

Abstract
Epithelial cells can acquire invasive and tumorigenic capabilities through epithelial–mesenchymal-transition (EMT). The glycan-binding protein galectin-8 (Gal-8) activates selective β1-integrins involved in EMT and is overexpressed by certain carcinomas. Here we show that Gal-8 overexpression or exogenous addition promotes proliferation, migration, and invasion in nontumoral Madin–Darby canine kidney (MDCK) cells, involving focal-adhesion kinase (FAK)-mediated transactivation of the epidermal growth factor receptor (EGFR), likely triggered by α5β1integrin binding. Under subconfluent conditions, Gal-8–overexpressing MDCK cells (MDCK-Gal-8H) display hallmarks of EMT, including decreased E-cadherin and up-regulated expression of vimentin, fibronectin, and Snail, as well as increased β-catenin activity. Changes related to migration/invasion included higher expression of α5β1 integrin, extracellular matrix-degrading MMP13 and urokinase plasminogen activator/urokinase plasminogen activator receptor (uPA/uPAR) protease systems. Gal-8–stimulated FAK/EGFR pathway leads to proteasome overactivity characteristic of cancer cells. Yet MDCK-Gal-8H cells still develop apical/basolateral polarity reverting EMT markers and proteasome activity under confluence. This is due to the opposite segregation of Gal-8 secretion (apical) and β1-integrins distribution (basolateral). Strikingly, MDCK-Gal-8H cells acquired tumorigenic potential, as reflected in anchorage-independent growth in soft agar and tumor generation in immunodeficient NSG mice. Therefore, Gal-8 can promote oncogenic-like transformation of epithelial cells through partial and reversible EMT, accompanied by higher proliferation, migration/invasion, and tumorigenic properties.
INTRODUCTION
Most human cancers originate from epithelia (carcinomas), and their progression includes a process reminiscent of the epithelial–mesenchymal transition (EMT) that normally occurs during organogenesis, wound healing, and tissue repair (Bryant and Mostov, 2008; Nieto, 2011). EMT is also associated with organ fibrosis (Nieto, 2011). In opposition to the epithelial polarity program that generates and maintains epithelia differentiation and integrity (Tanos and Rodriguez-Boulan, 2008), EMT endows epithelial cells with capabilities to detach from neighbor cells, traverse the basement membrane, and move through the extracellular matrix (ECM), displaying migratory and invasive phenotypes (Bryant and Mostov, 2008; Nieto, 2011). Loss of apical/basolateral epithelial cell polarity and major changes in ECM-interacting integrins, ECM-degrading proteases, and motility properties occur during EMT with varied intensity (Zeisberg and Neilson, 2009; Sundararajan et al., 2015). Intermediate states of EMT, or partial EMT, can be reverted by a mesenchymal-epithelial transition (MET) process, as would be required for organogenesis and metastasis (Huang et al., 2012; Nieto, 2013). In carcinomas, EMT is considered a prelude not only of metastasis but also of associated malignant traits, such as the stage of tumor initiating cells or stemness (Ye et al., 2015) and chemoresistance (Fischer et al., 2015; Zheng et al., 2015). Indeed, the EMT program has to be strictly controlled; otherwise, serious pathogenic conditions may arise.
Inducers and modulators of the EMT program are intensively studied under different contexts both in nontumoral and carcinoma cells. Secreted factors within the cell microenvironment, including hepatocyte growth factor (HGF), Wnt proteins, fibroblast growh factor (FGF), epidermal growth factor (EGF), and transforming growth factor beta (TGF-β), play major roles in EMT acting as paracrine or autocrine stimuli of specific cell-surface receptors and signaling pathways (Thiery and Sleeman, 2006; Nieto, 2011). Extracellular matrix (ECM) elements can also induce EMT by triggering integrin-mediated signaling (Grande et al., 2015; Lovisa et al., 2015). EMT-prone signaling pathways converge on the activity of transcription factors such as Snail, Twist, and ZEB, which down-regulate epithelial markers (e.g., E-cadherin) and up-regulate mesenchymal markers (e.g., vimentin) (Zeisberg and Neilson, 2009; Nieto, 2011). The extent of EMT depends on the cooperation between signaling pathways emerging from different receptors (Zeisberg and Neilson, 2009; Nieto, 2011), among which the EGF receptor (EGFR) has a special interest.
The EGFR has a main role in the regulation of epithelial cell proliferation and differentiation, and its function is frequently exaggerated (oncogenic) in cancerous transformation and carcinoma progression (Mendelsohn and Baselga, 2006). EGFR activation mediates initial stages of EMT synergizing other EMT inducers such as the TGFβ system (Grande et al., 2002; Lindsey and Langhans, 2015). Heterologous stimuli emerging from other receptors (Carpenter, 1999) and integrins (Moro et al., 1998; Moro et al., 2002) can transactivate the EGFR. This typically occurs through pathways that promote matrix metalloproteinase (MMP)-mediated release of soluble growth factors from transmembrane precursors at the plasma membrane (Carpenter, 2000). Therefore, the EGFR constitutes a downstream nodal element of many other signaling pathways that emerge from the cell surface (Carpenter, 1999; Buvinic et al., 2007). This might expand the regulation of EMT by yet unsuspected factors. Evidence in Caenorhabditis elegans suggests that the EGFR contributes to modulate the ubiquitin-proteasome system (UPS), which controls protein homeostasis by degrading ubiquitin-tagged proteins (Liu et al., 2011). UPS is frequently overactivated in cancer cells and very likely participates in EMT (Adams, 2004; Voutsadakis, 2012a). An enhanced UPS can down-regulate signal-transduction elements that negatively control cell growth and cell viability and might also help to avoid deleterious accumulations of mutated proteins generated by the variety of genetic defects in cancer cells (Deshaies, 2014). New extracellular factors potentially driving EMT and the acquisition of tumoral traits by epithelial cells are certainly of great interest (Gopal et al., 2015, 2016).
Galectins are a family of 15 glycan-binding proteins characterized by their conserved carbohydrate-recognition domains (CRDs) for β-galactosides and their capability to modulate a variety of cellular processes, with physiological, pathogenic, and therapeutic implications (Kaltner and Gabius, 2012; Rabinovich and Croci, 2012; Nabi et al., 2015). Cellular activities and responses to extracellular stimuli are globally modulated by galectins exerting simultaneous control over several cell-surface glycoproteins and signaling pathways (Kaltner and Gabius, 2012; Rabinovich and Croci, 2012; Nabi et al., 2015). Although these lectins are synthesized as cytosolic proteins, most of their functions take place by interacting with glycans of cell surface and ECM proteins after unconventional secretion (Kaltner and Gabius, 2012; Rabinovich and Croci, 2012; Nabi et al., 2015). Galectins have long been involved in cancer-related processes (Rabinovich and Croci, 2012; Thijssen et al., 2015). However, their role in EMT remains mostly unknown and has just recently been analyzed in carcinoma cells (Rizqiawan et al., 2013; Wang et al., 2013; Bacigalupo et al., 2015) but not yet in nontumoral epithelial cells from which carcinomas originate.
The role of galectins in epithelial cells has been little explored and can vary depending on the galectin and cellular contexts. For instance, the endogenous expression of galectin-3 (Gal-3), Gal-9, and Gal-4 in epithelial cells mediates the apical distribution of specific proteins (Delacour et al., 2005; Mo et al., 2012; Perez Bay et al., 2014), contributing to the execution of the apical/basolateral polarity program, in contraposition to EMT (Bryant and Mostov, 2008; Tanos and Rodriguez-Boulan, 2008; Nieto, 2013). However, Gal-1 and Gal-3 have also been shown to promote EMT in squamous cell carcinomas (Rizqiawan et al., 2013; Wang et al., 2013) and also Gal-1 in hepatocellular carcinoma (Bacigalupo et al., 2015), seemingly engaging the TGF-β and Wnt signaling pathways. In contrast, Gal-9, instead of inducing EMT, has deleterious effects in nontumoral MDCK cells and K-Ras mutants of colorectal carcinoma cells (Wiersma et al., 2015). It is important to define whether other galectins currently associated with cancer, such as Gal-8, can promote EMT in nontumoral epithelial cells and whether other signaling pathways contribute to this process.
Gal-8 is one of the most widely expressed galectins in human tissues and carcinomas (Elola et al., 2014). This is a tandem-repeat galectin consisting of N- and C-terminal CRDs with different glycan selectivity and joined by a linker peptide, whose length variation gives rise to short and long isoforms. The N-terminal CRD preferentially binds to α2,3-sialylated glycans, a unique specificity among galectins, thus endowing Gal-8 with special functional properties (Ideo et al., 2011; Elola et al., 2014). Gal-8 can have antitumoral effects, inducing growth arrest and apoptosis (Hadari et al., 2000; Arbel-Goren et al., 2005) or inhibiting migration and tumoral growth (Nagy et al., 2002), which might explain its down-regulation in certain carcinomas, such as colon cancer (Nagy et al., 2002; Elola et al., 2014). However, Gal-8 can also promote tumorigenesis by stimulating angiogenesis (Delgado et al., 2010; Troncoso et al., 2014) and integrin-mediated cell adhesion to the ECM during metastasis (Reticker-Flynn et al., 2012). A recent study in glioblastoma cells that tests the effects of Gal-8 silencing demonstrates that endogenous Gal-8 expression can promote cell proliferation and survival in certain cancers (Metz et al., 2016). Gal-8, similarly to other galectins, is overexpressed and associated with malignancy in several carcinomas, suggesting a gain of function that so far remains unknown (Rabinovich and Croci, 2012; Elola et al., 2014; Thijssen et al., 2015).
Integrins are major counterreceptors of Gal-8 (Hadari et al., 2000; Levy et al., 2001; Carcamo et al., 2006). Selected β1-integrins have been involved in Gal-8-mediated cell adhesion and signaling toward activation of focal-adhesion kinase (FAK) (Hadari et al., 2000; Levy et al., 2001; Carcamo et al., 2006) and mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) (Carcamo et al., 2006; Norambuena et al., 2009). Interestingly, integrins are intimately intertwined with EGFR function, forming complexes as signaling units sensitive to ECM stimulation (Miyamoto et al., 1996; Moro et al., 1998; Sieg et al., 2000; Desgrosellier and Cheresh, 2010) and together with EGFR signaling pathways are important mediators of EMT (Thiery, 2002; Tanos and Rodriguez-Boulan, 2008; Frisch et al., 2013). All these data suggest a pathway through which Gal-8 might modulate epithelial cell processes associated with tumorigenesis.
Herein, we study the effects of Gal-8 overexpression or its exogenous addition in nontumoral MDCK cells, widely used in studies of cell transformation (Stiles et al., 1976; U et al., 1985) and EMT (Batlle et al., 2000; Cano et al., 2000). Our results reveal that epithelial cells overexposed to Gal-8 can undergo partial and reversible EMT associated with tumorigenic transformation and invasive properties. Furthermore, we show that Gal-8 induces integrin-mediated transactivation of EGFR, leading to proteasome activation in a manner restricted by apical/basolateral polarity.
RESULTS
Gal-8 increases the rate of proliferation in MDCK cells acting extracellularly on cell-surface glycans
To study the effects of Gal-8 overexpression on epithelial cells, we transfected MDCK cells and selected two colonies expressing relatively low (MDCK-Gal-8L) and high (MDCK-Gal-8H) Gal-8 levels. Reverse transcriptase-PCR (RT-PCR), immunoprecipitation, and immunoblot assays characterize the Gal-8 expression of these two colonies (Figure 1A). Immunoblot of conditioned media indicated that MDCK-Gal-8H cells secrete twice as much Gal-8 as MDCK-Gal-8L cells (Figure 1A). Both MDCK-Gal-8L and MDCK-Gal-8H cells displayed higher proliferation rates, incorporating two- and fourfold more [3H]thymidine, respectively, than MDCK cells (Figure 1B). Cells transfected with an empty vector behave similarly to wild-type MDCK cells (unpublished data), and thus we will refer to control cells as MDCK cells. Doubling times of proliferation decreased from 20.1 h in MDCK cells to 16.3 h in MDCK-Gal-8H cells (Figure 1C). These results indicate that the levels of Gal-8 overexpression impinge on the Gal-8 secreted levels, which in turn increases the proliferation rate of MDCK cells acting on cell-surface glycans, as shown below.
