

Abstract
BCR-ABL1-targeting tyrosine kinase inhibitors (TKIs) have revolutionized treatment of Philadelphia chromosome-positive (Ph+) hematologic neoplasms. Nevertheless, acquired TKI resistance remains a major problem in chronic myeloid leukemia (CML), and TKIs are less effective against Ph+ B-cell acute lymphoblastic leukemia (B-ALL). GAB2, a scaffolding adaptor that binds and activates SHP2, is essential for leukemogenesis by BCR-ABL1, and a GAB2 mutant lacking SHP2 binding cannot mediate leukemogenesis. Using a genetic loss-of-function approach and bone marrow transplantation (BMT) models for CML and BCR-ABL1+ B-ALL, we show that SHP2 is required for BCR-ABL1-evoked myeloid and lymphoid neoplasia. Ptpn11 deletion impairs initiation and maintenance of CML-like myeloproliferative neoplasm, and compromises induction of BCR-ABL1+ B-ALL. SHP2, and specifically, its SH2 domains, PTP activity and C-terminal tyrosines, is essential for BCR-ABL1+, but not WT, pre-B cell proliferation. The MEK/ERK pathway is regulated by SHP2 in WT and BCR-ABL1+ pre-B cells, but is only required for the proliferation of BCR-ABL1+ cells. SHP2 is required for SRC family kinase (SFK) activation only in BCR-ABL1+ pre-B cells. RNAseq reveals distinct SHP2-dependent transcriptional programs in BCR-ABL1+ and WT pre-B cells. Our results suggest that SHP2, via SFKs and ERK, represses MXD3/4 to facilitate a MYC-dependent proliferation program in BCR-ABL1-transformed pre-B cells.
Introduction
The Philadelphia (Ph+) chromosome translocation t(9, 22) generates the oncogene BCR-ABL1, encoding a constitutively activated tyrosine kinase. Different BCR-ABL1 isoforms cause chronic myeloid leukemia (CML) and Ph+ B-cell lymphoblastic leukemia (B-ALL).1,2 ABL1 tyrosine kinase inhibitors (TKIs), such as imatinib, have revolutionized the treatment of Ph+ hematopoietic neoplasia. Nevertheless, >40% of CML patients are PCR-positive after 10 years of imatinib treatment and remain at risk of relapse,3 perhaps because CML stem cells (CML-SCs) are insensitive to BCR-ABL1 inhibitors.4–6 The response to TKIs (combined with chemotherapy) is poorer in Ph+ B-ALL,7 with several TKI resistance mechanisms proposed, including BCR-ABL1 mutation/amplification, elevated drug exporters, and upregulation of other oncogenic pathways.8–10 Therefore, new approaches are needed to eradicate BCR-ABL1+ neoplasia.
CML-like MPN can be reproduced in mice by retroviral transduction of BCR-ABL1 into hematopoietic stem cell (HSC)-enriched bone marrow (BM) cells in the presence of myeloid cytokines, followed by transplantation into irradiated recipients.11–13 B-ALL can be induced by transducing bulk BM cells in the presence of interleukin-7 (IL-7) before transplantation.12 In Ph+ cell lines and mouse leukemia models, BCR-ABL1 is phosphorylated on Y177, which recruits the adaptor GRB2 and, thereby, the scaffolding adaptor GAB2.14,15 Consequently, GAB2 is constitutively tyrosyl-phosphorylated and binds SHP2 and the p85 subunit of PI3K to activate the MEK/ERK and PI3K/AKT pathways, respectively.16,17 Y177F mutation compromises myeloid transformation and leukemogenesis,18–20 and GAB2 is required both for BCR-ABL1-induced myeloid and lymphoid leukemogenesis.21 At present, it is not feasible to pharmacologically target GAB2, rendering it essential to identify and validate GAB2-interacting proteins that mediate leukemogenesis. Reconstituting Gab2−/− donor cells with a GAB2 mutant lacking its SHP2 binding sites does not restore myeloid or lymphoid leukemogenesis, suggesting that SHP2 is required for these diseases.21 Nevertheless, the functions of SHP2 are not mediated solely through GAB2, and its role in BCR-ABL1-induced neoplasia remains undefined.
SHP2, encoded by PTPN11, is a ubiquitously expressed non-receptor protein-tyrosine phosphatase (PTP) required for full RAS-ERK pathway activation in response to growth factors and cytokines. SHP2 also modulates the PI3K-AKT and JAK-STAT pathways.17,22–25 SHP2 catalytic activity is suppressed by intra-molecular interaction of its N-SH2 and PTP domains, preventing substrate binding and catalysis.26,27 Following agonist stimulation, auto-inhibition is relieved by binding of pY peptides (such as in phospho-GAB2) to the SHP2 SH2 domains, leading to altered subcellular localization, PTP domain exposure, and enzyme activation.17,23,26–29
SHP2 has critical roles in hematopoiesis and leukemogenesis. Myeloid and erythroid differentiation are impaired in embryonic stem cells (ESCs) expressing mutant SHP2.30 SHP2 deficiency leads to defective growth factor- and cytokine-evoked ERK and AKT activation, resulting in impaired self-renewal and apoptosis of adult HSCs.31,32 Moreover, somatic gain-of-function alleles of PTPN11 cause >30% of juvenile myelomonocytic leukemia (JMML) cases, are found in ~5% of AML and ALL patients, and can cooperate with Tet2 deficiency and AML1-ETO to generate AML in mice.33–37
Multiple studies implicate SHP2 in BCR-ABL1-induced pathogenesis. SHP2 is constitutively phosphorylated in BCR-ABL1-transformed cells,38,39 interacts with GAB2,16,21 and is required for BCR-ABL1-evoked transformation of a yolk sac cell line.40 However, the role of SHP2 in adult Ph+ hematopoietic neoplasia remains elusive. Here, we utilize mouse models to address this issue, report a critical role for SHP2 in myeloid and lymphoid Ph+ neoplasia, and uncover a differential requirement for SHP2 in normal versus leukemic pre-B cells.
Materials and Methods
Mice
Ptpn11flox/flox mice41,42 were bred to Mx1-Cre or Rosa26-creERT2 mice (The Jackson Laboratory) in the C57BL/6 background. Genotyping was performed as described.31
Virus production
Replication-defective ecotropic retroviral stocks of BCR-ABL1-expressing p210MIGFP, p210MIGFPCre, and p210MINVneo16,43,44 were generated by transient transfection of 293T cells.21 Viral supernatants were collected 48 and 72 hours post-transfection and stored at −80°C.
Mouse models of CML and B-ALL
For CML-like MPN, BM was flushed from femurs and tibiae. Red blood cells (RBCs) were lysed in 0.16M NH4Cl, and RBC-depleted BM cells were incubated with rat anti-mouse lineage (Lin) antibodies (CD3, CD19, Gr1, and Ter119 (BioLegend) and CD4, CD8α, CD127, and B220 (eBioscience)) for 30 minutes. Lin+ cells were depleted with sheep anti-rat Dynabeads (Invitrogen) for 1 hour, and the remaining cells were pre-stimulated overnight in IMDM-15%FBS, supplemented with IL-3 (6ng/ml), IL-6 (10ng/ml), and SCF (20ng/ml). On each of the following two days, pre-stimulated Lin− cells were spin-infected, and on the third day, cells were harvested, resuspended in cold PBS, and injected intravenously (IV) into 6-Gy irradiated syngeneic recipients.45
For B-lymphoid leukemogenesis, RBC-depleted BM was resuspended in IMDM-15%FBS, supplemented with IL-7 (10ng/ml), and infected as above. After infection, cells were cultured at 37°C for 4 hours, resuspended in cold PBS, and injected IV into 11-Gy irradiated syngeneic recipients.12
B-lymphoid progenitor cultures
RBC-depleted BM cells were incubated for 30 minutes with rat anti-CD4, -CD8, -Gr1, -Mac-1 and -Ter119 antibodies, followed by sheep-anti-rat Dynabeads for 1 hour. Following magnetic separation, the remaining cells were cultured in 24-well plates in OptiMEM-10%FBS containing 5ng/ml IL-7 and 50μM β-mercaptoethanol.46
Flow cytometry
All studies used an LSRII flow cytometer. RBC-depleted cells from peripheral blood, spleen, and/or bone marrow were labeled with antibodies to myeloid (Gr-1, Mac-1), B lymphoid (B220, CD19), and T lymphoid (CD4, CD8) markers. To assess apoptosis, cells were washed in cold PBS, resuspended in Annexin V staining buffer (BD Biosciences), and incubated with Annexin V (PE- or FITC-conjugated; 1:300) and Sytox Blue (1:1000) for 20 minutes at room temperature in the dark. Samples were analyzed within 1 hour, with low-FSC apoptotic bodies gated. For cell cycle analysis, cells were resuspended in OptiMEM-10%FBS, supplemented with 5 ng/ml IL-7 containing 10 μM Hoechst-33342. After 30 minutes at 37°C, Pyronin-Y was added (2.5 μg/ml), and cells were incubated for another 15 minutes prior to analysis.
