

Abstract
Myelocytomatosis proto-oncogene transcription factor (Myc) is an intrinsically disordered protein with critical roles in cellular homeostasis and neoplastic transformation. It is tightly regulated in the cell, with Myc phosphorylation playing a major role. In addition to the well-described tandem phosphorylation of Thr-52 and Ser-62 in the Myc transactivation domain linked to its degradation, P21 (RAC1)–activated kinase 2 (PAK2)–mediated phosphorylation of serine and threonine residues in the C-terminal basic helix–loop–helix leucine zipper (bHLH-LZ) region regulates Myc transcriptional activity. Here we report that PAK2 preferentially phosphorylates Myc twice, at Thr-358 and Ser-373, with only a minor fraction being modified at the previously identified Thr-400 site. For transcriptional activity, Myc binds E-box DNA elements, requiring its heterodimerization with Myc-associated factor X (Max) via the bHLH-LZ regions. Using isothermal calorimetry (ITC), we found that Myc phosphorylation destabilizes this ternary protein–DNA complex by decreasing Myc's affinity for Max by 2 orders of magnitude, suggesting a major effect of phosphorylation on this complex. Phosphomimetic substitutions revealed that Ser-373 dominates the effect on Myc–Max heterodimerization. Moreover, a T400D substitution disrupted Myc's affinity for Max. ITC, NMR, and CD analyses of several Myc variants suggested that the effect of phosphorylation on the Myc–Max interaction is caused by secondary structure disruption during heterodimerization rather than by a change in the structurally disordered state of Myc or by phosphorylation-induced electrostatic repulsion in the heterodimer. Our findings provide critical insights into the effects of PAK2-catalyzed phosphorylation of Myc on its interactions with Max and DNA.
Keywords: intrinsically disordered protein, Myc (c-Myc), protein phosphorylation, protein dynamic, protein–protein interaction, biophysics
Introduction
The myelocystomatosis proto-oncogene encodes a transcription factor, Myc, that is a central regulator of metabolism, proliferation, apoptosis, and other key cellular processes (1). Myc is well-recognized for its role in maintaining cellular homeostasis and neoplastic transformation. It is deregulated via chromosomal translocations, insertional mutagenesis, and gene amplification and is the most common gene amplification in human cancers (2, 3). In the absence of binding partners, Myc protein is intrinsically disordered. In particular, the bHLH4 domain (Fig. 1, A and B) is unstructured but forms a helical structure when in complex with Max, allowing the heterodimer to interact with E-box DNA and Myc to transactivate or repress genes (4). The transactivation complex is then assembled via recruitment of multiple coactivators to the highly conserved “MYC box” elements in the N-terminal transactivation domain (Fig. 1B). Because of its oncogenic potential, the Myc gene is tightly regulated at transcriptional (5–7), posttranscriptional (8, 9), and posttranslational (10) levels.
Figure 1.

Myc structure and phosphorylation. A, crystal structure of the heterodimer formed by the bHLH-LZ domains of Myc (blue) and Max (orange) in complex with E-box DNA (PDB code 1NKP), showing potential phosphorylation sites as red space-filling spheres. B, domain structure of Myc protein (Mb–Myc box). C, a sequence logo representing Myc bHLH-LZ based on a hidden Markov model. Higher stacks represent increased conservation of a residue. D, secondary structure of Max-bound Myc, including the phosphorylation sites detected in this study. Relative phosphorylation of different sites is qualitatively represented with red filling. E, 1H-15N HSQC spectrum fingerprint of unphosphorylated (black, ID 27414) and phosphorylated (green, BMRB ID 27422) Myc bHLH-LZ (fully assigned in Fig. S4) with marked phosphorylation sites. F, 1D slices (in 15N) of 1H15N HSQC peaks for Thr-358, Ser-373, and Thr-400 in unmodified (black) Myc and Myc doubly phosphorylated by PAK2 (blue).
The close links between Myc and oncogenesis make it an attractive therapeutic target. There have been numerous reports of the identification of small molecule ligands that bind to the bHLH-LZ domains of Myc and disrupt the interaction with Max (11–14). Compounds working via this mechanism represent the most direct way of modulating the activity of Myc and offer the potential to block Myc action in cancer. However, there have been no reports of such compounds being optimized to demonstrate in vivo efficacy in relevant models. Significant in vivo efficacy targeting Myc has only been achieved through transient expression of an engineered Myc variant termed Omomyc, but a detailed biophysical characterization of the interaction with Myc or Max has not been described (15, 16).
The development of Myc-targeted therapeutics is further complicated by posttranslational modifications of Myc, including phosphorylation (17, 18), ubiquitination (19–21), and acetylation (22, 23). Phosphorylation of Thr-58 and Ser-62 in the transactivation domain has been of particular interest because of their conservation across species and, along with adjacent amino acids, their location in a hot spot frequently mutated in Burkitt's lymphoma (24). Interestingly, the phosphorylation of these two residues is interdependent, with the phosphorylation of Ser-62 required prior to Thr-58 (18), which then triggers the proteasomal degradation of Myc (25). Other Myc phosphorylation sites have also been characterized (26–28), including phosphorylation of Thr-358, Ser-373, and Thr-400 by PAK2 (29), which again are highly conserved residues (Fig. 1C). Thr-358 is located in the basic region of the bHLH domain, which interacts directly with DNA, whereas Ser-373 and Thr-400 are located in the adjacent helix–loop–helix region, which participates in the interaction with Max (Fig. 1, A and D). Phosphorylation of these residues has been shown qualitatively to interfere with formation of the Myc–Max–DNA ternary complex and promote the E-box-independent regulation of transcription and differentiation (30).
