

Abstract
Purpose
The retinoblastoma tumor suppressor (RB), a key regulator of cell cycle progression and proliferation, is functionally suppressed in up to 50% of non-small cell lung cancer (NSCLC). RB function is exquisitely controlled by a series of proteins including the CyclinD-CDK4/6 complex. In the current study, we interrogated the capacity of a CDK4/6 inhibitor, palbociclib, to activate RB function.
Experimental Design and Results
We employed multiple isogenic RB proficient and deficient NSCLC lines to interrogate the cytostatic and cytotoxic capacity of CDK 4/6 inhibition in vitro and in vivo. We demonstrate that while short term exposure to palbociclib induces cellular senescence, prolonged exposure results in inhibition of tumor growth. Mechanistically, CDK 4/6 inhibition induces a pro-apoptotic transcriptional program through suppression of IAPs FOXM1 and Survivin, while simultaneously augmenting expression of SMAC and Caspase 3 in an RB-dependent manner.
Conclusions
This study uncovers a novel function of RB activation to induce cellular apoptosis through therapeutic administration of a palbociclib and provides a rationale for the clinical evaluation of CDK 4/6 inhibitors in the treatment of NSCLC patients.
Keywords: Retinoblastoma Protein, palbociclib, SMAC, Apoptosis and FOXM1
Introduction
Cell cycle progression from G1 to S phase is tightly regulated via phosphorylation of the retinoblastoma tumor suppressor (RB) by the CDK4/6-cyclin D complex. It is well established that p16 inhibits the catalytic activity of the CDK4/6-cyclin D complex resulting in cell cycle arrest. Expression of RB and p16 are inversely correlated across multiple cancers; thus RB-positive tumors tend to have low levels of p16 and active CDK4/6-cyclin D. Within the context of non-small cell lung cancer (NSCLC) p16 is mutated, deleted, or silenced via methylation of CpG islands, in 10-50% of cases, permitting constitutive phosphorylation of RB and cell cycle progression (1). p16 function can be mimicked pharmacologically using a CDK4/6 inhibitor (2).
Recently, a CDK4/6 specific inhibitor, PD-0332991, palbociclib, was FDA-approved for the management of women with breast cancer (2). Clinically, this drug is delivered for the first 21 days of a 28-day cycle and there are currently over 10 active clinical trials testing CDK4/6 inhibitors in patients with NSCLC. Management of NSCLC has been dramatically enhanced through targeting unique mutations such as EGFR and ALK (3). We sought to elucidate mechanistically the functional consequence of CDK4/6 inhibition on both RB proficient and RB deficient NSCLC.
In this study, we utilized two isogenic RB proficient and deficient NSCLC cell lines. As expected, RB activation led to cell cycle arrest and cellular senescence. However, surprisingly, in vivo analysis demonstrated that RB activation via CDK 4/6 inhibition resulted in apoptosis. Mechanistically, we demonstrate that intact RB represses the expression of two inhibitors of apoptosis proteins (IAPs), FOXM1 and Survivin, which enable SMAC and cytochrome C to activate cleaved caspase 3 and promote the apoptotic pathway. In the absence of functional RB, FOXM1 and Survivin form a protein complex with Caspase 3, inhibiting the apoptotic machinery. This novel finding suggests a more potent role of CDK4/6 inhibitors in cancer management.
Materials and Methods
Cell Culture
Parental KRAS wild type H1299 and KRAS mutant H460 or A459 cells, were provided by Dr. Bo Lu and Dr. Sunday Shoyele (Department of Radiation Oncology and Dept. of Pharmacology and Experimental Therapeutics, Thomas Jefferson University, Philadelphia). Cell lines were authenticated by DDS Medical. shCon H1299, shRB H1299, shCon H460, shRB H460, shCon and shSMAC cells were maintained in improved minimum essential medium (IMEM) supplemented with 10% FBS (heat-inactivated FBS) and maintained at 37°C in a humidified 5% CO2 incubator.
Genetic modulation of RB or FOXM1 or Survivin/BIRC5 or SMAC in NSCLC Cells with Luciferase Expression
Stable Knockdown of RB or FOXM1, or Survivin or SMAC was carried out as previously described (4,5). RB deficient lines were generated using retroviral infection, while SMAC, FOXM1 and Survivin stable knockdown was performed with lentiviral constructs (Santa Cruz, California). shRB, shFOXM1, shSmac and shSurvivin stable polyclonal populations were puromycin selected and knockdown was verified using qRT-PCR or immunoblotting as previously described (4,5). shRNA nucleotide sequences are provided in Supplemental Table 1. Further, RB proficient and deficient cells were infected with lentiviral constructs coding luciferase and selected using G418 antibiotic (Thermo Fisher Scientific, Waltham, MA).
RNA Analysis
Total RNA was isolated from RB-proficient and RB-deficient H1299 and H460 cells treated with PD 0332991 (500 nM) using Trizol reagent (Invitrogen). The concentration and quality of RNA was analyzed using a Nanodrop. Total RNA was reverse transcribed and subjected to semi-quantitative PCR or real time PCR. Real time PCR was performed with an ABI Step-One apparatus using the Power SYBR Green Master Mix. Target mRNA primers for RB, PCNA, CycinA, and GAPDH were used. The signals were normalized with an internal control GAPDH and quantitated by ΔΔCT values. The primers are presented in the supportive information, Supplemental Table 2.
Human Transcriptome Array Profiling and Identification of E2F Regulated Signatures involved in apoptosis signaling
RNA was isolated from RB-proficient H1299 cells after three-week treatment with PD 0332991 (500 nM). Human transcriptome array (HTA) 2.0 was used and the microarray analysis and gene signatures were performed using GeneSpring v14.5 and Ingenuity Pathway Analysis software and GSEA were used to identify disease function, senescence and apoptosis, RB/E2F gene signatures. Microarray data were deposited at Gene Expression Omnibus (GEO): GSE87879 (H1299). Targets were validated via qRT-PCR using SYBR Green in StepOne Plus PCR Thermocycler (Applied Biosystems). The signals were normalized with respective GAPDH control signals and quantitated using ΔΔCT values, as described (5).
Immunoblot Analysis
Briefly, shCon and shRB cells treated with PD 0332991 (500 nM) for three weeks and were harvested by trypsinization, and cell lysis was carried out in radio-immunoprecipitation assay (RIPA) buffer [(150 mmol/L NaCl, 1% NP40, 0.5% deoxycholate, 0.1% SDS, 50 mmol/L Tris (pH, 8.0)] supplemented with protease inhibitors, phosphatase inhibitors, and phenyl methylsulfonyl fluoride. After sonication, lysates were clarified, and protein concentrations were determined using Bio-Rad Protein Assay Reagent. Protein was subjected to SDS-PAGE and transferred onto Immobilin-P PVDF transfer membranes (Millipore Corp).
The membranes were immunoblotted for RB (BD Sciences, USA), phospho-RB (phospho-serine 780), PCNA, CDK4, CDK6, CyclinA, Caspase3, Cleaved caspase3, SMAC, FOXM1, Survivin/BIRC5, LaminB and GAPDH (Santa Cruz Inc., USA), p16 antibody from Proteintech (USA), Annexin V from GeneTex (Irvine, CA, USA). Protein signals were visualized via X-ray film using enhanced Western lightening chemiluminescence (Perkin-Elmer Life Sciences) and normalized with LaminB or GAPDH loading control.
Co-Immunoprecipitation Assay
Total cell lysates from RB deficient H1299 and H460 cells overexpressing Flag tagged human-SMAC (Vigene Biosciences, MD, USA) or human FOXM1 or human Survivin cDNA’s from (Origene, Rockville, MD, USA), were immunoprecipitated with either FOXM1 or Survivin or SMAC antibody and immunoblotted with FOXM1, Survivin (BIRC5), Caspase 3, Cleaved Caspase 3, LaminB, or GAPDH (Santa Cruz Inc., Sigma Inc. and Cell Signal). Signals were normalized with LaminB or GAPDH internal controls.
