

Abstract
Purpose
We investigated the mechanisms of how TGF-β pathway is activated by chemotherapeutics and whether a novel TGF-β trap called RER can block chemotherapeutics-induced TGF-β pathway activation and enhance their anti-tumor activity in gynecological cancer.
Experimental Design
An unbiased bioinformatic analyses of differentially expressed genes in thirty-one ovarian cases due to chemotherapy was used to identify altered master regulators. Phosphorylated Smad2 was determined in thirty paired cervical cancer using immunohistochemistry. Furthermore, the effects of chemotherapeutics on TGF-β signaling and function, and the effects of RER on chemotherapy-induced TGF-β signaling were determined in gynecological cancer cells.
Results
Chemotherapy-induced transcriptome alteration in ovarian cancer was significantly associated with TGF-β signaling activation. Chemotherapy was found to activate TGF-β signaling as indicated by phosphorylated Smad2 in paired cervical tumor samples (pre- and post-chemotherapy). Similar to TGF-β1, chemotherapeutics were found to stimulate Smad2/3 phosphorylation, cell migration, and markers related to epithelial-mesenchymal transition (EMT) and cancer stem cells (CSC). These TGF-β-like effects were due to the stimulation of TGF-β1 expression and secretion, and could all be abrogated by TGF-β inhibitors including a novel TGF-β trap protein called RER both in vitro and in vivo. Importantly, combination treatment with RER and cisplatin showed a higher tumor inhibitory activity than either agent alone in a xenograft model of ovarian cancer.
Conclusions
Chemotherapeutics can stimulate TGF-β1 production and consequently enhance TGF-β signaling, EMT, and CSC features resulting in reduced chemo-sensitivity. Combination therapy with a TGF-β inhibitor should alleviate this unintended side effect of chemotherapeutics and enhance their therapeutic efficacy.
Keywords: TGF-β, inhibitors, chemotherapeutics, ovarian cancer, cervical cancer
Introduction
Gynecological cancers, including cervical cancer and ovarian cancer, are leading causes of cancer-related death in women worldwide. Ovarian cancer is the most lethal gynecologic malignancy with an estimated 238,700 new cases and 151,900 deaths in 2012 worldwide (1). Cervical cancer is the fourth most commonly diagnosed cancer and the fourth leading cause of cancer death among women worldwide, which accounts for nearly 3.7% of the total newly diagnosed cancer cases and 3.3% of the total cancer deaths in 2012 (1). Chemotherapy remains the main treatment option for ovarian cancer and advanced/relapsed cervical cancer. However, the response to chemotherapeutic treatment is often inadequate, with a subpopulation of tumor cells surviving chemotherapy. Consequently, relapse is a common event and these recurrent tumors are associated with increased aggressiveness, resistance to various chemotherapeutics, and a high mortality rate. Thus, chemotherapy resistance remains a major therapeutic hurdle in the management of gynecologic malignancies and novel strategies to enhance antitumor activity of chemotherapeutics agents are urgently needed.
TGF-β belongs to a family of homodimeric peptide growth factors that regulate a wide variety of cellular processes, including proliferation, differentiation, invasion, immunosurveillance and stem cell maintenance (2). There are three mammalian isoforms of TGF-β ligand, named TGF-β1, -β2, and -β3 with significant homology and similarities in function. All three isoforms are secreted in a latent form and are activated via various mechanisms (3). Active TGF-β binds to three different cell surface receptors called type I (TβRI), type II (TβRII), and type III (TβRIII) receptors (4). TβRI and TβRII are serine/threonine kinase receptors, whereas TβRIII, also known as betaglycan (BG), serves as an accessory ligand-binding receptor. TGF-β ligand signals through TβRII, which recruits and activates TβRI kinase through transphosphorylation. The activated TβRI phosphorylates intracellular Smad2 and Smad3, which then interact with Smad4 protein to regulate gene expression in the nucleus (5). Over-expression of TGF-β1 has been reported in various gynecological malignancies including ovarian cancer and cervical cancer (6-9). In patients with ovarian cancer and cervical cancer, elevated levels of TGF-β1 were associated with tumor progression, treatment resistance, and poor outcome (6, 9, 10). These pro-malignant functions of TGF-β can be collectively attributed to its unique abilities in modulating tumor microenvironment and stimulating stem cell-like features in tumor cells (8, 11).
Given its multifaceted role in driving malignant progression, it is important to identify modalities that can activate TGF-β signaling and to develop effective means for the blockade of TGF-β signaling. We and others have previously shown that some chemotherapeutic agents, such as doxorubicin, paclitaxel, and irradiation can activate TGF-β signaling, which in turn causes therapy resistance (12-14),(15). As a result, administration of TβRI kinase inhibitor or TGF-β neutralizing antibody significantly enhanced anti-tumor activity of chemo- and radiation therapy in mouse models of breast cancer (12, 13, 15). However, the underlying mechanisms of how TGF-β pathway is activated by chemotherapeutics causing chemo-resistance are not well understood. Furthermore, whether the stimulation of TGF-β signaling during chemotherapy is unique to certain drugs such as doxorubicin and paclitaxel or more universal to various classes of chemotherapeutic drugs remains to be determined.
In the current study, we investigated the effects of four chemotherapeutic agents commonly used in gynecological cancers, including cisplatin, paclitaxel, doxorubicin, and camptothecin, on TGF-β signaling in cervical and ovarian cancer cells because the TGF-β signaling pathway was found to be strongly activated pathways by chemotherapy in ovarian cancer and cervical cancer. Using a novel TGF-β trap protein, we demonstrate that these drugs activate TGF-β signaling by stimulating TGF-β1 production via transcriptional and/or post-transcriptional mechanisms and the sequestration of TGF-β by the novel TGF-β trap protein was highly effective in blocking the TGF-β-like activities of the drugs in vitro and in vivo.
Materials and Methods
Ethics statement
This study was approved by the ethical committee of the Second Affiliated Hospital of Wenzhou Medical University and conducted according to the Helsinki declaration. Informed consent was obtained from all subjects prior to participation in the study. Cervical tissue sections used for this study were cut from leftover tissue blocks from consented patients, who were treated with neoadjuvant chemotherapy. All animal experiments were conducted following appropriate guidelines. They were approved by the Institutional Animal Care and Use Committee and monitored by the Department of Laboratory Animal Resources at the University of Texas Health Science Center at San Antonio.
Patients and tissue specimens
Patients with Stage IB2 or IIA2 (bulky, primary tumor >4cm in diameter) cervical cancer seen at the Second Affiliated Hospital of Wenzhou Medical University, Wenzhou, China, between January 2007 and August 2014, were recruited for a pilot study aimed to identify predictive biomarkers for responses to neoadjuvant chemotherapy. A total of 30 subjects were enrolled with a median age of 44 years (range, 25-59 years). None of the patients had received any anti-tumor therapy before the specimen collection. Paired tumor samples from each patient were obtained during cervical biopsy (pre-chemotherapy) or surgery (post-chemotherapy).
All eligible patients received one or two courses of cisplatin-based neoadjuvant chemotherapy, as previous described (16): cisplatin, 60 mg/m2 on day 1; 5-fluorouracil, 750 mg/m2 on day 1; mitomycin (8 mg/m2) on day 1 for one or two courses, every 28 days. All of these chemotherapeutics were administrated via uterine artery injection. All patients were treated with radial hysterectomy and bilateral pelvic lymphadenectomy two to three weeks after completion of the neoadjuvant chemotherapy regimen as described previously (17).