The thermodynamics and kinetics of Myc–Max–DNA ternary complex formation have been characterized previously (31–35), but the underlying mechanism for perturbation of the complex following phosphorylation of residues in the Myc bHLH-LZ region is unknown (29). In this work, ITC, NMR, and CD have been used to characterize the effects of these phosphorylation events on the structure of Myc and the affinity of the interaction with Max. The results show that the residual structure in the Myc bHLH-LZ domains in the absence of Max is relatively unperturbed by phosphorylation, but the influence on heterodimer formation is more significant. These observations provide insight to assist with the identification of new therapeutic leads that interfere with the formation of the Myc–Max–DNA ternary complex.
Results
Effects of Myc phosphorylation on Max and DNA binding
The Myc bHLH-LZ (MycWT) sequence (Fig. 1C) is generally well-conserved among species, including the three previously identified Ser/Thr residues. These amino acids are all preceded by positively charged residues, although the Ser-373 site does not quite match the (K/R)RX(S/T) consensus sequence of PAK2. In vitro phosphorylation of MycWT by PAK2 results primarily in a dual phosphorylation event, where Myc is predominantly modified at Thr-358 and Ser-373 (MycWT-2P) (Fig. 1D and Figs. S1–S3 and Table S1). This behavior is apparent from the positions of the Thr-358 and Ser-373 in the NMR spectrum of MycWT-2P compared with the position of these residues in the NMR spectrum of MycWT (Fig. 1, E and F). Phosphorylation of Thr-358 and Ser-373 also induces a shift of additional resonances belonging to residues close to the phosphorylation sites (Fig. S4). Phosphorylation of the Thr-400 site is not detectable in the NMR spectra, but MS phosphorylation profiling detected low-level phosphorylation on this residue and at Tyr-402, Ser-405, and Ser-437 (Table S1 and Figs. S1–S3).
The effect of dual phosphorylation of Myc bHLH-LZ on the stability of the Myc–Max–DNA ternary complex was measured using a size exclusion DNA peak shift assay. Compared with MycWT, MycWT-2P is impaired in the ability of its heterodimer with Max to bind to E-box DNA (Fig. 2A). The MycWT–Max heterodimer binds tightly to E-box DNA, shifting the size exclusion peak to a shorter elution time, whereas the purified MycWT-2P leads to a mixture of free and bound DNA (Fig. 2A and Figs. S5 and S6). The presence of free and bound DNA arises from the fact that, although MycWT-2P has a reduced ability to form the MycWT-2P–Max heterodimer, Max bHLH-LZ itself can form a dimer, bind to the DNA E-Box, and induce a shift of DNA.
Figure 2.
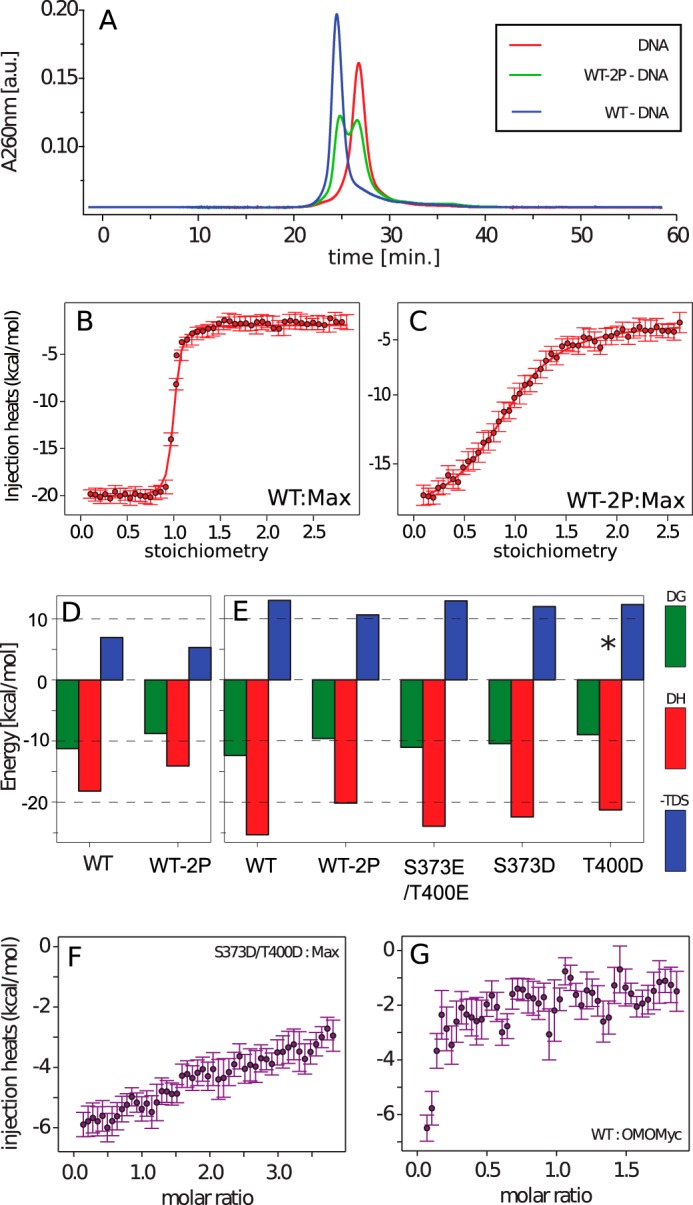
A, size exclusion profiles of E-box DNA in the absence (red) and presence of Max and unmodified (blue) and PAK2-phosphorylated (green) MycWT. a.u., arbitrary units. B and C, ITC titration curves of unphosphorylated (B) and phosphorylated (C) MycWT binding to Max at pH 7.4. D and E, ITC-derived thermodynamic parameters of Myc bHLH-LZ variants binding to Max at pH 7.4 (D) and pH 6.5 (E). The asterisk indicates 2:1 binding stoichiometry. F and G, flat profiles of ITC-integrated heat data of 8 μm MycS373D/T400D titration to 80μm Max (F) and 8 μm MycWT titration to 80 μm Omomyc (G), revealing no interaction between the proteins in vitro. All ITC experiments were performed with MycWT, MycWT-2P, Mycmutant, and Omomyc as titrants and Max as analyte.