Immunoblotting
Myeloid- or B lymphoid-enriched BM cells were transduced with p210MIGR1 or p210MINVneo viruses. Where indicated, Ptpn11 deletion was induced with poly I:C (125μg × 2 times) for in vivo experiments or 4-OH (1μM) for in vitro studies. Transduced cells were isolated by FACS (for p210MIGFP) or G418 selection (for p210MINVneo), starved for 2 hours in IMDM-2%FBS (myeloid cells) or OptiMEM-2%FBS (lymphoid cells), and lysed in RIPA buffer.47 Lysates were resolved by SDS-PAGE, and immunoblotting was performed as described.21 Anti-phospho-STAT5 (Tyr694), -phospho-CRKL (Tyr207), -phospho-(Ser473) and -total AKT, -phospho-(Thr202/Tyr204) and -total p44/42 ERK1/2, -phospho-S6 (S235/236), SRC, and -phospho-SRC (Y416), which cross-reacts with the other SFKs, were from Cell Signaling Technology. Anti-ABL1 and -SHP2 were from Santa Cruz Biotechnology. Antibodies were used at each manufacturer’s recommended concentrations.
RNA-seq
RNA (150 ng) was reverse-transcribed using the Illumina TruSeq Stranded mRNA kit. cDNA libraries were size-indexed and validated using an Agilent Bioanalyzer, and concentrations were confirmed by qPCR. For the 16 samples, each of the 4 libraries (1 WT SHP2+/+, 1 WT SHP2−/−, 1 BCR-ABL1 SHP2+/+, and 1 BCR-ABL1 SHP2−/−) was loaded onto an Illumina cBot for cluster generation, and the flow cell was subjected to 100 cycles of paired-end sequencing on an Illumina HiSeq 2000. Alignment was performed with Bowtie, using default parameters. Gene expression levels were estimated using Cufflinks and subjected to quantile normalization.48 Batch effect adjustment was performed by Combat.49 Differentially expressed genes between samples were identified by voom-limma.50 Enrichment analyses for GO terms or transcription factor effectors were performed with DAVID or Enrichr, respectively.51,52 GSEA was implemented using software from the Broad Institute.
Quantitative RT-PCR
Total RNA was prepared using the RNeasy minikit (Qiagen), and reverse-transcribed using SuperScriptIII (Invitrogen). SYBR Green-based gene expression analyses (BIO-RAD) were conducted according to the manufacturer’s instructions. Each sample was measured in triplicate, and relative expression was normalized to Tbp.
Statistics
Survival curves were compared by the log-rank test. Signaling intensities in myeloid and lymphoid cells were compared by two-sided t-tests. Corrections for multiple comparisons were performed by using the Benjamini-Hochberg procedure. Sample size was determined to ensure at least 80% power to detect the difference of interest, based on the empirically estimated variance in each group.
Study approval
All animal studies were approved by the University Health Network Animal Care Committee (Toronto, Ontario, Canada) and performed in accordance with the standards of the Canadian Council on Animal Care.
Results
SHP2 deficiency attenuates induction of CML-like MPN by BCR/ABL
BCR-ABL1 induces cytokine-independent myeloid progenitor colonies,53 and BCR-ABL1-transformed hematopoietic stem and progenitor cells (HSPCs), transplanted into irradiated syngeneic recipient mice, induce CML-like MPN.13 To address the role of SHP2 in BCR-ABL1-evoked myeloid disease, we transduced BM from Ptpn11fl/fl mice41 with p210MIGFP- or p210MIGFPCre-based retroviruses that co-express BCR-ABL1 with GFP or GFP-CRE, respectively. GFP-CRE induces floxed allele deletion, resulting in absence of SHP2. Lin− BM cells from WT or Ptpn11fl/fl mice were infected with parental pMIGR1, p210MIGFP, or p210MIGFPCre virus, GFP+ cells were isolated by FACS and seeded in methylcellulose-based media, and cytokine-independent colonies were counted 1 week later. As expected, p210MIGFP-infected WT and Ptpn11fl/fl BM cells yielded similar numbers of cytokine-independent myeloid colonies. By contrast, myeloid colony formation by p210MIGFPCre-infected Ptpn11fl/fl BM cells was impaired markedly, indicating that SHP2 is required for BCR-ABL1-induced myeloid transformation (Figure 1a). Notably, p210MIGFPCre-infected WT BM yielded fewer cytokine-independent colonies than did p210MIGFP-infected BM, suggesting that constitutive CRE expression might impair myeloid transformation, consistent with previous reports of CRE toxicity.54
Figure 1. SHP2 is required for BCR-ABL1-evoked myeloid transformation and initiation of CML-like MPN.
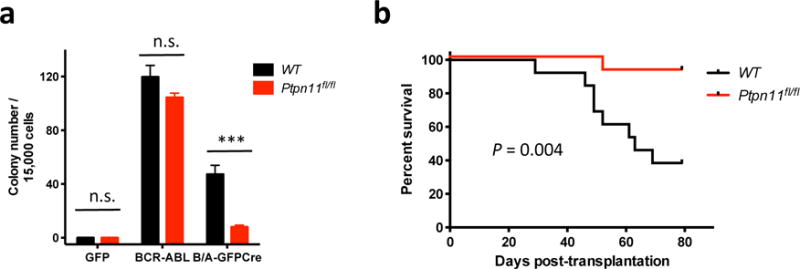
(a) Cytokine-independent colony formation of WT or Ptpn11fl/fl cells infected with pMIGR1 (GFP), p210MIGFP (BCR-ABL), or p210MIGFPCre (BCR-ABL-GFPCre) virus (n = 4). (n.s.=not significant; *** P < 0.001). WT cells infected with p210MIGFP yielded more cytokine-independent colonies than infected with p210MIGFPCre (P < 0.001). (b) Kaplan-Meier curve of recipients of WT or Ptpn11fl/fl donor cells infected with p210MIGFPCre virus (right, n = 13). Recipients of the Ptpn11fl/fl donor cells survived significantly longer (P = 0.004; log-rank test). A similar trend showing that p210GFPCre recipients survive longer than p210GFP recipients was reported by Walz et al..43
We next transplanted p210MIGFP-Cre transduced, Lin− BM cells from WT or Ptpn11fl/fl mice into sub-lethally irradiated WT recipients (Supplementary Figure S1a). Recipients of BCR-ABL1-expressing WT BM cells developed fatal, CML-like MPN (Figure 1b), characterized by leukocytosis with maturing myeloid cells, splenomegaly, and leukemic cell infiltrates in spleen and liver (Supplementary Figure S1b–g). Disease latency and histopathology were comparable to that reported previously.43 By contrast, the Ptpn11fl/fl cohort had markedly extended survival, reduced leukocytosis and splenomegaly, and diminished leukemic cell infiltration. Notably, 12 days post-transplant, peripheral blood and spleen leukemic cell number were much higher in recipients of WT BM cells, compared with Ptpn11fl/fl BM recipients (Supplementary Figure S1b,c), while BM engraftment was comparable (Supplementary Figure S1d). Therefore, SHP2 is required for initiation of CML, rather than homing to, or establishment in, the BM niche.
SHP2 is required for maintenance of BCR-ABL1-evoked CML-like MPN
To ask if SHP2 is required for CML maintenance, we crossed Ptpn11fl/fl mice to mice expressing Cre under the control of the type I interferon-inducible Mx promoter (MxCre), and infected BM cells from MxCre or MxCre;Ptpn11fl/fl donors with p210MIGFP virus. Between 11–15 days following transplantation of infected BM cells into WT recipients, two thirds of the mice in each cohort were injected with poly I:C to induce Cre expression in donor cells (Supplementary Figure S2a). All recipients of MxCre BM or MxCre;Ptpn11fl/fl BM that had not been poly I:C-treated succumbed to CML-like MPN within 30 days post-BM transplantation (BMT). Poly I:C-treated recipients of MxCre;Ptpn11fl/fl BM cells exhibited delayed/diminished disease and longer survival (Figure 2a). Leukemic cell levels in peripheral blood were reduced substantially (Supplementary Figure S2b,d), and WBC counts were normalized (Supplementary Figure S2c,e). All recipients that developed CML-like MPN despite poly I:C induction had incomplete Ptpn11 deletion in donor leukemic cells (Supplementary Figure S3). Hence, SHP2 is also necessary for CML-like MPN maintenance.
Figure 2. SHP2 is required for CML maintenance.
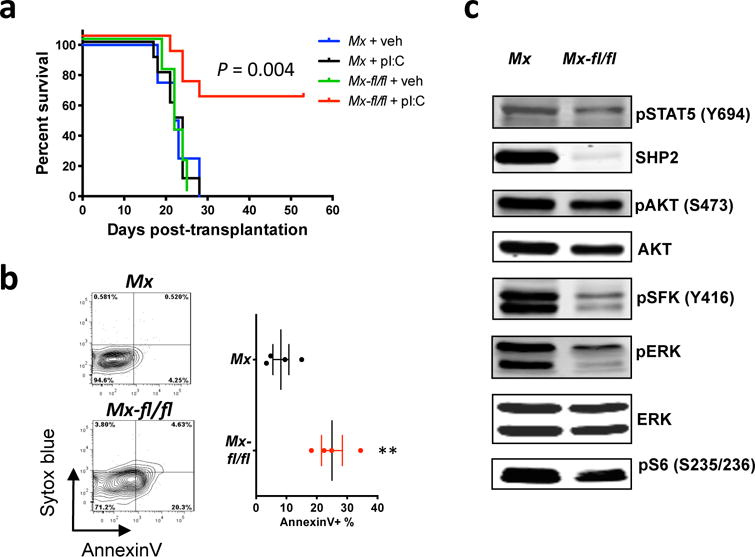
(a) Kaplan-Meier curve for recipients of MxCre (Mx) or MxCre;Ptpn11fl/fl (Mx-fl/fl) bone marrow infected with p210MIGFP virus, with (n = 10) or without (n = 5) poly I:C induction. The Mx-fl/fl + poly I:C group survived significantly longer than the other groups (P = 0.004; log-rank test). (b) GFP+Lin−Sca1+cKit+ splenocytes were isolated from poly I:C-induced recipients of Mx or Mx-fl/fl donor cells, and stained with Annexin V and Sytox blue (n = 4). Note that apoptosis is significantly higher in the Mx-fl/fl group than in Mx mice (P < 0.01). (c) Representative immunoblots of poly I:C-induced Mx or Mx-fl/fl BCR-ABL1+ myeloid cell lysates. Similar results were obtained in ≥5 biological replicates.