Gene expression profiles analysis
Gene expression profiles of malignant carcinoma samples from ovarian cancer patients were obtained from GEO (GSE7463) (18). Only the carcinoma samples were included in current study. Probes were normalized by quantile normalization with preprocess Core in R (19). Differential gene expression analysis was performed by comparing samples from ovarian cancer patients treated with neo-adjuvant chemotherapy (source_name_ch1 as Cancer) to samples from patients without chemotherapy treatment (source_name_ch1 as Carcinoma), controlling tumor stage and histology, by limma in R (20). Significantly differentially expressed genes were defined as those with FDR below 0.05. Upstream regulator prediction was analyzed through the use of Qiagen's Ingenuity Pathway Analysis (IPA, Qiagen, Redwood City,CA) with default setting. Gene Ontology of Biological Processes was analyzed in DAVID Bioinformatics Resources 6.7 (21, 22). Heatmap of significantly differentially expressed genes was plotted with heatmap.2 (gplotsin R) (23).
Cell cultures and reagents
Human cervical cancer cell lines (HeLa and C-4I) and ovarian cancer cell lines (OVCAR-3 and TOV-21G) were originally obtained from the American Type Culture Collection (ATCC). All four cell lines were authenticated with the DNA markers used by ATCC. Our cell lines were all stocked mycoplasma free in liquid nitrogen tanks and used for experiments for 4-5 months with at least one additional test for mycoplasma. The mycoplasma-free cultures were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) and 0.1% penicillin in 5% CO2 at 37°C. For drug treatment, cisplatin, doxorubicin, paclitaxel, and camptothecin were purchased from Sigma and dissolved in water (cisplatin) or dimethylsulfoxide (DMSO) (doxorubicin, paclitaxel and camptothecin). Their aliquots were stored at -80°C.
Preparation of TGF-β inhibitors
RER was designed and synthesized in our laboratory (24). Briefly, RER was produced by transient transfection of HEK293F cells grown in suspension in Freestyle 293 medium at 8% CO2, 80% humidity, and rotating at 80 rpm (Infors HT, Laurel, MD). The proteins were purified from the conditioned medium seven days post-transfection using a combination of metal affinity and size exclusion chromatography as previously described (24). HTS466284 (HTS) used in our study was reported previously to be an ATP-competitive inhibitor of the TGF-βRI kinase domain (25, 26). The chemical name of the compound is [3-(pyridine-2yl)-4-(4-quinonyl)]-1Hpyrazole, which was synthesized according to the procedure described by Sawyer et al (25).
Western immunoblotting analysis
Both cell and tissue samples were homogenized and lysed in Laemmli buffer with a cocktail of protease inhibitors. The total protein concentrations were quantified by the BCA protein assay (Thermo Scientific, Rockford, IL). Equal amounts of total protein were resolved by SDS PAGE, transferred to a nitrocellulose membrane under constant voltage and blocked with TBST containing 5% non-fat dried milk. Primary antibodies and secondary antibodies were diluted in TBST or 3% non-fat dried milk and applied with a washing step in between. Proteins were detected using the Amersham ECL Western blotting detection kit (GE Healthcare, Piscataway, NJ). Primary antibodies used including: phosphorylated Smad2 (P-Smad2) (Cell signaling, Danvers, MA), P-Smad3 (Abcam, Cambridge, MA), T-Smad2 (Cell signaling, Danvers, MA), T-Smad3 (BD Pharmingen, San Diego, CA), Snail (Santa Cruz Biotechnology, Santa Cruz, CA), and E-cadherin (Santa Cruz Biotechnology, Santa Cruz, CA).
RNA extraction and quantitative real-time PCR
RNA extracted from the cultured cells or xenograft tumors was treated with DNase1 (Invitrogen, Grand Island, NY) to remove genomic DNA contamination. Total RNA (2μg) was reverse-transcribed into cDNA using random primers and M-MLV reverse transcriptase from Invitrogen Life Technology (Grand Island, NY). Quantitative real-time PCR (qRT-PCR) was performed using Power SYBR Green PCR Mix from Life Technologies. All primers used in this study were designed by Primer Blast of NCBI and synthesized by Integrated DNA Technologies (Coralville, IA). Primer pair specificity was determined by generation of a single peak for dissociation curve (melting curve) at the end of RT-PCR cycling program. PRL27 was used as the internal control.
ELISA assay
OVCAR-3 cells were grown to confluence in the complete medium, washed twice with PBS, and then incubated with a serum-free medium containing 10nM paclitaxel (PTX), 250nM camptothecin (CPT), 10μM cispaltin (DDP), 100nM doxorubicin (Doxo), or no drug for 24h. The conditioned medium was collected, filtered using a 0.45-μm syringe filter, and frozen at -80°C until ready for use. Total and active TGF-β1 present in the conditioned medium was quantified using a sandwich ELISA kit from R&D Systems (Cambridge, MA) following the manufacturer's protocol.
Immunohistochemistry
Immunohistochemistry staining was performed on paraffin-embedded 4μm tissue sections and mounted on poly-L-lysine-coated slides. Briefly, after deparaffinization in xylenes and rehydration through graded ethanol solutions, antigen retrieval was performed by submerging the sections into a sodium citrate solution (10mM, pH6.0) or a EDTA solution (1mM, pH8.0) at 95°C for 15min or 30min, in a microwave oven. The tissue sections were then treated with 3% hydrogen peroxide in methanol to suppress the endogenous peroxidase activity. Tissue sections were then incubated with antibodies to P-Smad2 (Cell signaling, Danvers, MA), T-Smad2 (Cell signaling, Danvers, MA), slug (Cell signaling, Danvers, MA), CD133 (Miltenyi Biotec, San Diego, CA), and CD49f (LifeSpan Biosciences, Seattle, WA) at 4° overnight. After washing, the sections were incubated with pre-diluted secondary antibody (BD Pharmingen, San Diego, CA), followed by further incubation with 3,3-diaminobenzidine tetrahydrochloride (DAB). Finally, the slides were counterstained with hematoxylin and mounted in an aqueous mounting medium. Appropriate positive and negative controls were stained in parallel. For negative controls, primary antibodies were replaced with PBS.
Evaluation of immunoreactivity
Immunochemistry staining of total Smad2 and P-Smad2 was assessed by two independent observers, who were blinded to the study. Expression of the two markers was determined by an individual labeling score combining the percent stained cells and the staining intensity of positive cells (27). Intensity of stained cells was graded semi-quantitatively into four levels as following: 0 (no staining); 1 point (weak staining : light yellow); 2 points (moderate staining : yellow brown) and 3 points (strong staining: brown). The percentage was scored as following: 0 (0 to 5%), 1 point (6% to 24%), 2 points (25% to 49%), 3 points (50% to 74%), and 4 points (75% to 100%). The intensity score and the fraction of positive cell scores were multiplied for each marker to derive the immunoreactive score.