The effect of dual phosphorylation of MycWT on the interaction with Max in the absence of DNA was determined using ITC (Fig. 2, B–F, and Table 1). Max is known to form a homodimer at high concentration, and dilution of Max into buffer shows an ITC curve that is typical for dimer dissociation (Fig. S7), with a dissociation constant KD of 6 μm (Table 1). Therefore, ITC measurements for the Myc–Max interaction were performed with Max as the analyte and the Myc variants as the titrants (Figs. S8 and S9), which minimizes this dilution effect. Under all conditions measured, binding was exothermic with an unfavorable entropic contribution. In line with previous qualitative studies (29), MycWT-2P has a decreased affinity for Max by 100-fold compared with MycWT. The KD for binding of MycWT to Max was found to be pH-dependent, with a lower pH favoring binding (0.9 nm at pH 6.5 compared with 6 nm at pH 7.4). This effect is maintained on phosphorylation. The differences in binding affinities and in ΔHbind for MycWT and MycWT-2P binding to Max are the same (within error) at pH 6.5 and 7.4. This implies that an interaction with phosphorylation is responsible for the pH dependence and that the impact of phosphorylation is unrelated to the charge on the phosphate groups. The difference of Gibbs binding energy of Myc–Max heterodimerization upon phosphorylation (ΔΔGphos) is 2.5 ± 1 kcal mol−1 at pH 7.4 and 2.7 ± 0.8 kcal mol−1 at pH 6.5.
Table 1.
ITC-derived dissociation constants and thermodynamic parameters of Myc bHLH-LZ variants upon interaction with Max bHLH-LZ
| KD | ΔG | ΔH | −T·ΔS | |
|---|---|---|---|---|
| nm | kcal mol −1 | kcal mol −1 | kcal mol −1 | |
| pH 7.4 | ||||
| MycWT | 6 ± 1 | −11.2 ± 0.1 | −18.2 ± 0.4 | 7.0 ± 0.6 |
| MycWT-2P | 376 ± 7 | −8.77 ± 0.01 | −14.08 ± 0.01 | 5.31 ± 0.02 |
| pH 6.5 | ||||
| MycWT | 0.9 ± 0.5 | −12.4 ± 0.4 | −25.6 ± 0.6 | 13.3 ± 1.0 |
| MycWT-2P | 96.9 ± 0.1 | −9.6 ± 0.4 | −20.2 ± 0.1 | 10.6 ± 0.5 |
| MycT373E/T400E | 8.0 ± 1. 0 | −11.0 ± 0.1 | −24.0 ± 2.0 | 13.0 ± 2.0 |
| MycT373D/T400D | n/a | n/a | n/a | n/a |
| MycS373D | 23.0 ± 9.0 | −10.4 ± 0.2 | −22. ± 1.0 | 12.0 ± 1.0 |
| MycT400Da | 270.0 ± 50.0 | −9.0 ± 0.1 | −21.0 ± 2.0 | 12.0 ± 2.0 |
| Max dissociation | 5600 ± 600 | −7.2 ± 0.1 | 6000 ± 2000 | 6000 ± 2000 |
a The stoichiometry for this reaction is 0.5.
To provide a broad comparison for the Myc–Max interaction, we also assayed the equivalent interaction between MycWT and Omomyc, which is a Myc-derived bHLH-LZ protein that forms a stable homodimer (36) and is reported to interact with Myc in vivo (37). However, MycWT binding to Omomyc was too weak to be detected in the ITC experiment (Fig. 2G), suggesting that heterodimerization is not the primary effect.
Contributions of individual phosphorylation sites
Purified MycWT-2P is predominantly phosphorylated on Thr-358 and Ser-373 (Fig. 1F). Ser-373 is positioned at the interface between Myc and Max, whereas Thr-358 is distant from this interaction. Its phosphorylation is expected to influence DNA binding only (Fig. 1A). Therefore, to examine more closely the causes of the decreased affinity of the Myc–Max interaction following phosphorylation, Ser-373 was mutated to Glu and Asp. For comparison, mutation to Glu and Asp of one of the secondary sites of phosphorylation in the heterodimer interface was also carried out. Residue Thr-400 was chosen because this site is highly conserved (Fig. 1C) and is the closest of the secondary phosphorylation sites to Ser-373. The NMR spectra of the mutant proteins (MycS373D, MycT400D, MycS373D/T400D, and MycS373E,T400E) were compared with MycWT-2P, phosphorylated using PAK2, and showed no significant shifts, indicating that the overall conformation of the protein is maintained (Fig. S4). To compare the Asp and Glu mutations more accurately with MycWT-2P, the data recorded at pH 6.5 were used as the reference set, as this favors a single negative charge on the phosphate groups, to match the carboxylate side chains.
In agreement with the behavior of MycWT and MycWT-2P, ITC measurements at 298 K for MycS373D binding to Max gave exothermic binding isotherms with a stoichiometry of 1 (Table 1, Fig. 2E, and Fig. S9). The dissociation constant for Max binding (23 nm) is comparable with that of MycWT-2P (97 nm), and the small difference (approximately 4-fold) is consistent with a close but slightly imperfect mimicking of a phosphate group by Asp (1). In contrast, the T400D mutation perturbs the interaction much more significantly, altering the stoichiometry to 2:1 MycT400D:Max bHLH-LZ (Fig. S9) and increasing the KD to 270 nm. The combination of these mutations in a S373D/T400D construct would be expected to increase the KD for Max to approximately 6 mm if the effects at the two sites were independent and, consistent with this, no binding was detected in ITC experiments (Fig. 2F). The effect of mutating Ser-373 and Thr-400 to Glu was far less pronounced than the equivalent mutations to Asp. MycS373E/T400E binds 9-fold more weakly than MycWT to Max (KD = 8.2 nm) but 12-fold more strongly than MycWT-2P.