SHP2 is required for the survival of WT primitive hematopoietic cells,31 so we assessed its role in primitive CML cells. GFP+Lin−Sca1+cKit+ splenocytes from poly I:C-treated recipient mice were stained with Sytox blue and Annexin V, and analyzed by flow cytometry. Apoptosis was increased markedly in SHP2-deficient cells, compared with controls (Figure 2b).
To study the role of SHP2 in BCR-ABL1-evoked myeloid signaling, we infected SHP2-expressing or -deficient Lin− BM cells with p210MIGFP retrovirus, isolated infected cells, and performed immunoblotting. In the absence of SHP2, phospho-ERK1 and -ERK2 levels were reduced, as were phospho-S6-S235/236 (Figure 2c and Supplementary Figure S2f); the latter sites can be phosphorylated by the ERK-dependent kinase RSK2, as well as by mTORC1.55 These results are consistent with the requirement for RAS-ERK signaling in BCR-ABL1-induced CML.56–58 In addition, phospho-SFK-Y416, which marks the catalytically activated form of SFKs, was reduced in the absence of SHP2. By contrast, we did not see significant changes in phospho-AKT or phospho-STAT5 levels.
SHP2 is required for Ph+ B-ALL
As expected, BCR-ABL1-expressing, IL-7-stimulated bulk BM cells evoked B-ALL in lethally irradiated syngeneic recipients.12 To probe the role of SHP2 in Ph+ B-ALL initiation, we transplanted IL-7-stimulated BM from WT or Ptpn11fl/fl donors transduced with p210MIGFPCre virus into lethally irradiated syngeneic recipients. Compared with recipients of BCR-ABL1-infected WT BM, recipients of transduced Ptpn11fl/fl BM had much lower levels of BCR-ABL1+ B lineage engraftment in their peripheral blood (Figure 3a and Supplementary Figure S4a) and survived longer (Figure 3b). Hence, SHP2 is required for effective initiation of BCR-ABL1+ B-ALL. Consistent with the effects of GFP-CRE in myeloid cells, we observed less severe disease in recipients of p210MIGFPCre, compared with recipients of p210MIGFP, BM, possibly reflecting CRE toxicity (Figure 3a).
Figure 3. SHP2 is required for initiation, and most likely, for maintenance, of Ph+ B-ALL.
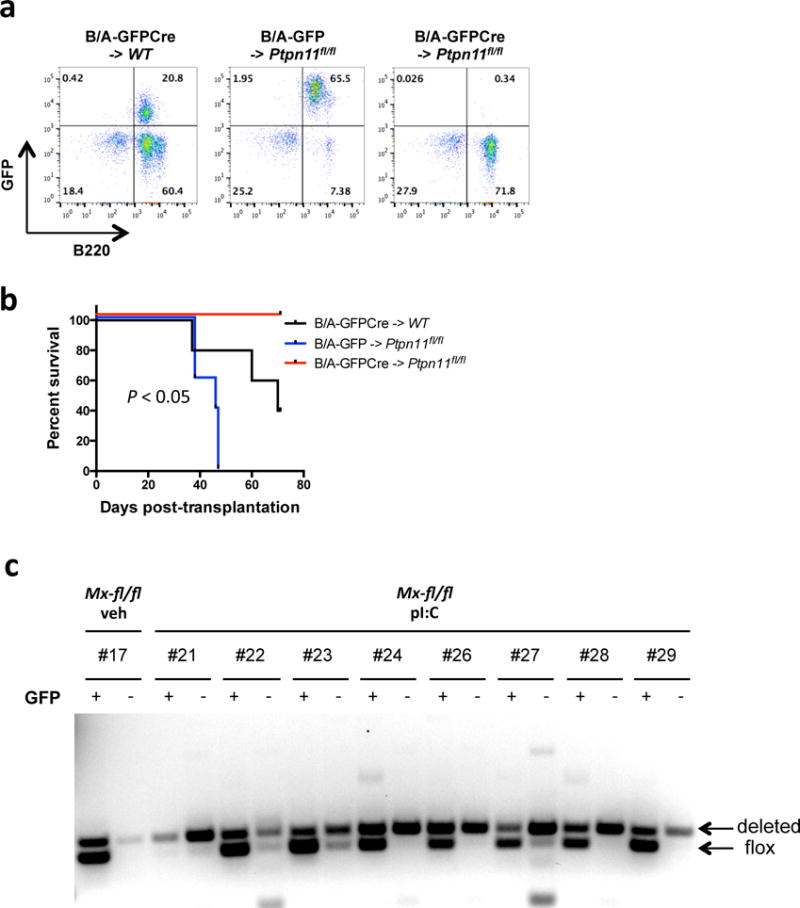
(a) Flow cytometric assessment of leukemic cell engraftment in recipients of WT or Ptpn11fl/fl donor cells infected with p210MIGFP (B/A-GFP) or p210MIGFPCre (B/A-GFPCre) retrovirus, assessed on day 20 post-transplantation. (b) Kaplan-Meier curve of recipients of WT or Ptpn11fl/fl donor cells infected with p210MIGFP (B/A-GFP) or p210MIGFPCre (B/A-GFPCre) retrovirus (n = 5). Survival was longer in B/A-GFPCre -> Ptpn11fl/fl mice (P < 0.05; log-rank test). (c) GFP+ (leukemic) or GFP− (WT) B-lymphoid cells were isolated from peripheral blood of recipients of Mx-fl/fl donor cells, treated with vehicle or poly I:C, as indicated. Ptpn11 deletion was assessed by PCR of genomic DNA. Deleted and floxed alleles are as indicated. Bands that are not “deleted” or “floxed” are likely non-specific. Note that recipients in the Mx-fl/fl + pI:C group exhibited more efficient deletion in GFP− than GFP+ cells.
We next attempted to assess the effect of Ptpn11 deletion on B-ALL maintenance. Bulk BM cells from MxCre or MxCre;Ptpn11fl/fl donors were infected with p210MIGFP virus, and transplanted into lethally irradiated, syngeneic recipients. After B-ALL developed, half of the recipients in each cohort were treated with poly I:C to induce Ptpn11 deletion. However, poly I:C alone significantly alleviated disease (Supplementary Figure S4b), consistent with an interferon-induced inhibitory effect against B-ALL. These effects prevented assessment of the role of SHP2 on established B-ALL. We also generated mice (Rosa26-CreER;Ptpn11fl/fl) expressing a CRE-ER fusion protein, and used tamoxifen to induce Ptpn11 deletion. Unfortunately, the floxed allele did not delete efficiently, and although tamoxifen-treated mice showed a small increase in survival, Ptpn11-undeleted BCR-ABL blasts rapidly outgrew (data not shown).
Instead, we compared the abundance of Ptpn11-replete and -deleted cells in the same mouse: if SHP2 is required for disease maintenance, then Ptpn11-replete leukemic cells should out-compete Ptpn11-deficient cells. Ptpn11 deletion was assessed by PCR of GFP+ (BCR-ABL1+) and GFP− (uninfected) peripheral blood cell DNA from poly I:C-induced MxCre;Ptpn11fl/fl recipients. Ptpn11 deletion was much more efficient in GFP−, than in GFP+, cells (Figure 3c), indicating that SHP2 is required specifically for BCR-ABL1+ B-lymphoid proliferation/survival.
SHP2 deficiency selectively affects cycling of BCR-ABL1+ pre-B cells
We compared the effects of SHP2 deficiency on WT and BCR-ABL1+ pre-B cell proliferation in vitro. Rosa26-CreER;Ptpn11fl/fl pre-B cells were cultured in IL7-supplemented OptiMEM-10%FBS, and Ptpn11 deletion was induced with 4-OH in half of the cells (Supplementary Figure S5a). Unexpectedly, SHP2-deficient WT cells proliferated at the same rate as untreated controls (Figure 4a and Supplementary Figure S5b,c); hence, SHP2 is dispensable for WT pre-B cell proliferation in vitro. Furthermore, Cd19tm1(cre)Cgn;Ptpn11fl/fl mice exhibited grossly normal B cell development and agonist responses (Supplementary Figure S6), and preliminary studies of Cd79atm1(cre)Reth;Ptpn11fl/fl mice yielded similar results. Next, we cultured BCR-ABL1+ Rosa26-CreER and BCR-ABL1+ Rosa26-CreER;Ptpn11fl/fl pre-B cells, and induced Ptpn11 deletion in the latter with 4-OH. Remarkably, SHP2 deficiency selectively abrogated the proliferation of BCR-ABL1+ pre-B cells (Figure 4a and Supplementary Figure S5c). The allosteric SHP2 inhibitor SHP09959 also markedly suppressed BCR-ABL1+ cell proliferation (Supplementary Figure S7). Hence, SHP2 is required for BCR-ABL1+-evoked, but not WT, pre-B cell proliferation. SHP2 deficiency did not induce apoptosis in BCR-ABL1+ cells (Figure 4b and Supplementary Figure S5d), but instead, caused increased G0 and decreased G1 and S-G2-M cells (Figure 4c and Supplementary Figure S5e).