FIGURE 1:

Gal-8 overexpression increases the proliferation of MDCK cells interacting with cell-surface glycans. (A) MDCK cells stably transfected with human recombinant Gal-8. RT-PCR, immunoprecipitation (IP) of cell extracts, and immunoblot (IB) of TCA precipitates from (24 h) conditioned media show Gal-8 expressed and secreted at relatively low (MDCK-Gal-8L) or high (MDCK-Gal-8H) levels. (B) MDCK-Gal-8L (n = 21 wells from seven experiments) and MDCK-Gal-8H (n = 35 wells, from 12 experiments) show higher rates of 24 h 3[H]thymidine incorporation than MDCK cells (n = 30 wells, from 10 experiments); mean ± SEM; *p < 0.05; **p < 0.005; one-way ANOVA followed by Tukey’s multiple comparisons test. (C) Comparison of cell growth rates and doubling times (dT) of MDCK and MDCK-Gal-8H cells (n = 6, from three experiments); mean ± SEM; *p <0.05; **p < 0.005; two-way ANOVA followed by Sidak’s multiple comparisons test. (D) TDG (20 mM) abrogates the increased proliferation of MDCK-Gal-8H cells. (E) Exogenous Gal-8 (GST-Gal-8 or Gal-8 proteolitically released from GST) increases MDCK cell proliferation (n = 12, from four experiments); mean ± SEM; *p < 0.05; **p < 0.005; one-way ANOVA adjusted by Tukey’s multiple comparisons test.
Gal-8, similarly to other galectins, exerts different functions interacting with intracellular or extracellular elements (Carcamo et al., 2006; Norambuena et al., 2009; Thurston et al., 2012). As galectins lack a signal peptide, their secretion occurs through an unconventional mechanism, presumably involving exosomes (Klibi et al., 2009; Barres et al., 2010). To determine whether Gal-8 overexpression exerts its proliferation effect acting on intracellular or cell-surface extracellular elements, we tested the effect of tiodigalactoside (TDG), which, similarly to lactose, blocks the binding of galectins to externally exposed carbohydrates (Carcamo et al., 2006; Norambuena et al., 2009). TDG almost completely abolished the increased proliferation of MDCK-Gal8H cells (Figure 1D), indicating an extracellular action of Gal-8 interacting with cell-surface glycans. Recombinant GST-Gal-8 and GST-free Gal-8 added to the medium also increased the incorporation of [3H]thymidine in MDCK cells. A concentration of 50 μg/ml Gal-8, close to that estimated for 24 h conditioned media, elicited a proliferative level similar to that of MDCK-Gal-8H cells (Figure 1E).
All these results, which include the distinct proliferation rates of MDCK-Gal-8L and MDCK-Gal-8H cells related to the levels of Gal-8 in conditioned media, the similar effects of endogenous and exogenous Gal-8 and the counteracting effect of TDG, indicate that MDCK cells increase their proliferation rate according to the level of Gal-8 stimulation at the cell surface. In principle, epithelial cells can be stimulated by Gal-8 both in autocrine and paracrine manners, that is, Gal-8 secreted by neighboring cells might induce proliferation.
MDCK-Gal-8H cell proliferation involves transactivation of the EGFR and ERK mitogenic signaling
We then studied whether the proliferative response to Gal-8 involves the mitogenic activity of the EGFR, which is a crucial regulator of epithelial cell proliferation (Mendelsohn and Baselga, 2006), including MDCK cells (Buvinic et al., 2007). MDCK-Gal-8H cells showed higher levels of EGFR tyrosine-phosphorylation than MDCK cells, without changing the receptor mass and thus reflecting EGFR activation (Figure 2A). To assess whether Gal-8 directly activates or indirectly transactivates the EGFR, we inhibited the MMP activity (Carpenter, 1999; Prenzel et al., 1999; Buvinic et al., 2007), which proteolitically release growth factors from cell-surface membrane precursors (Prenzel et al., 1999). GM6001 (GM), a general MMP inhibitor, decreased the level of EGFR tyrosine-phosphorylation in MDCK-Gal-8H cells, as assessed in immunoprecipitated EGFR (Figure 2A) or directly in cell extracts (Figure 2B). However, MDCK and MDCK-Gal-8H had similar mass of EGFR (Figure 2, A, bottom blot, and B, middle blot), indicating transactivation of EGFR in MDCK-Gal-8H cells.
FIGURE 2:

MMP-mediated transactivation of EGFR drives the higher proliferation of Gal-8–overexpressing MDCK-Gal-8H cells. (A) Immunoprecipitated (IP) EGFR analyzed by immunoblot (IB) for tyrosine-phosphorylation (p-Tyr, top blot) and total mass (EGFR, bottom blot); (B) Immunoblot of EGFR for Tyr1068 phosphorylation (p-EGFR1068; top blot) and total mass (EGFR; middle blot) and actin in 50 μg of proteins from cell extracts of MDCK-Gal-8H and MDCK cells incubated in the absence or presence of MMP inhibitor GM6001 (GM; 2.5 μM) for 4 h. MDCK-Gal-8H cells show higher activation but similar expression of EGFR compared with MDCK cells, and their EGFR activity decreased after treatment with GM. (C) Immunoblot analysis of phospho-ERK1/2 and total mass of ERK1/2 in cell extracts (50 μg) shows higher activity of ERK1/2 in MDCK-Gal-8H cells compared with MDCK cells, which is reduced by the MEK inhibitor PD98059 (PD), indicating activation of the EGFR/RAS/RAF/MEK/ERK pathway. (D) Inhibitors of EGFR tyrosine-kinase (0.1 μM AG1478; AG), MEK (25 μM PD98059; PD), and MMP (2.5 μM GM6001; GM) all abolish the increased proliferation of MDCK-Gal-8H cells (n = 12 wells from four experiments); mean ± SEM; **p < 0.005; one-way ANOVA, Tukey’s multiple comparisons test.
MDCK-Gal-8H cells also showed higher levels of ERK1/2 activation compared with MDCK cells (Figure 2C). Furthermore, inhibitors of MMP (GM6001), EGFR tyrosine-kinase (AG1478), and the downstream MEK kinase (PD98059) all decreased MDCK-Gal-8H cell proliferation to the levels of MDCK cells (Figure 2D). Therefore, EGFR transactivation and signaling via the Ras/Raf/MEK/ERK pathway mostly accounts for the mitogenic effect of Gal-8.
Gal-8 binds α5β1 integrin and activates FAK leading to EGFR transactivation
Previous studies in other cells show that Gal-8 interacts with selected β1-integrins, including α5β1 (Levy et al., 2001; Carcamo et al., 2006; Norambuena et al., 2009). Gal-8 has also been shown to activate FAK (Levy et al., 2001), a main integrin downstream signaling element (Mitra and Schlaepfer, 2006; Seguin et al., 2015). Our pull-down experiments with GST-Gal-8 detected the α5-integrin subunit, indicating that Gal-8 also interacts with this β1-integrin in MDCK cells (Figure 3A). GST-Gal-8 also pulled down the EGFR (Figure 3A) and lactose (50 mM), widely used to block galectin interactions with β-galactosides (Carcamo et al., 2006; Norambuena et al., 2009), and decreased the pull down of both proteins (Figure 3A), indicating a glycan-mediated interaction. As our previous results with MMP inhibitors favor transactivation instead of direct activation of the EGFR by Gal-8, we tested whether Gal-8 activates integrin signaling and whether this might account for EGFR transactivation (Moro et al., 1998). Accordingly, we observed FAK activation in MDCK-Gal-8H cells (Figure 3B), and the FAK specific inhibitor Y15 completely abrogated the EGFR activation induced by Gal-8 (Figure 3C). All these results are congruent with EGFR transactivation through α5β1 integrin-FAK pathway triggered by Gal-8.
FIGURE 3:

Gal-8 binds α5β1 integrin and activates FAK in the pathway of EGFR transactivation. (A) GST-Gal-8 pulls down α-5 integrin, β1 integrin, and EGFR (lane 1) from 500 μg of cell extract, which is blocked by lactose (50 mM) (lane 2), indicating dependency of Gal-8/glycan interaction. Lane 3 is empty to avoid spill-off from lane 4 showing the presence of analyzed proteins in 50 μg of total cell extract. (B) MDCK-Gal-8H cells have activated FAK as shown by immunoblot with anti-FAK (Tyr-397). (C) Gal-8 activates EGFR in a FAK-dependent manner. MDCK cells incubated with Gal-8 show an increased level of Tyr phosphorylation of EGFR (pEGFR), which is reduced by FAK inhibitor Y15.
Gal-8 increases migration and invasion in MDCK cells
As Gal-8 stimulated FAK and the EGFR, both involved in migration and invasion of carcinoma cells (Sieg et al., 2000; Long et al., 2010; Sulzmaier et al., 2014), we assessed two-dimensional migration in a wound closure assay, as well as three-dimensional migration/invasion across a matrigel-covered polycarbonate membrane of Transwell chambers. MDCK-Gal-8H cells and MDCK cells treated with exogenous Gal-8 both showed faster wound healing than untreated MDCK cells (Figure 4A). TDG decreased the migration of MDCK-Gal-8H cells, thus demonstrating an extracellular action of secreted Gal-8 on cell-surface glycans (Figure 4A). MDCK-Gal-8H cells also displayed sixfold-higher invasion activity compared with MDCK cells, which was counteracted by EGFR (AG1478) and MMP broad-spectrum inhibitors (GM-6001 and ONO-4817) (Figure 4B). Therefore, in addition to proliferation, Gal-8–induced EGFR transactivation stimulates MDCK cell migration and invasion.
FIGURE 4:

Gal-8 increases migration and invasion capabilities of MDCK cells. (A) Wound healing assay. A confluent monolayer of MDCK and MDCK-Gal-8H cells was scraped with a micromanipulator and pictured every 30 min for 2 h. The picture depicts wound healing at 120 min, while the graph illustrates healing progression under the indicated conditions, including the effects of TDG (20 mM) and exogenous Gal-8 (50 μg/ml) (n = 6 wounds from three experiments). Mean ± SEM; *p < 0.05; two-way ANOVA followed by Sidak’s multiple comparisons test; Scale bar = 40 μm. (B) Invasion assay. MDCK and MDCK-Gal-8H cells (5 × 104) were seeded in Transwell filters (8-μm pore) coated with Matrigel and incubated in the absence or presence of AG1478, GM6001, and ONO4817 for 24 h. Cells stained with crystal violet (arrowheads) were counted on bottom sides of the filter. Graph shows number of cells per field (n = 6 fields from three experiments) Mean ± SEM; *p < 0.05; **p < 0.005; one-way ANOVA, Tukey’s multiple comparisons test).
Gal-8 increases the expression of extracellular matrix–degrading proteases
As cell invasion relies on the capability of cells to degrade ECM, we assessed the secretion of the serine protease uPA (Smith and Marshall, 2010) and MMPs (Kessenbrock et al., 2010) involved in ECM degradation. Conditioned media from MDCK-Gal-8H cells showed higher uPA activity in agarose/casein gels containing plasminogen (Figure 5A). In addition, the analysis of conditioned media from MDCK-Gal-8H cells with zymographs of gelatin degradation revealed higher MMP activity attributable to MMP-13 (Figure 5B). RT-PCR showed higher levels of the corresponding transcripts of these proteins, as well as the uPA receptor (Figure 5C). In correlation with these observations, we found an increased mass of both uPA and MMP-13 proteins in cell extracts and media and the uPA receptor in cell extracts (Figure 5D). These results reveal an enhanced activity of the uPA/uPAR system and MMP-13 in MDCK-Gal-8H cells due to their increased expression levels, which indeed would contribute to the higher invasive properties of these Gal-8–overexpressing cells.