Conformational perturbation on phosphorylation
The perturbation of the Myc–Max interaction by phosphorylation can, in principle, result from a direct disruption of the Myc–Max interface or, because Myc is an intrinsically disordered protein in the absence of Max, through changing the structural propensity of the isolated Myc bHLH-LZ domain. Potential effects of phosphorylation on the structural propensity of Myc were investigated using a combination of NMR and CD spectroscopy. In the NMR spectra of MycWT, MycWT-2P (Fig. 1E), all of the Asp variants (S373D/T400D, S373D, and T400D), and MycS373E/T400E (Fig. S4), resonances for the C-terminal residues of the bHLH region and the leucine zipper (399–413) were broadened beyond detection, so measurements are only available for residues 352 to 398.
One reporter of changes in structural propensity is a change in fluctuations of individual residues, and heteronuclear 15N{1H} NOEs are sensitive reporters of such fluctuations on the picosecond–nanosecond timescale (38, 39). Most residues in the basic region and helix 1 exhibit intermediate 15N{1H} NOE values (≤0.6) (Fig. 3A). A fully folded protein of this size would be expected to have NOE values between 0.7 and 0.8, whereas a very dynamic region would be expected to have values between 0.4 and −1.5. The loop and helix 2 are slightly more dynamic than the basic region and helix 1, with 15N{1H} NOE values around 0.4. These measurements show that the domain is partially structured and does not behave as a completely disordered protein, although any structure in Myc bHLH-LZ is transient. Phosphorylation of MycWT with PAK2 moderately increases NOE values close to the phosphorylated residues, Thr-358 and Ser-373, indicative of a small increase in local structure. However, this small effect is not reproduced by substitutions with Asp or Glu residues.
Figure 3.

A–C, NMR derived picosecond–nanosecond timescale dynamics (A) and secondary structures (B and C) of Myc bHLH and its variants, including the schematics of Myc bHLH-LZ secondary structure. A, heteronuclear 15N-1H NOE of Myc bHLH-LZ and its variants. pThr-358 and pSer-373 phosphorylation sites are marked with arrows. B and C, secondary structure propensities of Myc bHLH and variants obtained from Cα and Cβ chemical shifts.
The conformational properties of the Myc variants in the absence of Max were assessed further by deriving secondary structure propensities (40) from 13Cα and 13Cβ chemical shifts. Up to 50% helical propensity is estimated in the basic region and helix 1 in MycWT, peaking around residues 363–365. In contrast, up to 20% β-strand propensity is estimated in the loop residues and in helix 2 (Fig. 3B). Phosphorylation of MycWT has a large effect on residues on the N-terminal side of the phosphorylation sites, where it decreases the α-helical propensity by up to 30% (Fig. 3B), in line with other systems (41). Downfield 1H chemical shift changes on phosphorylation indicate that the phosphate group forms a hydrogen bond with its backbone amide, which would compete with hydrogen bonding participating in secondary structure formation.
There is no evidence that phosphorylation leads to a stabilizing N-capping effect, according to the chemical shift changes of residues to the C-terminal side of the phosphorylation sites. The Asp variants mimicked the chemical shift changes observed on phosphorylation to a large extent, but the Glu variant behaved much more closely to unphosphorylated MycWT (Fig. 3B). The decrease in α-helical propensity caused by the S373D mutation is independent of the T400D mutation (Fig. 3C), suggesting that there is little long-range interaction between the sites.
CD spectroscopy was used in conjunction with the NMR measurements to assess the effects of phosphorylation (and mutation) on the secondary structure content of Myc because NMR approaches were unable to report on the C-terminal region, including residue Thr-400 (42). The Myc–Max heterodimer has ∼70% ± 9% α-helical content, with the remainder being random coil (19% ± 5%) or turn conformations (9% ± 3%). The isolated Myc variants all have approximately half the per-residue helical content of the heterodimer. All proteins show a Θ minimum at 205 nm resulting from a combination of α-helix, polyproline II helix, and random coil spectral fingerprints, with the amount of α-helix characterized by Θ at 222 nm (Fig. 4A) (42). The differences between the Myc variants are subtle, with only minor changes in the relative values of Θ205 and Θ222.
Figure 4.

Phosphorylation induces destabilization of the α-helical structure of Myc bHLH-LZ. A, CD spectra of Myc variants (blue) compared with the spectrum of the Myc–Max complex (black). B, relation between KD and relative helix content (Θ205/Θ222 ratio) and disordered structures obtained from CD. C, four-state model for folding and binding of the Myc–Max heterodimer. Phosphorylation of MycWT by PAK2 destabilizes α-helical structures of Myc bHLH-LZ (wide to narrow ribbon transition). Decreased sampling of α-helical structures shifts the Myc–Max equilibrium toward the free Myc bHLH-LZ. Binding of Myc bHLH-LZ to Max bHLH-LZ stabilizes the α-helical structures of both Myc and Max.