Figure 4. SHP2 is required for the proliferation of BCR/ABL1+ pre-B cells in vitro.
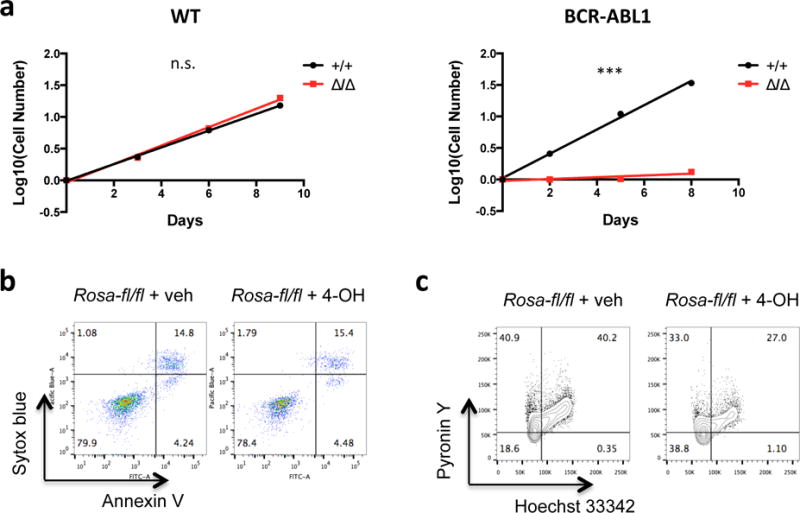
(a) WT Rosa26-CreER;Ptpn11fl/fl pre-B cells with intact (+/+) or deleted (Δ/Δ) Ptpn11 were cultured, and cell number was monitored (n = 4). Proliferation rates were not significantly different in +/+ and Δ/Δ cells (left). BCR-ABL1+ Rosa26-CreER;Ptpn11fl/fl pre-B cells with intact (+/+) or deleted (Δ/Δ) Ptpn11 were cultured, and cell number was monitored (n = 4). Note that the proliferation of SHP2-deficient BCR-ABL1+ pre-B cells was impaired (***P < 0.001; F test) (right). (b) BCR-ABL1+ Rosa26-CreER;Ptpn11fl/fl pre-B cells treated with vehicle or 4-OH were analyzed by flow cytometry for Annexin V and Sytox blue. Similar results were obtained in 4 biological replicates. (c) BCR-ABL1+ Rosa26-CreER;Ptpn11fl/fl pre-B cells treated with vehicle or 4-OH were stained with Hoechst 33342 and Pyronin Y. Similar results were obtained in 4 biological replicates.
SFK and MEK-ERK signaling contribute to the differential SHP2 requirement
We next assessed the effects of SHP2 on WT and BCR-ABL1+ pre-B cell signaling. In WT cells, SHP2 deficiency reduced phospho-ERK1 and phospho-ERK2 levels (Figure 5a and Supplementary Figure S8a), indicating that WT pre-B cells are insensitive to a reduction of ERK signaling of this magnitude. In WT pre-B cells, phospho-AKT (S473), phospho-STAT5 (Y694), and phospho-SFK (Y416) levels were not affected by SHP2 deficiency. In BCR-ABL1+ pre-B cells, Ptpn11 deletion also led to significantly lower levels of phospho-ERK1, phospho-ERK2, but comparable levels of phospho-AKT (S473) and phospho-STAT5 (Y694) (Figure 5a and Supplementary Figure S8a). However, Ptpn11 deletion also compromised SFK activation, as judged by phospho-SFK (Y416), only in BCR-ABL1+ pre-B cells (Figure 5a and Supplementary Figure S8a).
Figure 5. SFKs and ERK mediate the SHP2 requirement in BCR-ABL1+ pre-B cells.
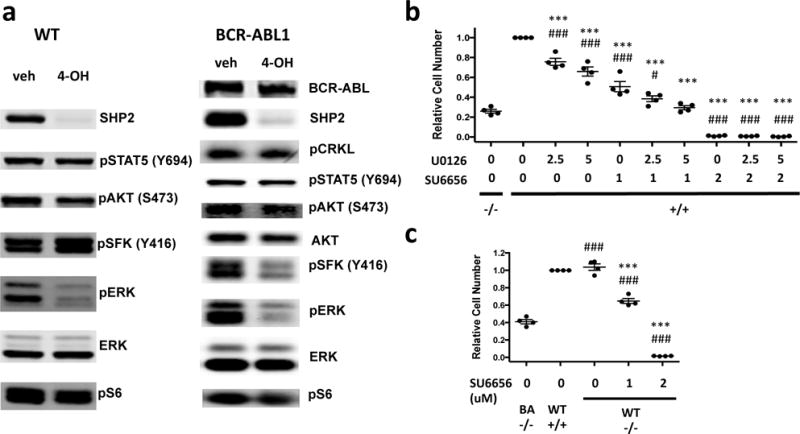
(a) Status of selected signaling molecules in WT or BCR-ABL1+ Rosa26-CreER;Ptpn11fl/fl pre-B cells treated with vehicle or 4-OH. Similar results were obtained in 4 biological replicates. (b) SHP2-replete BCR-ABL1+ pre-B cells were treated with the MEK/ERK inhibitor U0126 and/or the SFK inhibitor SU6656 at different concentrations. Viable cell numbers 3 days post-drug treatment were compared with SHP2-replete BCR-ABL1+ vehicle control (denoted by “*”) or SHP2-deficient control (denoted by “#”) groups. The significance of the cell number difference between combination treatment and SHP2 deficiency is decreased compared with the difference between single-agent treatment and SHP2 deficiency (mean ± s.e.m.; P < 0.05 between U0126(2.5μM) + SU6656(1μM) vs SHP2−/−) and the difference is not significant between U0126(5μM) + SU6656(1μM) vs SHP2−/−. (c) SHP2-deficient WT pre-B cells were treated with SU6656 (0, 1, 2μM). Viable cell numbers from SHP2-deficient WT groups were compared with SHP2-replete WT (denoted by “*”) or SHP2-deficient BCR-ABL1+ (denoted by “#”) groups (n = 4). 1μM SU6656 treatment recapitulates most of the proliferation inhibition by SHP2 deficiency, although the difference is still significant (0.65±0.06 vs 0.41±0.05, mean ± s.e.m). ***P < 0.001, #P < 0.05, ###P < 0.001; ANOVA with Bonferroni post-test multiple comparison.
To ask if decreased MEK/ERK and/or SFK activity contribute to the requirement for SHP2 for BCR-ABL1+ pre-B cell proliferation, we treated (SHP2-replete) BCR-ABL1+ pre-B cells with MEK/ERK (U0126) and/or SFK-selective (SU6656)60 inhibitors. At 1μM, SU6656 inhibited SFK activity (as assessed by phospho-SFK Y416) to levels similar to that caused by SHP2 deficiency (Supplementary Figure S8b). This dose also impaired BCR-ABL1+ pre-B cell proliferation, although not by as much as Ptpn11 deletion (Figure 5b). At 2.5μM, U0126 diminished phospho-ERK2 to levels comparable to those in SHP2-deficient cells, and had a smaller, but significant, effect on proliferation (Supplementary Figure S8b). Combined SU6656-U0126 treatment recapitulated the proliferation inhibition caused by SHP2 deficiency. Treatment with another selective SFK inhibitor, PP2, yielded similar results (data not shown). Therefore, decreased proliferation of SHP2-deficient, BCR-ABL1+ pre-B cells results from the combined effects of impaired SFK and ERK activation, with the former playing a more important role. SU6656 did not affect ERK activation, nor did U0126 affect SFK activation, so these pathways function independently downstream of SHP2 in pre-B cells.
The ability of WT pre-B cells to activate SFKs without SHP2 might explain why they do not require SHP2 to proliferate. Treatment of SHP2-deficient (BCR-ABL1-negative) pre-B cells with SU6656 (1μM) reduced phospho-SFK to levels slightly more than in SHP2-deficient BCR-ABL1+ cells, whereas 2μM SU6656 lowered p-SFK levels to a slightly greater extent (Supplementary Figure S8c). Remarkably, these doses of SU6656 inhibited SHP2-deficient WT pre-B cell proliferation to an extent slightly less than, or slightly more than, did SHP2-deficiency in BCR-ABL1+ pre-B cells (Figure 5c). Persistent SFK signaling probably explains why SHP2 is dispensable for WT pre-B cell proliferation.
Structural determinants of SHP2 required for BCR-ABL1+ pre-B cell proliferation
To identify sub-domains of SHP2 required for BCR-ABL1+ signaling in pre-B cells, we compared proliferation and signaling in SHP2-sufficient (veh) or -deficient BCR-ABL1+ pre-B cells reconstituted with empty vector (4-OH), WT SHP2 (WT), the phosphatase-inactive C459E mutant (CE), or a mutant lacking both C-terminal tyrosine residues, Y542F/Y580F (Y2F). WT SHP2 rescued proliferation and ERK and SFK activity. By contrast, the CE or Y2F mutant did not restore proliferation or signaling (Figure 6 and Supplementary Figure S9), even though each was expressed at levels similar to WT SHP2. SH2 domain engagement is also essential, given the effects of SHP099 (Supplementary Figure S7), which locks SHP2 in the closed form and prevents N-SH2/p-Tyr peptide binding.59,61
Figure 6. SHP2 domains required for signaling and proliferation.