FIGURE 5:

Gal-8 increases the secretion of extracellular matrix-degrading proteases. (A) Zymography of caseinolitic activity reveals higher uPA activity in 24 h conditioned media from MDCK-Gal-8H cells compared with MDCK cells. (B) Zymography of gelatin degradation and electrophoretic mobility of MMP-9 (92 kDa), MMP-2 (72 kDa), and MMP-13 (48 kDa). The increased gelatinolytic activity in 48 h conditioned media from MDCK-Gal-8H corresponds to MMP-13. (C) RT-PCR analysis showing higher levels of transcripts of uPA, uPAR, and MMP13 in MDCK-Gal-8H cells. (D) Immunoblot analysis of cell extracts (Cells) and 24 conditioned media (Media). MDCK-Gal-8H cells show increased levels of uPA uPAR and MMP-13 compared with MDCK cells. Graph shows the mean ratio of MDCK-Gal-8H vs. MDCK cells in arbitrary units of each band density relative to actin (n = three experiments). (E) Zymography of uPA activity shows sensitivity to inhibition by AG1478 (AG) and Y15 but not to PD98059 (PD).
To test whether the changes in serine protease activity has a similar dependence on the signaling pathways controlling proliferation of MDCK-Gal-8H cells, we assessed uPA activity under inhibitors of these pathways. AG1478 and Y15 inhibitors of EGFR and FAK, respectively, but not the inhibitor of ERK (PD98059), abrogated uPA activity detected in the media (Figure 5E). These results indicate that FAK-dependent EGFR transactivation regulates uPA activity involving a pathway distinct from ERK1/2.
Gal-8 promotes partial/reversible epithelial mesenchymal transition
Enhanced invasiveness of epithelial cells implies an ongoing EMT program, which has been proposed to be partial and reversible during embryonic development and metastatic processes (Nieto, 2013). The most characteristic hallmarks of EMT during organogenesis and tumorigenesis comprise decreased expression of E-cadherin due to higher expression of its negative transcriptional regulator, Snail, accompanied by reciprocal increments of vimentin and fibronectin expression (Zeisberg and Neilson, 2009; Nieto, 2013). EMT also involves an increased expression of α5β1 integrin (Zeisberg and Neilson, 2009). Accordingly, we found that MDCK-Gal-8H cells in subconfluent conditions displayed higher expression levels of fibronectin, α5β1, vimentin, and snail, as well as a decreased E-cadherin (Figure 6A). In contrast, confluent MDCK and MDCK-Gal-8H cells showed similar expression levels of all these proteins, even decreasing some of them (e.g., α5 integrin and Snail) (Figure 6A). We also found nuclear translocation reflecting activation of β-catenin (Figure 6B), another well-characterized EMT marker (Zeisberg and Neilson, 2009). However, despite expressing EMT hallmarks, MDCK-Gal-8H cells did not acquire a typical mesenchymal cell phenotype under subconfluent conditions (Figure 6C). Both MDCK and MDCK-Gal-8H cells showed a tendency to form clusters, indicating that Gal-8 does not abrogate cell–cell interactions (Figure 6C). The morphology of MDCK-Gal-8H cells frequently showed extensions resembling the lamellipodial and fusiform projections of migratory cells, but not a fibroblastic phenotype (Figure 6C), as can also be seen during wound healing (Figure 4A).
FIGURE 6:

Gal-8 promotes reversible EMT maintaining the capability to generate transepithelial resistance and apical (Ap)/basolateral (Bl) polarity. (A) Subconfluent MDCK-Gal-8H cells display hallmarks of EMT compared with MDCK cells, such as increased levels of fibronectin, α5-integrin, vimentin, and Snail, accompanied by lower levels of E-cadherin. These changes are reverted in confluent cells. Graph shows the mean ratio of MDCK-Gal-8H vs. MDCK cells in arbitrary units of each band density relative to actin in subconfluent and confluent cells (n = three experiments). (B) Higher nuclear distribution of β-catenin in subconfluent MDCK-Gal-8H cells compared with MDCK cells. Lamin A is used as a marker of nuclear fraction. Graph shows the average percentage of distribution (n = 3 experiments); (C) Subconfluent MDCK-Gal-8H cells show extensions opposing the mass of cell clusters compared with MDCK cells, without acquiring a complete mesenchymal phenotype; Scale bar = 10 μm. (D) MDCK-Gal8H grown to confluence on Transwell filters (0.4-μm pore) develop more rapidly and higher levels of TER than MDCK cells. (E) Domain-selective biotinylation shows similar basolateral distribution and cell-surface levels of E-cadherin in Transwell-seeded MDCK-Gal8H and MDCK cells. Total E-cadherin corresponds to 5% of cell extract (50 μg/protein). (F) Immunoblot in 18 h conditioned media shows apical secretion of Gal-8 and basolateral secretion of fibronectin. (G) Apical GP135 and basolateral E-cadherin immunofluorescence analyzed in a Leica SP8 scanning confocal microscope. Images of apical, basolateral, and X-Z sections are displayed. GP135 displays a clustered pattern in MDCK-Gal8H cells. Scale bar = 10 µm.
All of these observations prompt us to test whether MDCK-Gal-8H cells maintain the capability to form apical-basolateral polarized monolayers. MDCK-Gal-8H cells grown to confluence in Transwell chambers developed transepithelial resistance more rapidly and at higher levels than MDCK cells, very likely due to their higher proliferation rate (Figure 6D). Domain-specific biotinylation assays showed similar basolateral distributions and cell-surface levels of E-cadherin in both MDCK and MDCK-Gal-8H cells (Figure 6E). Interestingly, we detected Gal-8 exclusively secreted to the apical chamber, contrasting with the basolateral secretion of fibronectin (Figure 6F). Both GP135 and E-cadherin displayed the expected apical and basolateral distribution, respectively. The GP135 protein showed an apical clustered pattern in MDCK-Gal-8H (Figure 6G). Therefore, when cells reach confluence, the reversion of protein expression characteristic of EMT is accompanied by the acquisition of a polarized phenotype. These results indicate that Gal-8 promotes the expression of a partial and reversible EMT program, which provides higher proliferation, migration, and invasion properties while maintaining the capability of differentiation toward a polarized epithelial phenotype. Because β1-integrins and EGFR are basolaterally distributed in polarized MDCK cells (Schoenenberger et al., 1994; Kil et al., 1999), the apical secretion of Gal-8 might lead to a reverted EMT phenotype due to an impediment of its interaction with α5β1, which would be required for transactivating the EGFR.
Gal-8 increases proteasomal activity
The ubiquitin proteasome system (UPS) regulates many signaling elements crucial to both EMT and cancer (Adams, 2004; Voutsadakis, 2012a). Therefore, we studied whether Gal-8 overexpression or its exogenous addition affects proteasome activity. We first measured the chymotrypsin-like activity of the β5 subunits of the 20S proteasome and found 1.5-fold higher activity in MDCK-Gal-8H compared with MDCK cells (Figure 7A). The counteracting effect of lactacystin, a specific proteasome inhibitor, further demonstrated that such increased proteolysis corresponds to proteasome activity. Its inhibition with lactose, but not with sucrose, indicates dependency of this Gal-8–induced proteasome activity on interactions with cell-surface glycans (Figure 7A).
FIGURE 7:

Gal-8 increases the activity but not the expression of the proteasome through the FAK/EGFR pathway. (A) MDCK-Gal-8H cells show higher proteasome activity than MDCK cells, which is lowered by 20 mM β-lactose (but not by 20 mM sucrose) and by the proteasome inhibitor Lactacystin (1 μM). (B) Gal-8 (50 μg/ml) treatment for 4 h increases the chymotrypsin-, trypsin-, and caspase-like activity, corresponding to the proteasomal β5, β2, and β1 catalytic subunits. (C) Lactose (20 mM) (but not sucrose), and lactacystin counteract the Gal-8–induced increase of the proteasomal β5 catalytic activity. (D) Inhibition of EGFR (AG1478) and FAK (Y15) decrease the proteasome activity of MDCK-Gal-8H cells to the levels of MDCK cells (mean ± SEM; n = 9 wells from three experiments; *p < 0.05, **p < 0.005, one-way ANOVA). (E) Gal-8 does not change the proteasome expression. The α4 subunit of the 20S proteasome shows similar levels in MDCK-Gal-8H and MDCK, as well as in MDCK cells treated with 5, 10, 20, and 50 μg/ml Gal-8. Graphs show the mean ± SEM densitometric intensity from three experiments.
We then assessed the effect of exogenous Gal-8 on the proteasome activity of MDCK cells but extending the analysis to chymotrypsin-like, trypsin-like, and caspase-like activities, which correspond to proteasomal β5, β2, and β1 catalitic subunits, respectively. Gal-8 added to the media for just 4 h increased all of these proteasome activities (Figure 7B). Again, lactacystin and lactose (20 mM), but not sucrose (20 mM), inhibited this Gal-8 effect (Figure 7C).
Interestingly, inhibitors of EGFR tyrosine-kinase and FAK both abrogated the effects of Gal-8 on proteasome activity (Figure 7D). Immunoblot analysis showed similar expression levels of the proteasome α4 subunit in MDCK and MDCK-Gal-8H cells, as well as in MDCK cells exogenously incubated with 50 μg/ml Gal-8 for 4 h (Figure 7E). Therefore, the Gal-8–induced increase of proteasome activity requires EGFR and FAK activation and is not due to up-regulation of proteasome expression.
Gal-8 increases proteasomal activity in a polarized manner
We noticed that Gal-8–induced proteasome activity occurred in subconfluent but not in highly confluent MDCK cells (Figure 8A). As Gal-8 is apically secreted (see Figure 6A), this observation suggested that Gal-8 loses its effects in polarized epithelial cells due to an impeded interaction with basolateral integrins. In congruency with this possibility, Gal-8 added to the basolateral, but not to the apical, domain increased the proteasome activity (Figure 8B). Indeed, the domain-specific biotinylation showed the expected basolateral localization of its interacting protein β1-integrin, similarly to E-cadherin (Figure 8C). These results indicate that Gal-8 enhances proteasome activity in epithelial cells only under nonpolarized conditions or when Gal-8 secreted by other cells have access to the basolateral β1-integrins.