Full quantification of the differences between the Myc variants using deconvolution of CD spectra into a combination of secondary structure elements (43–45) was not possible, given the data quality below 190 nm and small uncertainties in protein concentrations because of the low ϵ280 of Myc HLH-LZ. However, using the ratio of Θ222 to Θ205, the α-helical content is ordered WT > S373D > S373E/T400E > S373D/T400D > WT-2P > T400D. The α-helical content of the Myc constructs mostly follows the trend seen for the binding affinities for Max, with only MycS373D/T400D a significant outlier from this relationship, as it has an undetectable binding affinity using ITC. Ignoring this variant, and expressing the α-helical content as an equilibrium constant relative to the helical content of the Myc–Max complex, gives a correlation coefficient of 0.8 and a slope of 0.08 ± 0.02 for the relationship with log(KD) (Fig. 4B).
Discussion
Several previous studies have focused on disentangling the kinetics and thermodynamics of Myc–Max dimerization or Myc(Max)–Max–DNA ternary complex formation (31–35). In this study, ITC is used to characterize the thermodynamics of Myc and Max bHLH-LZ binding. The measured KD = 6 nm at 25 °C is substantially lower than the KD = 167 nm obtained previously at 23 °C by fluorescence anisotropy (32). This difference could be accounted for by the fluorescence labeling of Max (32) or by the use of slightly different constructs (supporting information). Surprisingly, ITC did not reveal an interaction between Myc bHLH-LZ and Omomyc (Fig. 2D). Therefore, the cellular effect of Omomyc expression (16, 37) may not be caused by Myc sequestration by Omomyc (46). Rather, it appears that the cellular effect of Omomyc reflects the competition of Omomyc homodimers with the Myc–Max heterodimer for DNA binding sites instead of competition of Omomyc with Max for Myc (or with Myc for Max).
Phosphorylation of Myc bHLH-LZ by PAK2
The regulation of Myc activity by the action of PAK2 is predicted to be via the disruption of the interaction with E-box DNA, which can occur either directly through perturbation of the DNA binding sites or indirectly through disrupting the interaction with Max. Identification of the preferred phosphorylation sites can provide information regarding the relative significance of the two mechanisms. Using a combination of MS and NMR, it was shown that PAK2 phosphorylates Myc bHLH-LZ at multiple sites, but the major phosphorylation sites are at Thr-358 and Ser-373. In MycWT-2P, Thr-358 and Ser-373 are phosphorylated to the exclusion of all other residues. In the overall phosphorylation mixture, low phosphorylation levels of Thr-400 were detected, and, to a lesser extent, Tyr-402, Ser-405, and Ser-437, although the phosphorylation of the latter three sites was below the detection level of NMR. This pattern of phosphorylation is broadly in line with previous qualitative observations of Myc phosphorylation by PAK2 (29) but downplays the significance of phosphorylation of Thr-400.
Dual phosphorylation of Myc significantly affects Myc–Max heterodimer binding to DNA. The effect of phosphorylation of Thr-358 is readily rationalized from the structure of the Myc–Max–DNA ternary complex because this residue is part of the DNA-binding motif, and the phosphate group of pThr-358 would be juxtaposed with the phosphate diester backbone of the E-box DNA. The effect of phosphorylation of Ser-373 is less clear. Although part of the DNA-binding helix, Ser-373 is positioned away from the DNA but adjacent to the Myc–Max dimerization region, which suggests a perturbation of Myc–Max dimerization in the reduced stability of the Myc–Max–DNA ternary complex. The side-chain OG atom of Ser-373 is only 3 Å away from the OD1 atom of Asp-74 in the LZ region of Max, and so phosphorylation of Ser-373 has the potential to introduce charge repulsion to this part of the heterodimer interface. Deconvolution of the contributions of the equilibrium between MycWT-2P–Max, Max–Max–DNA, and a potentially weaker MycWT-2P–Max–DNA complex is not appropriate for a nonequilibrium technique such as size exclusion chromatography. Consequently, ITC was employed to directly determine the effects of the phosphorylation of Ser-373 on the association of Myc with Max in the absence of DNA.
Compared with MycWT, MycWT-2P has a 100-fold reduced affinity for Max. The S373D mutation reduces the affinity for Max by 20-fold, which potentially leaves a 5-fold (1 kcal mol−1) contribution from the phosphorylation of Thr-358. However, there is significant uncertainty in this value because S373D is not a perfect mimic of pSer-373. Because Thr-358 is distant from the Myc–Max heterodimer interface in the ternary complex, pThr-358 would only affect Max binding if there were some “non-native” intramolecular interactions in free Myc, and there is little evidence of long-range interactions in any of the NMR spectra.
Folded state effects
Charge repulsion between pSer-373 of Myc and Asp-74 of Max should have a pH dependence near to the pKa of the phosphate group. The pKa of pSer is usually 5.8–6.2 (41), but this is likely to be increased when pSer-373 is close to the carboxylate group of Asp-74 of Max. As a result, the interaction between MycWT-2P and Max is expected to be significantly affected between pH 6.5 and pH 7.4. There is an effect of pH on the Myc–Max binding thermodynamics in this pH range, but it occurs independently of the phosphorylation of Ser-373. Hence, the observed effect is most likely due to His-81 of Max, the imidazole NH groups of which are hydrogen-bonded by Glu-410 and Glu-417 of Myc in the heterodimer, which would require protonation of His-81 at pH 7.4. The lack of a phosphorylation-dependent pH effect is also unexpected because the specificity of leucine zipper interactions is dictated by the charged residues that flank the core hydrophobic interface (47), and the electrostatic environment of Myc is modified by the introduction of anionic phosphate groups. Consequently, the results suggest that repulsion between pSer-373 of Myc and Asp-74 of Max is not a main contributory factor to the reduction of affinity for Max caused by the dual phosphorylation of Myc. This conclusion is supported by the negligible effect the MycS373E/T400E variant has on the Myc–Max interaction, even though the Glu carboxylate oxygen atoms and the phosphate oxygens in pSer and pThr are separated by the same number of bonds from the backbone Cα.