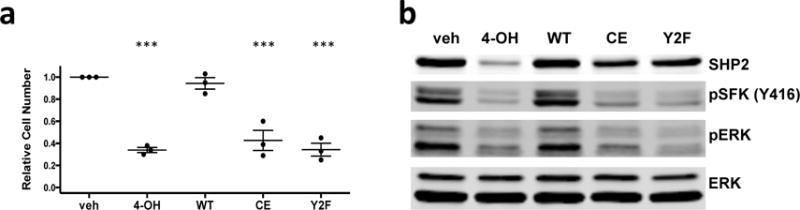
(a) Relative viable cell number after 2-day cultures, pooled from 3 biological replicates. The proliferation of Rosa26-CreER;Ptpn11fl/fl BCR-ABL1+ cells subjected to the indicated treatments was assessed: (1) empty vector infection followed by vehicle treatment (veh); (2) empty vector infection followed by 4-OH treatment (4-OH); (3–5) reconstitution with SHP2 variants (WT, C459E or Y542F/Y580F) followed by 4-OH treatment. Viable cell numbers were compared with the vehicle group. (n = 3; mean ± s.e.m.; ***P < 0.001; ANOVA with Bonferroni post-test) (b) Comparison of phospho-SFK(Y416) and phospho-ERK in Rosa26-CreER;Ptpn11fl/fl BCR-ABL1+ cells from experiments in panel (a). Similar results were obtained in 3 biological replicates.
Distinct effects of SHP2 on transcriptional profiles of BCR-ABL1+ and WT pre-B cells
To gain further insight into the role of SHP2 in WT and BCR-ABL1+ pre-B cells, we performed RNA-seq. Unsupervised hierarchical clustering and principal component analysis (PCA) revealed good separation between both WT and BCR-ABL1+ cells and between SHP2-deficient and -replete cells, respectively (Supplementary Figure S10a,b). As expected62,63, BCR-ABL1+ cells (compared with WT) showed up-regulation of ERK targets and down-regulation of interferon-alpha response genes (Supplementary Figure S11 and Supplementary Tables S1–S2). Supervised comparisons revealed that SHP2 significantly regulates the expression of ~770 genes in WT, and >1900 genes in BCR-ABL1+, pre-B cells (Supplementary Figure S10c). Also as expected, SHP2 deficiency decreased ERK-regulated gene expression in WT and BCR-ABL1+ samples (Supplementary Table S3). TLR-regulated genes also were decreased by SHP2-deficiency in both contexts. Shared up-regulated genes were not enriched for any curated Reactome pathway (Supplementary Table S4).
Next, we compared BCR-ABL1+ SHP2-deficient versus SHP2-expressing cells and WT SHP2-deficient versus SHP2-expressing cells, respectively. SHP2 deficiency in normal pre-B cells affected genes annotated by the terms metabolic process and glycolysis (Supplementary Figure S10d and Supplementary Table S5). In BCR-ABL1+ cells, SHP2-deficiency also affected genes for metabolic and biosynthetic processes; in addition, cell-proliferation genes were altered (Supplementary Figure S10d and Supplementary Table S6). We identified, and validated by qRT-PCR, differential effects of SHP2 deficiency on cell cycle regulators in WT and BCR-ABL1+ cells. Ccnd1 (encoding CYCLIN D1) expression was decreased, while genes encoding the CDK inhibitors p18ARF and p19ARF (Cdkn2c and Cdkn2d) were increased preferentially by SHP2 deficiency in BCR-ABL1+ cells (Figure 7a). These results comport with the cell cycle impairment seen in SHP2-deficient BCR-ABL1+ pre-B cells (Figure 4c).
Figure 7. SHP2 affects distinct transcriptional programs in WT and BCR-ABL1+ pre-B cells.
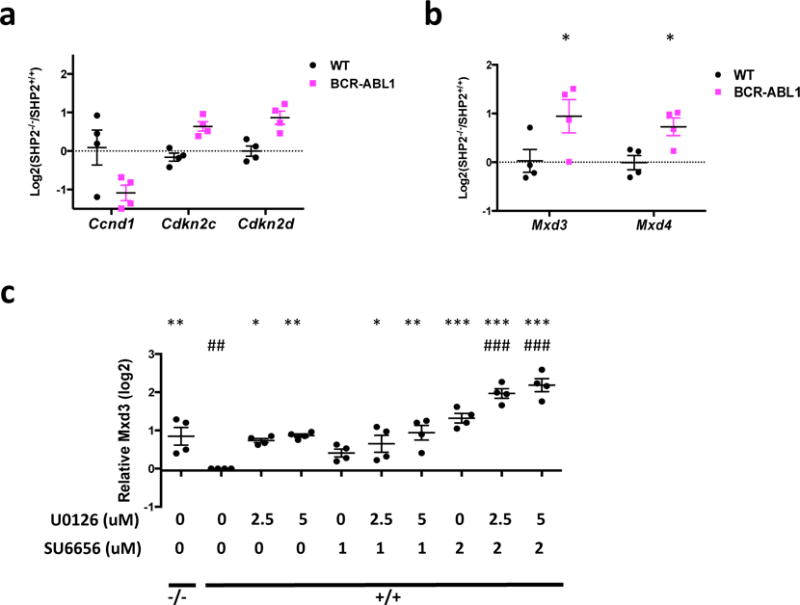
(a) Validation of differential expression of cell cycle regulatory genes by q-RT-PCR (n = 4). Ccnd1, Cdkn2c, and Cdkn2d are regulated by SHP2 in in BCR-ABL1+, but not WT, pre-B cells. (b) qRT-PCR validation of genes involved in regulation of MYC transcription (n = 4). The log ratios of Mxd3 or Mxd4 transcription levels in SHP2-deficient vs SHP2-replete cells are shown. Mxd3 and Mxd4 mRNA levels are not regulated by SHP2 deficiency in WT pre-B cells, but are significantly up-regulated in BCR-ABL1+ pre-B cells. (c) BCR-ABL1+ SHP2-deficient (−/−) or replete (+/+) pre-B cells were treated with U0126 and/or SU6656 at indicated concentrations for 2 days. Relative mRNA levels for Mxd3 were quantified (n = 4). Transcription levels were compared with SHP2-replete BCR-ABL1+ vehicle control (denoted by “*”) or SHP2-deficient control (denoted by “#”) groups. (*P < 0.05, **P < 0.01, ***P < 0.001, ##P < 0.01, ###P < 0.001; ANOVA with Bonferroni post-test).
Consistent with GAB2 regulating multiple signal relay molecules,15,64 more genes and gene sets were affected by GAB2 deletion21 than by SHP2 deficiency (Supplementary Figure S12a and Supplementary Tables S7–S8). Nevertheless, the top gene sets affected by either deficiency showed strong overlap and similar FDR q-values (Supplementary Figure S12b). Gene sets down-regulated only by GAB2 deficiency included multiple DNA repair/synthesis-related and metastasis-related gene sets, among others (Supplementary Tables S7–S8).
GSEA revealed down-regulation of MYC-induced signatures in SHP2- (and GAB2-) deficient, BCR-ABL1+ cells, but no significant alteration in SHP2-deficient WT cells (Supplementary Figure S13a and Supplementary Tables S7 & S9). Enrichr analysis showed that genes down-regulated by SHP2 deficiency in BCR-ABL1+ cells were highly enriched for binding sites for MYC and its co-factors (Supplementary Table S12). Genes down-regulated in SHP2-deficient WT cells also were enriched for MYC/co-factor binding sites, but with much lower overlap and significance (Supplementary Figure S13b and Supplementary Table S13). Intriguingly, multiple MYC targets were regulated in opposite directions in SHP2-deficient BCR-ABL1+ and WT pre-B cells. For example, glycolytic gene expression was increased in WT cells, but down-regulated in BCR-ABL1+ cells (Supplementary Figure S13c,d). Hence, SHP2 deficiency has quantitatively and qualitatively distinct effects on MYC-dependent transcriptome (“MYC regulome”) in BCR-ABL1+ (compared with WT) pre-B cells.
Gene sets up-regulated by SHP2 deficiency in BCR-ABL1+ and WT cells differed widely. Interferon response genes were among the top up-regulated sets in BCR-ABL1+ cells (Supplementary Figure S14 and Supplementary Table S10), whereas hypoxia response genes were the top up-regulated group in WT cells (Supplementary Table S11). Up-regulated genes in WT and BCR-ABL1 contexts were enriched comparably for binding sites for EP300, ETS1, and several other transcription factors/regulators (Supplementary Tables S14–S15). Genes with binding sites for CHD2 and ZKSCAN1 were enriched preferentially in BCR-ABL1+ cells, whereas those with RAD21 binding sites were enriched preferentially in WT cells (Supplementary Tables S14–S15).
Given the key role of MYC in cell proliferation,65 the selectively impaired proliferation of SHP2-deficient BCR-ABL1+ pre-B cells probably reflects alteration of the MYC regulome, at least in part. We asked why MYC target genes are regulated differentially in SHP2-deficient WT and BCR-ABL1+ cells. Myc mRNA levels were unaffected by SHP2-deficiency in WT or BCR-ABL1+ cells (Supplementary Figure S15a). MYC stability is regulated by ERK66 and, consistent with impaired ERK activation in SHP2-deficient WT and BCR-ABL1+ pre-B cells, MYC levels were decreased in both, but to similar extents (Supplementary Figure S15b). By contrast, Mxd3 and Mxd4 were up-regulated by SHP2 deficiency only in BCR-ABL1+, but not in WT cells (Figure 7b and Supplementary Figure S15c). These increases were recapitulated by co-inhibition of ERK and SFKs in SHP2-replete BCR-ABL1+ cells (Figure 7c and Supplementary Figure S15d). MXD3 and MXD4 compete with MYC for binding to MAX and inhibit transcription of a subset of MYC targets involved in proliferation and transformation.67 Failure to repress Mxd3 and Mxd4 transcription might explain the more profound impairment of MYC-dependent transcription in SHP2-deficient BCR-ABL1 pre-B cells.