FIGURE 8:
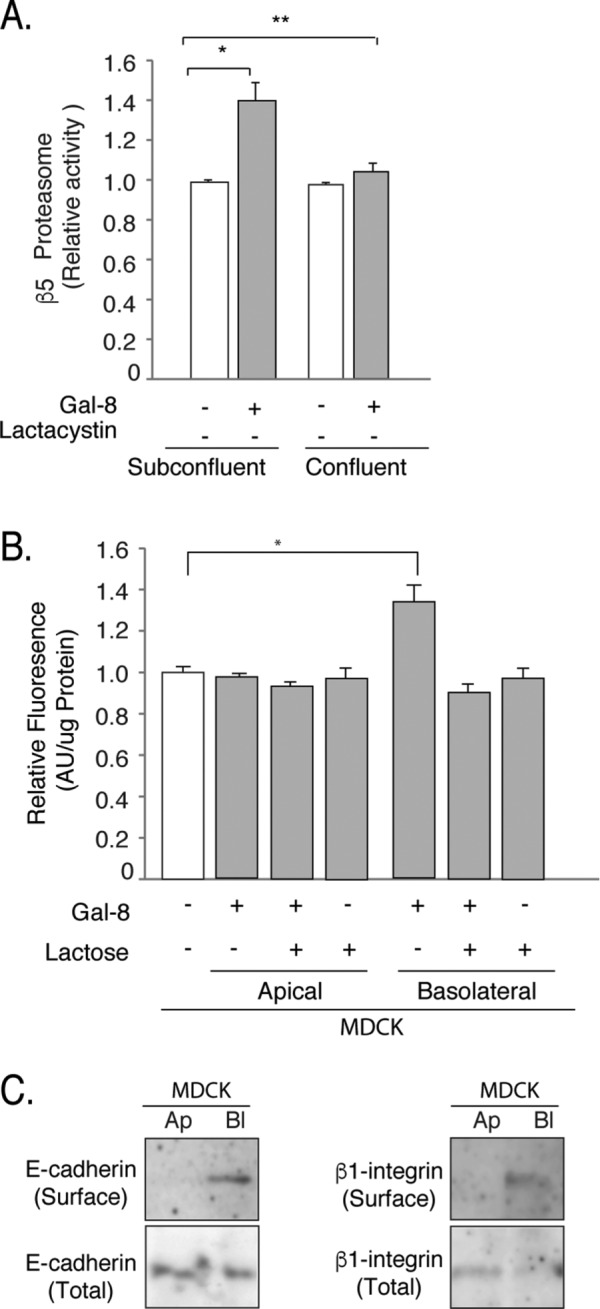
Gal-8 induces proteasome activation in a polarized manner. (A) Gal-8 induces proteasome activity in subconfluent but not in confluent MDCK cells. (B) MDCK cells grown in Transwell chambers increases proteasome activity in response to Gal-8 added to the basolateral but not to the apical compartment (mean ± SEM; n = 9 wells from three experiments; *p < 0.05, **p < 0.005, one-way ANOVA). (C) Domain-specific cell-surface biotinylation shows basolateral distribution of the Gal-8-interacting protein β1-integrin, similarly to E-cadherin. Total β1-integrin and E-cadherin correspond to 5% of cell extract (50 μg/protein).
Gal-8 stimulates anchorage-independent growth and invasive tumorigenesis
Another property acquired during cancer-associated EMT is the capability of anchorage-independent growth, which correlates with tumorigenicity and anoikis resistance when epithelial cells are released from cell–cell and cell–ECM interactions (Frisch et al., 2013; Paoli et al., 2013). We observed larger and increased numbers of colonies generated in soft agar by MDCK-Gal-8H cells than by MDCK cells (Figure 9A). Then we compared the tumorigenic potential of MDCK-Gal-8H and parental MDCK cells in immunosupressed NSG mice. Subcutaneous injection of 106 cells showed that MDCK cells only form small nodules of ∼1–1.5 mm (longest diameter), detectable after 8–10 wk. In contrast, the five mice injected with MDCK-Gal-8H developed larger tumor masses, in the range of 3–9 mm. The two biggest tumors showed intense confluence of blood vessels and signs of tumor infiltration into the adjacent skeletal muscle (Figure 9B, arrowhead). Histology analysis showed that MDCK-formed nodules have well-delineated borders and contain cuboidal cells organized in tubules, with abundant luminal contents of cell detritus and separated by dense collagen fibers (Figure 10, A, A′, and A″). Instead, MDCK-Gal-8H cells generated tumors of irregular contours, constituted by epithelial cell proliferation and variable morphology, including channels, papillae, and micropapillae (Figure 10, B, B′, B″, and C), as well as skeletal muscle infiltration at the periphery (Figure 10, B andB′), compromise of the perineural connective tissue (perineural invasion) (Figure 10, B, B″, and C), vascularized papillae (Figure 10D), focalized areas of poorly differentiated cells with a tumoral aspect (Figure 10E), and increased mitotic activity (unpublished data). (Supplemental Figure 1 shows higher magnifications of Figure 10, B′, B″, C, and D.) These features are typically observed in human malignant tumors. Furthermore, immunohistochemistry of cell proliferation marker Ki67 shows a low percentage of cell proliferation (<15%) in MDCK nodules contrasting with the high index of cell proliferation (60–70%) in the MDCK-Gal-8H tumor (Figure 10, F and G). Also, in contrast with MDCK that form nodules with basolateral distribution of E-cadherin (Figure 11A), MDCK-Gal-8H tumors are heterogeneous, showing areas of cells expressing E-cadherin with either epithelial morphology (Figure 11B′) or mesenchymal phenotypes (Figure 11B″), suggesting different grades of EMT. These images indicate that Gal-8 endows MDCK cells with invasive tumorigenic potential.
FIGURE 9:

Gal-8 overexpression promotes anchorage-independent growth in soft agar and tumor generation in immunodeficient NSG mice. (A) Anchored-independent growth. MDCK-Gal-8H cells (500 cells) seeded in soft agar generated an increased number of colonies, and larger, than MDCK cells. Analysis of 13 fields for each condition (number of colonies/field, mean ± SEM; **p < 0.005; two tailed unpaired t test with Welch´s correction. Areas of colonies from three experiments were measured with ImageJ software in 53 colonies of MDCK cells and 88 colonies of MDCK-Gal-8H cells (mean ± SEM; *p < 0.05 two-tailed unpaired t test with Welch´s correction). Scale bar, 500 μm. (B) Tumor generation in NSG mice. Cells (106) were subcutaneously injected in NSG mice, and the size of tumors was periodically assessed during 10 wk. The size of the nodules formed by MDCK cells remained similar from 2 to 10 wk, measuring around 1–1.5 mm in the killed animals (black arrow), whereas MDCK-Gal-8H cells formed tumors in the range of 3–9 mm (longest diameter: scale bar, 5 mm). The tumor depicted in the figure measured around 9 mm and shows a region infiltrating the muscle (white arrow). Graph shows each tumor volume (**p < 0.005; Mann-Whitney test, two tailed).
FIGURE 10:

Histology and proliferation activity of MDCK- and MDCK-Gal-8H-generated tumors. Tumors generated in NSG mice subcutaneously injected with 106 cells were analyzed after 10 wk. (A–E) MDCK tumor HE staining. (A) A well-defined nodule generated by MDCK cells showing an area with small and medium vascular structures in the peripheral pseudocapsule (enlarged in A′), and a region enriched in tubules formed by cuboidal cells of differentiated appearance, filled with cellular detritus and separated by ECM and interstitial cells (enlarged in A″). (B–E) MDCK-Gal-8H tumor HE staining. (B) A tumor generated by MDCK-Gal-8H cells is labeled at regions showing an invasive behavior revealed by muscle infiltration (enlarged in B′, arrow) and perineural invasion (enlarged in B″, arrowhead) with abundant micropapillae differentiation (B″, arrow). (C–E) Selected areas of other MDCK-Gal-8H-generated tumors showing malignant features: (C) muscle infiltration (asterisk), papillae differentiation (arrow), and perineural invasion (arrowhead). (D) A papillae with a vascular structure (arrow). See higher magnifications of B, B″, C, and D in Supplemental Figure 1. (E) Undifferentiated cells with categorical tumoral appearance. (F, G) Immunohistochemistry of cell proliferation marker Ki67 shows a low percentage of cell proliferation (<15%) in MDCK nodules contrasting with the high index of cell proliferation (60–70%) in the MDCK-Gal-8H tumor.
FIGURE 11:

Immunohistochemistry of E-cadherin of MDCK- and MDCK-Gal-8H-generated tumors. (A) MDCK-generated nodule shows epithelial cells organized in tubular structures with basolateral distribution of E-cadherin (arrows, A′). (B) MDCK-Gal8H-generated tumor shows regions with epithelial morphology and basolateral distribution of E-cadherin (**; arrows in B′) and poorly differentiated regions with cells displaying mesenchymal morphology and a more intracellular distribution of E-cadherin (*; arrows in B″).
DISCUSSION
This study demonstrates that Gal-8 overexpression can transform MDCK cells into highly proliferative, migratory/invasive, and tumorigenic cells, involving FAK-mediated EGFR transactivation, enhanced proteasomal activity, and partial/reversible EMT. Exogenously added Gal-8 also transactivates the EGFR with similar functional consequences. The epithelial polarity program impedes such EMT-prone changes compartmentalizing Gal-8 secretion to the apical domain while its effectors β1-integrins are basolaterally distributed. The results suggest that epithelial cells might undergo a tumorigenic EMT within a tumor microenvironment provided by Gal-8–overexpressing carcinoma cells.
More than one signaling pathway probably converges on Gal-8–induced tumorigenic EMT. Gal-8 can interact with several cell-surface receptors (Nishi et al., 2003; Carcamo et al., 2006; Vicuña et al., 2013), including uPAR (Vinik et al., 2015) and TGFβR (Sampson et al., 2016). Here we focused on EGFR due to its well-known contribution to tumorigenesis and intertwined function with β1-integrins (Miyamoto et al., 1996; Moro et al., 1998; Sieg et al., 2000; Desgrosellier and Cheresh, 2010), which are major counter-receptors of Gal-8 (Hadari et al., 2000; Levy et al., 2001; Carcamo et al., 2006) and mediate transactivation of the EGFR by the ECM (Moro et al., 2002). As reported in other cells (Levy et al., 2001), we show that Gal-8 binds α5β1 and activates FAK in MDCK cells. We also show that this Gal-8–induced FAK signaling transactivates the EGFR through the classical MMP pathway. The EGFR pulled down with GST-Gal-8 might be secondary to interaction with α5β1. The evidence indicates that MDCK cells under Gal-8 stimulation increase their proliferation and migration/invasion capabilities, requiring EGFR activity sustained by this FAK/MMP pathway. This pathway differs from the EGF-induced down-regulation of FAK activity required for EGF-mediated initial increase in tumor cell motility and invasion (Lu et al., 2001). FAK itself is involved in cell proliferation, migration, and invasion (Sieg et al., 2000; Seguin et al., 2015), as well as in EMT (Taliaferro-Smith et al., 2015). Therefore, FAK can contribute to EMT-related effects through additional routes beyond the EGFR. In addition, EGFR and TGFβR signaling are synergic in promoting EMT (Grande et al., 2002), and Gal-8 has recently been reported to activate TGFβR signaling (Sampson et al., 2016). Therefore, even though other converging EMT-driving pathways might participate in the tumorigenic EMT induced by Gal-8, at least, an enhanced FAK/EGFR signaling seems to be crucially involved.
MDCK-Gal-8H cells also show higher activity of MMP13 and uPA protease systems, as well as increased expression of uPAR, fibronectin, and α5β1 integrin. All these proteins can modulate EGFR activity (Jo et al., 2007; Meierjohann et al., 2010; Ye et al., 2014) and can underlie proliferation and invasiveness in a cooperative manner (Billottet et al., 2008; Yong et al., 2010). For instance, the uPA/uPAR system promotes directional invasion distributing to the leading edge of migrating cells (Blasi and Carmeliet, 2002; Smith and Marshall, 2010), while MMP13 is expressed in a broad range of primary malignant tumors and has been proposed as a potential biomarker for metastases (Balbin et al., 1999; Pivetta et al., 2011). Complexes containing EGFR, uPA/uPAR, α5β1-integrin, and fibronectin stimulate cell migration and tumor metastasis through converging signaling pathways (Aguirre-Ghiso et al., 1999; Monaghan-Benson and McKeown-Longo, 2006), including FAK (Sieg et al., 2000; Guo and Giancotti, 2004). Furthermore, EGFR signaling (Okada et al., 1997; Xue et al., 2006; Venkov et al., 2007), MMP13 (Billottet et al., 2008), uPA/uPAR (Lester et al., 2007), α5β1, and fibronectin have all been involved in EMT in different cellular contexts (Zeisberg and Neilson, 2009). Accordingly, MDCK-Gal-8H cells show characteristic hallmarks of EMT, including decreased expression of E-cadherin and increased levels of vimentin and Snail, as well as activated β-catenin (Zeisberg and Neilson, 2009). Therefore, Gal-8 emerges as a new and strong EMT-inducer, compromising a variety of regulation systems intertwined with EGFR function. The combined action of all these molecules can certainly support the exaggerated proliferation and migration/invasive activities of MDCK-Gal-8H cells.
