reagent was obtained through the NIH AIDS Reagent Program, AIDS Program, NIAID, NIH: HIV-1 V3 Peptides from the Division of AIDS, NIAID.
Isolation of plasma antibody species
Whole plasma IgG was purified on a Protein A or Protein A/G affinity chromatography column (GE Healthcare, Piscataway, NJ) according to the manufacturer’s instructions and dialyzed against PBS prior to use. Affinity chromatography columns were made with activated CH Sepharose beads (GE Healthcare, Piscataway, NJ) coupled to 2 mg of recombinant HIV-1 Ba-L gp120 (Fouts et al., 2000), as described previously (Guan et al., 2009). Beads specific for human IgG1, human κ chain, and human λ chain were purchased from Capture Select (Naarden, Netherlands). The columns were used to purify antigen-specific IgG (anti-gp120), fractionate IgG1 from whole IgG, or fractionate IgG into κ and λ fractions, as previously described (Guan et al., 2009; Sajadi et al., 2011). Briefly, IgG was incubated with beads at 37°C for one hour prior to extensive washing with PBS. Columns were eluted at room temperature with pH 2.8 0.2M glycine (for elution of κ antibodies pH 2.0 was used) and dialyzed against 4 liters PBS 3 times (a minimum of 24 hours total) prior to testing. Dedicated columns were used for each subject and antigen. IgG concentration was measured using an in-house quantitative ELISA as previously described (Guan et al., 2009). After a series of steps, the plasma was fractionated into IgG1κ and IgG1λ antibodies (plasma -> protein A column -> IgG1 column -> kappa and lambda columns), anti-gp120 κ and anti-gp120 λ antibodies (plasma -> protein A column -> gp120 column -> kappa and lambda columns), or anti-gp120 antibodies (plasma -> protein A column -> gp120 column).
Affinity purified and fractioned antibody was subjected to free flow electrophoresis on the BD Free Flow Electrophoresis System (BD, Franklin Lakes, NJ). The separation, stabilization and counter flow media was freshly prepared according to instructions of the manufacturer. The separation and counter flow media contained 0.2% hydroxypropyl methylcellulose (HPMC). The pH range of separation media was 0.88 to 12.8. The media flow rate in the separation chamber was 41 mL/hour. The antibodies (200 to 350 μg/ml) were introduced to separation chamber at the rate of 560 μl/h in the electrical field of 2300V/10 mA/24 W. IEF fractionated samples collected in a 96 deep-well polystyrene microtiter plate, with each well containing 1–2ml. Approximately half of these wells contained antibody fractionated based on pI. Fractionation was confirmed with pH reading of individual fractions (Figure S1), as well as an IEF gel. Separately, affinity purified antibody fractions (prior to FFE) were also run on IEF gels.
Neutralization assay
HIV-1 neutralization testing was performed using a luciferase-based assay in TZM.bl cells, as previously described (Sajadi et al., 2011). This assay measures the reduction in luciferase expression following a single round of virus infection. Stocks of Env-pseudotyped viruses were prepared by transfection of 293T/17 cells, as previously described (Sajadi et al., 2011). Unfractioned serum samples, affinity purified antibody, fractionated affinity purified IgG samples, and mAbs were tested against MuLV control and a panel of psuedoviruses. Three-fold serial dilutions of IgG were tested in duplicate (96-well flat bottom plate) in 10% D-MEM growth medium (100 ul/well). 200 TCID50 of pseudovirus was added to each well in a volume of 50 ul and the plates were incubated for 1 hour at 37° C. TZM.bl cells were then added (1x104/well in 100 ul volume) in 10% D-MEM growth medium containing DEAE-Dextran (Sigma, St. Louis, MO) at a final concentration of 11 ug/ml. The final volume for each well was 250ul. Assay controls included replicate wells of TZM.bl cells alone (cell control), TZM.bl cells with virus (virus control), and MuLV control. Following a 48 hour incubation at 37° C, 150 ul of assay medium was removed from each well and 100 ul of Bright-Glo luciferase reagent (Promega, Madison, WI) was added. The cells were allowed to lyse for 2 minutes, then 150 ul of the cell lysate was transferred to a 96-well black solid plate and luminescence was measured using a Victor 3 luminometer (Perkin Elmer, Waltham, MA).
ELISA
HIV-1 envelope capture ELISAs were performed as previously described (Guan et al., 2009) with various antigens (as indicated in the text) that were directly coated (YU2 gp120 core construct and YU2 gp120 core plus V3; 2 ug/ml) or captured (Bal-gp120 or FLSC at a concentration of 0.15ug/ml) by antibody D7324 or JR52 that had been adsorbed to the solid phase at 2 ug/ml. For IEF-fractionated affinity purified IgG, 5 ng from each fraction was tested in a total assay volume of 50 ul. All IgG preparations were incubated with antigens for 1 hour at 37 °C. Bound Abs were then detected with 1:1,000-diluted alkaline phosphatase (AP)-goat antihuman IgG (Southern Biotech; Birmingham, AL) and detected with Blue Phos Microwell Phosphatase Substrate System (KPL, Gaithersburg, MD). All assays were performed in duplicate or repeated several times. Negative control assays were carried out with secondary antibody; background values were subtracted from all test absorbance readings.
Single cell sorting
Single-cell sorting and sequencing was done at Atreca (Redwood City, CA) on PBMC plasmablasts and bone marrow plasma cells and patient-specific B cell databases generated. All paired chain antibody sequencing was carried out on IgG cells sorted into microtiter plates at one cell per well by FACS. IgG plasmablasts and bone marrow cells were enriched from cryopreserved peripheral blood mononuclear cells or bone marrow cells by gating for CD3-CD14-CD16-CD19+CD20-CD27+CD38hiIgA-IgM-IgD- cells. In some experiments, the bone marrow plasma cells (CD3-CD14-CD16-CD38hiIgA-IgM-IgD-) were further sorted and analyzed based on CD19 and CD138.
Paired chain antibody sequencing
Generation of barcoded cDNA, PCR amplification, and 454 sequencing of IgG were performed as described in Tan et al. 2014, with the following modifications: biotinylated Oligo (dT) and RT maxima H (Fisher Scientific Company) were used for reverse transcription, cDNA was extracted using Streptavidin C1 beads (Life Technologies), DNA concentrations were determined using qPCR (KAPA SYBR® FAST qPCR Kit for Titanium, Kapabiosystems), and amplicons were sequenced using Roche 454 Titanium sequencing.
Barcode assignment, sequence assembly, assignment of V(D)J and identification of mutations
These steps were performed as previously described (Tan et al., 2014), except for the following: a minimum coverage of 10 reads was required for each heavy and light chain assembly to be acceptable. Wells with more than one contig for a chain were rejected from consideration unless one of the contigs included at least 90% of the reads. V(D)J assignment and mutation identification was performed using a variant of SoDA (Volpe et al., 2006). Antibody amino acid sequences were aligned to heavy and light chain hidden Markov models using hmmalign (http://hmmer.org). The resulting multiple sequence alignments were used to generate a neighbor-joining tree with RapidNJ (Simonsen M, 2008), with branch lengths representing Kimura Protein Distance (Kimura, 1983).
Mass spectrometry preparation
Antibody species that were isolated to individual fractions were subjected to LC-MS/MS (in addition to FFE fractions, several experiments were carried out with affinity purified fractions or cut-out IEF bands from an IEF gel). Antibody was digested with trypsin, chymotrypsin, or Glu-C overnight at 37° C, the peptides evaporated to 15ul.