Conformational ensemble effects
The effects of dual phosphorylation are not limited to electrostatics but can also include the disruption of all equilibria that affect the overall thermodynamics. To simply describe the binding of Myc to Max, a four-state mechanism that separates the coupled binding and folding into two events was used as a thermodynamic model (Fig. 4C; note that our measurements do not report on the pathway for this interaction). In the absence of Max, an ensemble of partially disordered Myc conformers is in rapid equilibrium with fully disordered Myc (equilibrium 1), which is the equilibrium observed using NMR. Rarely (and thus invisible to NMR), the partially disordered ensemble will populate a fully folded, monomeric Myc (equilibrium 2). Association with Max allows stabilization of the fully folded Myc in the heterodimer (equilibrium 3). The arguments above concentrate on equilibrium 3.
Some effects of dual phosphorylation on equilibrium 1 are visible in the NMR-derived parameters. Normally, helical regions that are C-terminal of phosphorylation sites are stabilized through N-capping interactions, whereas helical regions N-terminal of phosphorylation sites are destabilized by disruption of the helix hydrogen bonding (41). Phosphorylation of Ser-373 leads to no measurable helicity at residue 373 and has a substantial destabilizing effect on both the N-terminal and C-terminal side. The S373D and S373D/T400D variants behave similar to each other, but the destabilizing effects are focused on the N-terminal side of the mutation and are significantly less than for pSer-373. Phosphorylation of Thr-358 leads to a response in its vicinity that is more reminiscent of the S373D mutation than Ser-373 phosphorylation. Helix propensity is lost completely on its N-terminal side (where it was already low) but is moderately unaffected at position 358 and on its C-terminal side, where the bulk of helicity associated with equilibrium 1 resides. Hence, the primary effect of phosphorylation on equilibrium 1 appears to result from local secondary structure perturbation by pSer-373. The similarity in behavior between the effects of pSer-373 and S373D mutations, but not S373E mutations, strongly implies that the competition between helix formation and side chain–backbone hydrogen bonding is the most significant factor in the perturbation of equilibrium 1.
Overall, the changes in helical propensities reported in the NMR data are small, which is consistent with the modest changes in CD spectra between the same species. Intriguingly though, there is a good correlation (Fig. 4B) between the perturbation of secondary structure formation in the monomer and the free energy of the Myc–Max binding interaction across the phosphorylated and mutated Myc species. However, this correlation accounts for less than 10% of the observed effects on binding. This leaves the strong implication that the perturbation of secondary structure formation in the monomer by phosphorylation is a reflection of a more major effect on equilibrium 2. This would mean that the ensemble of partially disordered Myc conformers formed in equilibrium 1 is more capable of accommodating the perturbation introduced by phosphorylation (as, for example, reported by the NMR data in the vicinity of pSer-373) than the more structured, extended helical conformation that is required to bind to Max in the WT mode (equilibrium 3).
Comparison with pThr-400
Thr-400 is a canonical PAK2 phosphorylation site, even though the pThr-400 modification is not prevalent in the purified MycWT-2P samples. Notably, the effects of introducing a charged group here are more significant than at Ser-373. Thr-400 is located near the C terminus of helix 2, which is adjacent to helix 1 of Max in the ternary complex. The T400D mutation has the largest effect on Max binding and on the conformational ensemble of unbound Myc (as detected by CD spectroscopy). Surprisingly though, the MycT400D–Max complex switches to become heterotrimeric. Although such leucine zipper complexes have been reported previously, and the rules governing homotrimeric structure are well-understood (48), it is not clear how this single point mutation alters the preference over dimer in this complex. The line broadening observed in the C-terminal region of the Myc bHLH region prevents a full comparison of the NMR and CD data, but it is apparent from the fact that the perturbations introduced by the T400D mutation are not widespread in equilibrium 1.
In summary, Myc is an intrinsically disordered protein that elicits its biological effect by adopting a more ordered structure that enables it to interact with Max and affect transcription at the DNA level. Posttranslational modifications of intrinsically disordered proteins are key mechanisms for regulating this class of proteins. The data presented here provide insight into the structural effect of PAK2-catalyzed phosphorylation of Myc. Further characterization of phosphorylated Myc could provide information to assist with the design of small-molecule ligands targeting this state. Stabilization of the phosphorylated form could be a mechanism for disruption of Myc function in cancer. Identification of suitable ligands targeting this mechanism remains a challenge.
Experimental procedures
All protein constructs and mutants (supplemental information) were generated by gene synthesis with an N-terminal hexahistidine tag and tobacco etch virus protease site. All were subcloned into a pET-9 vector for Escherichia coli expression. Protein expression was performed in BL21-Gold (DE3) (Novagen) E. coli cells induced using 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h at 37 °C.
Cells were resuspended in PBS buffer containing complete protease inhibitor tablets (Roche) and benzonase nuclease (2.5 units/ml) and lysed using a Constant Systems cell disruptor at 25 psi. Guanidium chloride was added to resuspended cells to 4 m final concentration and stirred for 1 h. Guanidium-containing lysate was further clarified by centrifugation at 75,000 × g for 1 h.
Proteins were purified from supernatant using nickel affinity chromatography followed by Superdex 75 gel filtration chromatography (50 mm HEPES (pH 7.5), 500 mm NaCl, 1 mm EDTA, and 1 mm DTT), and the His tag was cleaved by tobacco etch virus protease. Cleaved protein was separated under denaturing conditions (4 m guanidium chloride and 2 mm CaCl2 to chelate the EDTA) as flow-through on nickel chromatography. Protein was concentrated by ultrafiltration (3-kDa pore) and purified and buffer-exchanged on Superdex 75 gel filtration chromatography (50 mm potassium phosphate (pH 6.5), 500 mm ammonium chloride, 1 mm EDTA, and 1 mm DTT).