Flow Cytometry Analysis
Cancer cell proliferation assay was performed via BrdU incorporation and coupled with flow cytometry analysis as described previously (6,7). RB-proficient and deficient H1299 and H460 cells and SMAC overexpressing H1299 and H460 cells treated with either PD 0332991 or DMSO were pulse-labeled with BrdU for 2 hours prior to harvest. Cells were fixed in 75% ethanol, pelleted, re-suspended in 2N HCl + 0.5 mg/mL pepsin, and incubated at room temperature for 30 minutes. Sodium tetraborate (0.1 mol/L) was added to neutralize 2N HCl. The cell pellets were washed with IFA buffer, centrifuged, and then washed with IFA + 0.5% Tween 20 solution. The pellet was resuspended in IFA solution containing FITC conjugated anti-BrdU antibody (BD Bioscience), incubated in the dark at room temperature for 45 minutes, washed with IFA + 0.5% Tween-20 and pelleted. The pellet was then suspended in PBS containing propidium iodide (0.2 μg/μL) and analyzed for BrdU incorporation using Beckman Coulter Cell Lab Quanta SC flow cytometer. Flow cytometry analysis was performed using FlowJo software (GE Healthcare, Piscataway, NJ, USA).
Cellular Apoptosis and Reactive Oxygen Species (ROS) Measurements
RB proficient and deficient H1299 xenografts and H460 cells were exposed to CDK4/6 specific inhibitor (PD 0332991) for three weeks (500 nM in in vitro or 150 mg/kg in vivo). At the end of the experiment, total lysates were made. The total cell lysates were analyzed for apoptosis or cell death using human apoptosis antibody array (Abcam) or cell death detection ELISA (Roche) according to the manufacture’s protocol. ROS generation in shCon or shRB or or shFOXM1 or shSMAC or Adeno Flag-SMAC (Vgene, USA) overexpressing H1299, and H460 cells in response to vehicle or PD 0332991 were measured by Dichlorofluorescin fluorescence. Briefly, 1×106cells were treated with DCFDA (1μM) and incubated for 30 min in dark at 37°C. At the end of incubation, cells were washed once and re-suspended in phosphate buffered saline. DCF fluorescence was measured using 100μl of cell suspension in a plate reader at excitation 525 nm and emission at 575 nm (8).
Cellular Toxicity and Cell Viability Analysis
shCon or shRB or or shFOXM1 or shSMAC or Adeno Flag-SMAC were exposed to various conditions (Vehicle, PD 0332991, cisplatin (20 μM), 2Gy ionizing radiation [Pantak radiator, Thomas Jefferson University], or combination thereof) as indicated. Viable cells were counted using trypan blue exclusion assay.
Mapping of RB/E2F binding sites on FOXM1, Survivin/BIRC5, PCNA, and CyclinA2 Promotor and Chromatin Immunoprecipitation Assay
1KB human RHAMM promoter was analyzed putatively for transcription factor binding sites using bioinformatics tools and programs, including Match-Public (www.gene-regulation.com/cgi-bin/pub/programs/match/match.cgi), AliBaba2 (wwwiti.cs.uni-magdeburg.de/∼grabe/alibaba2), and transcription Element Search software that utilizes the Transcription Factor Database (bioinformer.ebi.ac.uk:80/newsletter/archives/2/tess.html). The affinity of RB on FOXM1 or BIRC5 or CCNA2, or PCNA promoters were determined by ChIP assay as described (5). Briefly, RB-proficient and RB-deficient H1299 or H460 cells were treated overnight with PD 0332991 (500 nM), cross-linked with formaldehyde and processed for chromatin immunoprecipitation analysis as described. Equal concentrations of chromatin from all treatment groups were pre-cleared with protein G or Protein A Dynabeads in the presence of bovine serum albumin to reduce the non-specific background. After removal of beads by centrifugation, 2 micrograms of RB antibody or ACH4 (BD Sciences, USA. Cell Signaling, USA) was added and kept overnight at 4°C on a rotary platform. The immunoprecipitated DNA was purified using PCR purification kit (Qiagen, Valencia, CA) and suspended in 50 μl of sterile water. Purified DNA was then amplified via semi- or real-time quantitative PCR to analyze RB bound on to the promoters of FOXM1, Survivin/BIRC5, CyclinA or PCNA promoters with their respective target primers. The primer sequences are presented in Supplemental Table 1.
Input DNA served as a positive control whereas mouse IgG, or rabbit mouse monoclonal anti-GFP served as a negative control. PCR amplified products were resolved on agarose gel, and the images were captured using BioRad HemiDoc Imager (BioRad Laboratories, Hercules, CA). All real-time PCR analysis was performed with SYBR Green in a StepOne Plus PCR Thermocycler (Applied Biosystems). The signals were normalized with respective IgG or input signals and quantitated using ΔΔCt values as described (5).
Docking of E2F with FOXM1, Survivin/BIRC5, CyclinA, and PCNA promoters
The potential RB/E2F transcription factor binding sites on FOXM1, Survivin/BIRC5, CyclinA, or PCNA promoter DNA binding site were modeled under program coot (8) using the crystal structure of an E2F4-DP2-DNA complex as a template (9). The resultant model was subjected to iterative energy minimization routines using macromolecular docking program HEX (10) to generate 15 docking models with low interaction energies and clash-free spatial conformations. The model with lowest interaction energy is displayed using molecular viewer program PyMOL.
β-Galactosidase Assay
Briefly, shCon, shRB H1299 and H460 cells were grown in the presence of DMSO or PD-0332991 (500 nM) for two weeks and were processed for β-galactosidase activity as described earlier. Staining was carried out according to manufacturers provided protocol (Cell Signaling Technology). β-Galactosidase-positive cells were scored and calculated as a percentage of total cell population.
In silico TCGA Analysis
RB1 GISTIC putative copy number estimates and mRNA RSEM expression estimates of selected target genes were obtained from the cBioPortal for Cancer Genomics using the MATLAB CGDS Cancer Genomics Tool box (1,11). Both RB1 GISTIC copy number estimates and RSEM mRNA expression values were available for 512 lung adenocarcinoma cases, and both were available for 501 lung squamous cell carcinoma cases. Box plots were created for select target gene mRNA expression as a function of RB1 GISTIC copy number score (including −2: homozygous loss, −1: heterozygous loss, 0: normal diploid, and ≤+1: copy gain). Differential expression p-values between the copy-number states were estimated using one-way ANOVA.
Mouse Xenograft and Immunohistochemistry
Mouse Xenografts were generated as described previously. RB-proficient or RB-deficient H1299 or H460 cells at a concentration of 2×106 or 5×105 were individually mixed (1:1) with matrigel in a 200μL volume (BD Biosciences), and then implanted subcutaneously into the flanks of 4- to 6-week-old female nude mice (Taconic, USA). Tumor growth was measured periodically using calipers or the IVIS imaging system with RediJect D-Luciferin as described (12). Once the tumors reached 100-120 mm3 volumes, animals were given PD 0332991 (150 mg/kg body weight in sodium lactate buffer, pH 4.0) or vehicle daily via oral gavage. Three weeks post PD 0332991 treatment for H1299 tumor bearing animals or 15 days for H460 tumor bearing animals were euthanized, tumors were excised, imaged and fixed in 10% neutral buffered formalin for 24 h followed by 70% ethanol and were then embedded in paraffin. All animal experiments were conducted in accordance with the NIH Guide for Care and Use of Laboratory Animals and were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee. Xenografts were analyzed for pRBS780, PCNA, MCM7 and LaminB. IHC analysis was carried out as described in (5). Paraffin embed shCon H1299 or shRB H1299 induced tumor xenografts were sectioned and stained for H&E. Tissue sections were scanned on a ScanScope™ XT, with an average scan time of 120 seconds (compression quality 70). For the nuclear analysis, the total area obtained from the cellblock was analyzed counting the number of cells, the average nuclear size, the cellularity ratio (cell count/area) and the intensity of the nuclear stain. For the cytoplasmic analysis areas of staining were color separated from hematoxylin counter-stained sections and the intensity of the staining was measured on a continuous 0 (black) to 255 (bright white) intensity scale.
Statistics
Statistical analyses were performed using GraphPad Prism (version 6.0) software (GraphPad PrismSoftware, Inc). All the data were analyzed for statistical significance using Student’s t-test/ one-way ANOVA. For all experiments, p < 0.05 was considered statistically significant.
Results
Activated RB Inhibits Cell Cycle Progression
The retinoblastoma tumor suppressor is altered in 4-30% of non-small cell lung cancers (1,11). In order to delineate the functional impact of RB loss, two isogenic cell lines (H1299 and H460) with stable knockdown of RB were generated. Genetically, these lines are distinct in that H1299 has homozygous deletion of p53 and wildtype KRAS, whereas H460 harbors mutant KRAS. RB knockdown was confirmed by mRNA and protein analysis (Figure 1A). Loss of RB allows E2F transcription factors to bind to promoters facilitating a transcriptional program, which enables G1/S cell cycle progression. The functional consequence of RB loss was verified using two established RB/E2F targets, CyclinA and PCNA. As compared to RB proficient cells, RB deficient cells demonstrated elevated basal transcripts (Figure 1B) and protein levels (Figure 1C) of CyclinA and PCNA. Further, activation of RB, via CDK4/6 inhibition, resulted in suppression of these targets in an RB-dependent manner (Figure 1B and C). CDK4/6 inhibition triggered hypophosphorylation of RB protein (pRB) as compared to vehicle treatment (Figure 1C).