Mass spectrometry analysis
The LC-MS system consisted of a Thermo Electron Orbitrap Velow ETD mass spectrometer with a Protana nanospray ion source interfaced with a Phenomenex Jupiter C18 reversed-phase capillary column. The peptide digest was fragmented with both CID and HCD. LC-MS/MS was performed at the University of Maryland School of Pharmacy and Northwestern Proteomics Center of Excellence, none of which were involved in the data analysis.
An array of whole IgG H and L amino acid sequences were translated from the patient-specific database obtained from the single cell PCR described above (bone marrow plasma cells/plasmablasts and circulating plasmablasts), and used as a basis for interpreting the peptide data. The LC-MS/MS derived spectra were searched against the databases independently using the following settings using PEAKS Studio (Bioinformatics Solutions Inc., Ontario, CA): Parent Mass Error Tolerance 5.0 ppm, Fragment Mass Error Tolerance 0.5 Da, Fixed modification of Carboxymethyl (58.01), False Discovery Rate for peptides 5%. This search yielded a list of potential antibody matches from the database. One caveat of the PEAKS search algorithm is that certain peptides (typically from framework regions) can redundantly align with multiple Ig H and L template pairs that share the same gene family usage. Thus, multiple antibodies within the same gene family can potentially match (yielding many false positives). To mitigate this, potential antibodies were further ranked based on the presence of unique peptides, which represent peptides that match only one particular antibody in the database and not any others. In this way, peptides that are shared among dissimilar antibodies (again, mostly in the framework regions) are segregated in the analysis from those that are clonal in origin. The total peptide coverage of the variable region (percent of amino acid coverage of an antibody in the database provided by peptides) was also considered. Antibodies that greater than 50% total variable region coverage and ≥ 4 unique peptides in at least one of the H and L chain of each pair (or with ≥ 4 unique peptides required in each H and L chain for the combined fractions) were chosen for further study.
Generation of plasma antibodies
The identified VH or VL region clones were cloned into an expression vector upstream to an human IgG1 constant domain sequence (CH1-3 from IGHG1*03), as described previously (Guan et al., 2013). Minipreps of these DNA pools, derived from suspension bacterial cultures, were used to transiently transfect 293 Freestyle cells. Transfectant supernatants containing recombinant antibodies were screened in ELISA and neutralization assays.
In the primary approach, sequencing of FFE fractions identified 8 paired H and L Ig genes encoding plasma mAbs N60P1.1, N60P22, N6025.1, N60P36, N60P38, N60P39.1, N60P35, and N60P37. The secondary approach of sequencing polyclonal anti-gp120 antibodies after isoelectric focusing on gels identified all but one of the H and L sequence pairs found in the primary approach as well as 4 additional ones: N60P2.1, N60P30, N60P31.1, N60P48.1, and N60P51. The final approach of sequencing directly from affinity-enriched anti-gp120 plasma antibodies and combining this information with the gel digests from the second approach (consisting of a total of 27 total separate digests) identified most of the same H and L sequence pairs found by the other approaches (missing 2 but identifying 1 additional mAb-N60P39). We identified one additional mAb that was not picked up with the above methods by a homology search of the bone marrow database. This mAb (N60P47) had no binding to gp120 on ELISA, and thus had either no binding to gp120, as in the case of antibodies targeted at the hybrid epitope of CD4 and gp120, or bound to gp120 so weakly that too little was recovered to identify correctly.
Crystallization
Initial crystal screens were done in robotic vapor-diffusion sitting drop trials using a Gryphon Protein Crystallization Robot (Art Robbins Instruments) with commercially available sparse matrix crystallization screens from Molecular Dimensions (Proplex and MacroSol), Emerald BioSystems (Precipitant Wizard Screen) and Emerald BioSystems (Synergy Screen). The screens were monitored periodically for protein crystals. Conditions that produced micro crystals were then reproduced and optimized using the hanging-drop, vapor diffusion method with drops of 0.5 μl protein and 0.5 μl precipitant solution. For the N60P23 complex conditions producing diffraction quality crystals came from 0.1 M Magnesium acetate hexahydrate, 0.065 M NaCl and 0.1 M MOPS pH 7.5 after incubation at 22°C. N49P7 complex crystals came from 10% PEG 5000 MME, 12% isopropanol, and 0.1 M MES pH 6.5. Crystals were frozen in liquid nitrogen after a brief soak in mother liquor supplemented with 20% MPD prior to being used for data collection.
Data collection and structure solution and refinement
Diffraction data for N60P23 Fab-and N49P7 Fab-gp12093TH057 coree complexes were collected at the Stanford Synchrotron Radiation Light Source (SSRL) at the beam line BL14-1 (N60P23) and BL12-2 (N49P7) equipped with Marmosaic 325 or Pilatus area detectors respectively. N60P23 crystals belong to a space group C2 with the unit-cell parameters a = 127.6, b = 68. 6, c = 119.4 Å and β = 111.4º with one N60P23 Fab-gp12093TH057 coree complex present in the asymmetric unit (ASU). N49P7 crystals belong to space group P212121 with the unit-cell parameters a = 61.4, b = 63.9, and c = 255.3 Å with one N49P7 Fab-gp12093TH057 coree complex present in the ASU. Data was processed and reduced with HKL2000, as previously described (Guan et al., 2013). The data for the N49P7 complex was highly anisotropic and was further processed using the STARANISO server (Global Phasing Ltd. [http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi]). The N60P23 structure was solved by molecular replacement with Phaser from the CCP4 suite based on the coordinates of gp120 (PDB: 3TGT) and the VRCO1 Fab (PDB: 4RFE) for the N6P23 Fab. N49P7 was solved using coordinates of gp120 (PDB: 3TGT) and N5-I5 Fab (PDB: 3TNN) for the N49P7 Fab. Refinement was done with Refmac and/or Phenix, coupled with manual refitting and rebuilding using COOT, as previously described (Guan et al., 2013). The N60P23 complex complex was refined to an R-factor of 0.214 and an R-free of 0.258 and the N49P7 complex was refined to an R-factor of 0.225 and R-free of 0.285. Data collection and refinement statistics are shown in (Table S5). In addition, experimental 2Fo-Fc electron density maps around the N49P7 Fab-gp120 interface of N49P7 Fab-gp12093TH057 coree complex are shown in (Figure S7).
Structure validation and analysis
The quality of the final refined model was monitored using the program MolProbity, as previously described (Guan et al., 2013). Structural alignments were performed using the Dali server and the program lsqkab from CCP4 suite. The PISA webserver was used to determine contact surfaces and interface residues. All illustrations were prepared with the PyMol Molecular Graphic suite (http://pymol.org) (DeLano Scientific, San Carlos, CA, USA).
Quantification and Statistical Analysis
The 50% inhibitory concentration (IC50) and 80% inhibitory concentration (IC80) titers were calculated as the immunoglobulin concentration that caused a 50% or 80% reduction in relative luminescence units (RLU) compared to the virus control wells after subtraction of cell control RLUs. ELISA AUC calculated with GraphPad Prism 5.0 (GraphPad, La Jolla, CA).