Discussion
Our genetic approach enabled us to assess the role of SHP2 in BCR-ABL1-induced neoplasia in well-characterized mouse models. We find that SHP2 is essential for initiation and maintenance of CML-like MPN, and initiation and, most likely, maintenance, of Ph+ B-ALL. Similar to its effects in WT HSCs,31,32 SHP2 is required for the survival of phenotypic CML-SCs. Intriguingly, SHP2, and in particular, SHP2 catalytic activity and its C-terminal tyrosine residues, is essential for BCR-ABL1+, but not WT, pre-B cell proliferation. SHP2 mediates ERK activation in WT and BCR-ABL+ cells, but its differential effect on proliferation correlates with, and is likely caused by, distinct effects on SFKs. Transcriptional profiling suggests that the combined effects of ERK and SFKs, perhaps by repressing Mxd3/4 transcription, are needed to fully activate the MYC regulome.
Previous studies implicated SHP2 in critical events downstream of BCR-ABL1, but none addressed its role directly in a pathologically relevant context.21,40 For example, PTPN11 knockdown reduces cytokine-independent colony formation by CD34+ CML cells in vitro,68 but its effects on CML and Ph+ B-ALL development remained unclear. SHP2 is required for BCR-ABL transformation of a yolk sac cell line,40 but BCR-ABL1-induced CML and B-ALL are typically adult diseases, and adult and embryonic hematopoiesis differ.69 GAB2, and its SHP2 binding sites, is essential for induction of Ph+ hematopoietic neoplasia in mice.21 Yet other signaling molecules might bind the same sites, and SHP2 uses other adaptors/interactors besides GAB2; hence, loss of GAB2/SHP2 binding might have underestimated the role of SHP2 in BCR-ABL1 disease.24,25 Notably, SHP2 binding sites on GAB2 are required for full activation of STAT5 and AKT,21 but SHP2 deficiency did not affect STAT5 or AKT activation in the current study. One potential explanation for this apparent discrepancy is that the SHP2 binding sites of GAB2 might be shared by other proteins. For example, SHP2 and SOCS3 both interact with pY759 of gp130.70,71 Likewise, other proteins might share the SHP2 binding sites of GAB2. Alternatively, whereas GAB2-bound SHP2 can activate AKT and STAT5, other signaling proteins might inhibit AKT and/or STAT5 via SHP2, such that the net effect of SHP2 depletion differs from disruption of GAB2-SHP2 interaction.
The relationship between SHP2 and the RAS-ERK pathway is well documented.24,25 In hematopoietic cells, gain-of-function PTPN11 mutants induce RAS-ERK hyper-activation and can cause JMML. Conversely, Ptpn11 deletion impairs ERK activation by growth factors and cytokines in HSCs and progenitors.31,32 SHP2 also is required for ERK activation in WT and BCR-ABL1 transformed myeloid and pre-B cells. Surprisingly, however, WT pre-B cells, unlike myeloid progenitors31,32 and multiple other proliferating cells,72,73 are agnostic to defective ERK activation. How pre-B cells avoid the requirement for ERK activation seen in almost all other proliferative cells and tissues awaits further investigation.
Regulation of SFKs by SHP2 also has been reported.41,74,75 For as yet unclear reasons, SHP2 is required for SFK activation in BCR-ABL1-transformed, but not WT pre-B cells. SHP2 also is required for BCR-ABL1+, but not WT, pre-B cell proliferation. Inhibitor studies suggest that SFKs (to a major extent) and ERK (to a lesser extent) contribute independently to this selective requirement. Likewise, SHP2-independent SFK activation in WT cells is likely a(the) major resason for the differential requirement for SHP2 in BCR-ABL1+ and WT pre-B cell proliferation. Consistent with our results, SFKs, although dispensable for CML, are essential for Ph+ B-ALL.76
Previous studies suggested that SHP2 catalytic activity is required for ERK and/or SFK activation.41,77 The C-terminal tyrosines (Y542 and Y580) modulate activation of ERK downstream of multiple RTKs,78 although their roles in SFK activation had not been defined. SH2 domain/pY peptide interaction also is required for myeloid transformation by leukemogenic SHP2 mutants.79 We found that SH2 domain engagement, PTP activity, and the C-terminal tyrosines are required for BCR-ABL1+ pre-B cell proliferation and full activation of ERK and SFKs by BCR-ABL1.
Our RNA-seq studies reveal transcriptional programs differentially regulated by SHP2 in WT and BCR-ABL1+ pre-B cells. GSEA and Enrichr strongly suggest that defective MYC-driven transcription is a major consequence of SHP2 deficiency and is affected differentially in BCR-ABL1+ and WT pre-B cells. Some MYC-regulated genes are affected by SHP2 deficiency in BCR-ABL1-transformed, but not WT B cells, whereas others, including glycolytic genes, are regulated in opposite directions. Failure to engage Warburg respiration, a common feature of malignant B cells and other tumor cells,80,81 might contribute to cell cycle arrest in SHP2-deficient BCR-ABL1+ pre-B cells. As MYC is a key transcriptional hub for B cell growth and proliferation control,82 differential MYC transcription is likely to be a(the) major reason for the impaired proliferation of BCR-ABL1+ SHP2-deficient B cells.
Down-regulated MYC activity also is likely mediated by decreased SFK and/or ERK activation. SFKs increase MYC mRNA/protein levels,60,83–85 and MYC is stabilized by ERK-mediated phosphorylation.66,73,84 Yet Myc upregulation/MYC stabilization does not explain the differential regulation of MYC-dependent transcription by SHP2. Instead, we find that SFKs and/or ERK regulate cofactors/regulators of MYC: SHP2 deficiency elevates Mxd3 and Mxd4 specifically in BCR-ABL1+ pre-B cells, and this regulation is recapitulated by co-inhibition of ERK and SFKs.
MXD3 and MXD4 can occupy the MYC E-box, inhibit MYC/MAX binding, and repress MYC-induced transformation.86 Serum or insulin induce phosphorylation-directed degradation of MXD1 in HeLa cells,87 but transcriptional regulation of MXDs has not been reported. Notably, inspection of RNA-seq data from PDGF-stimulated smooth muscle cells88 reveals that MXD1, MXD3, MXD4, and MNT, which encodes an MXD-like protein, are down-regulated. STK38 reportedly regulates MYC-dependent transcription in ST486 B cell lymphoma cells by affecting MYC turnover.89 In that dataset, however, MXD4 and MNT also were up-regulated by STK38 silencing. Re-analysis of other studies90,91 also reveals regulation of MXDs and/or MNT mRNA levels by various cytokines or growth factors. Hence, regulation of the MXD family67 might be a general, but under-appreciated, mechanism for modulating MYC-dependent transcription. MXD3/4 up-regulation by SHP2 deficiency in BCR-ABL1+ pre-B cells likely contributes to inhibition of MYC-mediated transcription.
Although altered MYC-dependent transcription probably is a major contributor to defective proliferation in SHP2-deficient BCR-ABL1 pre-B cells, Enrichr analysis suggests that several other transcription factors/regulators also are regulated differentially, including BHLHE40, ETS1, CHD1, and CHD2. Further studies are needed to determine whether these molecules contribute to the specific requirement of SHP2 in BCR-ABL1+ pre-B cells.
IFN-α responsive genes also were up-regulated by SHP2 deficiency specifically in BCR-ABL1+ cells. IFN-α has been used to treat Ph+ B-ALL (as well as CML) and improves survival.92,93 IFN-α also induces growth arrest, and sometimes differentiation, in human and murine B cell lines.94–96 We did not detect differential expression of drivers/markers of B cell differentiation, including Ikzf1, Spi1, Pax5, Ebf1, Tcf3, Tnf13, and Tnf13b97–99 (data not shown), so enhanced differentiation is unlikely to contribute to the effects of SHP2 deficiency in BCR-ABL1+ B-ALL.
In summary, we find that SHP2 is essential for the pathogenesis of BCR-ABL1-evoked myeloid and lymphoid neoplasia. SHP2 is required specifically for BCR-ABL1+, but not WT, pre-B cell proliferation, because it mediates SFK and ERK activation in BCR-ABL1+ pre-B cells, but only ERK activation in WT pre-B cells. In BCR-ABL1+ cells, SHP2 suppresses Mxd3 and Mxd4 through ERK and SFKs, likely leading to induction of select MYC targets, whereas in WT cells, SHP2 does not regulate the transcription of these genes. Our results suggest that SHP2 might be an alternative/additional target in BCR-ABL1-induced malignancies, a prospect made tangible with the recent development of bioavailable and highly selective SHP2 inhibitor.59
Supplementary Material
Acknowledgments
We thank the Princess Margaret Genomics Centre for RNA-seq, and the Sick Kids-UHN Flow Cytometry Facility for FACS. This work was supported by R35CA49152 to B.G.N., and NIH R01 CA090576 to R.A.V.. B.G.N. was a Canada Research Chair, Tier 1, and work in his laboratory was partially supported by the Princess Margaret Cancer Foundation and a grant from the Ontario Ministry of Health and Long Term Care.