Figure 1. RB activation via CDK4/6 inhibition restricts NSCLC growth.
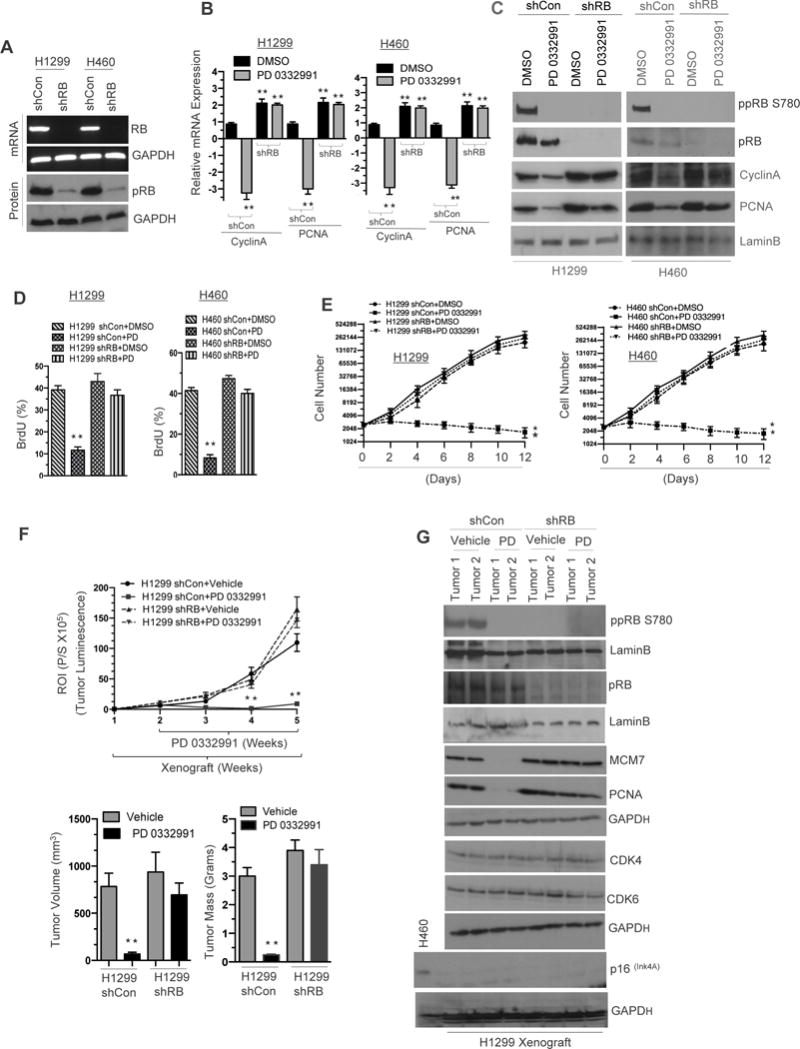
(A) RB mRNA and RB immune blot analysis in RB proficient and deficient H1299 and H460 non small cell lung cancer (NSCLC) cells and the loading controls GAPDH mRNA and LaminB. (B) qRT-PCR analysis of E2F targets CyclinA and PCNA in RB proficient and deficient H1299 and H460 cells (C) Immunoblot analysis of ppRB, S780, pRB, CyclinA, PCNA and a loading control LaminB in RB proficient and deficient H1299 and H460 cells. (D) Flow cytometric analysis of BrdU incorporation in RB proficient and deficient H1299 and H460 cells in response CDK4/6 inhibitor (500 nM). (E) Graphic representation of growth analysis in RB proficient and RB deficient H460 and H1299 (shCon and shRB) cells in response to CDK4/6 inhibitor (500 nM) for 12 days in culture. (F) Graphic representation (top panel) of IVIS derived tumor luminescence of shCon and shRB H1299 cell line induced xenografts in response to CDK4/6 inhibition (150mg/kg body wt) and a graphic representation of tumor volume and tumor mass of shCon and shRB H1299 cell line induced xenografts (bottom panel) in response to CDK4/6 inhibition for three weeks (150mg/kg body wt). (G) Immunoblotting analysis of pRB, ppRBS780, LaminB, MCM7, PCNA, CDK4, CDK6, p16 and GAPDH loading control in shCon and shRB H1299 xenografts in response to CDK4/6 inhibition for three weeks (150mg/kg body wt). Each data point is a mean ± SD 5 or more animals or three or more independent experiments. **p <0.05 were considered as statistically significant.
To ensure that repression of CyclinA and PCNA expression were mediated via canonical RB /E2F function, the RB-E2F consensus binding sites on their respective promoters were identified (Supplemental Figure 1A). Further chromatin immunoprecipitation indicated that upon CDK4/6 inhibition, RB binds robustly on the CyclinA and PCNA promoter sites with concurrent deacetylation of histone 4 (Supplemental Figure 1B), indicating that RB binding results in a closed chromatin state that represses cyclin A and PCNA transcription. Conversely, no binding was detected in the context of RB loss. Binding affinities on the promoters were further confirmed through an in silico-docking model (Supplemental Figure 1C). Interestingly, RB status did not alter cell cycle under basal conditions as measured by pulsed BrdU incorporation, however, exposure to the CDK4/6 inhibitor significantly reduced the proportion of cells within S-phase in the RB proficient setting only (Figure 1D). This translated into a statistically significant RB-dependent decrease of in vitro cell growth in both NSCLC (H1299 and H460) cell lines when exposed to a CDK 4/6 inhibitor (Figure 1E).
CDK4/6 Inhibition Restricts Tumor Growth and Tumor Burden
RB-proficient and RB-deficient luciferase expressing H1299 and H460 cells were implanted into nude mice. Upon reaching 100-120 mm3, CDK4/6 inhibitor, (palbociclib) was administrated orally for 21 days as per the FDA clinical indication. Weekly luciferase assessment of tumor growth demonstrated that CDK4/6 inhibition significantly reduced tumor burden in RB-proficient tumors, but did not impact growth on RB-deficient tumors (Figure 1F, top panel, Supplemental Figures 1D, 1E top panel, and 1F). In vivo tumor volume and mass correlated with luciferase activity (Figure 1F, bottom panel, Supplemental Figure 1E bottom panel). Xenografts were harvested and analyzed for RB phosphorylation status and RB target genes; protein analysis demonstrate that RB was activated via hypophosphorylation resulting in repression of RB/E2F target genes MCM7 and PCNA in RB-proficient tumors (Figure 1G, Supplemental Figure 1G) with no change in either CDK4 or CDK6 protein levels. These results demonstrate that CDK4/6 inhibition restricts the cell cycle in an RB-dependent manner and resulting in decreased tumor burden.
RB-Induced Cellular Senescence
Recent investigations have provided provocative evidence that CDK4/6 inhibition can trigger irreversible cell cycle withdrawal, a state referred to as senescence (13). To investigate whether CDK4/6 inhibition induced cellular senescence was dependent on functional RB, RB-proficient and RB-deficient H1299 and H460 cells were exposed to PD 0332991 for two weeks and analyzed for the expression of the senescence marker β-galactosidase activity. Interestingly, elevated levels of β-galactosidase activity were observed in RB-proficient cells in response to PD 0332991, while RB-deficient cells failed to induce β-galactosidase, suggesting an RB-dependent senescence program (Supplemental Figure 2A). Microarray data derived from RB-proficient H1299 cells under vehicle and CDK4/6 inhibitor (PD 0332991) treated conditions were subjected to Ingenuity Pathway Analysis (IPA) pathway analysis and PD 0332991 treated cells demonstrated an enhanced cellular senescence via differentially regulated gene signatures (Supplemental Figure 2B). Further, Gene Set Enrichment Analysis (GSEA) of array data indicates de-enrichment of senescence-related gene sets contained within the Molecular Signatures Database (MSigDB) (Supplemental Figure 2B, left and right panel). The network demonstrates that senescence involved signature interactions (Supplemental Figure 2C). These findings confirm that RB is a key regulator of cellular senescence in NSCLC and may result in the RB-dependent inhibition of cellular growth.