Data and Software Availability
Sequences for N60 P series and N49 P series mAbs have been deposited in GenBank (see Tables S2 and S4, or Key Resources Table, for accession numbers). Coordinates of N60P23 Fab-gp12093TH057 coree complex and N49P7 Fab-gp12093TH057 coree complex were deposited in the Protein Data Bank under accession codes PDB 5WB9 and PDB 6BCK, respectively.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| D7324 | Aalto Bio | Cat#D7324 |
| JR52 | Dr. James Robinson (Robinson et al., 2016) | N/A |
| Anti-human Goat IgG-AP | Southern Biotech | Cat#2040-04 |
| Anti-human CD3-FITC | BioLegend | Cat#300405 |
| Anti-human CD38-A647 | BioLegend | Cat#303514 |
| Anti-human CD45-BV510 | BioLegend | Cat#304141 |
| Anti-human CD19-BV421 | BioLegend | Cat#302233 |
| Anti-human CD14-FITC | BioLegend | Cat#325603 |
| Anti-human IgD-A488 | BioLegend | Cat#348215 |
| Anti-human IgM-FITC | BioLegend | Cat#314506 |
| Anti-human IgA-FITC | Miltenyi | Cat#130-099-107 |
| Anti-human CD138-PE | BeckmanCoulter | Cat# MA1-81808 |
| N60P1.1 Heavy Chain | This study | Ref#MG791872 |
| N60P1.1 Light Chain | This study | Ref#MG819662 |
| N60P2.1 Heavy Chain | This study | Ref#MG819649 |
| N60P2.1 Light Chain | This study | Ref#MG819677 |
| N60P22 Heavy Chain | This study | Ref#MG819659 |
| N60P22 Light Chain | This study | Ref#MG819666 |
| N60P23 Heavy Chain | This study | Ref#MG819661 |
| N60P23 Light Chain | This study | Ref#MG819663 |
| N60P25.1 Heavy Chain | This study | Ref#MG819648 |
| N60P25.1 Light Chain | This study | Ref#MG819664 |
| N60P30 Heavy Chain | This study | Ref#MG819660 |
| N60P30 Light Chain | This study | Ref#MG819670 |
| N60P31.1 Heavy Chain | This study | Ref#MG819647 |
| N60P31.1 Light Chain | This study | Ref#MG819665 |
| N60P35 Heavy Chain | This study | Ref#MG819650 |
| N60P35 Light Chain | This study | Ref#MG819668 |
| N60P36 Heavy Chain | This study | Ref#MG819651 |
| N60P36 Light Chain | This study | Ref#MG819671 |
| N60P37 Heavy Chain | This study | Ref#MG819652 |
| N60P37 Light Chain | This study | Ref#MG819669 |
| N60P38 Heavy Chain | This study | Ref#MG819653 |
| N60P38 Light Chain | This study | Ref#MG819667 |
| N60P39 Heavy Chain | This study | Ref#MG819654 |
| N60P39 Light Chain | This study | Ref#MG819672 |
| N60P39.1 Heavy Chain | This study | Ref#MG819656 |
| N60P39.1 Light Chain | This study | Ref#MG819673 |
| N60P47 Heavy Chain | This study | Ref#MG819655 |
| N60P47 Light Chain | This study | Ref#MG819674 |
| N60P48 Heavy Chain | This study | Ref#MG819657 |
| N60P48 Light Chain | This study | Ref#MG819675 |
| N60P51 Heavy Chain | This study | Ref#MG819658 |
| N60P51 Light Chain | This study | Ref#MG819676 |
| N49P6 Heavy Chain | This study | Ref#MG819637 |
| N49P6 Light Chain | This study | Ref#MG819642 |
| N40P7 Heavy Chain | This study | Ref#MG819638 |
| N49P7 Light Chain | This study | Ref#MG819643 |
| N49P7.1 Heavy Chain | This study | Ref#MG819639 |
| N49P7.1 Light Chain | This study | Ref#MG819644 |
| N40P9 Heavy Chain | This study | Ref#MG819641 |
| N49P9 Light Chain | This study | Ref#MG819646 |
| N49P11 Heavy Chain | This study | Ref#MG819640 |
| N49P11 Light Chain | This study | Ref#MG819645 |
| Bacterial and Virus Strains | ||
| 117 HIV-1 pseudovirus “global panel” | (Seaman et al., 2010); this study | N/A |
| 53 pseudovirus expanded panel | (Huang et al. 2016) | N/A |
| Biological Samples | ||
| Human Plasma | University of Maryland School of Medicine | N/A |
| Human Bone Marrow Cells | University of Maryland School of Medicine | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ba-L gp120 | (Fouts et al., 2000); this study | N/A |
| Single chain gp120-CD4 complex (FLSC) | (Fouts et al., 2000); this study | N/A |
| YU2 gp120 core | (Wu et al., 1996); this study | N/A |
| YU2 gp120 core + V3 | (Wu et al., 1996); this study | N/A |
| D368R Ba-L gp120 | (Li et al., 2007); this study | N/A |
| HIV-1 V3 Peptides | NIH AIDS Reagent Program | Cat#1840 |
| Critical Commercial Assays | ||
| Protein A agarose | GE Healthcare | Cat#20333 |
| Protein A/G agarose | GE Healthcare | Cat#21186 |
| IgG1 (Hu) Affinity Matrix | Thermo Scientific | Cat#191303005 |
| LC-lambda (Hu) Affinity Matrix | Thermo Scientific | Cat#084905 |
| KappaXL Affinity Matrix | Thermo Scientific | Cat#19432101L |
| KAPA SYBR® FAST qPCR Kit | Kapa biosystems | Cat# KK4600 |
| Maxima Reverse Transcriptase | Thermo Scientific | Cat# EP0741 |
| Deposited Data | ||
| N60P series mAbs | This study | Table S2 |
| N49P series mAbs | This study | Table S4 |
| N60P23 Fab-gp12093TH057 coree complex | This study | Ref#PDB 5WB9 |
| N49P7 Fab-gp12093TH057 coree complex | This study | Ref#PDB 6BCK |
| Experimental Models: Cell Lines | ||
| HEK293T cells | ATCC | Cat#CRL-3216 |
| TZM-bl cells | NIH AIDS Reagent Program | Cat#8129 |
| Freestyle 293-F cells | Thermo Scientific | Cat#R79007 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Promega™ Biotinylated Oligo(dT) Probe | Fisher Scientific | Cat#PR-Z5261 |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Prism (v5.0) | GraphPad | http://www.graphpad.com |
| PEAKS Studio (v7.5) | Bioinformatics Solutions Inc. | www.bioinfor.com |
| hmmalign | HMMER | http://hmmer.org |
| RapidNJ | Aaarhus University | http://birc.au.dk/software/rapidnj |
| HKL2000 | HKL Research | www.hkl-xray.com/download-instructions-hkl-2000 |
| COOT | University of Cambridge | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| STARANISO server | Global Phasing Limited | http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi |
| PISA webserver | Protein Data Bank in Europe | www.ebi.ac.uk/pdbe/pisa |
| MolProbity | Duke University | molprobity.biochem.duke.edu |
| CCP4 Suite | Science and Technology Facilities Council | www.ccp4.ac.uk/ |
| PyMol Molecular Graphic suite | DeLano Scientific | http://pymol.org |
| Other | ||
Supplementary Material
Anti-gp120 IgG1 kappa (panel A) and lambda (panel B) were fractionated by FFE (see Materials and Methods). Fractions pH ranges from approximately 6.5 (fraction 25) and increasing to 10 (fraction 68) (not shown on the graph). Aliquots (0.05 ug) of IgG from each fraction was tested by ELISA for reactivity against the indicated HIV antigens: (BaL-gp120 monomer, BaL-gp120 monomer with the D368R mutation to abrogate CD4bs binding, Yu2 gp120 core with V3 loop, and Yu2 gp120 core, and full length single chain (FLSC), presenting a full length CD4-induced gp120 structure in which the CD4 binding site is occupied). The X axis represents the IEF fractions (spanning a pH gradient of 6 to 10 from left to right). The Y axis represents ELISA signals expressed as background-corrected OD450 readings/ug IgG. The right Y axis shows IgG concentration of each fraction (ug/ml). Assays were repeated at least twice. Areas of broad and limited neutralization previously identified based on neutralization (ability to neutralize Tier 2 viruses at <10ug/ml of affinity purified antibody). Each fraction contains antibodies that can distinguish single epitopes. In panel A, Fractions 55–68 do not bind to D368R envelope mutants but do to the wild-type virus (BaL-gp120). Likewise, Fractions 25–30 and 35–40 in Panel A bind to FLSC (fusion protein between CD4 and gp120) but not monomeric gp120. This strongly suggests antibodies targeting a single epitope (CD4-binding site antibodies and CoReceptor binding site, respectively), as a mixed population of CD4-binding site and non-CD4 binding site antibodies would show some binding to the D368R mutant, and a mixed population of CoReceptor binding site and non-CoReceptor binding site would show binding to the monomer. In Panel B, when the fraction IgG concentrations are compared, the IgG1 anti-gp120 lambda response is almost entirely limited to fractions 28–32, suggesting that one or few antibodies are responsible for the lambda fraction (and by extension up to 50% of the total anti-gp120 response).