Flow Cytometry
OVCAR-3 cells were treated with DDP (1μM) in the presence or absence of RER (40nM) for 4 days. The medium was changed every alternate day with the addition of DDP and/or RER. The harvested control and treated cells, after trypsinization, were stained for flow cytometry at a concentration of 100,000 cells per 100μl of buffer (PBS pH 7.4, 2% PBS, 2mM EDTA) containing conjugated antibodies (2mg/100 cells) against CD133 (MiltenyiBiotec, San Diego, CA) and CD44 (BD Biosciences, San Diego, CA) at room temperature for 1hr. The analysis of stained cells was carried out using FACSAria flow cytometer (Becton Dickinson) at the core imaging facility of the UT Health Science Center at San Antonio, Texas.
Cell migration assay
Cell migration assays were performed in 24-well transwells with 8-μm pore polycarbonate membranes (BD Biosciences, San Diego, CA). Cells at a density of 20,000∼40,000 cells/well in serum-free medium with or without treatment were seeded in the upper insert in triplicates. Complete medium with or without treatment was added in the lower chamber. After 18h for OVCAR-3 cell and 6h for HeLa cell, the cells that did not migrate across the membrane were removed with a cotton swab and the migrated cells were stained with the Hema 3 Stain 18 kit (Fisher Scientific, Waltham, MA) according to the manufacturer's protocol. Migrated cells were counted under a microscope with 100× magnification.
Animal studies
Four-week-old female nude mice were used for in vivo animal experiments. The animals were housed under specific pathogen free condition. Exponentially growing OVCAR-3 cells (5 × 106 cells/120 μl/mouse) suspended in 50% Matrigel (Corning Life Sciences,Tewksbury, MA) in cold PBS were injected subcutaneously into the back of the mice. After tumor cell inoculation for one week, growing tumors were observed and their size was recorded twice a week. The length and width of each tumor were measured using a caliper, and the volumes were calculated by the following formula: volume (mm3) = length × width × width/2. After another 2-3 weeks, mice with tumor burden≥100mm3 in volume were ranked and divided into 6 groups (5 mice for each group) with matched mean tumor volumes and treated as follows: control (normal saline), RER (5mg/kg), low dose of DDP (2.5mg/kg), high dose of DDP (5mg/kg), low dose of DDP (2.5mg/kg) and RER (5mg/kg), high dose of DDP (5mg/kg) and RER (5mg/kg). RER was administered daily and DDP was given once a week by intraperitoneal injection. After treatment for 29 days, xenograft tumors were isolated from mice. A portion of the tumors tissue was fixed in 4% paraformaldehyde for histological study, and the rest were frozen for other experiments.
Statistical analyses
Two-tailed Student's t-test was used to compare the means of two groups. One-way analysis of variance with Tukey-Kramer post hoc test was used for analyzing data when means from more than two groups were compared. Results are expressed as mean ± sem. P < 0.05 was considered to be statistically significant.
Results
Chemotherapy-altered transcriptomes in ovarian cancer is associated with TGF-β pathway activation
Gene expression profiles of malignant carcinoma samples from ovarian cancer patients were obtained from GEO (GSE7463) (18). Comparison of samples from ovarian cancer patients with chemotherapy treatment to samples without chemotherapy treatment identified a total of 790 upregulated and 929 downregulated probeSets (Table S1). These differential expression probes correctly cluster patients based on whether they have undergone chemotherapy or not, except for two patients treated with chemotherapy being clustered into patients without chemotherapy (Figure 1A). These genes are significantly enriched in Gene Ontology (GO) terms associated with cell cycle regulation (Figure 1B, Table S2), which is the expected effect of chemotherapy. Next we examined the potential upstream regulators of these differentially expressed genes to identify potential master regulators mediating the effects of chemotherapy, using Upstream Regulator Prediction from Qiagen's Ingenuity Pathway Analysis (IPA, Qiagen, Redwood City, CA). As indicated by the results in Figure 1C and Table S3, it is not a surprise that TP53 was the top activated upstream regulator in response to chemotherapy with a significant positive z-score and lowest P value, which is consistent with the observation made by Morenoand colleagues (18). Regulators associated with the estrogen pathway, including beta-estradiol and ESR1, in the Table S3, were predicted to be most significantly inhibited with negative z-scores, suggesting a unique and interesting response to chemotherapy in ovarian cancer. Relevant to this study, TGF-β1 was the second most significantly activated regulator upon chemotherapy (Figure 1C and Table S3). Out of the genes corresponding to the 1,719 probeSets, which are significantly altered by chemotherapy, 98.57% of them are regulated by TGF-β1 in various cellular compartments as shown in Supplementary Figure 1, suggesting TGF-β signaling pathway is an important master regulator in chemotherapy response.
Figure 1.
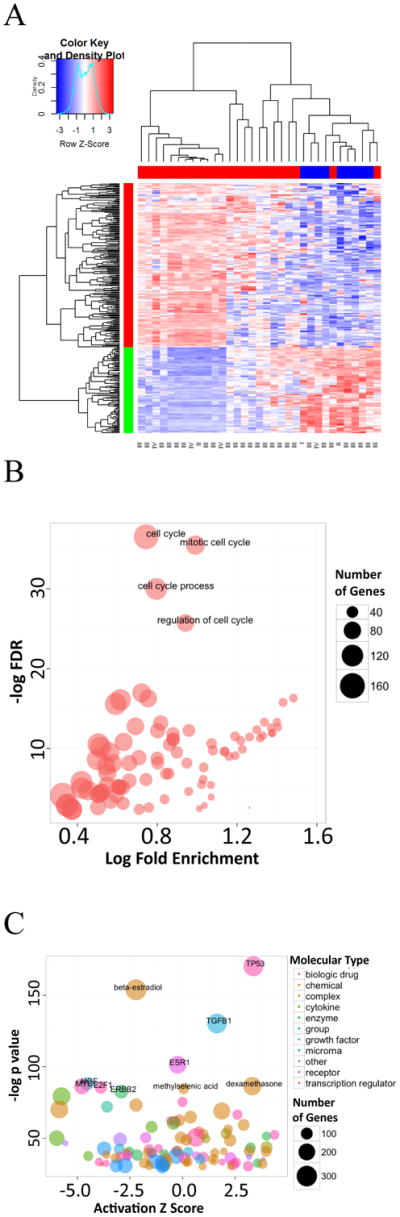
(A) Heatmap of relative expression of differentially expressed genes comparing 24 chemotherapy treated patients (labeled as “Cancer” in original GEO dataset) to 9 non-treated patients (labeled as “Carcinoma” in original GEO dataset). The color bar on the top indicates sample types (red for chemotherapy treated and blue for non-treated). The color bar on the left indicates genes that are upregulated (red) or downregulated (green) comparing chemotherapy treated to non-treated. The stages of each tumor sample are labeled at the bottom of the heatmap. The red color bars on the right indicate the genes that are regulated by TGF-β. (B) Gene Ontology analyses on Biological Processes that are enriched in chemotherapy response. Only terms with FDR<0.1 are shown in the plot. The size of circle represents the number of differentially expressed in genes that are enriched in this term. (C) Upstream regulators prediction from IPA demonstrates key regulators in chemotherapy response. Only regulators with −log p value above 30 are shown in the plot. A regulator with a positive activation z score suggests it is being activated by chemotherapy and positively correlated with phenotype, vice versa. The size of circle represents the number of differentially expressed genes that are enriched in this term.