CDK 4/6 Inhibition Impacts on Cellular Apoptosis
Immunohistochemical analysis of tumor sections revealed an unexpected finding; an apoptotic as opposed to senescent mechanism for RB-dependent tumor inhibition. The role of RB in promoting or inhibiting apoptosis depends considerably on the context and apoptotic cues (14). While, RB-proficient cells treated with vehicle (Figure 2A) showed clear mitotic figures, those cells exposed to the CDK4/6 inhibitor exhibited smaller cell membranes with wrinkled nuclei; a hallmark of apoptosis. In contrast, RB-deficient tumors, regardless of treatment condition, maintained mitotic figures, indicating that the apoptotic response to CDK 4/6 inhibition in the context of NSCLC is an RB-dependent process (Figure 2A). To further validate that apoptosis was a driving mechanism within this system, RB-proficient and RB-deficient xenografts exposed to CDK4/6 inhibitor were analyzed for apoptotic protein markers cytochrome C and cleaved caspase 3. Only RB-proficient xenografts treated with the CDK 4/6 inhibitor expressed high levels of cytochrome C and cleaved caspase 3 (Figure 2B). In addition, RB specificity was demonstrated using a cytoplasmic histone-associated DNA fragments after induced cell death immunoassay (Figure 2C). Concomittantly, elevated levels of p27, and Smac were observed (Figure 2D). Given that apoptosis is a complex biological process, we performed a human apoptosis antibody array to determine which components of the apoptotic pathway were critical for CDK 4/6 inhibition induced apoptosis. We noted that the following cellular apoptosis markers SMAC, p27, HTRA, TNFR-1, and Caspase 3 were strongly induced following therapeutic challenge of RB proficient tumors with a CDK4/6 inhibitor in both NSCLC cell lines (Figure 2E). Confocal microscopy performed on RB-proficient H1299 and H460 cells exposed to CDK4/6 inhibitor illustrated pronounced fragmented (apoptotic) nuclei and fragmented F-actin; in contrast, RB-deficient H1299 and H460 cells displayed intact nuclei and active F-actin fibers (Supplemental Figure 3A). Apoptosis markers including SMAC, p27, HTRA, TNFR-1, and Caspase 3 were strongly induced following therapeutic challenge of RB proficient tumors with a CDK4/6 inhibitor in vitro (Supplemental Figure 3B and 3C). With induction of apoptosis, there was a concomitant increase in reactive oxygen species noted with CDK 4/6 inhibition in the RB proficient setting (Supplemental Figure 3D). An in silico analysis of human lung adeno carcinoma gene expression data revealed a direct correlation of the apoptotic marker TNFR-1 with RB1 copy number (Supplemental Figure 3E). These collective findings nominate RB as a key modulator of CDK 4/6 inhibition induced cellular apoptosis in NSCLC.
Figure 2. RB Promotes In Vivo Cellular Apoptosis in NSCLC.
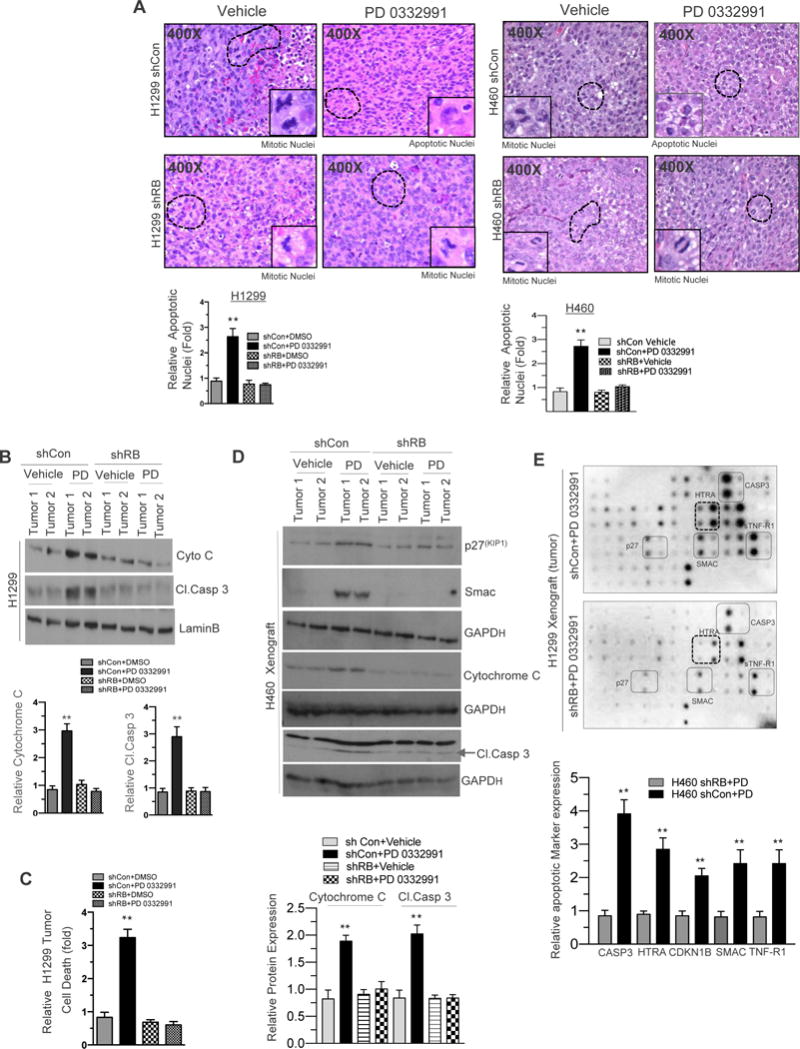
(A) H&E staining of shCon and shRB H1299 and H460 cell line induced xenografts (400×) in response to CDK4/6 inhibition (150 mg/kg body wt) and a graphic representation of apoptosis positive nuclei in RB proficient and deficient H1299 and H460 cell line induced xenografts in response to CDK4/6 inhibition (left and right panel). (B) Immunoblotting analysis of cytochrome C, Cleaved caspase3 and LaminB in shCon and shRB H1299 cell line induced xenografts in response to CDK4/6 inhibition (150 mg/kg body wt) and a graphic representation of cytochrome C and cleaved caspase 3 expression in shCon and shRB tumor lysates. (C) Graphic representation of cell death analysis via photometric enzyme immunoassay in shCon and shRB H1299 cells in response to CDK4/6 inhibition (150-mg/kg body wt). (D). Immunoblotting analysis of p27, Smac Cytochrome c, cleaved caspase 3 and GAPD in shCon and shRB H460 xenograft in response to CDK4/6 inhibitor (150 mg/kg body weight). (E) Apoptotic array (protein array) images from shCon and shRB H1299 cells induced xenografts in response CDK4/6 inhibition (150-mg/kg body wt) and a graphic representation of apoptotic markers and a graphic representation of Caspase3, HTRA, p27, Smac and TNFR-1 expression in shCon and shRB tumor lysates. In each group 5 or more animals were used and each data point is a mean ± SD from 5 or more animals or three or more independent experiments. **p <0.05 were considered as statistically significant.
SMAC Activation Promotes Cellular Apoptosis
Cellular apoptosis can be activated via either the extrinsic death receptor pathway or intrinsic mitochondrial pathway (15). Given the elevation of cytochrome C, a key factor in the mitochondrial pathway, when CDK4/6 inhibition was introduced into the RB proficient setting (Supplemental Figure 3B), we interrogated the role of SMAC, which was elevated on our array (Figure 2E, Supplemental 3C) and is induced with cytochrome C to release from the mitochondria (16) in mediating RB activated apoptosis. Protein analysis demonstrated that RB activation led to elevated cleaved caspase 3 via increased SMAC levels (Figure 3A). Generation of ectopic expression of Flag-SMAC increased cleaved caspase 3 in both in vitro NSCLC models (Figure 3B), repressed growth independent of RB status (Figure 3C, Supplemental 4A and 4B) and increased the ROS levels (Figure 3D). Interestingly, knockdown of SMAC inhibited cleaved caspase 3 activation and Annexin V, despite CDK4/6 inhibitor treatment (Figure 3E, top panel, Supplemental 4C and D), increased the cell growth (Figure 3E, bottom panel, Supplemental 4C and D) and maintained cell viability despite genotoxic stress (Figure 3F and supplemental Figure 4E (top and bottom panel). Thus, SMAC is a critical mediator of RB-activated apoptosis.