A) Individual dendrograms for all lineages. Seven neutralizing lineages described in the text are indicated with color-coded numbers matching their branch color. Bar indicates amino acid sequence distance for panel A calculated using the Kimura protein distance method (Kimura, 1983). B) Lineage 1 phylogeny. Lineage 1 represents the broadly neutralizing CD4-binding site antibodies.
A) Elisa reactivity of N60 P series. B) Elisa reactivity of N49 P series. Dilutions of each mAb was tested by ELISA (starting at 0.8ug/ml) for reactivity against the indicated HIV antigens: gp120 (BaL monomer), FLSC (full length CD4-induced BaL gp120 structure in which the CD4bs is occupied), D368R (BaL-gp120 monomer with the D368R mutation to abrogate CD4bs binding), Core (Yu2 gp120), Core/V3 (Yu2 gp120 core with V3 attached). Area under the curve (AUC) were calculated and are reported as a color as indicated by the key (the single key is for both panels).
The binding kinetics for BaL-gp120 or D368R with mAb captured on Protein A-coated chips are shown. Data sets with significant dose response were globally fit to a 1:1 binding model to obtain the kinetic parameters of the binding. All mAbs tested bound strongly to gp120 monomer but exhibited weak binding to D368R.
A panel of 53 HIV-1 psuedovirus Tier 1–3 isolates (individual viruses listed on the left column) were tested against N49P7. IC50 values are color-coded according to the color key on the left: the greater the neutralization, the darker red the color; white represents no neutralization (IC50>50ug/ml). IC50=Inhibitory Concentration 50 in ug/ml. NC=not classified
(A) Ribbon diagram of N60P23 Fab-gp12093TH057 coree complex with light and heavy chains of Fab shown in light and dark green, respectively, and the complementarity-determining regions (CDRs) shown in blue (CDR L1), black (CDR L2), orange (CDR L3), pink (CDR H1), green (CDR H2), and yellow (CDR H3). The gp120 is colored in white. The D (S274 – T283), V5 (T455-N465) and the CD4 binding (Q362-G372) loops are colored in cyan, violet and magenta, respectively. (B) Structural comparison of N60P23 Fab-gp12093TH057 coree complex and VRCO1-gp12093TH057 coree (PDB code 3NGB) complexes. Complexes were aligned based on the gp120 and are shown as the ribbon diagrams. The light and heavy chains of VRCO1 Fab are shown in light and dark cyan and the CDRs are colored as in N60P23 complex. (C) N60P23 and VRCO1 epitope footprints. N60P23/VRCO1 contacts on gp12093TH057 coree are highlighted in light green/cyan (light chain) and dark green/cyan (heavy and both chains) on the gp120 surface. (D) N60P23 and VRCO1 Fabs and gp12093TH057 coree contact residues on the primary sequence of the Fabs and gp12093TH057 coree, respectively. Residues contributing to the Fabs and gp12093TH057 coree are highlighted and contacts as defined by a 5 Å cutoff are marked above the sequence. Side chain (+) and main chain (−) contacts are colored based on contact type; hydrophobic in blue, hydrophilic in green, or both in black. Framework and complementary-determining regions are as indicated in the alignment. N60P23 binds within the CD4BS of gp120 engaging the CD4-binding loop as well as loops D and V5 also known to interact with the CD4 receptor and the CD4-binding site antibody VRC01. It recognizes the gp120 antigen with striking similarities to VRC01 with epitope footprint almost entirely overlapping the gp120 surface involved in VRC01 Fab-CD4-gp12093TH057 coree complex and utilizing the same CDR contacts. The close structural similarity of the complex interfaces is reflected in relatively low root mean square deviation (RMSD) value of 0.7 Å for the Cα atoms of the Fab variable domains and the gp120 coree.