TGF-β signaling is activated after chemotherapy in cervical cancer
Due to the lack of a similar gene expression profiling study in cervical cancer, We explored the effect of chemotherapy on TGF-β signaling in cervical cancer by examining phosphorylated Smad2 (P-Smad2) and total Smad2 (T-Smad2) levels in 30 matched primary cervical cancer specimens before and after cisplatin-based neoadjuvant chemotherapy using immunohistochemistry. As shown in Figure 2, P-Smad2 and T-Smad2 protein expression in the tumor tissues were detected mainly in the nucleus with relatively weaker staining in cytoplasm. Pre-chemotherapy cervical cancer tissues consistently showed weak positive staining of P-Smad2, while post-chemotherapy tissue consistently showed moderate or intense positive staining. Using a Wilcoxon test, the immunoreactive score, which reflects total staining extent and intensity in both nuclei and cytoplasm, for P-Smad2 expression was significantly increased in post-chemotherapy samples compared with pre-chemotherapy samples (P < 0.05). With regard to T-Smad2, no significant difference was observed between pre-chemotherapy and post-chemotherapy cervical samples. These data suggest that TGF-β signaling is activated after chemotherapy in cervical cancer.
Figure 2.

Immunochemistry staining of P-Smad2 and T-Smad2 in paired pre- and post-chemotherapy samples of cervical tumors (×400). P-Smad2 and T-Smad2 proteins are primarily localized in nucleus. Their staining intensity and frequency in each tissue slide were scored as described in Materials and Methods and plotted. Scale bar = 200 μm.
Chemotherapeutic agents activate TGF-β signaling in human ovarian and cervical cancer cells
To confirm the role of TGF-β signaling in response to chemotherapy, we initially investigated TGF-β signaling activity in cancer cell lines treated with various chemotherapeutic agents in vitro. Two cervical cancer cell lines, HeLa and C-4I, and two ovarian cancer cell lines, OVCAR-3 and TOV-21G, were used. Four chemotherapeutic agents, cisplatin (DDP), paclitaxel (PTX), doxorubicin (Doxo) and camptothecin (CPT), which are commonly used as therapeutics for gynecological malignancy, were investigated in this study. The four cell lines were treated with 10 μM DDP, 10 nM PTX, 100 nM Doxo, or 250 nM CPT for 1h, 6h, or 24h. Because TGF-β signal is mediated through the phosphorylation of intracellular Smad2 and Smad3 proteins thereby affecting gene expression in the nucleus, we measured the levels of phosphorylated Smad2 and Smad3 to evaluate the activation of TGF-β signaling pathway by the drugs. As showed in Figure 3, all four drugs stimulated phosphorylation of Smad2 and Smad3 with varying efficacy and time kinetics in cervical cancer cells (Figure 3A, 3B) and ovarian cancer cells (Figure 3C, 3D). The density of each P-Smad2 or P-Smad3 was normalized to its corresponding T-Smad2 or T-Smad3, respectively, and presented in Supplementary Figure 2.
Figure 3.

Activation of TGF-β signaling by chemotherapeutic agents in human cervical cancer cells and ovarian cancer cells. Cervical cancer cell lines HeLa (A) and C-4I cell (B), and ovarian cancer cell lines OVCAR-3 (C) and TOV-21G (D) were treated with 10μM cisplatin (DDP), 10nM paclitaxel (PTX), 100nMdoxorubicin (Doxo) or 250nMcamptothecin(CPT) for 1h, 6h or 24h. The cells were harvested and their extracts were used for western immunoblotting for the detection of phosphorylated Smad2 (P-Smad2) and of phosphorylated Smad3 (P-Smad3). Total Smad2 (T-Smad2) and total Smad3 (T-Smad3) were used to verify equal sample loading.
TGF-β inhibitors block chemotherapeutics-induced phosphorylation of Smad2/3
Next, we set out to determine whether chemotherapeutics-activated Smad2 and Smad3 phosphorylation can be blocked by TGF-β inhibitors. Two TGF-β inhibitors, HTS466284 (HTS) and RER, were used. HTS466284 (HTS) is an ATP competitive inhibitor of TGF-β type I receptor kinase (25, 26). RER, a novel recombinant trivalent TGF-β trap protein comprised of the endoglin (E) domain of TβRIII flanked by the extracellular domain of TβRII (R), is designed and synthesized in our laboratory (24). RER showed similar or more potent activity than the kinase inhibitor in blocking TGF-β1-induced Smad2 and Smad3 phosphorylation in HeLa and C-4I cells at the concentrations used (Figure 4A and Supplementary Figure 3). All four cell lines shown in Figure 4 were pre-treated with or without HTS (100 nM) or RER (40 nM) for 1h followed by treatment with 10 μM DDP, 10 nM PTX, 100 nM Doxo or 25 0nM CPT for 6h. Consistent with the data in Figure 3, the levels of phosphorylated Smad2 and/or Smad3 protein were in general increased after treatment with the indicated drugs. This action was blocked by both HTS and RER in most cases, suggesting that the drugs appeared to increase extracellular active TGF-β levels, which was neutralized by RER (Figure 4).
Figure 4.

Blockade of chemotherapeutics-induced phosphorylation of Smad2 and Smad3 by TGF-β inhibitors. Four cell lines were pre-treated with or without RER (40nM) or HTS (100nM) for 1h followed by the treatment with 10μM cispaltin (DDP) (A), 10nM paclitaxel (PTX) (B), 100nM doxorubicin (Doxo) (C), or 250nM camptothecin (CPT) (D) for 6h in HeLa, C-41 and TOV-21G cells, or 24h in OVCAR-3 cells. The cell lysates were used for western immunoblotting for the detection of the levels of P-Smad2 and P-Smad3. T-Smad2 and T-Smad3 were used to verify equal sample loading.
Chemotherapeutics increase cancer stem cell markers and population
Because TGF-β signaling has been widely reported to increase cancer stem cell (CSC) population, we hypothesized that chemotherapeutics-induced TGF-β signaling might also lead to increased CSCs. CD133, CD44, CD49f and ABCG were previously reported as CSC markers in ovarian cancer and CD133+CD49f+ cells sorted from the OVCAR-3 ovarian cancer cell line were shown to have CSC features (28-30). Upon DDP treatment, OVCAR-3 cells displayed higher mRNA levels of stem cell markers including CD44, CD133 and ABCG, as compared to untreated cells, which were dampened by RER or HTS treatment (Figure 5A). We confirmed the increase of stem cell population with DDP treatment by quantifying CD133+CD44+ OVCAR-3 cells with flow cytometry. Interestingly, the majority of our OVCAR-3 cells express CD133, but not CD44 (Figure 5B). Treatment with 1 μM DDP increased CD133+CD44+ cells by 20-fold in comparison to the untreated control. Addition of RER to the DDP treatment decreased CD133+CD44+ cells from 15.6% to 3.1%. A live/dead gate was presented in Supplementary Figure 4. These data indicate that DDP treatment can increase stemness properties of OVCAR-3 cells and this induction is partly dependent on TGF-β signaling.
Figure 5.