MDCK-Gal-8H cells acquired higher capability for anchorage-independent growth in soft agar, indicating resistance to anoikis, a property usually correlated with oncogenic EMT and required for metastasis (Frisch et al., 2013; Nieto, 2013; Paoli et al., 2013). These MDCK-Gal-8H cells subcutaneously injected in NSG mice generate tumors contrasting with their MDCK counterpart. Some of the tumors show clear signs of invasive behavior, such as perineural invasion and skeletal muscle infiltration. Confluence of blood vessels toward the tumor and vascularized papillae suggest ongoing angiogenesis, a process that can be stimulated by Gal-8 (Delgado et al., 2010; Troncoso et al., 2014). Previous studies reported MDCK cells as nontumorigenic in adult nude mice (Stiles et al., 1976; U et al., 1985; Kadono et al., 1998) and NOD/Scid mice (Gopal et al., 2015), becoming tumorigenic after transformation with the Moloney sarcoma virus (U et al., 1985), v-src (Kadono et al., 1998), oncogenic H-Ras, or YBX1 transcription factor (Gopal et al., 2015). Our results suggest that long exposure to high levels of Gal-8 can promote a similar oncogenic-like tumorigenic phenotype in MDCK cells.
EMT frequently generates intermediate phenotypes (partial EMT) between the extreme epithelial and mesenchymal phenotypes (Zeisberg and Neilson, 2009; Nieto, 2013). This typically occurs during collective migration, when epithelial cells move together without losing adhesions between them (Nieto, 2013). MDCK-Gal-8H cells at subconfluence maintain most of the cobblestone appearance of parental MDCK cells, with motile projections but contrasting with the spindle shape, the mesenchymal phenotype of oncogen-transformed MDCK cells (Grande et al., 2002). As in collective migration, few MDCK-Gal-8H cells detach from the main epithelial mass during the wound-healing assay. We show that once reaching confluence, these cells lose EMT hallmarks and form monolayers with transepithelial resistance and apical/basolateral polarity, segregating Gal-8 secretion to the apical domain while β1-integrins are distributed basolaterally. Partial EMT and reversion also seems to occur in vivo. We show that tumors generated by MDCK-Gal-8H cells are heterogeneous, displaying regions of cells expressing E-cadherin with a mesenchymal phenotype, adjacent to epithelial structures with basolaterally distributed E-cadherin, suggesting different levels of EMT.
Enhanced proteasome activity is very likely instrumental to the Gal-8–driven EMT. Proteasome-mediated degradation of ubiquitinylated proteins regulates several transcriptional and signaling elements involved in EMT (Voutsadakis, 2012a, b). Changes in proteasome activity have been associated with either induction or repression of EMT, depending on the cellular context. For instance, hepatocarcinoma cell lines develop EMT under the expression of tetraspanin TM4SF5, involving down-regulation of proteasome expression, while in the absence of TM4SF5 these cells respond to proteasome inhibitors and develop EMT (Kim et al., 2011). Mammary epithelial cells down-regulate proteasome subunits during EMT and long-term inhibition of proteasome activity induces EMT associated with a stem-cell phenotype (Banno et al., 2016). On the contrary, in prostate cancer cells, proteasome inhibition abrogates EMT by decreasing NFκB activation, leading to down-regulation of Snail expression (Baritaki et al., 2009). Esophageal cancer cells also respond to proteasome inhibition by attenuating EMT (Taylor et al., 2010). MDCK cells treated with HGF or expressing v-Src tyrosine kinase undergo EMT clearly reflected in cell scattering, which can be blocked by proteasome inhibitors that avoid Rac1 down-regulation (Tsukamoto and Nigam, 1999; Lynch et al., 2006). Indeed, lowering proteolytic basal activity by direct proteasome inhibitors have pleiotropic effects that might hinder the interpretation of the proteasomal role in such a complex process as EMT (Voutsadakis, 2012a, b). Instead, here we show that Gal-8 increases proteasome activity through a FAK/EGFR pathway (Figure 8), whose inhibition not only abrogates the Gal-8–induced proteasome overactivity (Figure 8) but also the higher migration/invasion capabilities (Figure 4) and uPA activity (Figure 5E), which are characteristics of EMT. Furthermore, Gal-8–overexpressing MDCK cells, once reaching confluence, revert not only their EMT traits (Figure 6) but also their proteasome overactivity (Figure 9). Neither the inhibitors of the FAK/EGFR pathway nor the confluent condition decrease proteasome activity under the basal level of untreated MDCK cells. All this, together with previous observations in other cellular systems (Voutsadakis 2012a, b), strongly suggests that Gal-8 promotes partial EMT involving overactivation of a FAK/EGFR/proteasome pathway. To our knowledge, there is no precedent of any galectin- or FAK-mediated regulation of proteasome activity. Furthermore, the only reported example of EGFR-stimulated UPS is in C. elegans, but this involves up-regulation of genes for ubiquitin ligase elements related with lifespan rather than direct stimulation of proteasome activity (Liu et al., 2011). Our results indicate that Gal-8 elevates proteasome activity through a FAK/EGFR pathway that does not increase the mass of the proteasome and thus could involve a yet undefined post-translational modification.
Previous studies showed that the chimera type Gal-3 forms large lattices with cell-surface elements and facilitates ligand-induced EGFR activity, counteracting in this way the interactions of the activated EGFR with both the endocytic machinery and the tumor suppressor caveolin-1 (Partridge et al., 2004; Boscher and Nabi, 2013). In contrast, the tandem repeat Gal-9 reduces EGFR activation determining decreased proliferation in cholangiocarcinoma cell lines (Kobayashi et al., 2015) and induces apoptosis, loss in cell viability, and reduced colony formation in MDCK cells (Wiersma et al., 2015). Therefore, our finding that Gal-8 transactivates the EGFR suggests that different galectins can modulate EGFR function and epithelial biology in complementary or antagonistic manners.
The opposite segregation of Gal-8 and its interacting cell-surface receptor β1-integrin in polarized MDCK cells reveals a galectin-mediated cell regulation that depends on the asymmetric compartmentalization of the epithelial cell surface. Sorting of growth factors and their corresponding receptors to opposite cell-surface domains has previously been proposed as an important regulation system in polarized epithelial cells (Tanos and Rodriguez-Boulan, 2008). In the respiratory epithelium, apical secretion of heregulin and basolateral distribution of ErbB receptors, including EGFR, constitutes a paradigmatic example of a mechanism by which injury-driven loss of polarity allows for the productive interaction of a growth factor with its cognate signaling receptors, leading in this case to cell proliferation and wound healing (Vermeer et al., 2003). Here we show that Gal-8 is apically secreted, whereas β1-integrins are basolaterally distributed in polarized MDCK-Gal-8H cells. Tight junctions indeed prevent diffusion of Gal-8 to the basolateral side, precluding its interaction with β1-integrins and the consequent activation of the FAK/EGFR pathway. Accordingly, we show that Gal-8 only activates the proteasome from the basolateral side in polarized MDCK cells. This would explain why EMT traits, including proteasome activity, manifest only under subconfluent conditions and tend to dissipate when MDCK-Gal-8H cells reach confluence. Continuous Gal-8 interaction with β1-integrins would be required to maintain EMT in these cells.
Taken together, all these data provide new clues to understand and to counteract Gal-8–mediated cancer pathogenicity. Gal-8 is overexpressed in several carcinomas (Elola et al., 2014), including renal carcinomas associated with the worst prognosis (Liu et al., 2015). We recently described that Gal-8 silencing reduces proliferation and survival of glioblastoma cells, indicating an additional role in the malignancy of other cancerous cells (Metz et al., 2016). Carcinoma cells frequently coexist with differentiated epithelial cells in tumors (Nieto, 2013), and therefore they can generate a Gal-8 enriched microenvironment not only favorable for their own growth but also for tumorigenicity in neighboring epithelial cells.
MATERIALS AND METHODS
Antibodies, reagents, and plasmids
Companies and antibodies against the indicated proteins: Santa Cruz Biotechnology, Santa Cruz, CA ( α5-integrin, H104; uPA, H140; uPAR, FL-290; MMP13, H-230; β-actin C4; Lamin A; β-catenin); Millipore (Darmstadt, Germany) (phospho-Tyrosine 4G10); Sigma (St. Louis, MO) (fibronectin, #3648); BD Transduction Lab (Lexington, KY) (E-cadherin, BD610181; FAK, BD610088); ECM Biosciences (pFAK Tyr397, FM1211); Cell Signaling Technology (Danvers, MA) (vimentin #5741; E-cadherin, #); Abcam (Cambridge, UK) (Snail, ab180714); American Type Culture Collection (ATCC) (Manassas, VA) (EGFR, hybridoma HB8506); Bio SB (Santa Barbara, CA) (Ki67); Rockland (Boyertown, PA) (horseradish peroxidase [HRP]-conjugated secondary antibodies). Ulrike Kuckelkorn (Humboldt Universität zu Berlin, Germany) kindly provided proteasomal 20S α4 subunit antibodies, and Enrique Rodriguez-Boulan (Weill Cornell Medical College, New York, NY) the mouse monoclonal anti-GP135. EGFR984 antibodies were generated as described (Salazar and González, 2002). Human recombinant Gal-8 produced in Esherichia coli (Carcamo et al., 2006) was used for cell treatment and generation of rabbit polyclonal antibodies (anti-Gal-8C4). Reagents were purchased from Calbiochem (La Jolla, CA) (GM6001, PD98059, AG1478, ONO4817, Lactacystin, and Fluorogenic proteasome Substrates II, III, and VI); Santa Cruz Biotechnology, (Santa Cruz, CA) (FAK inhibitor Y15); Sigma (St. Louis, MO) (β-lactose, DMEM containing high glucose, protein-A-Sepharose); Life Technologies (Grand Island, NY) (fetal bovine serum [FBS]); Invitrogen (Carlsbad, CA) (cell-culture reagents); and Amersham (Piscataway, NJ) ([3H]thymidine and the enhanced chemiluminescence [ECL] system).
Cell culture, transfection, and proliferation
We used MDCK cells previously characterized for cell polarity, proliferation, and EGFR function (Burgos et al., 2004; Buvinic et al., 2007). Most of the results were also reproduced in MDCK cells from the ATCC provided by Enrique Rodriguez-Boulan (Weill Cornell Medical College, New York, NY) (Meiss et al., 1982). Cells were routinely analyzed by RT-PCR for mycoplasm contamination and cultured in DMEM supplemented with 7.5% FBS and antibiotics (100 U/ml penicillin, 100 mg/ml streptomycin, and 5 μg/ml Plasmocin). Permanently transfected cells with pcDNA3-myc-Gal-8 or pcDNA3-myc empty vector (MOCK) were generated using Lipofectamine 2000 and selection media containing 0.8 mg/ml geneticin sulfate (G418) and were maintained in 0.4 mg/ml G418, as described (Burgos et al., 2004; Soza et al., 2004; Buvinic et al., 2007). Cell proliferation was assessed by 3[H]thymidine incorporation (Buvinic et al., 2007), simultaneously adding TDG (10 mM), lactose (20 mM), sucrose (20 mM), or exogenous Gal-8 (GST-Gal-8 or as Gal-8 proteolitically released from GST) for 24 h.