Figure 3. Mitochondrial protein SMAC regulates NSCLC apoptosis.
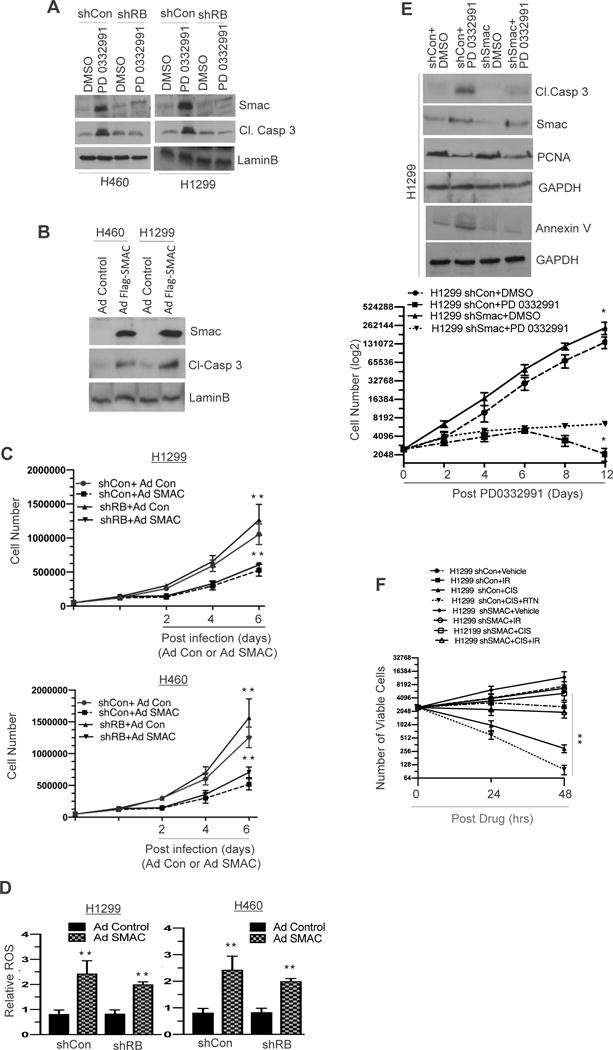
(A) Immunoblotting analysis of SMAC, cleaved caspase 3 and a loading control LaminB in RB proficient and RB deficient H1299 and H460 cells (shCon and shRB) in response to PD 0332991 (500 nM). (B) Immunoblotting analysis of SMAC, cleaved caspase 3 and a loading control LaminB in control and SMAC overexpressing H1299 and H460 cells. (C) A graphic representation of cell growth analysis in control and SMAC overexpressing H1299 and H460 cells. (D) A graphic representation of ROS (reactive oxygen species) analysis in control and SMAC overexpressing shCon and shRB H1299 and H460 cells. (E) Immunoblotting analysis of cleaved caspase 3, SMAC, PCNA, annexin V and a loading control GAPDH in SMAC proficient (shCon) and SMAC deficient (shSMAC) in H1299 cells (top panel) and a graphic representation of cell growth in SMAC proficient (shCon) and SMAC deficient (shSMAC) in H1299 cells (bottom panel) in response to PD 0332991 (500 nM). (F) A graphic representation of cell viability in SMAC proficient (shCon) and SMAC deficient (shSMAC) in H1299 cells in response to cisplatin (20 μM) or radiation (2Gy) or a combination of cisplatin and radiation. Each data point is a mean ± SD from three or more independent experiments. **p <0.05 were considered as statistically significant.
Reactivated RB Represses IAPs (FOXM1 and Survivin/BIRC5) in NSCLC
As RB mediates its canonical function through inhibition of E2F target signatures (17–19), we utilized an unbiased genome wide microarray approach to examine which biological pathways and gene products are impinged on via CDK 4/6 inhibition. Pathway analysis of human transcriptome array via gene ontology identified that the apoptosis pathway genes were the most perturbed in the setting of CDK 4/6 inhibition (Figure 4A). Further, the top two inhibitors of apoptosis genes (IAPs), which were repressed via RB activation included FOXM1 and Survivin, which are key inhibitors of apoptosis proteins (IAPs) (Figure 4B, Supplemental 5A and B). FOXM1 and Survivin were validated by real time RT-PCR and protein analysis in RB proficient and deficient H1299 cells in response to the CDK4/6 inhibitor (Figure 4C). In addition, an in-silico analysis of human lung cancer data revealed that RB copy number gain inversely correlated with the RB loss signature, FOXM1 and Survivin (Figure 4D) suggesting that RB is necessary to invoke apoptosis.
Figure 4. Transcriptome Array Profiling and Identification RB/E2F Targets (FOXM1 and Survivin) in NSCLC.
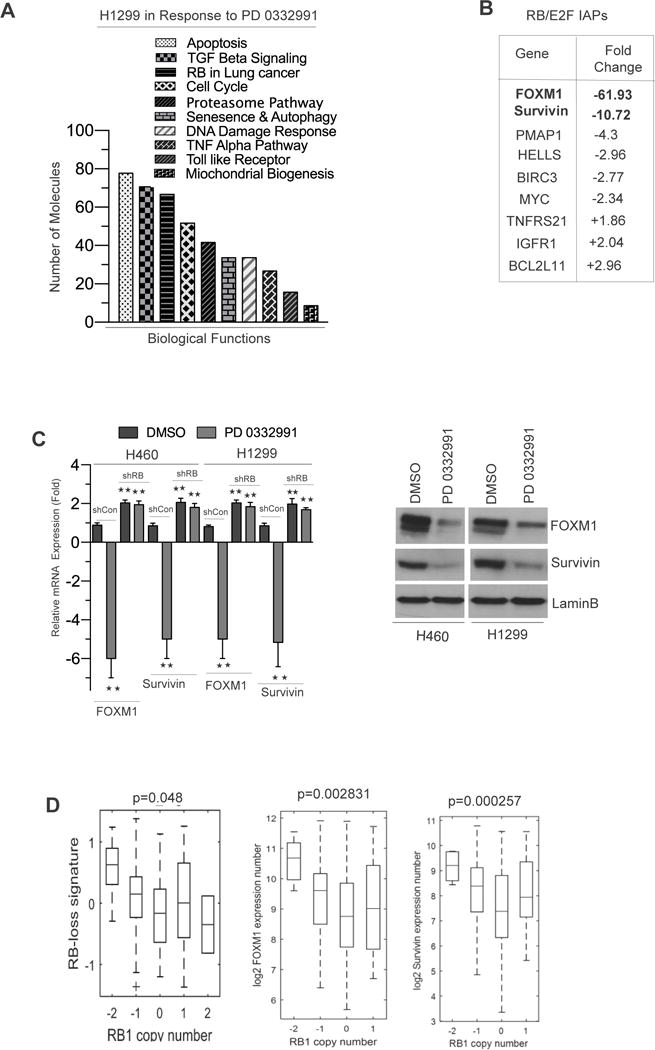
(A) Gene ontology analysis in H1299 microarray data via IPA software in response to CDK4/6 inhibition for 21 days. (B) Microarray analysis derived RB/E2F signatures involved in apoptosis signaling. (C) qRT-PCR and protein validation of FOXM1, Survivin in H1299 cells in response to CDK4/6 inhibition for 21 days. (D) In silico analysis and box plot representation of RB loss signatures, FOXM1 and Survivin transcript expression in relation to RB copy number in human lung adenocarcinoma. Each data point is a mean ± SD from three or more independent experiments. **p <0.05 were considered as statistically significant.
RB/E2F Governs FOXM1 and Survivin Transcription
RB/E2F complexes regulate transcription primarily by binding to promoter regions and altering chromatin structure (20). An in-silico analysis of RB/E2F putative binding sites within 1Kb of the FOXM1 and Survivin start site revealed several potential binding sites (Figure 5A). Binding affinities on the promoters were further confirmed through an in silico-docking model (Supplemental Figure 5C). Further chromatin immunoprecipitation indicated that upon CDK4/6 inhibition, RB binds robustly on the RB/E2F site II (Figures 5B and C) with consonant deacetylation of histone 4 (Figures 5B and C) in both H1299 and H460 cells, indicating that RB binding results in a closed chromatin state that inhibits transcription. Conversely, no binding was detected in the context of RB loss. Furthermore, RB failed to associate with any other putative/predicted RB-E2F binding site. Ectopic expression of adenovirus harboring E2F1, E2F2, and E2F3 cDNAs demonstrated that inhibitor of apoptosis proteins FOXM1 and Survivin (IAPs) are RB/E2F regulated genes (Figure 5D). Thus, these results confirm that transcriptional regulation of these IAPs are mediated through the RB/E2F signaling cascade.