Increased stem cell markers and population in OVCAR-3 cell line by DDP and induction of promoted migration and epithelial-mesenchymal transition (EMT) by chemotherapeutics. (A) OVCAR-3 cells were treated with TGF-β1 (80pM), DDP (1μM), HTS (100nM) and RER (40nM) alone or in combination for 4 days. Total RNA extracted from the cells was used for the measurements of CD44, CD133 and ABCG transcript levels by qRT-PCR. Relative mRNA level was obtained by normalizing the Ct value of each gene transcript with the Ct value of RPL27 transcript. Data are presented as mean±SEM. *P< 0.05; ** P< 0.01; *** P< 0.001 with one-way ANOVA and Tukey-Kramer post hoc test. (B) OVCAR-3 cells were treated with DDP (1 μM) and RER (40 nM) alone or in combination for 4 days. Cells were trypsinized and analyzed by FACS for CD133/CD44 expression. (C) HeLa cells (20,000 cells/insert) and OVCAR-3 cells (30,000 cells/insert) were plated in 24-well cell migration inserts and then treated with 1 μM DDP, 100 nM Doxo, 250 nM CPT or 5 nM PTX in the left panel. In separate experiments, the cells were also treated with DDP or Doxo with or without HTS (100 nM) and RER (40 nM) shown in the right panels. Migration assay was terminated after 6h for HeLa cell and 18h for OVCAR-3 cell. Migrated cells in each insert were counted under microscope. Data presented are mean±SEM from triplicate wells. *P< 0.05; ** P< 0.01; *** P< 0.001 with one-way ANOVA and Tukey-Kramer post hoc test. (D) HeLa and C-41cells were treated with TGF-β1 (80 pM), DDP (1 μM), HTS (100 nM) and RER (40 nM) alone or in combination for 4 days. The cells were harvested and their extracts were used for Western immunoblotting for the detection of Snail and E-cadherin. GAPDH expression level was used to validate equal sample loading.
Chemotherapeutics induce EMT markers and promote migration
Epithelial to mesenchymal transition (EMT) is associated with acquisition of tumor stem-like properties (31-33). Furthermore, TGF-β is a potent inducer of EMT and plays an important role in tumor cell motility and migration (34). These raised the possibility that chemotherapeutics may act like TGF-β and have the potential to promote tumor metastasis by stimulating tumor cell motility and invasion via EMT. Therefore, we tested the effect of the drugs on the migration and EMT of the cervical cancer HeLa cell line and ovarian cancer OVCAR-3 cell line. DDP and Doxo treatment significantly promoted migration in both cell lines, which was blocked by TGF-β inhibitor RER and HTS (Figure 5C). CPT significantly promoted migration in HeLa cells, but not in OVCAR-3 cells. On the contrary, PTX inhibited migration in these two indicated cancer cell lines, likely due to its role in disrupting microtubule dynamics and consequently cell migration. With respect to EMT, we observed a decreased expression of the epithelial cell marker E-cadherin protein after TGF-β1 and DDP treatment, which was reversed by the addition of HTS or RER in HeLa and C-4I cell lines (Figure 5D). On the other hand, the mesenchymal marker snail was increased after the treatment of TGF-β1 or DDP in these cancer cell lines (Figure 5D). Like TGF-β1, DDP appears to transcriptionally stimulate E-cadherin and repress Snail as their mRNA levels were increased or decreased, respectively, by DDP in OVCAR-3 cells, which were again reversed by RER or HTS (Supplementary Figure 5). These results indicate that chemotherapeutics, especially DDP and doxorubicin, can stimulate EMT and tumor cell migration in a TGF-β dependent manner.
Chemotherapeutics stimulate TGF-β1 expression and production
Because RER blocks TGF-β signaling by neutralizing extracellular TGF-βs, the blockade of TGF-β-like activities of chemotherapeutics by RER indicated that the drugs likely increased extracellular TGF-β levels. To explore the mechanism of activation of TGF-β pathway by chemotherapeutics, two assays were performed. Using quantitative real-time PCR, we initially investigated the effect of the drugs on TGF-β1 and TGF-β2 mRNA expression in the OVCAR-3 cell line treated with the four drugs for 24h. The result showed DDP, Doxo, and CPT increased TGF-β1 transcript with no or very moderate effect on TGF-β2 transcript (Figure 6A). In contrast, PTX treatment showed no effect on either TGF-β1 or TGF-β2 transcript level. To determine whether chemotherapeutics stimulated TGF-β1 production and secretion or induced extracellular TGF-β1 activation, we initially treated confluent cultures of OVCAR-3 cell lines with or without the indicated drugs for 24 hours in a serum-free medium. The medium was then collected for the measurements of total and active forms of TGF-β1 with a sandwich ELISA kit from R&D Systems. As shown in Figure 6B, the secreted active and total TGF-β1 were significantly increased by all four chemotherapeutics. Thus, PTX increases TGF-β1 expression in a post-transcriptional manner while the other three drugs appear to increase extracellular TGF-β1 via both transcription and post-transcriptional mechanisms.
Figure 6.

Stimulation of TGF-β1 production and secretion in OVCAR-3 cells by chemotherapeutics. (A) qRT-PCR analysis for TGF-β1 and TGF-β2 mRNA expression in OVCAR-3 cell line after treatment with 10 μM DDP, 10 nM PTX, 100 nM Doxo, or 250 nM CPT for 24h. (B) ELISA assay of total TGF-β1 and active TGF-β1 secreted in OVCAR-3 cell conditioned medium. OVCAR-3 cell lines were plated in 6-well plates and treated with or without the indicated drugs for 24 hours in a serum-free medium. The medium was then collected for the measurements of total and active forms of TGF-β1 with a sandwich ELISA kit from R&D Systems. Data presented are mean ±SEM. *P<0.05; ** P< 0.01; *** P< 0.001 with one-way ANOVA and Tukey-Kramer post hoc test.
RER enhances anticancer effect of cisplatin in OVCAR-3 xenograft mouse model
Our in vitro studies demonstrated that chemotherapeutics increased extracellular TGF-β1 level and consequently activated TGF-β signaling and CSC enrichment in human cervical cancer, as well as ovarian cancer cells. To confirm these observations in vivo and further investigate whether blockade of TGF-β singling with the novel TGF-β trap, RER, can enhance cisplatin anti-tumor activity in vivo, a nude mouse xenograft model was established using OVCAR-3 cells. Mice bearing growing OVCAR-3 xenografts were divided into six groups with similar mean tumor volumes of greater than 100mm3 and were then treated with a low (2.5mg/kg weekly) or a high dose (5mg/kg weekly) of DDP, or RER (5 mg/kg daily), as single agent or in combination for 29 days. As showed in Figure 7A and 7B, while the treatment with RER alone showed no effect on tumor growth, the relative tumor volumes of the other treatment groups were significantly lower than that of the control at the end of the experiment. Treatment with RER and low dose DDP was more effective than the treatment with low dose DDP alone in inhibiting tumor growth (Figure 7A, B) and the relative tumor volumes at the last two measurements were statistically different (Figure 7A). Similar to the >50% reduction of the terminal tumor volume, we also observed >50% reduction of the terminal tumor weight (Figure 7B). However, the reduction in tumor weight was not statistically significant at P<0.05, which was apparently due to their larger coefficient of variation, possibly related to the difficulty in our ability to accurately separate tumors from their surrounding tissues resulting in the large variation in tumor weights. Similarly, RER appeared to also enhance the tumor inhibitory activity of the high dose DDP resulting in complete regression of two tumors although the tumor volumes between the two treatment groups were not statistically different (Figure 7A, B). These data suggested that RER appeared to enhance the anti-tumor activity of DDP by neutralizing DDP-induced pro-tumor activity of TGF-β. Indeed, low dose DDP treatment significantly increased the mRNA and active forms of TGF-β1 levels in the xenograft tumors, were reduced by the combination treatment with RER (Figure 7C). Consistently, RER treatment also reduced DDP-induced TGF-β signaling in tumors as shown by the reduction of phosphorylated Smad2 levels of tumor specimens from DDP-treated mice with Western immunoblotting analysis (Figure 7D). There were not enough tumor tissues from the high dose DDP group for us to perform these assays.