Footnotes
Authorship Contributions
S.G. and B.G.N. designed the experiments. S.G., G.C., and W.Y. performed the experiments. S.G., A.Z., Z.L., and C.V. analyzed RNA-seq data. R.A.V. provided essential advice. S.G. and B.G.N. wrote and all authors edited the manuscript.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
- 1.Fainstein E, Marcelle C, Rosner A, Canaani E, Gale RP, Dreazen O, et al. A new fused transcript in Philadelphia chromosome positive acute lymphocytic leukaemia. Nature. 1987;330:386–8. doi: 10.1038/330386a0. [DOI] [PubMed] [Google Scholar]
- 2.Nowell PC, Hungerford DA. A minute chromosome in chronic granulocytic leukemia. Science. 1960;132:1488–501. [Google Scholar]
- 3.Kalmanti L, Saussele S, Lauseker M, Müller MC, Dietz CT, Heinrich L, et al. Safety and efficacy of imatinib in CML over a period of 10 years: data from the randomized CML-study IV. Leukemia. 2015;29:1123–32. doi: 10.1038/leu.2015.36. [DOI] [PubMed] [Google Scholar]
- 4.Graham SM, Jørgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–25. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 5.Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926–35. doi: 10.1038/sj.leu.2404609. [DOI] [PubMed] [Google Scholar]
- 6.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121:396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanguy-Schmidt A, Rousselot P, Chalandon Y, Cayuela J-M, Hayette S, Vekemans M-C, et al. Long-term follow-up of the imatinib GRAAPH-2003 study in newly diagnosed patients with de novo Philadelphia chromosome-positive acute lymphoblastic leukemia: a GRAALL study. Biol Blood Marrow Transplant. 2013;19:150–5. doi: 10.1016/j.bbmt.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 8.Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12:7374–9. doi: 10.1158/1078-0432.CCR-06-1516. [DOI] [PubMed] [Google Scholar]
- 9.Pfeifer H, Wassmann B, Pavlova A, Wunderle L, Oldenburg J, Binckebanck A, et al. Kinase domain mutations of BCR-ABL frequently precede imatinib-based therapy and give rise to relapse in patients with de novo Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL) Blood. 2007;110:727–34. doi: 10.1182/blood-2006-11-052373. [DOI] [PubMed] [Google Scholar]
- 10.Duy C, Hurtz C, Shojaee S, Cerchietti L, Geng H, Swaminathan S, et al. BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR-ABL1 kinase inhibition. Nature. 2011;473:384–8. doi: 10.1038/nature09883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daley GQ, Van Etten RA, Baltimore D. Blast crisis in a murine model of chronic myelogenous leukemia. Proc Natl Acad Sci U S A. 1991;88:11335–8. doi: 10.1073/pnas.88.24.11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li S, Ilaria RL, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med. 1999;189:1399–412. doi: 10.1084/jem.189.9.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- 14.Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75:175–85. [PubMed] [Google Scholar]
- 15.Ahmed W, Van Etten RA. Signal transduction in the chronic leukemias: implications for targeted therapies. Curr Hematol Malig Rep. 2013;8:71–80. doi: 10.1007/s11899-012-0150-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sattler M, Mohi MG, Pride YB, Quinnan LR, Malouf Na, Podar K, et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell. 2002;1:479–92. doi: 10.1016/s1535-6108(02)00074-0. [DOI] [PubMed] [Google Scholar]
- 17.Gu H, Pratt JC, Burakoff SJ, Neel BG. Cloning of p97/Gab2, the major SHP2-binding protein in hematopoietic cells, reveals a novel pathway for cytokine-induced gene activation. Mol Cell. 1998;2:729–40. doi: 10.1016/s1097-2765(00)80288-9. [DOI] [PubMed] [Google Scholar]
- 18.Million RP, Van Etten RA. The Grb2 binding site is required for the induction of chronic myeloid leukemia-like disease in mice by the Bcr/Abl tyrosine kinase. Blood. 2000;96:664–70. [PubMed] [Google Scholar]
- 19.Zhang X, Subrahmanyam R, Wong R, Gross AW, Ren R. The NH(2)-terminal coiled-coil domain and tyrosine 177 play important roles in induction of a myeloproliferative disease in mice by Bcr-Abl. Mol Cell Biol. 2001;21:840–53. doi: 10.1128/MCB.21.3.840-853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He Y, Wertheim JA, Xu L, Miller JP, Karnell FG, Choi JK, et al. The coiled-coil domain and Tyr177 of bcr are required to induce a murine chronic myelogenous leukemia-like disease by bcr/abl. Blood. 2002;99:2957–68. doi: 10.1182/blood.v99.8.2957. [DOI] [PubMed] [Google Scholar]
- 21.Gu S, Chan WW, Mohi G, Rosenbaum J, Sayad A, Lu Z, et al. Distinct GAB2 signaling pathways are essential for myeloid and lymphoid transformation and leukemogenesis by BCR-ABL1. Blood. 2016;127:1803–1813. doi: 10.1182/blood-2015-06-653006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noguchi T, Matozaki T, Horita K, Fujioka Y, Kasuga M. Role of SH-PTP2, a protein-tyrosine phosphatase with Src homology 2 domains, in insulin-stimulated Ras activation. Mol Cell Biol. 1994;14:6674–82. doi: 10.1128/mcb.14.10.6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–93. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 24.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27:179–92. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 25.Grossmann KS, Rosário M, Birchmeier C, Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res. 2010;106:53–89. doi: 10.1016/S0065-230X(10)06002-1. [DOI] [PubMed] [Google Scholar]
- 26.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–50. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 27.Barford D, Neel BG. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 1998;6:249–54. doi: 10.1016/s0969-2126(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 28.Lechleider RJ, Sugimoto S, Bennett AM, Kashishian AS, Cooper JA, Shoelson SE, et al. Activation of the SH2-containing phosphotyrosine phosphatase SH-PTP2 by its binding site, phosphotyrosine 1009, on the human platelet-derived growth factor receptor. J Biol Chem. 1993;268:21478–81. [PubMed] [Google Scholar]
- 29.Tsutsumi R, Masoudi M, Takahashi A, Fujii Y, Hayashi T, Kikuchi I, et al. YAP and TAZ, Hippo signaling targets, act as a rheostat for nuclear SHP2 function. Dev Cell. 2013;26:658–65. doi: 10.1016/j.devcel.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 30.Qu CK, Shi ZQ, Shen R, Tsai FY, Orkin SH, Feng GS. A deletion mutation in the SH2-N domain of Shp-2 severely suppresses hematopoietic cell development. Mol Cell Biol. 1997;17:5499–507. doi: 10.1128/mcb.17.9.5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan G, Cheung LS, Yang W, Milyavsky M, Sanders AD, Gu S, et al. Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood. 2011;117:4253–61. doi: 10.1182/blood-2010-11-319517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu HH, Ji K, Alderson N, He Z, Li S, Liu W, et al. Kit-Shp2-Kit signaling acts to maintain a functional hematopoietic stem and progenitor cell pool. Blood. 2011;117:5350–61. doi: 10.1182/blood-2011-01-333476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 34.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103:2325–31. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 35.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–20. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 36.Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004;104:307–13. doi: 10.1182/blood-2003-11-3876. [DOI] [PubMed] [Google Scholar]
- 37.Hatlen MA, Arora K, Vacic V, Grabowska EA, Liao W, Riley-Gillis B, et al. Integrative genetic analysis of mouse and human AML identifies cooperating disease alleles. J Exp Med. 2015 doi: 10.1084/jem.20150524. jem.20150524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu H, Griffin JD, Neel BG. Characterization of two SHP-2-associated binding proteins and potential substrates in hematopoietic cells. J Biol Chem. 1997;272:16421–30. doi: 10.1074/jbc.272.26.16421. [DOI] [PubMed] [Google Scholar]
- 39.Tauchi T, Feng GS, Shen R, Song HY, Donner D, Pawson T, et al. SH2-containing phosphotyrosine phosphatase Syp is a target of p210bcr-abl tyrosine kinase. J Biol Chem. 1994;269:15381–7. [PubMed] [Google Scholar]
- 40.Chen J, Yu W-M, Daino H, Broxmeyer HE, Druker BJ, Qu C-K. SHP-2 phosphatase is required for hematopoietic cell transformation by Bcr-Abl. Blood. 2007;109:778–85. doi: 10.1182/blood-2006-04-019141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, et al. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol Cell. 2004;13:341–55. doi: 10.1016/s1097-2765(04)00050-4. [DOI] [PubMed] [Google Scholar]
- 42.Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature. 2013;499:491–5. doi: 10.1038/nature12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walz C, Ahmed W, Lazarides K, Betancur M, Patel N, Hennighausen L, et al. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood. 2012;119:3550–60. doi: 10.1182/blood-2011-12-397554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Million RP, Harakawa N, Roumiantsev S, Varticovski L, Van Etten RA. A direct binding site for Grb2 contributes to transformation and leukemogenesis by the Tel-Abl (ETV6-Abl) tyrosine kinase. Mol Cell Biol. 2004;24:4685–95. doi: 10.1128/MCB.24.11.4685-4695.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neering SJ, Bushnell T, Sozer S, Ashton J, Rossi RM, Wang P-Y, et al. Leukemia stem cells in a genetically defined murine model of blast-crisis CML. Blood. 2007;110:2578–85. doi: 10.1182/blood-2007-02-073031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corfe Sa, Gray AP, Paige CJ. Generation and characterization of stromal cell independent IL-7 dependent B cell lines. J Immunol Methods. 2007;325:9–19. doi: 10.1016/j.jim.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 47.Harlow E, Lane D. Using antibodies: a laboratory manual. 1999 [Google Scholar]