Figure 5. RB Mediates FOXM1 and Survivin Transcription in NSCLC.
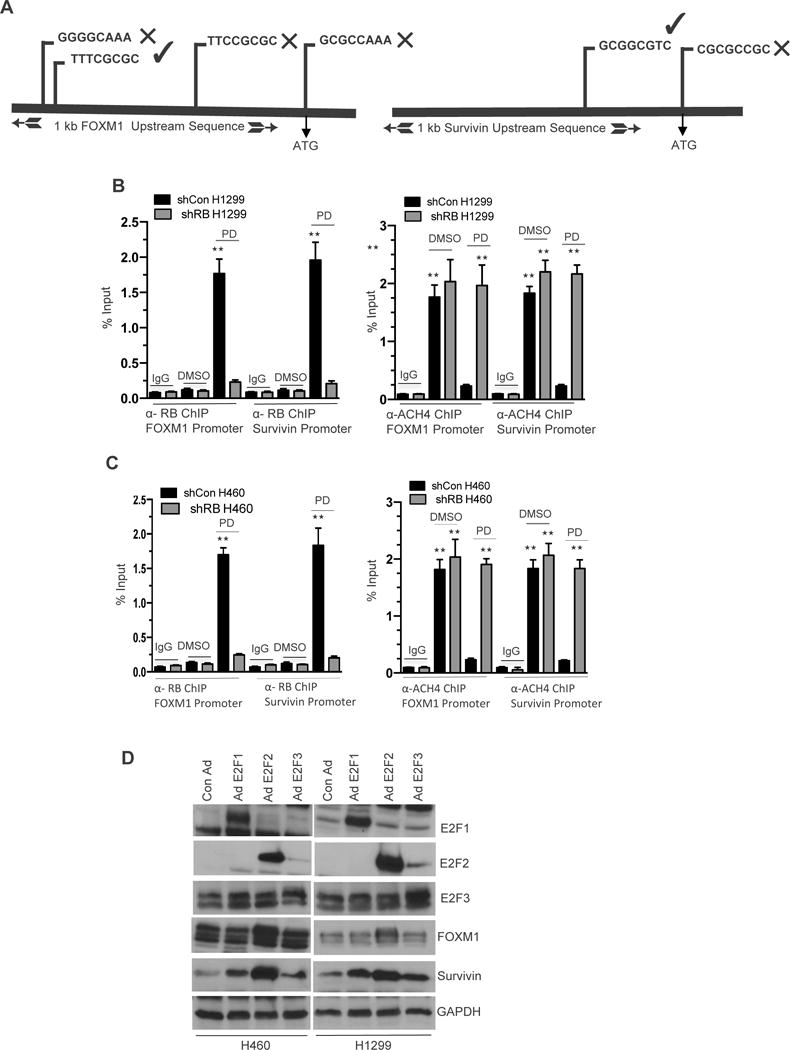
(A) Schematic illustration shows the location of three putative RB/E2F binding sites on FOXM1 and Survivin upstream sequence (promoters). (B & C left panels) Real-time PCR analysis of α-RB Chromatin immunoprecipitation assay shows the recruitment of RB on RB/E2F binding site II on FOXM1 and Survivin promoter, mouse α-GFP served as a negative control and input served as a positive control in shCon and shRB H1299 cells in response CDK4/6 inhibition (500 nM). (B & C right panels) PCR analysis of α-ACH4 chromatin immunoprecipitation assay shows that RB fails to recruit on RB/E2F binding site II on acetylated FOXM1 and Survivin promoter. Rabbit IgG served as a negative control and input served as a positive control in shCon and shRB cells in response to CDK4/6 inhibition (500 nM). (D) Immunoblotting analysis of FOXM1 Survivin, E2F1, E2F2, E2F3 and LaminB in ectopically expressed adenovirus harboring E2F1, E2F2, E2F3 cDNA in H1299 and H460 cells. Each data point is a mean ± SD from three or more independent experiments. **p <0.05 were considered as statistically significant.
RB Activated Apoptosis is mediated via FOXM1/SURVIVIN-SMAC Balance
IAP family members contain baculoviral IAP repeat (BIR) domains capable of inhibiting caspases (21). Inhibition of FOXM1 or Survivin function was achieved through stable knockdown (Figure 6A, left panel). FOXM1 and Survivin are key regulators of cell cycle (Figure 6A, right panel) and proliferation (Figure 6B). Further, inhibition of FOXM1 and Survivin led to increased levels of reactive oxygen species (Figure 6C), mimicking the function of activated RB. SMAC is known to interact with IAPs via the BIR domain (22). In our models, FOXM1 physically interacts with both Survivin and Caspase 3 preventing cleavage of Caspase 3 and the downstream apoptotic pathway (Figure 6D). Overexpression of either FOXM1 or Survivin inhibits the ability of palbociclib to alter cellular viability (Figure 6E and 6F). Interestingly, ectopic expression of Flag-SMAC enabled binding of SMAC with Caspase 3 and cleaved caspase (Figure 6G), yet did not perturb the interaction of FOXM1 and Survivin (Figure 6H). In summation, activated RB represses FOXM1 and Survivin expression with concurrent up-regulation of SMAC, leading to ROS generation and inducing cell death as shown in our working model (Supplemental Figure 6).
Figure 6. Genetic ablation of FOXM1 and Survivin negatively regulates NSCLC apoptosis via caspase 3 interactions.
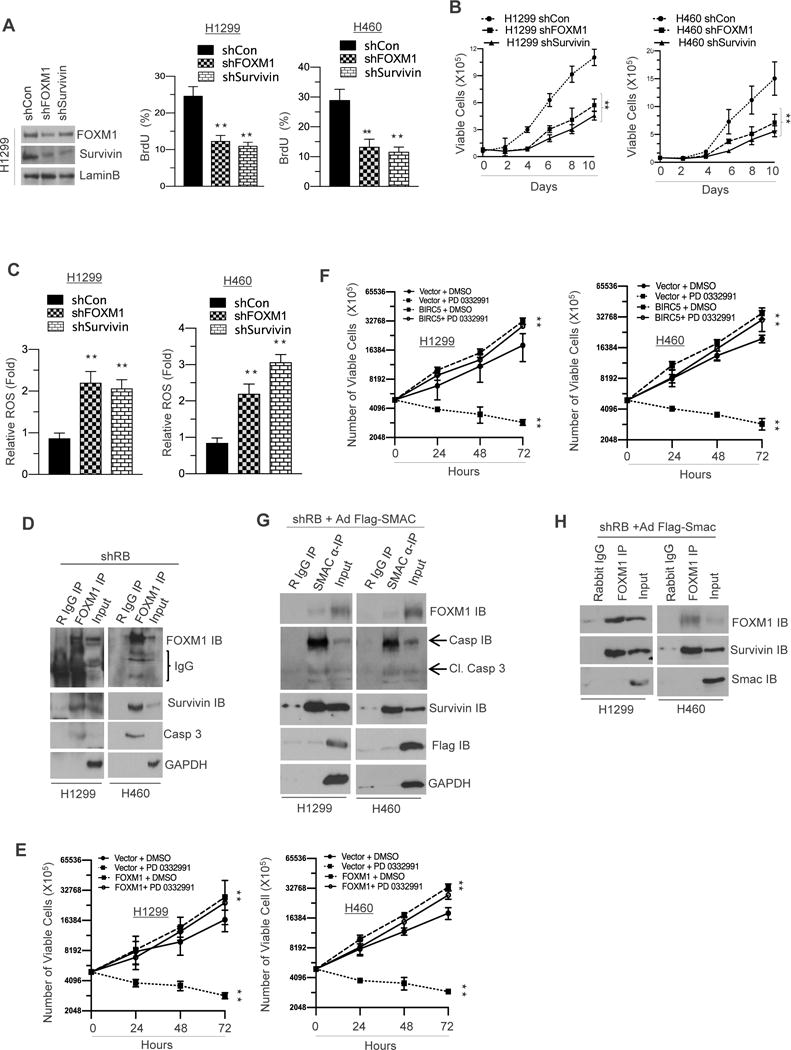
(A) Immunoblotting analysis of FOXM1, Survivin and a loading control in FOXM1 and Survivin proficient and deficient H1299 and H460 cells (left panel) and a flow cytometric analysis of BrdU incorporation in FOXM1 and Survivin proficient and deficient H1299 and H460 cells (right panel). (B) A graphic representation of cell survival analysis in FOXM1 and Survivin proficient and deficient H1299 and H460 cells (left and right panel). (C) A graphic representation of ROS (reactive oxygen species) analysis in FOXM1 and Survivin proficient and deficient H1299 and H460 cells (left and right panel). (D) Co-immunoprecipitation of FOXM1 and immune blotting analysis of FOXM1, Survivin and Caspase3 and a GAPDH loading control in shRB H1299 cells. (E) A graphic representation of cell viability analysis in FOXM1 overexpressing H1299 and H460 cells in response to PD 0332991 (left and right panel) (F) A graphic representation of cell viability analysis in Survivin overexpressing H1299 and H460 cells (left and right panel). (G) Co-immunoprecipitation of SMAC and immunoblotting analysis of FOXM1, Survivin, SMAC, Caspase 3, Cleaved Caspase3 and LaminB loading control in SMAC overexpressing shRB H1299 and H460 cells. (H) Co-immunoprecipitation of FOXM1 and immunoblotting analysis of FOXM1, Survivin and SMAC in SMAC overexpressing shRB H1299 and H460 cells. Each data point is a mean ± SD from three more independent experiments. **p <0.05 were considered as statistically significant.