Figure 7.

Enhanced anticancer effect of cisplatin in OVCAR-3 xenograft mouse model by RER. (A) Tumor volumes were calculated using the formula: v=length×width×width×0.5. The volume of each tumor on each day of assessment was divided by the volume of the same tumor on the day of the initiation of the treatment to obtain the relative tumor volume. Each data bar represents the mean±SEM of five tumors. (B) Tumor volumes at the end of treatment. Data presented are mean±SEM. (C) qRT-PCR analysis of mRNA levels of TGF-β1 in xenograft tumor tissues is shown in the left panel. The data represent mean ±SEM of four tumors. ELISA data of active TGF-β1 levels in xenograft tumor tissue extracts are shown in the right panel. The data represent mean ±SEM of four tumors. (D) Western immunoblotting analysis of P-Smad2 and T-Smad2 in tumor tissues from three mice in each treatment group as indicated. The bar plots show the mean±SEM of T-Smad2-normalized P-Smad2 band intensity in the three tumors for each group. *P< 0.05; ** P< 0.01; *** P< 0.001. NS: not statistically significant (P>0.05). (E) IHC staining for slug, CD133 and CD49f in xenograft tumor sections of the experimental mice. The representative picture was randomly taken for each staining from tissue sections of three mice in each group. Scale bar, 200 μm. (F) qRT-PCR detects the relative abundance of snail, CD133, CD44 and ABCG in xenograft tumor tissues of the experimental mice. Mean ±SEM, n=3 or 4. *P< 0.05; **P< 0.01; *** P< 0.001.
RER blocks cisplatin-induced EMT and CSCs population in OVCAR-3 xenograft mouse model
EMT has been associated with increased malignancy, chemotherapy resistance, and poor prognosis in ovarian cancer (35). Recent reports indicated that EMT is involved in the maintenance and formation of stem-like cancer cells in ovarian cancer (31, 36). We next examined whether DDP treatment induced EMT and CSC markers, which can be inhibited by RER in the OVCAR-3 xenograft tumors. Immunohistochemical staining showed that DDP increased the expression of mesenchymal marker protein slug and stem cell markers, including CD49f and CD133 (Figure 7E). qRT-PCR data showed that the transcript levels of snail, CD44, CD133 and ABCG were increased by DDP treatment, and this could be attenuated by RER (Figure 7F). Thus, DDP increased the expression of EMT and stem cell markers, which was blocked by RER. These results indicate a beneficial combination effect on reducing aggressive cancer cell population in the xenograft tumors.
Discussion
In the present study, our results showed, for the first time, that chemotherapeutics agents stimulate TGF-β1 production and secretion, and consequently activate the TGF-β pathway, EMT, and CSC features resulting in reduced chemo-sensitivity in gynecological cancer cells and xenograft tumors. Additionally, we also provide evidence that TGF-β signaling blocked by a novel trivalent TGF-β receptor trap, RER, is an effective treatment approach and can enhance the efficacy of chemotherapeutics in vivo.
Since its discovery in the early 1980s, TGF-β signaling pathway has been extensively investigated as a key regulator in carcinogenesis (37, 38). Abnormal TGF-β signaling activation has been frequently observed in a variety of human malignancies, including cervical cancer and ovarian cancer, and shown to promote tumor progression and regulate chemo-sensitivity (6, 7, 9). Our results indicated that ovarian cancer displayed an increased RNA transcript of genes associated with TGF-β signaling after chemotherapy. Additionally, the expression of P-Smad2 was significantly up-regulated in post-chemotherapy cervical cancer tissues compared with pre-chemotherapy samples. These results suggest TGF-β signaling pathway plays an important role in response of chemotherapy. Our result is consistent with the study of Marchiniand colleagues (35). They reported that TGF-β signaling pathway was significantly up-regulated in the chemo-resistant/relapsing ovarian cancers compared to chemo-sensitive ovarian tumors. Along similar lines, using reverse phase protein array, Careyand and colleagues identified TGF-β signaling as an indicator for primary chemotherapy response in patients with advanced serous ovarian cancer (39). Collectively, these results support the notion that aberrant activation of TGF-β pathway is likely a potent mediator of chemo-resistance in ovarian cancer.
TGF-β1 is secreted in a latent form and activated via various mechanisms (3). The activation of TGF-β pathway often leads to tumor progression and drug resistance. Our previous studies showed that doxorubicin, an anthracycline drug widely used for breast cancer, activated TGF-β signaling in human breast cancer MDA-MB-231 and murine mammary cancer 4T1 cell (12). Similarly, Bhola et al reported chemotherapeutic drug paclitaxel increased TGF-β signaling in breast cancer cells (13). Nevertheless, whether these observations are universal in various types of cancer remains to be determined. Thus, four drugs commonly used for gynecological cancers, including paclitaxel, camptothecin, cisplatin, and doxorubicin, were included in the present study. We observed all of these four chemotherapeutics activated TGF-β pathway by stimulating phosphorylation of Smad2 and Smad3, which is consistent with the published data in breast cancer, indicating chemotherapy-associated activation of TGF-β pathway is a common event.
More recently, several lines of evidence suggest that TGF-β-mediated drug resistance may be largely due to its induction of cancer stem-like properties in carcinoma cells (13, 14). CSCs, also known as “tumor-initiating cells”, represent a small proportion of cancer cells, with the properties involved in drug resistance, metastasis and relapse of cancers (40). Ovarian cancer has been described as a stem cell disease recently (41). An increasing body of data has demonstrated a subpopulation of CSCs in ovarian cancer, which contributes to chemo-resistance and tumor relapse (29, 30, 41-44). We show here that chemotherapeutics treatment stimulated CSC-like properties as evidenced by increased expression of stem cell markers, such as CD44, CD133 and ABCG, when compared to untreated control ovarian cancer cells both in vitro and in vivo. In addition, chemotherapeutics increased the expression of EMT markers and this transformation has also been associated with acquisition of tumor stem-like properties (31-33). Since metastasis is an important characteristic feature of CSCs, chemotherapeutics-promoted migration provided additional evidence that chemotherapeutics increased CSC population. These results are consistent with the finding by Wiechert et al. that cisplatin was able to induce the CSC state as indicated by a GFP reporter driven by a NANOG-promoter in ovarian cancer (29).