- 48.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–3. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 52.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gishizky ML, Witte ON. BCR/ABL enhances growth of multipotent progenitor cells but does not block their differentiation potential in vitro. Curr Top Microbiol Immunol. 1992;182:65–72. doi: 10.1007/978-3-642-77633-5_8. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol. 2007;8:665–8. doi: 10.1038/ni0707-665. [DOI] [PubMed] [Google Scholar]
- 55.Mahoney SJ, Dempsey JM, Blenis J. Chapter 2 Cell Signaling in Protein Synthesis. Progress in molecular biology and translational science. 2009:53–107. doi: 10.1016/S1877-1173(09)90002-3. [DOI] [PubMed] [Google Scholar]
- 56.Albers C, Illert AL, Miething C, Leischner H, Thiede M, Peschel C, et al. An RNAi-based system for loss-of-function analysis identifies Raf1 as a crucial mediator of BCR-ABL-driven leukemogenesis. Blood. 2011;118:2200–10. doi: 10.1182/blood-2010-10-309583. [DOI] [PubMed] [Google Scholar]
- 57.Packer LM, Rana S, Hayward R, O’Hare T, Eide Ca, Rebocho A, et al. Nilotinib and MEK Inhibitors Induce Synthetic Lethality through Paradoxical Activation of RAF in Drug-Resistant Chronic Myeloid Leukemia. Cancer Cell. 2011;20:715–727. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mohi MG, Boulton C, Gu T-L, Sternberg DW, Neuberg D, Griffin JD, et al. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004;101:3130–5. doi: 10.1073/pnas.0400063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen Y-NP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535:148–52. doi: 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- 60.Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, et al. SU6656, a Selective Src Family Kinase Inhibitor, Used To Probe Growth Factor Signaling. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ran H, Tsutsumi R, Araki T, Neel BG. Sticking It to Cancer with Molecular Glue for SHP2. Cancer Cell. 2016;30:194–6. doi: 10.1016/j.ccell.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cortez D, Reuther G, Pendergast AM. The Bcr-Abl tyrosine kinase activates mitogenic signaling pathways and stimulates G1-to-S phase transition in hematopoietic cells. Oncogene. 1997;15:2333–42. doi: 10.1038/sj.onc.1201400. [DOI] [PubMed] [Google Scholar]
- 63.Katsoulidis E, Sassano A, Majchrzak-Kita B, Carayol N, Yoon P, Jordan A, et al. Suppression of interferon (IFN)-inducible genes and IFN-mediated functional responses in BCR-ABL-expressing cells. J Biol Chem. 2008;283:10793–803. doi: 10.1074/jbc.M706816200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ding C, Yu W, Feng J, Luo J. Structure and function of Gab2 and its role in cancer (Review) Mol Med Rep. 2015;12:4007–14. doi: 10.3892/mmr.2015.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. 2000;16:653–99. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 66.Sears R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Diolaiti D, McFerrin L, Carroll PA, Eisenman RN. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim Biophys Acta. 2015;1849:484–500. doi: 10.1016/j.bbagrm.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scherr M, Chaturvedi A, Battmer K, Dallmann I, Schultheis B, Ganser A, et al. Enhanced sensitivity to inhibition of SHP2, STAT5, and Gab2 expression in chronic myeloid leukemia (CML) Blood. 2006;107:3279–87. doi: 10.1182/blood-2005-08-3087. [DOI] [PubMed] [Google Scholar]
- 69.Babovic S, Eaves CJ. Hierarchical organization of fetal and adult hematopoietic stem cells. Exp Cell Res. 2014;329:185–91. doi: 10.1016/j.yexcr.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 70.Schmitz J, Weissenbach M, Haan S, Heinrich PC, Schaper F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J Biol Chem. 2000;275:12848–56. doi: 10.1074/jbc.275.17.12848. [DOI] [PubMed] [Google Scholar]
- 71.Nicholson SE, De Souza D, Fabri LJ, Corbin J, Willson TA, Zhang JG, et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc Natl Acad Sci U S A. 2000;97:6493–8. doi: 10.1073/pnas.100135197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Geest CR, Coffer PJ. MAPK signaling pathways in the regulation of hematopoiesis. J Leukoc Biol. 2009;86:237–50. doi: 10.1189/jlb.0209097. [DOI] [PubMed] [Google Scholar]
- 73.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 74.Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, et al. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell. 2006;10:317–27. doi: 10.1016/j.devcel.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 75.Bertotti A, Comoglio PM, Trusolino L. Beta4 integrin activates a Shp2-Src signaling pathway that sustains HGF-induced anchorage-independent growth. J Cell Biol. 2006;175:993–1003. doi: 10.1083/jcb.200605114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36:453–61. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- 77.Stewart RA, Sanda T, Widlund HR, Zhu S, Swanson KD, Hurley AD, et al. Phosphatase-dependent and -independent functions of Shp2 in neural crest cells underlie LEOPARD syndrome pathogenesis. Dev Cell. 2010;18:750–62. doi: 10.1016/j.devcel.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Araki T, Nawa H, Neel BG. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J Biol Chem. 2003;278:41677–84. doi: 10.1074/jbc.M306461200. [DOI] [PubMed] [Google Scholar]
- 79.Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S, et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005;7:179–191. doi: 10.1016/j.ccr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 80.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 82.Zeller KI, Zhao X, Lee CWH, Chiu KP, Yao F, Yustein JT, et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A. 2006;103:17834–9. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chang Y-M, Bai L, Liu S, Yang JC, Kung H-J, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008;27:6365–75. doi: 10.1038/onc.2008.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ren Y, Chen Z, Chen L, Fang B, Win-Piazza H, Haura E, et al. Critical role of Shp2 in tumor growth involving regulation of c-Myc. Genes Cancer. 2010;1:994–1007. doi: 10.1177/1947601910395582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, et al. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A. 2001;98:7319–24. doi: 10.1073/pnas.131568898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hurlin PJ, Quéva C, Koskinen PJ, Steingrímsson E, Ayer DE, Copeland NG, et al. Mad3 and Mad4: novel Max-interacting transcriptional repressors that suppress c-myc dependent transformation and are expressed during neural and epidermal differentiation. EMBO J. 1995;14:5646–59. doi: 10.1002/j.1460-2075.1995.tb00252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci. 2008;105:6584–6589. doi: 10.1073/pnas.0802785105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang W, Ramachandran A, You S, Jeong H, Morley S, Mulone MD, et al. Integration of proteomic and transcriptomic profiles identifies a novel PDGF-MYC network in human smooth muscle cells. Cell Commun Signal. 2014;12:44. doi: 10.1186/s12964-014-0044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bisikirska BC, Adam SJ, Alvarez MJ, Rajbhandari P, Cox R, Lefebvre C, et al. STK38 is a critical upstream regulator of MYC’s oncogenic activity in human B-cell lymphoma. Oncogene. 2013;32:5283–5291. doi: 10.1038/onc.2012.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moodley D, Yoshida H, Mostafavi S, Asinovski N, Ortiz-Lopez A, Symanowicz P, et al. Network pharmacology of JAK inhibitors. Proc Natl Acad Sci. 2016;113:9852–9857. doi: 10.1073/pnas.1610253113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bradley G. Gene Expression Omnibus GSE60880. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60880. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60880 (accessed 13 Jan2017)
- 92.Wassmann B, Scheuring U, Pfeifer H, Binckebanck a, Käbisch a, Lübbert M, et al. Efficacy and safety of imatinib mesylate (Glivec) in combination with interferon-alpha (IFN-alpha) in Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ALL) Leuk Off J Leuk Soc Am Leuk Res Fund, UK. 2003;17:1919–24. doi: 10.1038/sj.leu.2403093. [DOI] [PubMed] [Google Scholar]
- 93.Visani G, Martinelli G, Piccaluga P, Tosi P, Amabile M, Pastano R, et al. Alpha-interferon improves survival and remission duration in P-190BCR-ABL positive adult acute lymphoblastic leukemia. Leukemia. 2000;14:22–7. doi: 10.1038/sj.leu.2401641. [DOI] [PubMed] [Google Scholar]
- 94.Thomas NS, Pizzey AR, Tiwari S, Williams CD, Yang J. p130, p107, and pRb are differentially regulated in proliferating cells and during cell cycle arrest by alpha-interferon. J Biol Chem. 1998;273:23659–67. doi: 10.1074/jbc.273.37.23659. [DOI] [PubMed] [Google Scholar]
- 95.Muromoto R, Ishida M, Sugiyama K, Sekine Y, Oritani K, Shimoda K, et al. Sumoylation of Daxx regulates IFN-induced growth suppression of B lymphocytes and the hormone receptor-mediated transactivation. J Immunol. 2006;177:1160–70. doi: 10.4049/jimmunol.177.2.1160. [DOI] [PubMed] [Google Scholar]
- 96.Subramaniam PS, Cruz PE, Hobeika AC, Johnson HM. Type I interferon induction of the Cdk-inhibitor p21WAF1 is accompanied by ordered G1 arrest, differentiation and apoptosis of the Daudi B-cell line. Oncogene. 1998;16:1885–1890. doi: 10.1038/sj.onc.1201712. [DOI] [PubMed] [Google Scholar]
- 97.Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: A Tutorial on B Cell Survival. Annu Rev Immunol. 2003;21:231–264. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 98.Ramírez J, Lukin K, Hagman J. From hematopoietic progenitors to B cells: mechanisms of lineage restriction and commitment. Curr Opin Immunol. 2010;22:177–184. doi: 10.1016/j.coi.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nutt SL, Kee BL. The Transcriptional Regulation of B Cell Lineage Commitment. Immunity. 2007;26:715–725. doi: 10.1016/j.immuni.2007.05.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.