A) The overall structure of the N49P7 Fab-gp12093TH057 coree complex is shown in the center with the molecular surface displayed over the Fab molecule to highlight the shape of the formed antigen binding site. The complementarity-determining regions (CDRs) are shown in green (CDR L1), blue (CDR L3), black (CDR H1), yellow (CDR H2), and red (CDR H3). The gp120 outer and inner domains are colored in black and gray, respectively. The D (S274 – T283), E5 (F353 – T358), V5 (T455 – N465) and the CD4 binding (Q362 – G372) loops are colored in cyan, orange, violet and magenta, respectively. The unique features of the gp120 antigen recognition mediated by N49P7 are shown (from 1 to 6) and discussed in details in the blow up view figures: (1) ‘Super short’ CDR L1- The CDR L1 of N49P7 consists of 8 amino acids (aa), 1 and 3 aa shorter than the CDRL1 of VRC01 and N6, respectively. The short CDR L1 avoids steric clashes with loop D and Loop E and permits the accommodation of complex and bulky glycans linked to N276 and N355 of gp120. The figure shows the overlay of the N49P7 Fab-gp12093TH057 coree and the N6 Fab-gp12093TH057 coree complex (Huang et al., 2016). Structures were superimposed based upon gp120 to show differences in the length and position of CDR L1 relative to the gp120 antigen (colored dark green for N49P7 and light green for N6) (2) Unpaired cysteine (C36) in a framework 2 (FW2) of the light chain. Both κ (as seen in N6) and λ light chains have a tyrosine at position of 36 of FW2. Replacement of this bulky residue by cysteine changes the packing of the hydrophobic core formed at the variable light (VL) and heavy (VH) domain interface of N49P7 as compared to N6. The result of the unique packing is a difference in the relative orientations (rotation/tilting) of VL and VH observed in N49P7 relative to N6. The figure shows the VL - VH core packing of N49P7 and N6. Fabs were superimposed based of VH domain. N6 is colored light and dark blue for light and heavy chain, respectively (CD1 L1 as in panel 1). (3) Rotation/tilting of the light chain- the assembly of VL and VH domains of N49P7 forms an asymmetric antigen binding side, wildly open from the VL side and protruding from the VH side. The open access from the VL side is due to the rotation/tilting of the light chain relative to the heavy chain as described in (2). The figure shows the same superimposition as in panel 2. The VL of N49P7 is rotated approximately 12 degrees (measured at the VL N-termini) away from Loops D and V5 of gp120. This generates a 5Å distance between the VL N-termini of N49P7 and N6. The wild opening of the VL side of the P7 antigen binding site combined with short CDRL1 allows N49P7 to accommodate different lengths of the highly variable loops D, E and V5. Changes in the length of gp120 loop V5 and the length (and glycosylation status) of loop E that cause steric clashes with an antibody CDRL1 were described previously as mechanisms of HIV-1 resistance to VRC01-class antibodies. (4) Normal length CDRH3 that contacts to Loop D and the inner domain of gp120. The CDRH3 of N49P7 consists of 19 aa that contact the conserved Loop D of gp120 and reach deeply into the gp120 inner domain (compared to the 13 aa –long CDRH3 of N6). The CDRH3 contributes 274 Å2 BSA to the complex interface (28.6% of the BSA of the whole Fab). A network of interactions is formed which includes hydrogen bonds formed by Lys of CDRH3 (of a S100GK motif) and residues of the α1 helix of Layer 2 and the α7-helix of Layer 3 of the inner domain. (5) ‘Bypassing’ the Phe43 cavity. The CDRH2 of N49P7 is a major anchor point in binding to the gp120 antigen contributing 489 Å2 BSA to the complex interface (51% of the BSA of the whole Fab) but it has a Glycine at position 54 (Tyrosine in N6) thus it is unable to anchor deeply inside the Phe43 cavity of the CD4BS. Instead N49P7 makes important contacts to inner domain mediated trough M53 and Q56 which compensate for lack of direct contacts to residues of the Phe43 cavity. Overall N49P7 contributes 207 Å2 of its buried surface area (BSA) to the gp120 inner domain which is the highest among N6 and the VRC01 Abs class (Table S6). The figure shows a blow up view of the CDRH2 –gp120 interface of N49P7 and N6. CDRH2 is colored light and dark yellow for N49P7 and N6, respectively. (6) The P60W motif in CDRH2. N49P7 is able accommodate changes in length and conformation of the gp120 V5 loop through a P60W motif of its CDRH2 which forms the framework for an interaction network with the base of the V5 loop. Figure shows a close-up view of the interaction of CDRH2 of N49P7 and N6 with Loop V5. The P60W (N49P7) and G60GG (N6) motifs are highlighted. B) 2Fo-Fc electron density map around gp120 inner domain contacting CDR H1-3 of N49P7 Fab contoured at 1.0σ. View and residues shown are as in Figure 6, panel B.
Highlights.
Plasma anti-gp120 response deconvolution is used to understand how the human body neutralizes HIV
Near pan-neutralizing activity is traced to one IgG lineage, targeting the CD4 binding site on gp120
Plasma broadly neutralizing Abs persist and function as a minor fraction of the polyclonal response
Plasma mAbs targeting the CD4 binding have unique features and near-pan neutralizing properties
Acknowledgments
We would like to thank members of the NVS Study and other research volunteers.This research was supported by NIH 1R01AI110259-01A1 and 1I01BX002358-01A1 to MMS, NIH 1R01AI116274 and 1R01AI129769 to MP, and NIH R01AI124912 to GKL. MSS and the Comprehensive Antibody Vaccine Immune Monitoring Consortium supported by the Bill & Melinda Gates Foundation Grant ID 1032144.
Footnotes
Author contributions
M.M.S., G.K.L., A.L.D. each contributed to the design of the study, analysis of the data, and preparation of this manuscript. M.M.S., A.D., Z.R.T., and X.O. affinity purification of antibody, free flow electrophoresis. X.O. performed antibody digestion. G.C. and D.K. planned and performed single-cell sequencing experiments. A.D., Z.R.T., and X.O. performed antibody cloning, epitope mapping assays. M.S.S. performed neutralization assays. W.D.T., N.G., and M.P. crystallographic analysis. M.M.S. and R.R.R. led the clinical care of the patients, and J.Y. performed bone marrow aspirates.
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- Boutz DR, Horton AP, Wine Y, Lavinder JJ, Georgiou G, Marcotte EM. Proteomic identification of monoclonal antibodies from serum. Analytical chemistry. 2014;86:4758–4766. doi: 10.1021/ac4037679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briney BS, Willis JR, Finn JA, McKinney BA, Crowe JE., Jr Tissue-specific expressed antibody variable gene repertoires. PLoS One. 2014;9:e100839. doi: 10.1371/journal.pone.0100839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT. Free R value: Cross-validation in crystallography. Method Enzymol. 1997;277:366–396. doi: 10.1016/s0076-6879(97)77021-6. [DOI] [PubMed] [Google Scholar]