Phosphorylation by PAK2 T402E (Dundee University) was carried out at room temperature in a mixture of Myc 0.5 mg/ml, PAK2 3 μg/ml, and ATP 300 μm, each in phosphorylation buffer (60 mm HEPES (pH 7.5), 3 mm MgCl2, 3 mm MnCl2, and 1.2 mm DTT). The phosphorylation was carried out until the level of dual phosphorylation was higher than 80%, as quality-controlled by MS (C18 reverse phase LC-ESI-Q-TOF). The reaction was quality-controlled every 24 h, and when the level of double phosphorylation was unsatisfactory, an additional 3 μg/ml of PAK2 and 0.3 μmol/ml ATP were added.
Dual-phosphorylated Myc was further purified on cation exchange. ResourceS 1.0-ml columns were equilibrated with 60 mm HEPES (pH 7.5) and eluted over 20 column volumes with 60 mm HEPES (pH 7.5) and 1 m NaCl. The fractions containing the dual-phosphorylated Myc were identified by MS (Fig. S6), pooled, and buffer-exchanged to 50 mm potassium phosphate (pH 6.5), 500 mm ammonium chloride, 1 mm EDTA, and 1 mm DTT.
Gel filtration mobility shift assays were performed using a Superdex 75 3.2/300 column equilibrated in 50 mm potassium phosphate (pH 6.5), 500 mm ammonium chloride, 1 mm EDTA, and 1 mm DTT using 5-μl injections of sample comprising 10 μm E-Box DNA alone or in combination with 10 μm Myc and/or 10 μm Max. The detector was set to λ = 260 nm to detect DNA. E-Box DNA was prepared by annealing of chemically synthetized oligonucleotides (supplemental information).
All ITC experiments were performed on a VP-ITC instrument at 25 °C. In all titration experiments, 20 μm, 40 μm, and 80 μm Myc bHLH-LZ was titrated into Max bHLH-LZ at 2 μm, 4 μm, and 8 μm concentrations. The concentrations of Myc and Max bHLH-LZ were initially determined by amino acid analysis and interpolated to a Bradford assay. All experiments were performed in 50 mm potassium phosphate (pH 6.5 or pH 7.4), 500 mm ammonium chloride, 1 mm EDTA, and 1 mm DTT. Max bHLH-LZ dissociation titration was obtained by titrating 160 μm Max bHLH-LZ into buffer. All experiments were analyzed using NITPIC (49) and SEDPHAT/ITCsy (50, 51). The ITC titration parameters were obtained from a simultaneous fit of titrations at 20 μm, 40 μm, and, when available, 80 μm Myc bHLH-LZ.
All NMR experiments were performed at 4 °C. All heteronuclear 15N{1H}NOE spectra and assignment spectra of MycWT and MycWT-2P were acquired on a Bruker Avance III at 800 MHz, whereas assignment spectra of MycS373E/T400E, MycS373D/T400D, MycS373D, and MycT400D were acquired using a Bruker Avance III at 600 MHz. Both spectrometers were equipped with a 5-mm z-gradient 1H/13C/15N TCI probe. The heteronuclear 15N{1H}NOE experiments were acquired using the hsqcnoef3gpsi pulse sequence with a 7-s relaxation delay. BEST type HNCA, HN(CO)CA, HNCACB, and HN(CO)CACB experiments (52, 53) were used for assignment. Secondary structure propensities were obtained by SSP (40) using Cα and Cβ only with internal SSP chemical shift referencing.
For CD experiments, all proteins were buffer-exchanged into 50 mm potassium phosphate (pH 6.5) or 500 mm potassium fluoride (pH 7.4). CD spectra were recorded at 293 K in a Jasco J-810 spectrometer equipped with a Peltier temperature controller in a 0.1-cm path length quartz cuvette containing protein at 0.3 mg/ml. Blank spectra were recorded with the same buffer in the absence of protein and subtracted from the protein spectra. All measurements for CD spectra were taken in triplicate with a response of 2 s, 0.1-nm data pitch, 1-nm bandwidth, and a scanning speed of 20 nm/s from 260–185 nm. To obtain the secondary structure composition, data were analyzed using the Contin-LL method (44) using reference set 7 (54).
MS analysis of the MycWT-2P phosphorylation pattern was performed on SDS-PAGE with gel bands excised from a Coomassie-stained gel and subjected to reduction, alkylation, and digestion with trypsin (55). For ESI MS-MS using the Qstar Elite (ABSciex) mass spectrometer, 5 μl of the sample was chromatographed using the U3000 Ultimate (Thermo) nanoflow chromatography system, and the outlet flow was run directly into the Qstar Elite for analysis via the nanoflow probe at a flow rate of 300 nl/min. A 20-min reverse phase gradient was run using a 300-μm inner diameter × 5 mm C18 PepMap trapping precolumn and a 75 μm inner diameter × 15 cm C18 PepMap analytical column. The Qstar collected data in positive ion mode, and an autoswitching setup was initiated with automatic precursor selection based on peak intensity and charge state. The collision energies were automatically adjusted based on the precursor. Nitrogen was used as the collision gas. The subsequent data files generated were searched against in the Myc sequences using the Mascot Daemon software. The searches were then manually verified.
Author contributions
P. M. and M. J. C. data curation; P. M., M. J. C., and S. P. formal analysis; P. M., M. J. C., K. J. E., G. A. H., J. W. M. N., and R. A. D. supervision; P. M. validation; P. M., M. J. C., G. A. H., and S. P. investigation; P. M., M. J. C., K. J. E., J. W. M. N., J. P. W., and R. A. D. methodology; P. M. and M. J. C. writing-original draft; P. M., J. P. W., and R. A. D. project administration; P. M., M. J.C., K. J. E., G. A. H., J. W. M. N., J. P. W., and R. A. D. writing-review and editing; M. J. C. visualization; J. W. M. N., J. P. W., and R. A. D. conceptualization; J. P. W. resources; R. A. D. funding acquisition.