Immunoprecipitation and immunoblotting
The EGFR was immunoprecipitated with the monoclonal HB8506 antibody, resolved by SDS–PAGE, and immunoblotted with anti-phosphotyrosine monoclonal antibody (mAb) 4G10 (1:1000), while EGFR mass was assessed by immunoblot with anti-EGFR984 (1:500), as described (Salazar and González, 2002; Buvinic et al., 2007; Shaughnessy et al., 2014). Gal-8 was immunoprecipited with anti-Gal-8C4 (produced in our laboratory) from cell extracts prepared in lysing buffer A (50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM, ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA), 2 mM MgCl2, 10% glycerol, and 1% Triton X-100, supplemented with 0.5 mM PMSF, 0.1 mM Pepstatin, and 0.1 mM Leupeptin antiproteases) and was then immunoblotted with the same antibody. To analyze protein secretion, trichloroacetic acid (TCA)-precipitated proteins from 48 h serum-free conditioned media was immunoblotted with anti-Gal-8 (1:500), anti-uPA (1:1000), and anti-MMP13 (1:1000). To assess the β-catenin distribution between cytosolic and nuclear fractions, the cells were suspended in lysing buffer B (10 mM HEPES, pH 8.0, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol [DTT], 10% NP-40, and antiproteases), vortexed for 10 s, and centrifuged for 30 s. The supernatant corresponds to the cytosolic fraction. The pellet nuclear fraction was lysed in Buffer C (20 mM HEPES, pH 8, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and antiproteases).
Immunofluorescence analysis
Cells grown on Transwell polycarbonate filters of 6.5 mm, pore diameter of 0.4 μm (Corning), were fixed in 4% paraformaldehyde and 4% sucrose, permeabilized with 0.2% Triton X-100, and treated for double indirect immunofluorescence. Primary antibodies: rabbit anti-E-cadherin (Cell Signalling), mouse anti-GP135 (provided by Enrique Rodriguez-Boulan, Cornell University), and Hoechst 33342 stain (Thermo Fisher Scientific). Secondary antibodies: anti-mouse Alexa-555 and anti-rabbit Alexa-488. The images were collected at 2048 × 2048 resolution in Z stacks of 300-nm steps using a Leica SP8 confocal microscope and a 63× oil immersion 1.4 ma lens.
RT-PCR
Transcripts of the following proteins were detected with the indicated primers:
Human Gal-8 (Forward 5′-ATACTCTGCTCTATGGCCAC-3′ Reverse 5′-TGGCATTTGCATTCACTTCT-3′)
Canine Gal-8 (Forward 5′-GACCTCAATCAACAATAAGG-3′ Reverse 5′-CTACCAGCTCCTTACTTCCA-3′).
Canine actin (Forward 5′-GATGACCCAGATCATGTTTG-3′ Reverse 5′-GTCAGGATCTTCATGAGGTA-3′);
Canine uPA (Forward: 5′-AAGCAGAGTTTCAATGTGGC-3′ Reverse: 5′-TGGGAATTAATGAAGCAGTGTG-3′);
Canine uPAR (Forward: 5′-TATGGGAAGATGGTGATGAGC-3′; Reverse: 5′-CGCAGGAAACACATTCTAGG-3′);
Canine MMP13 (Forward 5′-TTTGGAACTAAAGAGCATGGG-3′; Reverse: 5′-TCATCGGGAAGCATAAAGTG-3′).
Anchored-independent growth
Assays were performed in 12-well plates with 500 cells/well layered as a single cell suspension in 0.3% agar on top of 0.5% agar, as described (Lu et al., 2014; Shaughnessy et al., 2014). After 14 d, colonies were manually counted under a 10× objective in at least three wells per condition in each experiment. Colony areas were quantified with ImageJ software.
Zymograms
Zymography of urokinase and gelatinase activity was performed as described (Kleiner and Stetler-Stevenson, 1994; Santibanez et al., 1995). Proteins present in conditioned media were resolved by SDS–PAGE. To detect urokinase activity, the gel was then washed with 2.5% Triton X-100, placed over 1% agarose gel prepared in 50 mM Tris-HCl, pH 8, 20 mM CaCl2, and 0.05 μg/ml casein, and incubated for 24 h at 37°C to achieve a visible level of casein degradation. The reaction was stopped using 2% CuSO4. For detection of gelatinase activity, the samples were resolved in 10% SDS–PAGE containing 10 mg/ml gelatin, and the gel was incubated for 24 h at 37°C in 100 mM Tris-HCl, pH 8, and 20 mM CaCl2, stained with Coomassie blue.
Wound closure, invasion, and polarity assays
Cells (1 × 105) grown to 80% confluence on coverslips in medium with 5% FBS were deprived of serum for 16 h before wounding the monolayer with a micromanipulator, mounted on thermoregulated plate (Zeiss) at 37°C in DMEM-HEPES, pH 7.5, for time-lapse imaging in vivo. Digital images were taken at 30-min intervals with a 10× objective in a Zeiss Axiophot microscope using a 14-bit Zeiss Axiocam camera and Axiovision imaging software (Zeiss). The wound areas were quantified with MetaMorph imaging software (Universal Imaging, West Chester, PA). Treatments with Gal-8 or TDG were initiated 1 h before wounding. For invasion assays (Guerrero et al., 2004), Transwell polycarbonate filters of 6.5 mm, pore diameter of 8.0 μm (Corning), and coated with Matrigel solution (1 μg/ml) for 4 h at 37 °C were seeded with 5 × 104 cells and then incubated for 24 h, fixed, and stained with 0.2% crystal violet in 10% ethanol to assess the number of invading cells that traverse the filter. A domain-specific biotinylation assay was carried out in MDCK cells grown to confluence in Transwell polycarbonate filters of 24 mm diameter with a pore size of 0.4 μm. Apical and basolateral conditioned medium were collected separately for 18 h, precipitated with TCA, and analyzed by immunoblot. Biotinylated proteins were precipitated with neutroavidin-Sepharose followed by SDS–PAGE, immunoblot, and ECL detection (Thermo Scientific), as described (Burgos et al., 2004; Soza et al., 2004).
Proteasome activity assay
Cells (200,000/well) grown to confluence and left overnight in the absence of FBS were lysed in buffer A. Proteolysis of fluorogenic substrates (50 mM) for chymotrypsin-like (β5), trypsin-like (β2), and caspase-like (β1) activity of the 20S proteasome was measured in the cell lysates with a BioTek Synergy HT Fluorimeter using excitation and emission wavelengths of 390 and 460 nm, respectively (Mlynarczuk-Bialy et al., 2014).
Tumorigenesis in NOD scid gamma (NSG) mice
NSG mice (males, 25 g, 10 wk) were subcutaneously inoculated with 106 cells in the flanks, and tumors were allowed to develop for 10 wk. The mice were killed, and the tumors were removed, weighed, and measured, calculating their volume with the following formula: width2 × length × π/6, where length is the longest diameter and width is the perpendicular diameter of the tumor. All experimental protocols were performed in accordance with the United States National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee at Pontificia Universidad Católica de Chile.
Tumor histology and immunohistochemistry
Histology analysis and immunohistochemistry were performed in Formalin-fixed, paraffin-embedded sections of tumors, using primary antibodies diluted 1:200 in 10 mM Tris-HCl buffer (pH 7.8) and 1% bovine serum albumin (wt/vol), and secondary HRP-conjugated anti-rabbit IgG at 1:1000 dilution, following established protocols, as described (Espinoza et al., 2016).
Statistical analysis
The software GraphPad PRISM Version 6.0c (San Diego, CA) was used for statistical analysis. Data were analyzed with normality tests (D’Agostino and Pearson omnibus normality test, Shapiro-Wilk test, or Kolmogorov-Smirnov test) showing a normal distribution. Data are presented as the mean ± SEM values and differences were analyzed with the Student’s t test, one-way analysis of variance (ANOVA), or two-way ANOVA, and Mann-Whitney test, two-tailed, as indicated. Statistical significances correspond to *p < 0.05, **p < 0.005.
Supplementary Material
Acknowledgments
We thank Ulrike Kuckelkorn (Humboldt Universität zu Berlin, Germany) for antibodies against the proteasome α4 subunit and Enrique Rodriguez-Boulan (Weill Cornell Medical College) for monoclonal anti-GP135. This work received financial support from grants CONICYT PFB12/2007, Fondecyt 1141127 (A.G.), Fondecyt 1131122 (A.S.), Fondap-Conicyt 15130011, and Fondecyt 1130204 (J.C.R.). Postgraduate fellowships from CONICYT-Nacional are as follows: Doctorado 2006, Folio: 21060721, and Doctorado Complementary Support Folio 23090143 to C.O. and Doctorado 2013, Folio: 21130970, to R.S.
Abbreviations used:
- ECM
extracellular matrix
- EGFR
epidermal growth factor receptor
- EMT
epithelial–mesenchymal transition
- ERK
extracellular signal-regulated kinase
- FAK
focal adhesion kinase
- MDCK
Madin–Darby canine kidney
- MMP
matrix metalloproteinase
- NSG mice
NOD scid gamma mice
- TGF-β
transforming growth factor beta
- uPA
urokinase plasminogen activator
- uPAR
urokinase plasminogen activator receptor
- UPS
ubiquitin-proteasome system.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-05-0301) on January 3, 2018.