Discussion
The role of RB as a regulator of cell cycle and progression is well established (23), and numerous studies have established that pharmacologic inhibition of the CDK4/6 complex results in cell cycle arrest through activation of RB (24–29). Recently, RB has been recognized to function in many other key biological processes including apoptosis (30). RB has been demonstrated to either promote or inhibit apoptosis depending on the context, apoptotic cues and its own functional status. In the current study, we discovered for the first time a novel apoptotic role of RB beyond its transcriptional regulatory function. Our data strongly support the hypothesis that CDK4/6 inhibition activates the RB pathway and orchestrates cellular apoptosis through repression of IAPs and subsequent enhancement of pro-apoptotic signaling via SMAC and cleaved Caspase 3. This conclusion is supported by several key findings 1) Activated RB via CDK4/6 inhibition represses cellular proliferation and tumor burden in vivo; 2) induces cellular apoptosis and production of reactive oxygen species; 3) drastically activates/stabilizes pro-apoptotic mitochondrial proteins SMAC, Cytochrome C, HTRA, p27, TNFR-1; 4) suppresses transcription of FOXM1 and Survivin through promoter binding thus allowing SMAC to cleave Caspase 3. Based on these discoveries, we propose a novel role for CDK4/6 inhibitors in management of RB-proficient NSCLC and an important role of the RB/E2F signaling outside of canonical transcriptional control.
Apoptosis is often accompanied by a shift from the hyperphosphorylated (inactive) to the hypophosphorylated form of RB (31). Phosphatase activity directed toward RB appears to be necessary for induction of apoptosis in different cancer types (32–34), while RB hyperphosphorylation is linked to resistance to apoptotic therapy (35,36). Studies in promyelocytic leukemia and breast cancer suggest that dephosphorylation is required for caspase cleavage (37) and accumulation of the active hypophosphorylated form of RB triggers apoptosis (38) in the context of prostate cancer. Our study further supports the role of RB in promoting apoptosis whereby RB suppresses transcription of the IAP family members, FOXM1 and Survivin, while inducing SMAC.
SMAC is a mitochondrial protein that interacts with the Inhibitor of Apoptosis Proteins (IAPs) and upon apoptotic stimuli is released into the cytoplasm to inhibit IAP binding to caspase (39). SMAC binds to baculoviral IAP repeats (BIR), which directly prevents IAP-caspase binding enabling apoptosis. While translocation of JNK from the cytosol to the mitochondria (40) and downregulation of c-Myc (41) have been demonstrated to cause release of SMAC from the mitochondria, our study is the first to implicate RB in mediating this process. We demonstrate that SMAC is a key regulator of activated RB induced apoptosis, as SMAC knockdown prevented caspase cleavage despite presence of CDK4/6 inhibition.
Our study demonstrates a new potential treatment paradigm for lung cancer, CDK4/6 inhibitor monotherapy. Within the context of NSCLC, studies have demonstrated an antagonistic interaction of CDK4/6 inhibition and cisplatin response (42), however, in the setting of esophageal cancer, combination therapy was synergistic (43). This may be cell context dependent, perhaps reliant on the necessity of CDK4 vs. CDK6 function for different cancer types. Multiple combination trials with palbociclib, ribociclib, and abemaciclib are currently accruing; while findings may be generalizable within this class of agents, difference in patient response could be due to unique targeting of each agent to distinct kinases (44). Our study suggests efficacy of single agent use regardless of KRAS or p53 status. This is supported by reports that one patient, who had previously been treated with gefitinib, exhibited clinical remission after treatment with PD 0332991 (45). This demonstrates our study finding which implicates conversion of a presumed “cytostatic” agent into a cytotoxic therapeutic.
Supplementary Material
Statement of Translational Relevance.
Lung cancer remains the most common cause of cancer related death in the United States despite recent advances in targeted agents and immunotherapy. We describe the preclinical efficacy of palbociclib (PD 0332991) in RB-proficient and deficient non-small cell lung cancer (NSCLC) when dosed per the current FDA-indication. Our study demonstrates that CDK 4/6 inhibition results in apoptosis specifically in RB-proficient models. This mechanism is mediated through upregulation of SMAC leading to cleaved Caspase 3 with concurrent repression of inhibitors of apoptosis: FOXM1 and Survivin. The novelty of our finding suggests that “cytostatic” drugs may have cytocidal properties with prolonged exposure positing a new therapeutic strategy in the management of patients with RB positive NSCLC.
Acknowledgments
The study was supported by a Young Investigator Award from the Prostate Cancer Foundation (to RBD), a Physician Research Training Award from the Department of Defense Grant (PC101841 to RBD), Grant #IRG 08-060-04 from the American Cancer Society, a PA Cure Award (to KEK), NIH grants (R01 CA099996 to KEK), and PCF Challenge Award (to KEK), (R01 DK100483 to EB). The authors thank members of the K. Knudsen laboratory for input and comments.
Footnotes
Conflict of Interest: The authors have no conflict of interest
Author’s Contributions:
Conception and design: C. Thangavel, K.E. Knudsen, R.B. Den.
Development of methodology: C. Thangavel, Y. Liu, E. Boopathi, A. Haber, K.E. Knudsen, R.B. Den, R. Birbe, M. Perepelyuk, S. Addya, A. Bhardwaj, A. Ertel, S. Shoyele
Reagents and materials: JM. Salvino
Acquisition of data: C. Thangavel, Y. Liu, E. Boopathi, A. Haber, R.B. Den.
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis: C. Thangavel, E. Boopathi, A. Ertel, A. Bhardwaj, S. Addya, C. McNair, R. B. Den.
Writing, review, and/or revision of the manuscript: C. Thangavel, K.E. Knudsen, A. P. Dicker, R.B. Den.
Study supervision: C. Thangavel and R.B. Den.