Our finding that chemotherapeutics-induced CSC properties was associated with enhanced TGF-β signaling and could be abrogated by TGF-β inhibitors indicates that TGF-β signaling mediates this unwanted side effect of chemotherapeutics. In the present study, we report, for the first time, that chemotherapeutics agents stimulated TGF-β1 production and secretion in ovarian cancer cells. These data support our hypothesis that common chemotherapeutics may active TGF-β pathway by stimulating TGF-β1 production and secretion resulting in EMT and CSC expansion to cause drug resistance. Thus, combination therapy with a TGF-β inhibitor and chemotherapy should block drugs/TGF-β-induced EMT and CSC formation leading to enhanced inhibition of tumor growth and metastasis. While our study has shown that the chemotherapeutics can stimulate TGF-β1 expression at transcriptional and/or post-transcriptional levels, detailed mechanisms by which the different types of chemotherapeutics stimulate active TGF-β1 production remains to be elucidated. Given that TGF-β1 mRNA and/or protein levels were significantly increased with just 24 hours of drug treatment, we believe the drugs directly regulated TGF-β1 transcript and/or protein production instead of selecting cancer stem-like cells with higher levels of TGF-β1 transcript and/or protein, which was a mechanism suggested by Bhola and co-workers (13).
Over the past decade, various components of TGF-β signaling pathway have been explored for the inhibition of this pathway, including both intracellular and extracellular targets. TGF-β receptor kinase inhibitors are the most commonly used TGF-β inhibitor for preclinical and clinical studies. Several published studies have shown efficacy of TβRI kinase inhibitors in attenuating malignant properties of cancer cells in vitro and in vivo (45-48). However, these inhibitors have the possibility of inhibiting other kinases, which may result in undesirable “off-target” side effects. In this study, we report a novel trivalent TGF-β trap, RER, which sequesters extracellular active TGF-β. This trap was shown to potently block TGF-β binding to TβRII and antagonize TGF-β signaling in cultured epithelial cells at picomolar concentrations, and it showed better anti- TGF-β activities than a pan-TGF-β neutralizing antibody and the TβRI kinase inhibitor HTS in prostate cancer cells (24). The data in the present study showed that RER effectively blocked TGF-β/chemotherapeutics-induced Smad2/3 phosphorylation and inhibited TGF-β1/chemotherapeutics-stimulated EMT and CSCs expansion. Moreover, RER was in general more effective at blocking TGF-β/chemotherapeutics-induced Smad2/3 phosphorylation when compared with the TβRI kinase inhibitor. In addition, our in vivo data showed that the combination of cisplatin and RER enhanced the efficacy of cisplatin in inhibiting tumor growth in the OVCAR-3 xenograft model in comparison to single cisplatin treatment.
Collectively, the data presented in this study demonstrate that chemotherapeutics can activate TGF-β signaling by stimulating TGF-β1 production and secretion, resulting in EMT and CSC enrichment and decreased chemo-sensitivity in human ovarian cancer and cervical cancer cells. Combination therapy with the novel TGF-β trap RER and cisplatin neutralized cisplatin-stimulated TGF-β1 leading to more efficacious inhibition of ovarian cancer growth. Our studies shed light on an underlying mechanism of chemoresistance and potential utility of TGF-β traps for the treatment of gynecologic malignancies.
Supplementary Material
Translational relevance.
An unbiased bioinformatic analyses of a published gene expression study led us to identify TGF-β as the second most activated upstream regulator, after TP53, of differentially expressed genes after chemotherapy in patients with ovarian cancer. However, the mechanisms of how the TGF-β pathway is activated by chemotherapeutics and regulates chemo-sensitivity are not well understood. Therefore, we investigated the effects of a panel of chemotherapeutics on TGF-β signaling and function in several ovarian and cervical cancer cell lines. In the submitted manuscript, we report, for the first time, that chemotherapeutics can activate TGF-β signaling by stimulating TGF-β1 expression and secretion both in vitro and in vivo. Consequently, the chemo drugs induced EMT and cancer stem cell enrichment and decreased chemo-sensitivity in human ovarian cancer and cervical cancer cells both in vitro and in vivo. We also report, for the first time, that combination therapy with a novel TGF-β trap RER and cisplatin neutralized cisplatin-stimulated TGF-β1 leading to more efficacious inhibition of ovarian cancer growth. These results shed light on an underlying mechanism of chemoresistance and potential utility of TGF-β traps for the treatment of gynecologic malignancies.
Acknowledgments
This study was in part supported by NIH R01CA172886 awarded to L-Z.S. and A.H., and National Natural Science Foundation of China (81602305) and Medical and Technology Project of Zhejiang Province (No. 2016KYA141) to H.Z. Additional support was provided by the Flow Cytometry Shared Resource of the Cancer Therapy and Research Center, which is supported by the NIH NCI Cancer Center Support Grant P30 CA054174-17. H.Z. was supported by a fellowship from the Second Affiliated Hospital of Wenzhou Medical University. L.X. was in part supported by a fellowship from Xiangya School of Medicine, Central South University, Hunan, China. X.G. was in part supported by Cancer Research Training Program grants RP140105 and RP170345 from Cancer Prevention and Research Institute of Texas (CPRIT).
Footnotes
Conflict of interest: A.P. Hink, L. Sun, and C. Zwieb are listed as co-inventors on an issued patent on TGFb type II–type III receptor fusions that is owned by the University of Texas Health Science Center at San Antonio. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 3.Robertson IB, Rifkin DB. Unchaining the beast; insights from structural and evolutionary studies on TGFbeta secretion, sequestration, and activation. Cytokine Growth Factor Rev. 2013;24:355–72. doi: 10.1016/j.cytogfr.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–7. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 5.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–54. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamura S, Matsumura N, Mandai M, Huang Z, Oura T, Baba T, et al. The activated transforming growth factor-beta signaling pathway in peritoneal metastases is a potential therapeutic target in ovarian cancer. Int J Cancer. 2012;130:20–8. doi: 10.1002/ijc.25961. [DOI] [PubMed] [Google Scholar]
- 7.Hou F, Li Z, Ma D, Zhang W, Zhang Y, Zhang T, et al. Distribution of Th17 cells and Foxp3-expressing T cells in tumor-infiltrating lymphocytes in patients with uterine cervical cancer. Clin Chim Acta. 2012;413:1848–54. doi: 10.1016/j.cca.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Zhu H, Luo H, Shen Z, Hu X, Sun L, Zhu X. Transforming growth factor-beta1 in carcinogenesis, progression, and therapy in cervical cancer. Tumour Biol. 2016;37:7075–83. doi: 10.1007/s13277-016-5028-8. [DOI] [PubMed] [Google Scholar]
- 9.Fan DM, Tian XY, Wang RF, Yu JJ. The prognosis significance of TGF-beta1 and ER protein in cervical adenocarcinoma patients with stage Ib∼IIa. Tumour Biol. 2014;35:11237–42. doi: 10.1007/s13277-014-2110-y. [DOI] [PubMed] [Google Scholar]
- 10.Dickson J, Davidson SE, Hunter RD, West CM. Pretreatment plasma TGF beta 1 levels are prognostic for survival but not morbidity following radiation therapy of carcinoma of the cervix. Int J Radiat Oncol Biol Phys. 2000;48:991–5. doi: 10.1016/s0360-3016(00)00729-x. [DOI] [PubMed] [Google Scholar]