REFERENCES
- Adams J. (2004). The proteasome: a suitable antineoplastic target. Nat Rev Cancer , 349-360. [DOI] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Frankel P, Farias EF, Lu Z, Jiang H, Olsen A, Feig LA, de Kier Joffe EB, Foster DA. (1999). RalA requirement for v-Src- and v-Ras-induced tumorigenicity and overproduction of urokinase-type plasminogen activator: involvement of metalloproteases. Oncogene , 4718-4725. [DOI] [PubMed] [Google Scholar]
- Arbel-Goren R, Levy Y, Ronen D, Zick Y. (2005). Cyclin-dependent kinase inhibitors and JNK act as molecular switches, regulating the choice between growth arrest and apoptosis induced by galectin-8. J Biol Chem , 19105-19114. [DOI] [PubMed] [Google Scholar]
- Bacigalupo ML, Manzi M, Espelt MV, Gentilini LD, Compagno D, Laderach DJ, Wolfenstein-Todel C, Rabinovich GA, Troncoso MF. (2015). Galectin-1 triggers epithelial-mesenchymal transition in human hepatocellular carcinoma cells. J Cell Physiol , 1298-1309. [DOI] [PubMed] [Google Scholar]
- Balbin M, Pendas AM, Uria JA, Jimenez MG, Freije JP, Lopez-Otin C. (1999). Expression and regulation of collagenase-3 (MMP-13) in human malignant tumors. APMIS , 45-53. [DOI] [PubMed] [Google Scholar]
- Banno A, Garcia DA, van Baarsel ED, Metz PJ, Fisch K, Widjaja CE, Kim SH, Lopez J, Chang AN, Geurink PP, et al. (2016). Downregulation of 26S proteasome catalytic activity promotes epithelial-mesenchymal transition. Oncotarget , 21527-21541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baritaki S, Chapman A, Yeung K, Spandidos DA, Palladino M, Bonavida B. (2009). Inhibition of epithelial to mesenchymal transition in metastatic prostate cancer cells by the novel proteasome inhibitor, NPI-0052: pivotal roles of Snail repression and RKIP induction. Oncogene , 3573-3585. [DOI] [PubMed] [Google Scholar]
- Barres C, Blanc L, Bette-Bobillo P, Andre S, Mamoun R, Gabius HJ, Vidal M. (2010). Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood , 696-705. [DOI] [PubMed] [Google Scholar]
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. (2000). The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol , 84-89. [DOI] [PubMed] [Google Scholar]
- Billottet C, Tuefferd M, Gentien D, Rapinat A, Thiery JP, Broet P, Jouanneau J. (2008). Modulation of several waves of gene expression during FGF-1 induced epithelial-mesenchymal transition of carcinoma cells. J Cell Biochem , 826-839. [DOI] [PubMed] [Google Scholar]
- Blasi F, Carmeliet P. (2002). uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol , 932-943. [DOI] [PubMed] [Google Scholar]
- Boscher C, Nabi IR. (2013). Galectin-3- and phospho-caveolin-1-dependent outside-in integrin signaling mediates the EGF motogenic response in mammary cancer cells. Mol Biol Cell , 2134-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, Mostov KE. (2008). From cells to organs: building polarized tissue. Nat Rev Mol Cell Biol , 887-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos PV, Klattenhoff C, de la Fuente E, Rigotti A, Gonzalez A. (2004). Cholesterol depletion induces PKA-mediated basolateral-to-apical transcytosis of the scavenger receptor class B type I in MDCK cells. Proc Natl Acad Sci USA , 3845-3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buvinic S, Bravo-Zehnder M, Boyer JL, Huidobro-Toro JP, Gonzalez A. (2007). Nucleotide P2Y1 receptor regulates EGF receptor mitogenic signaling and expression in epithelial cells. J Cell Sci , 4289-4301. [DOI] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. (2000). The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol , 76-83. [DOI] [PubMed] [Google Scholar]
- Carcamo C, Pardo E, Oyanadel C, Bravo-Zehnder M, Bull P, Caceres M, Martinez J, Massardo L, Jacobelli S, Gonzalez A, Soza A. (2006). Galectin-8 binds specific beta1 integrins and induces polarized spreading highlighted by asymmetric lamellipodia in Jurkat T cells. Exp Cell Res , 374-386. [DOI] [PubMed] [Google Scholar]
- Carpenter G. (1999). Employment of the epidermal growth factor receptor in growth factor-independent signaling pathways. J Cell Biol , 697-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter G. (2000). The EGF receptor: a nexus for trafficking and signaling. Bioessays , 697-707. [DOI] [PubMed] [Google Scholar]
- Delacour D, Cramm-Behrens CI, Drobecq H, Le Bivic A, Naim HY, Jacob R. (2006). Requirement for galectin-3 in apical protein sorting. Curr Biol , 408-414. [DOI] [PubMed] [Google Scholar]
- Delacour D, Gouyer V, Zanetta JP, Drobecq H, Leteurtre E, Grard G, Moreau-Hannedouche O, Maes E, Pons A, Andre S, et al. (2005). Galectin-4 and sulfatides in apical membrane trafficking in enterocyte-like cells. J Cell Biol , 491-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado VM, Nugnes LG, Colombo LL, Troncoso MF, Fernandez MM, Malchiodi EL, Frahm I, Croci DO, Compagno D, Rabinovich GA, et al. (2010). Modulation of endothelial cell migration and angiogenesis: a novel function for the “tandem-repeat” lectin galectin-8. FASEB J , 242-254. [DOI] [PubMed] [Google Scholar]
- Desgrosellier JS, Cheresh DA. (2010). Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer , 9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ. (2014). Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol , 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elola MT, Ferragut F, Cardenas Delgado VM, Nugnes LG, Gentilini L, Laderach D, Troncoso MF, Compagno D, Wolfenstein-Todel C, Rabinovich GA. (2014). Expression, localization and function of galectin-8, a tandem-repeat lectin, in human tumors. Histol Histopathol , 1093-1105. [DOI] [PubMed] [Google Scholar]
- Espinoza JA, Garcia P, Bizama C, Leal JL, Riquelme I, Weber H, Macanas P, Aguayo G, Vinuela E, Roa JC, Nervi B. (2016). Low expression of equilibrative nucleoside transporter 1 is associated with poor prognosis in chemotherapy-naive pT2 gallbladder adenocarcinoma patients. Histopathology , 722-728. [DOI] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al. (2015). Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature , 472-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Schaller M, Cieply B. (2013). Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci , 21-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal SK, Greening DW, Hanssen EG, Zhu HJ, Simpson RJ, Mathias RA. (2016). Oncogenic epithelial cell-derived exosomes containing Rac1 and PAK2 induce angiogenesis in recipient endothelial cells. Oncotarget , 19709-19722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal SK, Greening DW, Mathias RA, Ji H, Rai A, Chen M, Zhu HJ, Simpson RJ. (2015). YBX1/YB-1 induces partial EMT and tumourigenicity through secretion of angiogenic factors into the extracellular microenvironment. Oncotarget , 13718-13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande M, Franzen A, Karlsson JO, Ericson LE, Heldin NE, Nilsson M. (2002). Transforming growth factor-beta and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J Cell Sci , 4227-4236. [DOI] [PubMed] [Google Scholar]
- Grande MT, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M, Rowe RG, Weiss SJ, Lopez-Novoa JM, Nieto MA. (2015). Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med , 989-997. [DOI] [PubMed] [Google Scholar]
- Guerrero J, Santibanez JF, Gonzalez A, Martinez J. (2004). EGF receptor transactivation by urokinase receptor stimulus through a mechanism involving Src and matrix metalloproteinases. Exp Cell Res , 201-208. [DOI] [PubMed] [Google Scholar]
- Guo W, Giancotti FG. (2004). Integrin signalling during tumour progression. Nat Rev Mol Cell Biol , 816-826. [DOI] [PubMed] [Google Scholar]
- Hadari YR, Arbel-Goren R, Levy Y, Amsterdam A, Alon R, Zakut R, Zick Y. (2000). Galectin-8 binding to integrins inhibits cell adhesion and induces apoptosis. J Cell Sci , 2385-2397. [DOI] [PubMed] [Google Scholar]
- Huang RY, Guilford P, Thiery JP. (2012). Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J Cell Sci , 4417-4422. [DOI] [PubMed] [Google Scholar]
- Ideo H, Matsuzaka T, Nonaka T, Seko A, Yamashita K. (2011). Galectin-8-N-domain recognition mechanism for sialylated and sulfated glycans. J Biol Chem , 11346-11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo M, Thomas KS, Takimoto S, Gaultier A, Hsieh EH, Lester RD, Gonias SL. (2007). Urokinase receptor primes cells to proliferate in response to epidermal growth factor. Oncogene , 2585-2594. [DOI] [PubMed] [Google Scholar]
- Kadono Y, Okada Y, Namiki M, Seiki M, Sato H. (1998). Transformation of epithelial Madin-Darby canine kidney cells with p60(v-src) induces expression of membrane-type 1 matrix metalloproteinase and invasiveness. Cancer Res , 2240-2244. [PubMed] [Google Scholar]
- Kaltner H, Gabius HJ. (2012). A toolbox of lectins for translating the sugar code: the galectin network in phylogenesis and tumors. Histol Histopathol , 397-416. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, Werb Z. (2010). Matrix metalloproteinases: regulators of the tumor microenvironment. Cell , 52-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kil SJ, Hobert M, Carlin C. (1999). A leucine-based determinant in the epidermal growth factor receptor juxtamembrane domain is required for the efficient transport of ligand- receptor complexes to lysosomes. J Biol Chem , 3141-3150. [DOI] [PubMed] [Google Scholar]
- Kim JY, Nam JK, Lee SA, Lee MS, Cho SK, Park ZY, Lee JW, Cho M. (2011). Proteasome inhibition causes epithelial-mesenchymal transition upon TM4SF5 expression. J Cell Biochem , 782-792. [DOI] [PubMed] [Google Scholar]
- Kleiner DE, Stetler-Stevenson WG. (1994). Quantitative zymography: detection of picogram quantities of gelatinases. Anal Biochem , 325-329. [DOI] [PubMed] [Google Scholar]
- Klibi J, Niki T, Riedel A, Pioche-Durieu C, Souquere S, Rubinstein E, Le Moulec S, Guigay J, Hirashima M, Guemira F, et al. (2009). Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood , 1957-1966. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Morishita A, Iwama H, Fujita K, Okura R, Fujihara S, Yamashita T, Fujimori T, Kato K, Kamada H, et al. (2015). Galectin-9 suppresses cholangiocarcinoma cell proliferation by inducing apoptosis but not cell cycle arrest. Oncol Rep , 1761-1770. [DOI] [PubMed] [Google Scholar]
- Lester RD, Jo M, Montel V, Takimoto S, Gonias SL. (2007). uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol , 425-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy Y, Arbel-Goren R, Hadari YR, Eshhar S, Ronen D, Elhanany E, Geiger B, Zick Y. (2001). Galectin-8 functions as a matricellular modulator of cell adhesion. J Biol Chem , 31285-31295. [DOI] [PubMed] [Google Scholar]
- Levy Y, Ronen D, Bershadsky AD, Zick Y. (2003). Sustained induction of ERK, protein kinase B, and p70 S6 kinase regulates cell spreading and formation of F-actin microspikes upon ligation of integrins by galectin-8, a mammalian lectin. J Biol Chem , 14533-14542. [DOI] [PubMed] [Google Scholar]
- Lindsey S, Langhans SA. (2015). Epidermal growth factor signaling in transformed cells. Int Rev Cell Mol Biol , 1-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Rogers J, Murphy CT, Rongo C. (2011). EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J , 2990-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Xu L, Zhu Y, Zhang W, Liu W, Liu H, Xu J. (2015). Galectin-8 predicts postoperative recurrence of patients with localized T1 clear cell renal cell carcinoma. Urol Oncol , 112, e111–118. [DOI] [PubMed] [Google Scholar]
- Long W, Yi P, Amazit L, LaMarca HL, Ashcroft F, Kumar R, Mancini MA, Tsai SY, Tsai MJ, O’Malley BW. (2010). SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol Cell , 321-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, et al. (2015). Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med , 998-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Wang X, Urvalek AM, Li T, Xie H, Yu L, Zhao J. (2014). Transformation of human ovarian surface epithelial cells by Kruppel-like factor 8. Oncogene , 10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Jiang G, Blume-Jensen P, Hunter T. (2001). Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol Cell Biol , 4016-4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch EA, Stall J, Schmidt G, Chavrier P, D’Souza-Schorey C. (2006). Proteasome-mediated degradation of Rac1-GTP during epithelial cell scattering. Mol Biol Cell , 2236-2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meierjohann S, Hufnagel A, Wende E, Kleinschmidt MA, Wolf K, Friedl P, Gaubatz S, Schartl M. (2010). MMP13 mediates cell cycle progression in melanocytes and melanoma cells: in vitro studies of migration and proliferation. Mol Cancer , 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiss HK, Green RF, Rodriguez-Boulan E. (1982). Lectin-resistant mutants of polarized epithelial cells. Mol Cell Biol , 1287-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J. (2006). Epidermal growth factor receptor targeting in cancer. Semin Oncol , 369-385. [DOI] [PubMed] [Google Scholar]