- de Taeye SW, Ozorowski G, Torrents de la Pena A, Guttman M, Julien JP, van den Kerkhof TL, Burger JA, Pritchard LK, Pugach P, Yasmeen A, et al. Immunogenicity of Stabilized HIV-1 Envelope Trimers with Reduced Exposure of Non-neutralizing Epitopes. Cell. 2015;163:1702–1715. doi: 10.1016/j.cell.2015.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey B, Berger EA. Blocking HIV-1 gp120 at the Phe43 cavity: if the extension fits. Structure. 2013;21:871–872. doi: 10.1016/j.str.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon AK, Donners H, Pantophlet R, Johnson WE, Decker JM, Shaw GM, Lee FH, Richman DD, Doms RW, Vanham G, et al. Dissecting the neutralizing antibody specificities of broadly neutralizing sera from human immunodeficiency virus type 1-infected donors. J Virol. 2007;81:6548–6562. doi: 10.1128/JVI.02749-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin R, Scheid JF, Marcovecchio PM, West AP, Jr, Klein F, Gao H, Gnanapragasam PN, Abadir A, Seaman MS, Nussenzweig MC, et al. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science. 2011;334:1289–1293. doi: 10.1126/science.1213782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria-Rose NA, Schramm CA, Gorman J, Moore PL, Bhiman JN, Dekosky BJ, Ernandes MJ, Georgiev IS, Kim HJ, Pancera M, et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature. 2014;509:55–62. doi: 10.1038/nature13036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugast AS, Arnold K, Lofano G, Moore S, Hoffner M, Simek M, Poignard P, Seaman M, Suscovich TJ, Pereyra F, et al. Virus-driven Inflammation Is Associated With the Development of bNAbs in Spontaneous Controllers of HIV. Clin Infect Dis. 2017;64:1098–1104. doi: 10.1093/cid/cix057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol Cell. 2010;37:656–667. doi: 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouts TR, Tuskan R, Godfrey K, Reitz M, Hone D, Lewis GK, DeVico AL. Expression and characterization of a single-chain polypeptide analogue of the human immunodeficiency virus type 1 gp120-CD4 receptor complex. J Virol. 2000;74:11427–11436. doi: 10.1128/jvi.74.24.11427-11436.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev IS, Joyce MG, Yang Y, Sastry M, Zhang B, Baxa U, Chen RE, Druz A, Lees CR, Narpala S, et al. Single-Chain Soluble BG505.SOSIP gp140 Trimers as Structural and Antigenic Mimics of Mature Closed HIV-1. Env J Virol. 2015;89:5318–5329. doi: 10.1128/JVI.03451-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, Lovo E, Wu X, Robinson JE, Seaman MS, et al. Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc Natl Acad Sci U S A. 2013;110:E69–78. doi: 10.1073/pnas.1217609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Sajadi MM, Kamin-Lewis R, Fouts TR, Dimitrov A, Zhang Z, Redfield RR, DeVico AL, Gallo RC, Lewis GK. Discordant memory B cell and circulating anti-Env antibody responses in HIV-1 infection. Proc Natl Acad Sci U S A. 2009;106:3952–3957. doi: 10.1073/pnas.0813392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, Gregoretti IV, Popova L, Kaminiski D, Fucile CF, Albizua I, et al. Long-Lived Plasma Cells Are Contained within the CD19(-)CD38(hi)CD138(+) Subset in Human Bone Marrow. Immunity. 2015;43:132–145. doi: 10.1016/j.immuni.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes BF, Mascola JR. The quest for an antibody-based HIV vaccine. Immunol Rev. 2017;275:5–10. doi: 10.1111/imr.12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kang BH, Ishida E, Zhou T, Griesman T, Sheng Z, Wu F, Doria-Rose NA, Zhang B, McKee K, et al. Identification of a CD4-Binding-Site Antibody to HIV that Evolved Near-Pan Neutralization Breadth. Immunity. 2016;45:1108–1121. doi: 10.1016/j.immuni.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kang BH, Pancera M, Lee JH, Tong T, Feng Y, Imamichi H, Georgiev IS, Chuang GY, Druz A, et al. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature. 2014;515:138–142. doi: 10.1038/nature13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine JG, Kulp DW, Havenar-Daughton C, Sarkar A, Briney B, Sok D, Sesterhenn F, Ereno-Orbea J, Kalyuzhniy O, Deresa I, et al. HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science. 2016;351:1458–1463. doi: 10.1126/science.aad9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaherian K, Langlois AJ, McDanal C, Ross KL, Eckler LI, Jellis CL, Profy AT, Rusche JR, Bolognesi DP, Putney SD, et al. Principal neutralizing domain of the human immunodeficiency virus type 1 envelope protein. Proc Natl Acad Sci U S A. 1989;86:6768–6772. doi: 10.1073/pnas.86.17.6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University Press; 1983. [Google Scholar]
- Kwong PD, Mascola JR. Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity. 2012;37:412–425. doi: 10.1016/j.immuni.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRosa GJ, Davide JP, Weinhold K, Waterbury JA, Profy AT, Lewis JA, Langlois AJ, Dreesman GR, Boswell RN, Shadduck P, et al. Conserved sequence and structural elements in the HIV-1 principal neutralizing determinant. Science. 1990;249:932–935. doi: 10.1126/science.2392685. [DOI] [PubMed] [Google Scholar]
- Lavinder JJ, Wine Y, Giesecke C, Ippolito GC, Horton AP, Lungu OI, Hoi KH, DeKosky BJ, Murrin EM, Wirth MM, et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc Natl Acad Sci U S A. 2014;111:2259–2264. doi: 10.1073/pnas.1317793111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Migueles SA, Welcher B, Svehla K, Phogat A, Louder MK, Wu X, Shaw GM, Connors M, Wyatt RT, et al. Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat Med. 2007;13:1032–1034. doi: 10.1038/nm1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Svehla K, Louder MK, Wycuff D, Phogat S, Tang M, Migueles SA, Wu X, Phogat A, Shaw GM, et al. Analysis of neutralization specificities in polyclonal sera derived from human immunodeficiency virus type 1-infected individuals. J Virol. 2009;83:1045–1059. doi: 10.1128/JVI.01992-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch RM, Wong P, Tran L, O’Dell S, Nason MC, Li Y, Wu X, Mascola JR. HIV-1 fitness cost associated with escape from the VRC01 class of CD4 binding site neutralizing antibodies. J Virol. 2015;89:4201–4213. doi: 10.1128/JVI.03608-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montezuma-Rusca JM, Moir S, Kardava L, Buckner CM, Louie A, Kim LJ, Santich BH, Wang W, Fankuchen OR, Diaz G, et al. Bone marrow plasma cells are a primary source of serum HIV-1-specific antibodies in chronically infected individuals. J Immunol. 2015;194:2561–2568. doi: 10.4049/jimmunol.1402424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov AN, Bourenkov GP. Choice of data-collection parameters based on statistic modelling. Acta Crystallogr D. 2003;59:1145–1153. doi: 10.1107/s0907444903008163. [DOI] [PubMed] [Google Scholar]
- Ranasinghe S, Soghoian DZ, Lindqvist M, Ghebremichael M, Donaghey F, Carrington M, Seaman MS, Kaufmann DE, Walker BD, Porichis F. HIV-1 Antibody Neutralization Breadth Is Associated with Enhanced HIV-Specific CD4+ T Cell Responses. J Virol. 2015;90:2208–2220. doi: 10.1128/JVI.02278-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JE, Hastie KM, Cross RW, Yenni RE, Elliott DH, Rouelle JA, Kannadka CB, Smira AA, Garry CE, Bradley BT, et al. Most neutralizing human monoclonal antibodies target novel epitopes requiring both Lassa virus glycoprotein subunits. Nat Commun. 2016;7:11544. doi: 10.1038/ncomms11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusert P, Kouyos RD, Kadelka C, Ebner H, Schanz M, Huber M, Braun DL, Hoze N, Scherrer A, Magnus C, et al. Determinants of HIV-1 broadly neutralizing antibody induction. Nat Med. 2016;22:1260–1267. doi: 10.1038/nm.4187. [DOI] [PubMed] [Google Scholar]
- Sajadi MM, Constantine NT, Mann DL, Charurat M, Dadzan E, Kadlecik P, Redfield RR. Epidemiologic characteristics and natural history of HIV-1 natural viral suppressors. J Acquir Immune Defic Syndr. 2009;50:403–408. doi: 10.1097/QAI.0b013e3181945f1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadi MM, Farshidpour M, Brown EP, Ouyang X, Seaman MS, Pazgier M, Ackerman ME, Robinson H, Tomaras G, Parsons MS, et al. lambda Light Chain Bias Associated With Enhanced Binding and Function of Anti-HIV Env Glycoprotein Antibodies. J Infect Dis. 2016;213:156–164. doi: 10.1093/infdis/jiv448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadi MM, Guan Y, DeVico AL, Seaman MS, Hossain M, Lewis GK, Redfield RR. Correlation between circulating HIV-1 RNA and broad HIV-1 neutralizing antibody activity. J Acquir Immune Defic Syndr. 2011;57:9–15. doi: 10.1097/QAI.0b013e3182100c1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadi MM, Heredia A, Le N, Constantine NT, Redfield RR. HIV-1 natural viral suppressors: control of viral replication in the absence of therapy. AIDS. 2007;21:517–519. doi: 10.1097/QAD.0b013e328013d9eb. [DOI] [PubMed] [Google Scholar]