References
- 1.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2015;373(3):209–19. doi: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- 3.Rocco G, Morabito A, Leone A, Muto P, Fiore F, Budillon A. Management of non-small cell lung cancer in the era of personalized medicine. The international journal of biochemistry & cell biology. 2016;78:173–9. doi: 10.1016/j.biocel.2016.07.011. [DOI] [PubMed] [Google Scholar]
- 4.Sharma A, Comstock CE, Knudsen ES, Cao KH, Hess-Wilson JK, Morey LM, et al. Retinoblastoma tumor suppressor status is a critical determinant of therapeutic response in prostate cancer cells. Cancer Res. 2007;67(13):6192–203. doi: 10.1158/0008-5472.CAN-06-4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thangavel C, Boopathi E, Ciment S, Liu Y, R ON, Sharma A, et al. The retinoblastoma tumor suppressor modulates DNA repair and radioresponsiveness. Clin Cancer Res. 2014;20(21):5468–82. doi: 10.1158/1078-0432.CCR-14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balasubramaniam S, Comstock CE, Ertel A, Jeong KW, Stallcup MR, Addya S, et al. Aberrant BAF57 signaling facilitates prometastatic phenotypes. Clin Cancer Res. 2013;19(10):2657–67. doi: 10.1158/1078-0432.CCR-12-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Augello MA, Burd CJ, Birbe R, McNair C, Ertel A, Magee MS, et al. Convergence of oncogenic and hormone receptor pathways promotes metastatic phenotypes. J Clin Invest. 2013;123(1):493–508. doi: 10.1172/JCI64750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 9.Zheng N, Fraenkel E, Pabo CO, Pavletich NP. Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes Dev. 1999;13(6):666–74. doi: 10.1101/gad.13.6.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie DW. HexServer: an FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010;38:W445–9. doi: 10.1093/nar/gkq311. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269) doi: 10.1126/scisignal.2004088. pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodwin JF, Kothari V, Drake JM, Zhao S, Dylgjeri E, Dean JL, et al. DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. Cancer Cell. 2015;28(1):97–113. doi: 10.1016/j.ccell.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20(5):620–34. doi: 10.1016/j.ccr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Indovina P, Pentimalli F, Casini N, Vocca I, Giordano A. RB1 dual role in proliferation and apoptosis: cell fate control and implications for cancer therapy. Oncotarget. 2015;6(20):17873–90. doi: 10.18632/oncotarget.4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peter ME. Programmed cell death: Apoptosis meets necrosis. Nature. 2011;471(7338):310–2. doi: 10.1038/471310a. [DOI] [PubMed] [Google Scholar]
- 16.Shivapurkar N, Reddy J, Chaudhary PM, Gazdar AF. Apoptosis and lung cancer: a review. J Cell Biochem. 2003;88(5):885–98. doi: 10.1002/jcb.10440. [DOI] [PubMed] [Google Scholar]
- 17.Wierstra I, Alves J. Transcription factor FOXM1c is repressed by RB and activated by cyclin D1/Cdk4. Biol Chem. 2006;387(7):949–62. doi: 10.1515/BC.2006.119. [DOI] [PubMed] [Google Scholar]
- 18.Sang XB, Zong ZH, Wang LL, Wu DD, Chen S, Liu BL, et al. E2F-1 targets miR-519d to regulate the expression of the ras homolog gene family member C. Oncotarget. 2017;8(9):14777–93. doi: 10.18632/oncotarget.14833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, et al. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle. 2010;9(20):4153–63. doi: 10.4161/cc.9.20.13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brehm A, Kouzarides T. Retinoblastoma protein meets chromatin. Trends in biochemical sciences. 1999;24(4):142–5. doi: 10.1016/s0968-0004(99)01368-7. [DOI] [PubMed] [Google Scholar]
- 21.Deveraux QL, Reed JC. IAP family proteins–suppressors of apoptosis. Genes Dev. 1999;13(3):239–52. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 22.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408(6815):1008–12. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 23.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81(3):323–30. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Yang J, Zhang Y, Fang Y, Wang F, Wang J, et al. A systematic study on drug-response associated genes using baseline gene expressions of the Cancer Cell Line Encyclopedia. Sci Rep. 2016;6:22811. doi: 10.1038/srep22811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson J, Thijssen B, McDermott U, Garnett M, Wessels LF, Bernards R. Targeting the RB-E2F pathway in breast cancer. Oncogene. 2016 doi: 10.1038/onc.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spring L, Bardia A, Modi S. Targeting the cyclin D-cyclin-dependent kinase (CDK) 4/6-retinoblastoma pathway with selective CDK 4/6 inhibitors in hormone receptor-positive breast cancer: rationale, current status, and future directions. Discov Med. 2016;21(113):65–74. [PMC free article] [PubMed] [Google Scholar]
- 27.Finn RS, Aleshin A, Slamon DJ. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016;18(1):17. doi: 10.1186/s13058-015-0661-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikolai BC, Lanz RB, York B, Dasgupta S, Mitsiades N, Creighton CJ, et al. HER2 Signaling Drives DNA Anabolism and Proliferation through SRC-3 Phosphorylation and E2F1-Regulated Genes. Cancer Res. 2016;76(6):1463–75. doi: 10.1158/0008-5472.CAN-15-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tao Z, Le Blanc JM, Wang C, Zhan T, Zhuang H, Wang P, et al. Coadministration of Trametinib and Palbociclib Radiosensitizes KRAS-Mutant Non-Small Cell Lung Cancers In Vitro and In Vivo. Clin Cancer Res. 2016;22(1):122–33. doi: 10.1158/1078-0432.CCR-15-0589. [DOI] [PubMed] [Google Scholar]
- 30.Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14(5):297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang RH, Liu CW, Avramis VI, Berndt N. Protein phosphatase 1alpha-mediated stimulation of apoptosis is associated with dephosphorylation of the retinoblastoma protein. Oncogene. 2001;20(43):6111–22. doi: 10.1038/sj.onc.1204829. [DOI] [PubMed] [Google Scholar]
- 32.Dou QP, An B, Will PL. Induction of a retinoblastoma phosphatase activity by anticancer drugs accompanies p53-independent G1 arrest and apoptosis. Proc Natl Acad Sci U S A. 1995;92(20):9019–23. doi: 10.1073/pnas.92.20.9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glozak MA, Rogers MB. Retinoic acid- and bone morphogenetic protein 4-induced apoptosis in P19 embryonal carcinoma cells requires p27. Exp Cell Res. 2001;268(2):128–38. doi: 10.1006/excr.2001.5281. [DOI] [PubMed] [Google Scholar]
- 34.Popowski M, Ferguson HA, Sion AM, Koller E, Knudsen E, Van Den Berg CL. Stress and IGF-I differentially control cell fate through mammalian target of rapamycin (mTOR) and retinoblastoma protein (pRB) J Biol Chem. 2008;283(42):28265–73. doi: 10.1074/jbc.M805724200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dou QP, Lui VW. Failure to dephosphorylate retinoblastoma protein in drug-resistant cells. Cancer Res. 1995;55(22):5222–5. [PubMed] [Google Scholar]
- 36.Wallace M, Coates PJ, Wright EG, Ball KL. Differential post-translational modification of the tumour suppressor proteins Rb and p53 modulate the rates of radiation-induced apoptosis in vivo. Oncogene. 2001;20(28):3597–608. doi: 10.1038/sj.onc.1204496. [DOI] [PubMed] [Google Scholar]
- 37.Tan X, Wang JY. The caspase-RB connection in cell death. Trends Cell Biol. 1998;8(3):116–20. doi: 10.1016/s0962-8924(97)01208-7. [DOI] [PubMed] [Google Scholar]
- 38.Day ML, Foster RG, Day KC, Zhao X, Humphrey P, Swanson P, et al. Cell anchorage regulates apoptosis through the retinoblastoma tumor suppressor/E2F pathway. J Biol Chem. 1997;272(13):8125–8. doi: 10.1074/jbc.272.13.8125. [DOI] [PubMed] [Google Scholar]
- 39.Qin S, Yang C, Li S, Xu C, Zhao Y, Ren H. Smac: Its role in apoptosis induction and use in lung cancer diagnosis and treatment. Cancer Lett. 2012;318(1):9–13. doi: 10.1016/j.canlet.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 40.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27(48):6245–51. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amendola D, De Salvo M, Marchese R, Verga Falzacappa C, Stigliano A, Carico E, et al. Myc down-regulation affects cyclin D1/cdk4 activity and induces apoptosis via Smac/Diablo pathway in an astrocytoma cell line. Cell Prolif. 2009;42(1):94–109. doi: 10.1111/j.1365-2184.2008.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bar J, Gorn-Hondermann I, Moretto P, Perkins TJ, Niknejad N, Stewart DJ, et al. miR Profiling Identifies Cyclin-Dependent Kinase 6 Downregulation as a Potential Mechanism of Acquired Cisplatin Resistance in Non-Small-Cell Lung Carcinoma. Clin Lung Cancer. 2015;16(6):e121–9. doi: 10.1016/j.cllc.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 43.Chen L, Pan J. Dual cyclin-dependent kinase 4/6 inhibition by PD-0332991 induces apoptosis and senescence in oesophageal squamous cell carcinoma cells. Br J Pharmacol. 2017 doi: 10.1111/bph.13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sumi NJ, Kuenzi BM, Knezevic CE, Remsing Rix LL, Rix U. Chemoproteomics Reveals Novel Protein and Lipid Kinase Targets of Clinical CDK4/6 Inhibitors in Lung Cancer. ACS Chem Biol. 2015;10(12):2680–6. doi: 10.1021/acschembio.5b00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu M, Xu S, Wang Y, Li Y, Li Y, Zhang H, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, sensitizes lung cancer cells to treatment with epidermal growth factor receptor tyrosine kinase inhibitors. Oncotarget. 2016;7(51):84951–64. doi: 10.18632/oncotarget.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.