- Metz C, Doger R, Riquelme E, Cortes P, Holmes C, Shaughnessy R, Oyanadel C, Grabowski C, Gonzalez A, Soza A. (2016). Galectin-8 promotes migration and proliferation and prevents apoptosis in U87 glioblastoma cells. Biol Res , 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Schlaepfer DD. (2006). Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol , 516-523. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Teramoto H, Gutkind JS, Yamada KM. (1996). Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J Cell Biol , 1633-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlynarczuk-Bialy I, Doeppner TR, Golab J, Nowis D, Wilczynski GM, Parobczak K, Wigand ME, Hajdamowicz M, Bialy LP, Aniolek O, et al. (2014). Biodistribution and efficacy studies of the proteasome inhibitor BSc2118 in a mouse melanoma model. Transl Oncol , 570-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo D, Costa SA, Ihrke G, Youker RT, Pastor-Soler N, Hughey RP, Weisz OA. (2012). Sialylation of N-linked glycans mediates apical delivery of endolyn in MDCK cells via a galectin-9-dependent mechanism. Mol Biol Cell , 3636-3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan-Benson E, McKeown-Longo PJ. (2006). Urokinase-type plasminogen activator receptor regulates a novel pathway of fibronectin matrix assembly requiring Src-dependent transactivation of epidermal growth factor receptor. J Biol Chem , 9450-9459. [DOI] [PubMed] [Google Scholar]
- Moro L, Dolce L, Cabodi S, Bergatto E, Boeri Erba E, Smeriglio M, Turco E, Retta SF, Giuffrida MG, Venturino M, et al. (2002). Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J Biol Chem , 9405-9414. [DOI] [PubMed] [Google Scholar]
- Moro L, Venturino M, Bozzo C, Silengo L, Altruda F, Beguinot L, Tarone G, Defilippi P. (1998). Integrins induce activation of EGF receptor: role in MAP kinase induction and adhesion-dependent cell survival. EMBO J , 6622-6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabi IR, Shankar J, Dennis JW. (2015). The galectin lattice at a glance. J Cell Sci , 2213-2219. [DOI] [PubMed] [Google Scholar]
- Nagy N, Bronckart Y, Camby I, Legendre H, Lahm H, Kaltner H, Hadari Y, Van Ham P, Yeaton P, Pector JC, et al. (2002). Galectin-8 expression decreases in cancer compared with normal and dysplastic human colon tissue and acts significantly on human colon cancer cell migration as a suppressor. Gut , 392-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA. (2011). The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol , 347-376. [DOI] [PubMed] [Google Scholar]
- Nieto MA. (2013). Epithelial plasticity: a common theme in embryonic and cancer cells. Science , 1234850. [DOI] [PubMed] [Google Scholar]
- Nishi N, Shoji H, Seki M, Itoh A, Miyanaka H, Yuube K, Hirashima M, Nakamura T. (2003). Galectin-8 modulates neutrophil function via interaction with integrin alphaM. Glycobiology , 755-763. [DOI] [PubMed] [Google Scholar]
- Norambuena A, Metz C, Vicuna L, Silva A, Pardo E, Oyanadel C, Massardo L, Gonzalez A, Soza A. (2009). Galectin-8 induces apoptosis in Jurkat T cells by phosphatidic acid-mediated ERK1/2 activation supported by protein kinase A down-regulation. J Biol Chem , 12670-12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H, Danoff TM, Kalluri R, Neilson EG. (1997). Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol , F563-F574. [DOI] [PubMed] [Google Scholar]
- Paoli P, Giannoni E, Chiarugi P. (2013). Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta , 3481-3498. [DOI] [PubMed] [Google Scholar]
- Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, Dennis JW. (2004). Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science , 120-124. [DOI] [PubMed] [Google Scholar]
- Perez Bay AE, Schreiner R, Benedicto I, Rodriguez-Boulan EJ. (2014). Galectin-4-mediated transcytosis of transferrin receptor. J Cell Sci , 4457-4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivetta E, Scapolan M, Pecolo M, Wassermann B, Abu-Rumeileh I, Balestreri L, Borsatti E, Tripodo C, Colombatti A, Spessotto P. (2011). MMP-13 stimulates osteoclast differentiation and activation in tumour breast bone metastases. Breast Cancer Res , R105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. (1999). EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature , 884-888. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Croci DO. (2012). Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity , 322-335. [DOI] [PubMed] [Google Scholar]
- Reticker-Flynn NE, Malta DF, Winslow MM, Lamar JM, Xu MJ, Underhill GH, Hynes RO, Jacks TE, Bhatia SN. (2012). A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nat Commun , 1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizqiawan A, Tobiume K, Okui G, Yamamoto K, Shigeishi H, Ono S, Shimasue H, Takechi M, Higashikawa K, Kamata N. (2013). Autocrine galectin-1 promotes collective cell migration of squamous cell carcinoma cells through up-regulation of distinct integrins. Biochem Biophys Res Commun , 904-910. [DOI] [PubMed] [Google Scholar]
- Salazar G, González A. (2002). Novel mechanism for regulation of epidermal growth factor receptor endocytosis revealed by protein kinase A inhibition. Mol Biol Cell , 1677-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JF, Suryawanshi A, Chen WS, Rabinovich GA, Panjwani N. (2016). Galectin-8 promotes regulatory T-cell differentiation by modulating IL-2 and TGFbeta signaling. Immunol Cell Biol , 213-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santibanez JF, Maccioni RB, Martinez J. (1995). The secretion of urokinase-like plasminogen activator is inhibited by microtubule-interacting drugs. Cell Biochem Funct , 217-225. [DOI] [PubMed] [Google Scholar]
- Schoenenberger CA, Zuk A, Zinkl GM, Kendall D, Matlin KS. (1994). Integrin expression and localization in normal MDCK cells and transformed MDCK cells lacking apical polarity. J Cell Sci (Pt 2), 527-541. [DOI] [PubMed] [Google Scholar]
- Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. (2015). Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol , 234-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaughnessy R, Retamal C, Oyanadel C, Norambuena A, Lopez A, Bravo-Zehnder M, Montecino FJ, Metz C, Soza A, Gonzalez A. (2014). Epidermal growth factor receptor endocytic traffic perturbation by phosphatidate phosphohydrolase inhibition: new strategy against cancer. FEBS J , 2172-2189. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. (2000). FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol , 249-256. [DOI] [PubMed] [Google Scholar]
- Smith HW, Marshall CJ. (2010). Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol , 23-36. [DOI] [PubMed] [Google Scholar]
- Soza A, Norambuena A, Cancino J, de la Fuente E, Henklein P, Gonzalez A. (2004). Sorting competition with membrane-permeable peptides in intact epithelial cells revealed discrimination of transmembrane proteins not only at the trans-Golgi network but also at pre-Golgi stages. J Biol Chem , 17376-17383. [DOI] [PubMed] [Google Scholar]
- Stiles CD, Desmond W, Chuman LM, Sato G, Saier MH. (1976). Growth control of heterologous tissue culture cells in the congenitally athymic nude mouse. Cancer Res , 1353-1360. [PubMed] [Google Scholar]
- Sulzmaier FJ, Jean C, Schlaepfer DD. (2014). FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer , 598-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan V, Gengenbacher N, Stemmler MP, Kleemann JA, Brabletz T, Brabletz S. (2015). The ZEB1/miR-200c feedback loop regulates invasion via actin interacting proteins MYLK and TKS5. Oncotarget , 27083-27096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaferro-Smith L, Oberlick E, Liu T, McGlothen T, Alcaide T, Tobin R, Donnelly S, Commander R, Kline E, Nagaraju GP, et al. (2015). FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget , 4757-4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanos B, Rodriguez-Boulan E. (2008). The epithelial polarity program: machineries involved and their hijacking by cancer. Oncogene , 6939-6957. [DOI] [PubMed] [Google Scholar]
- Taylor MD, Liu Y, Nagji AS, Theodosakis N, Jones DR. (2010). Combined proteasome and histone deacetylase inhibition attenuates epithelial-mesenchymal transition through E-cadherin in esophageal cancer cells. J Thorac Cardiovasc Surg , 1224 1232.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP. (2002). Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer , 442-454. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. (2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol , 131-142. [DOI] [PubMed] [Google Scholar]
- Thijssen VL, Heusschen R, Caers J, Griffioen AW. (2015). Galectin expression in cancer diagnosis and prognosis: A systematic review. Biochim Biophys Acta , 235-247. [DOI] [PubMed] [Google Scholar]
- Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature , 414-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troncoso MF, Ferragut F, Bacigalupo ML, Cardenas Delgado VM, Nugnes LG, Gentilini L, Laderach D, Wolfenstein-Todel C, Compagno D, Rabinovich GA, Elola MT. (2014). Galectin-8: A matricellular lectin with key roles in angiogenesis. Glycobiology , 907-914. [DOI] [PubMed] [Google Scholar]
- Tsukamoto T, Nigam SK. (1999). Cell-cell dissociation upon epithelial cell scattering requires a step mediated by the proteasome. J Biol Chem , 24579-24584. [DOI] [PubMed] [Google Scholar]
- U HS, Boerner P, Rindler MJ, Chuman L, Saier MH., Jr (1985). Characterization of chemically and virally transformed variants of Madin-Darby canine kidney (MDCK) epithelial cells. J Cell Physiol , 299-307. [DOI] [PubMed] [Google Scholar]
- Venkov CD, Link AJ, Jennings JL, Plieth D, Inoue T, Nagai K, Xu C, Dimitrova YN, Rauscher FJ, Neilson EG. (2007). A proximal activator of transcription in epithelial-mesenchymal transition. J Clin Invest , 482-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeer PD, Einwalter LA, Moninger TO, Rokhlina T, Kern JA, Zabner J, Welsh MJ. (2003). Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature , 322-326. [DOI] [PubMed] [Google Scholar]
- Vicuña L, Pardo E, Curkovic C, Doger R, Oyanadel C, Metz C, Massardo L, Gonzalez A, Soza A. (2013). Galectin-8 binds to LFA-1, blocks its interaction with ICAM-1 and is counteracted by anti-Gal-8 autoantibodies isolated from lupus patients. Biol Res , 275-280. [DOI] [PubMed] [Google Scholar]
- Vinik Y, Shatz-Azoulay H, Vivanti A, Hever N, Levy Y, Karmona R, Brumfeld V, Baraghithy S, Attar-Lamdar M, Boura-Halfon S, et al. (2015). The mammalian lectin galectin-8 induces RANKL expression, osteoclastogenesis, and bone mass reduction in mice. eLife , e05914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voutsadakis IA. (2012a). The ubiquitin-proteasome system and signal transduction pathways regulating epithelial mesenchymal transition of cancer. J Biomed Sci , 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voutsadakis IA. (2012b). Ubiquitination and the Ubiquitin-Proteasome System as regulators of transcription and transcription factors in epithelial mesenchymal transition of cancer. Tumour Biol , 897-910. [DOI] [PubMed] [Google Scholar]
- Wang LP, Chen SW, Zhuang SM, Li H, Song M. (2013). Galectin-3 accelerates the progression of oral tongue squamous cell carcinoma via a Wnt/beta-catenin-dependent pathway. Pathol Oncol Res , 461-474. [DOI] [PubMed] [Google Scholar]
- Wiersma VR, de Bruyn M, Wei Y, van Ginkel RJ, Hirashima M, Niki T, Nishi N, Zhou J, Pouwels SD, Samplonius DF, et al. (2015). The epithelial polarity regulator LGALS9/galectin-9 induces fatal frustrated autophagy in KRAS mutant colon carcinoma that depends on elevated basal autophagic flux. Autophagy , 1373-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C, Wyckoff J, Liang F, Sidani M, Violini S, Tsai KL, Zhang ZY, Sahai E, Condeelis J, Segall JE. (2006). Epidermal growth factor receptor overexpression results in increased tumor cell motility in vivo coordinately with enhanced intravasation and metastasis. Cancer Res , 192-197. [DOI] [PubMed] [Google Scholar]
- Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, Weinberg RA. (2015). Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature , 256-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Zhou X, Li X, Tang Y, Sun Y, Fang J. (2014). Inhibition of epidermal growth factor receptor signaling prohibits metastasis of gastric cancer via downregulation of MMP7 and MMP13. Tumour Biol , 10891-10896. [DOI] [PubMed] [Google Scholar]
- Yong HY, Kim IY, Kim JS, Moon A. (2010). ErbB2-enhanced invasiveness of H-Ras MCF10A breast cells requires MMP-13 and uPA upregulation via p38 MAPK signaling. Int J Oncol , 501-507. [PubMed] [Google Scholar]
- Zeisberg M, Neilson EG. (2009). Biomarkers for epithelial-mesenchymal transitions. J Clin Invest , 1429-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. (2015). Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature , 525-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.