- Sajadi MM, Lewis GK, Seaman MS, Guan Y, Redfield RR, DeVico AL. Signature biochemical properties of broadly cross-reactive HIV-1 neutralizing antibodies in human plasma. J Virol. 2012;86:5014–5025. doi: 10.1128/JVI.06547-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RW, Derking R, Cupo A, Julien JP, Yasmeen A, de Val N, Kim HJ, Blattner C, de la Pena AT, Korzun J, et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013;9:e1003618. doi: 10.1371/journal.ppat.1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather DN, Stamatatos L. Epitope specificities of broadly neutralizing plasmas from HIV-1 infected subjects. Vaccine. 2010;28(Suppl 2):B8–12. doi: 10.1016/j.vaccine.2009.07.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, Pietzsch J, Ott RG, Anthony RM, Zebroski H, Hurley A, et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature. 2009;458:636–640. doi: 10.1038/nature07930. [DOI] [PubMed] [Google Scholar]
- Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C, Giri A, Coffey RT, Harris L, Wood B, Daniels MG, et al. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol. 2010;84:1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simek MD, Rida W, Priddy FH, Pung P, Carrow E, Laufer DS, Lehrman JK, Boaz M, Tarragona-Fiol T, Miiro G, et al. Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol. 2009;83:7337–7348. doi: 10.1128/JVI.00110-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen MMT, Pedersen CNS. Rapid neighbor-joining. In: Crandall LJ, KA, editors. WABI 2008. Heidelberg: Springer; 2008. pp. 113–122. [Google Scholar]
- Tan YC, Blum LK, Kongpachith S, Ju CH, Cai X, Lindstrom TM, Sokolove J, Robinson WH. High-throughput sequencing of natively paired antibody chains provides evidence for original antigenic sin shaping the antibody response to influenza vaccination. Clin Immunol. 2014;151:55–65. doi: 10.1016/j.clim.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trkola A, Pomales AB, Yuan H, Korber B, Maddon PJ, Allaway GP, Katinger H, Barbas CF, Burton DR, Ho DD, et al. Cross-clade neutralization of primary isolates of human immunodeficiency virus type 1 by human monoclonal antibodies and tetrameric CD4-IgG. J Virol. 1995;69:6609–6617. doi: 10.1128/jvi.69.11.6609-6617.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel T, Kurth R, Norley S. The majority of neutralizing Abs in HIV-1-infected patients recognize linear V3 loop sequences. Studies using HIV-1MN multiple antigenic peptides. J Immunol. 1994;153:1895–1904. [PubMed] [Google Scholar]
- Volpe JM, Cowell LG, Kepler TB. SoDA: implementation of a 3D alignment algorithm for inference of antigen receptor recombinations. Bioinformatics. 2006;22:438–444. doi: 10.1093/bioinformatics/btk004. [DOI] [PubMed] [Google Scholar]
- Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Simek MD, Priddy F, Gach JS, Wagner D, Zwick MB, Phogat SK, Poignard P, Burton DR. A limited number of antibody specificities mediate broad and potent serum neutralization in selected HIV-1 infected individuals. PLoS Pathog. 2010;6:e1001028. doi: 10.1371/journal.ppat.1001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MS. Global indicators of X-ray data quality. J Appl Crystallogr. 2001;34:130–135. [Google Scholar]
- West AP, Jr, Diskin R, Nussenzweig MC, Bjorkman PJ. Structural basis for germ-line gene usage of a potent class of antibodies targeting the CD4-binding site of HIV-1 gp120. Proc Natl Acad Sci U S A. 2012;109:E2083–2090. doi: 10.1073/pnas.1208984109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LD, Ofek G, Schatzle S, McDaniel JR, Lu X, Nicely NI, Wu L, Lougheed CS, Bradley T, Louder MK, et al. Potent and broad HIV-neutralizing antibodies in memory B cells and plasma. Science Immunology. 2017;2:eaal2200. doi: 10.1126/sciimmunol.aal2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine Y, Boutz DR, Lavinder JJ, Miklos AE, Hughes RA, Hoi KH, Jung ST, Horton AP, Murrin EM, Ellington AD, et al. Molecular deconvolution of the monoclonal antibodies that comprise the polyclonal serum response. Proc Natl Acad Sci U S A. 2013;110:2993–2998. doi: 10.1073/pnas.1213737110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine Y, Horton AP, Ippolito GC, Georgiou G. Serology in the 21st century: the molecular-level analysis of the serum antibody repertoire. Current opinion in immunology. 2015;35:89–97. doi: 10.1016/j.coi.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, et al. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, Kwon YD, Scheid JF, Shi W, Xu L, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329:811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Anti-gp120 IgG1 kappa (panel A) and lambda (panel B) were fractionated by FFE (see Materials and Methods). Fractions pH ranges from approximately 6.5 (fraction 25) and increasing to 10 (fraction 68) (not shown on the graph). Aliquots (0.05 ug) of IgG from each fraction was tested by ELISA for reactivity against the indicated HIV antigens: (BaL-gp120 monomer, BaL-gp120 monomer with the D368R mutation to abrogate CD4bs binding, Yu2 gp120 core with V3 loop, and Yu2 gp120 core, and full length single chain (FLSC), presenting a full length CD4-induced gp120 structure in which the CD4 binding site is occupied). The X axis represents the IEF fractions (spanning a pH gradient of 6 to 10 from left to right). The Y axis represents ELISA signals expressed as background-corrected OD450 readings/ug IgG. The right Y axis shows IgG concentration of each fraction (ug/ml). Assays were repeated at least twice. Areas of broad and limited neutralization previously identified based on neutralization (ability to neutralize Tier 2 viruses at <10ug/ml of affinity purified antibody). Each fraction contains antibodies that can distinguish single epitopes. In panel A, Fractions 55–68 do not bind to D368R envelope mutants but do to the wild-type virus (BaL-gp120). Likewise, Fractions 25–30 and 35–40 in Panel A bind to FLSC (fusion protein between CD4 and gp120) but not monomeric gp120. This strongly suggests antibodies targeting a single epitope (CD4-binding site antibodies and CoReceptor binding site, respectively), as a mixed population of CD4-binding site and non-CD4 binding site antibodies would show some binding to the D368R mutant, and a mixed population of CoReceptor binding site and non-CoReceptor binding site would show binding to the monomer. In Panel B, when the fraction IgG concentrations are compared, the IgG1 anti-gp120 lambda response is almost entirely limited to fractions 28–32, suggesting that one or few antibodies are responsible for the lambda fraction (and by extension up to 50% of the total anti-gp120 response).
A) Individual dendrograms for all lineages. Seven neutralizing lineages described in the text are indicated with color-coded numbers matching their branch color. Bar indicates amino acid sequence distance for panel A calculated using the Kimura protein distance method (Kimura, 1983). B) Lineage 1 phylogeny. Lineage 1 represents the broadly neutralizing CD4-binding site antibodies.
A) Elisa reactivity of N60 P series. B) Elisa reactivity of N49 P series. Dilutions of each mAb was tested by ELISA (starting at 0.8ug/ml) for reactivity against the indicated HIV antigens: gp120 (BaL monomer), FLSC (full length CD4-induced BaL gp120 structure in which the CD4bs is occupied), D368R (BaL-gp120 monomer with the D368R mutation to abrogate CD4bs binding), Core (Yu2 gp120), Core/V3 (Yu2 gp120 core with V3 attached). Area under the curve (AUC) were calculated and are reported as a color as indicated by the key (the single key is for both panels).
The binding kinetics for BaL-gp120 or D368R with mAb captured on Protein A-coated chips are shown. Data sets with significant dose response were globally fit to a 1:1 binding model to obtain the kinetic parameters of the binding. All mAbs tested bound strongly to gp120 monomer but exhibited weak binding to D368R.