- 11.Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, et al. TGF-beta: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst. 2014;106:djt369. doi: 10.1093/jnci/djt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bandyopadhyay A, Wang L, Agyin J, Tang Y, Lin S, Yeh IT, et al. Doxorubicin in combination with a small TGFbeta inhibitor: a potential novel therapy for metastatic breast cancer in mouse models. PLoS One. 2010;5:e10365. doi: 10.1371/journal.pone.0010365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, et al. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123:1348–58. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oshimori N, Oristian D, Fuchs E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015;160:963–76. doi: 10.1016/j.cell.2015.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biswas S, Guix M, Rinehart C, Dugger TC, Chytil A, Moses HL, et al. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117:1305–13. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu H, Wu J, Zhang W, Luo H, Shen Z, Cheng H, et al. PKM2 enhances chemosensitivity to cisplatin through interaction with the mTOR pathway in cervical cancer. Sci Rep. 2016;6:30788. doi: 10.1038/srep30788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin L, Shen Q, Ding S, Jiang W, Jiang L, Zhu X. Immunohistochemical expression of Annexin A2 and S100A proteins in patients with bulky stage IB-IIA cervical cancer treated with neoadjuvant chemotherapy. Gynecol Oncol. 2012;126:140–6. doi: 10.1016/j.ygyno.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Moreno CS, Matyunina L, Dickerson EB, Schubert N, Bowen NJ, Logani S, et al. Evidence that p53-mediated cell-cycle-arrest inhibits chemotherapeutic treatment of ovarian carcinomas. PLoS One. 2007;2:e441. doi: 10.1371/journal.pone.0000441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.B BM. preprocessCore: A collection of pre-processing functions. R package version 1320 [Google Scholar]
- 20.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 22.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warnes Gregory R, B B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B. gplots: Various R programming tools for plotting data. R package version 2(4) [Google Scholar]
- 24.Qin Tai, B L, Xia Lu, Huang Haojie, Villarreal Maria M, Zwaagstra John, Collins Cathy, Yang Junhua, Zwieb Christian, Kodali Ravindra, Hinck Cynthia S, Kim Sun Kyung, Reddick Robert L, Shu Chang, O'Connor-McCourt Maureen D, Hinck Andrew P, Sun Lu-Zhe. A novel highly potent trivalent TGF-β receptor trap inhibits early-stage tumorigenesis and tumor cell invasion in murine Pten-deficient prostate glands. Oncotarget. 2016 doi: 10.18632/oncotarget.13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawyer JS, Anderson BD, Beight DW, Campbell RM, Jones ML, Herron DK, et al. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J Med Chem. 2003;46:3953–6. doi: 10.1021/jm0205705. [DOI] [PubMed] [Google Scholar]
- 26.Singh J, Chuaqui CE, Boriack-Sjodin PA, Lee WC, Pontz T, Corbley MJ, et al. Successful shape-based virtual screening: the discovery of a potent inhibitor of the type I TGFbeta receptor kinase (TbetaRI) Bioorg Med Chem Lett. 2003;13:4355–9. doi: 10.1016/j.bmcl.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 27.Zhu X, Jin L, Zou S, Shen Q, Jiang W, Lin W, et al. Immunohistochemical expression of RAGE and its ligand (S100A9) in cervical lesions. Cell Biochem Biophys. 2013;66:843–50. doi: 10.1007/s12013-013-9515-x. [DOI] [PubMed] [Google Scholar]
- 28.Ip CK, Li SS, Tang MY, Sy SK, Ren Y, Shum HC, et al. Stemness and chemoresistance in epithelial ovarian carcinoma cells under shear stress. Sci Rep. 2016;6:26788. doi: 10.1038/srep26788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiechert A, Saygin C, Thiagarajan PS, Rao VS, Hale JS, Gupta N, et al. Cisplatin induces stemness in ovarian cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruan Z, Liu J, Kuang Y. Isolation and characterization of side population cells from the human ovarian cancer cell line SK-OV-3. Exp Ther Med. 2015;10:2071–8. doi: 10.3892/etm.2015.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Latifi A, Abubaker K, Castrechini N, Ward AC, Liongue C, Dobill F, et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J Cell Biochem. 2011;112:2850–64. doi: 10.1002/jcb.23199. [DOI] [PubMed] [Google Scholar]
- 32.May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011;13:202. doi: 10.1186/bcr2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, et al. TGF-beta Tumor Suppression through a Lethal EMT. Cell. 2016;164:1015–30. doi: 10.1016/j.cell.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchini S, Fruscio R, Clivio L, Beltrame L, Porcu L, Fuso Nerini I, et al. Resistance to platinum-based chemotherapy is associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Eur J Cancer. 2013;49:520–30. doi: 10.1016/j.ejca.2012.06.026. [DOI] [PubMed] [Google Scholar]
- 36.Deng J, Wang L, Chen H, Hao J, Ni J, Chang L, et al. Targeting epithelial-mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts AB, Anzano MA, Lamb LC, Smith JM, Sporn MB. New class of transforming growth factors potentiated by epidermal growth factor: isolation from non-neoplastic tissues. Proc Natl Acad Sci U S A. 1981;78:5339–43. doi: 10.1073/pnas.78.9.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giannelli G, Villa E, Lahn M. Transforming growth factor-beta as a therapeutic target in hepatocellular carcinoma. Cancer Res. 2014;74:1890–4. doi: 10.1158/0008-5472.CAN-14-0243. [DOI] [PubMed] [Google Scholar]
- 39.Carey MS, Agarwal R, Gilks B, Swenerton K, Kalloger S, Santos J, et al. Functional proteomic analysis of advanced serous ovarian cancer using reverse phase protein array: TGF-beta pathway signaling indicates response to primary chemotherapy. Clin Cancer Res. 2010;16:2852–60. doi: 10.1158/1078-0432.CCR-09-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond) 2012;7:597–615. doi: 10.2217/nnm.12.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahmed N, Abubaker K, Findlay JK. Ovarian cancer stem cells: Molecular concepts and relevance as therapeutic targets. Mol Aspects Med. 2014;39:110–25. doi: 10.1016/j.mam.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Liao J, Qian F, Tchabo N, Mhawech-Fauceglia P, Beck A, Qian Z, et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS One. 2014;9:e84941. doi: 10.1371/journal.pone.0084941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuroda T, Hirohashi Y, Torigoe T, Yasuda K, Takahashi A, Asanuma H, et al. ALDH1-high ovarian cancer stem-like cells can be isolated from serous and clear cell adenocarcinoma cells, and ALDH1 high expression is associated with poor prognosis. PLoS One. 2013;8:e65158. doi: 10.1371/journal.pone.0065158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizuno T, Suzuki N, Makino H, Furui T, Morii E, Aoki H, et al. Cancer stem-like cells of ovarian clear cell carcinoma are enriched in the ALDH-high population associated with an accelerated scavenging system in reactive oxygen species. Gynecol Oncol. 2015;137:299–305. doi: 10.1016/j.ygyno.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Rodon J, Carducci MA, Sepulveda-Sanchez JM, Azaro A, Calvo E, Seoane J, et al. First-in-human dose study of the novel transforming growth factor-beta receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res. 2015;21:553–60. doi: 10.1158/1078-0432.CCR-14-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM, et al. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res. 2012;72:4119–29. doi: 10.1158/0008-5472.CAN-12-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang M, Kleber S, Rohrich M, Timke C, Han N, Tuettenberg J, et al. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011;71:7155–67. doi: 10.1158/0008-5472.CAN-11-1212. [DOI] [PubMed] [Google Scholar]
- 48.Mohammad KS, Javelaud D, Fournier PG, Niewolna M, McKenna CR, Peng XH, et al. TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011;71:175–84. doi: 10.1158/0008-5472.CAN-10-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.