A panel of 53 HIV-1 psuedovirus Tier 1–3 isolates (individual viruses listed on the left column) were tested against N49P7. IC50 values are color-coded according to the color key on the left: the greater the neutralization, the darker red the color; white represents no neutralization (IC50>50ug/ml). IC50=Inhibitory Concentration 50 in ug/ml. NC=not classified
(A) Ribbon diagram of N60P23 Fab-gp12093TH057 coree complex with light and heavy chains of Fab shown in light and dark green, respectively, and the complementarity-determining regions (CDRs) shown in blue (CDR L1), black (CDR L2), orange (CDR L3), pink (CDR H1), green (CDR H2), and yellow (CDR H3). The gp120 is colored in white. The D (S274 – T283), V5 (T455-N465) and the CD4 binding (Q362-G372) loops are colored in cyan, violet and magenta, respectively. (B) Structural comparison of N60P23 Fab-gp12093TH057 coree complex and VRCO1-gp12093TH057 coree (PDB code 3NGB) complexes. Complexes were aligned based on the gp120 and are shown as the ribbon diagrams. The light and heavy chains of VRCO1 Fab are shown in light and dark cyan and the CDRs are colored as in N60P23 complex. (C) N60P23 and VRCO1 epitope footprints. N60P23/VRCO1 contacts on gp12093TH057 coree are highlighted in light green/cyan (light chain) and dark green/cyan (heavy and both chains) on the gp120 surface. (D) N60P23 and VRCO1 Fabs and gp12093TH057 coree contact residues on the primary sequence of the Fabs and gp12093TH057 coree, respectively. Residues contributing to the Fabs and gp12093TH057 coree are highlighted and contacts as defined by a 5 Å cutoff are marked above the sequence. Side chain (+) and main chain (−) contacts are colored based on contact type; hydrophobic in blue, hydrophilic in green, or both in black. Framework and complementary-determining regions are as indicated in the alignment. N60P23 binds within the CD4BS of gp120 engaging the CD4-binding loop as well as loops D and V5 also known to interact with the CD4 receptor and the CD4-binding site antibody VRC01. It recognizes the gp120 antigen with striking similarities to VRC01 with epitope footprint almost entirely overlapping the gp120 surface involved in VRC01 Fab-CD4-gp12093TH057 coree complex and utilizing the same CDR contacts. The close structural similarity of the complex interfaces is reflected in relatively low root mean square deviation (RMSD) value of 0.7 Å for the Cα atoms of the Fab variable domains and the gp120 coree.
A) The overall structure of the N49P7 Fab-gp12093TH057 coree complex is shown in the center with the molecular surface displayed over the Fab molecule to highlight the shape of the formed antigen binding site. The complementarity-determining regions (CDRs) are shown in green (CDR L1), blue (CDR L3), black (CDR H1), yellow (CDR H2), and red (CDR H3). The gp120 outer and inner domains are colored in black and gray, respectively. The D (S274 – T283), E5 (F353 – T358), V5 (T455 – N465) and the CD4 binding (Q362 – G372) loops are colored in cyan, orange, violet and magenta, respectively. The unique features of the gp120 antigen recognition mediated by N49P7 are shown (from 1 to 6) and discussed in details in the blow up view figures: (1) ‘Super short’ CDR L1- The CDR L1 of N49P7 consists of 8 amino acids (aa), 1 and 3 aa shorter than the CDRL1 of VRC01 and N6, respectively. The short CDR L1 avoids steric clashes with loop D and Loop E and permits the accommodation of complex and bulky glycans linked to N276 and N355 of gp120. The figure shows the overlay of the N49P7 Fab-gp12093TH057 coree and the N6 Fab-gp12093TH057 coree complex (Huang et al., 2016). Structures were superimposed based upon gp120 to show differences in the length and position of CDR L1 relative to the gp120 antigen (colored dark green for N49P7 and light green for N6) (2) Unpaired cysteine (C36) in a framework 2 (FW2) of the light chain. Both κ (as seen in N6) and λ light chains have a tyrosine at position of 36 of FW2. Replacement of this bulky residue by cysteine changes the packing of the hydrophobic core formed at the variable light (VL) and heavy (VH) domain interface of N49P7 as compared to N6. The result of the unique packing is a difference in the relative orientations (rotation/tilting) of VL and VH observed in N49P7 relative to N6. The figure shows the VL - VH core packing of N49P7 and N6. Fabs were superimposed based of VH domain. N6 is colored light and dark blue for light and heavy chain, respectively (CD1 L1 as in panel 1). (3) Rotation/tilting of the light chain- the assembly of VL and VH domains of N49P7 forms an asymmetric antigen binding side, wildly open from the VL side and protruding from the VH side. The open access from the VL side is due to the rotation/tilting of the light chain relative to the heavy chain as described in (2). The figure shows the same superimposition as in panel 2. The VL of N49P7 is rotated approximately 12 degrees (measured at the VL N-termini) away from Loops D and V5 of gp120. This generates a 5Å distance between the VL N-termini of N49P7 and N6. The wild opening of the VL side of the P7 antigen binding site combined with short CDRL1 allows N49P7 to accommodate different lengths of the highly variable loops D, E and V5. Changes in the length of gp120 loop V5 and the length (and glycosylation status) of loop E that cause steric clashes with an antibody CDRL1 were described previously as mechanisms of HIV-1 resistance to VRC01-class antibodies. (4) Normal length CDRH3 that contacts to Loop D and the inner domain of gp120. The CDRH3 of N49P7 consists of 19 aa that contact the conserved Loop D of gp120 and reach deeply into the gp120 inner domain (compared to the 13 aa –long CDRH3 of N6). The CDRH3 contributes 274 Å2 BSA to the complex interface (28.6% of the BSA of the whole Fab). A network of interactions is formed which includes hydrogen bonds formed by Lys of CDRH3 (of a S100GK motif) and residues of the α1 helix of Layer 2 and the α7-helix of Layer 3 of the inner domain. (5) ‘Bypassing’ the Phe43 cavity. The CDRH2 of N49P7 is a major anchor point in binding to the gp120 antigen contributing 489 Å2 BSA to the complex interface (51% of the BSA of the whole Fab) but it has a Glycine at position 54 (Tyrosine in N6) thus it is unable to anchor deeply inside the Phe43 cavity of the CD4BS. Instead N49P7 makes important contacts to inner domain mediated trough M53 and Q56 which compensate for lack of direct contacts to residues of the Phe43 cavity. Overall N49P7 contributes 207 Å2 of its buried surface area (BSA) to the gp120 inner domain which is the highest among N6 and the VRC01 Abs class (Table S6). The figure shows a blow up view of the CDRH2 –gp120 interface of N49P7 and N6. CDRH2 is colored light and dark yellow for N49P7 and N6, respectively. (6) The P60W motif in CDRH2. N49P7 is able accommodate changes in length and conformation of the gp120 V5 loop through a P60W motif of its CDRH2 which forms the framework for an interaction network with the base of the V5 loop. Figure shows a close-up view of the interaction of CDRH2 of N49P7 and N6 with Loop V5. The P60W (N49P7) and G60GG (N6) motifs are highlighted. B) 2Fo-Fc electron density map around gp120 inner domain contacting CDR H1-3 of N49P7 Fab contoured at 1.0σ. View and residues shown are as in Figure 6, panel B.