Supplementary Material
Acknowledgments
We thank Rachel Rowlinson for the generation of MS data on Myc protein samples and Jonathan Renshaw for practical support with protein science.
This work was supported by a Discovery Sciences grant from the AstraZeneca Post-Doctoral Programme (to P. M.) and a student fellowship from Bruker BioSpin (to S. P.). The work described here was initiated as a project within AstraZeneca. K. J. E., R. A. D., G. A. H., and J. W. M. N. are current employees of AstraZeneca and may hold stock in the company. P. M. is a former employee of AstraZeneca and may hold stock in the company.
This article contains Figures S1–S10, Table S1, supplemental protein constructs, and E-box DNA.
The NMR chemical shift data reported in this paper have been submitted to the Biological Magnetic Resonance Data Bank (BMRB) under accession numbers 27418, 27416, 27421, 27422, 27419, and 27414.
- bHLH
- basic helix–loop–helix
- LZ
- leucine zipper
- ITC
- isothermal calorimetry
- ESI
- electrospray ionization
- HSQC
- heteronuclear single quantum coherence.
References
- 1. Dang, C. V. (1999) c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell Biol. 19, 1–11 10.1128/MCB.19.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zack, T. I., Schumacher, S. E., Carter, S. L., Cherniack, A. D., Saksena, G., Tabak, B., Lawrence, M. S., Zhang, C.-Z., Wala, J., Mermel, C. H., Sougnez, C., Gabriel, S. B., Hernandez, B., Shen, H., Laird, P. W., et al. (2013) Pan-cancer patterns of somatic copy number alteration. Nat. Publ. Group 45, 1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beroukhim, R., Mermel, C. H., Porter, D., Wei, G., Raychaudhuri, S., Donovan, J., Barretina, J., Boehm, J. S., Dobson, J., Urashima, M., Mc Henry, K. T., Pinchback, R. M., Ligon, A. H., Cho, Y.-J., Haery, L., et al. (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 10.1038/nature08822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nair, S. K., and Burley, S. K. (2003) X-ray structures of Myc-Max and Mad-Max recognizing DNA: molecular bases of regulation by proto-oncogenic transcription factors. Cell 112, 193–205 10.1016/S0092-8674(02)01284-9 [DOI] [PubMed] [Google Scholar]
- 5. Bentley, D. L., and Groudine, M. (1986) A block to elongation is largely responsible for decreased transcription of c-myc in differentiated HL60 cells. Nature 321, 702–706 10.1038/321702a0 [DOI] [PubMed] [Google Scholar]
- 6. Bentley, D. L., and Groudine, M. (1988) Sequence requirements for premature termination of transcription in the human c-myc gene. Cell 53, 245–256 10.1016/0092-8674(88)90386-8 [DOI] [PubMed] [Google Scholar]
- 7. Lang, J. C., Whitelaw, B., Talbot, S., and Wilkie, N. M. (1988) Transcriptional regulation of the human c-myc gene. Br. J. Cancer Suppl. 9, 62–66 [PMC free article] [PubMed] [Google Scholar]
- 8. Sampson, V. B., Rong, N. H., Han, J., Yang, Q., Aris, V., Soteropoulos, P., Petrelli, N. J., Dunn, S. P., and Krueger, L. J. (2007) MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 67, 9762–9770 10.1158/0008-5472.CAN-07-2462 [DOI] [PubMed] [Google Scholar]
- 9. Lal, A., Navarro, F., Maher, C. A., Maliszewski, L. E., Yan, N., O'Day, E., Chowdhury, D., Dykxhoorn, D. M., Tsai, P., Hofmann, O., Becker, K. G., Gorospe, M., Hide, W., and Lieberman, J. (2009) miR-24 inhibits cell proliferation by targeting E2F2, MYC, and other cell cycle genes via binding to “seedless” 3′UTR microRNA recognition elements. Mol. Cell 35, 610–625 10.1016/j.molcel.2009.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vervoorts, J., Lüscher-Firzlaff, J., and Lüscher, B. (2006) The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 281, 34725–34729 10.1074/jbc.R600017200 [DOI] [PubMed] [Google Scholar]
- 11. Berg, T., Cohen, S. B., Desharnais, J., Sonderegger, C., Maslyar, D. J., Goldberg, J., Boger, D. L., and Vogt, P. K. (2002) Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 99, 3830–3835 10.1073/pnas.062036999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang, H., Hammoudeh, D. I., Follis, A. V., Reese, B. E., Lazo, J. S., Metallo, S. J., and Prochownik, E. V. (2007) Improved low molecular weight Myc-Max inhibitors. Mol. Cancer Ther. 6, 2399–2408 10.1158/1535-7163.MCT-07-0005 [DOI] [PubMed] [Google Scholar]
- 13. Shi, J., Stover, J. S., Whitby, L. R., Vogt, P. K., and Boger, D. L. (2009) Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg. Med. Chem. Lett. 19, 6038–6041 10.1016/j.bmcl.2009.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hart, J. R., Garner, A. L., Yu, J., Ito, Y., Sun, M., Ueno, L., Rhee, J.-K., Baksh, M. M., Stefan, E., Hartl, M., Bister, K., Vogt, P. K., and Janda, K. D. (2014) Inhibitor of MYC identified in a Kröhnke pyridine library. Proc. Natl. Acad. Sci. U.S.A. 111, 12556–12561 10.1073/pnas.1319488111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soucek, L., Helmer-Citterich, M., Sacco, A., Jucker, R., Cesareni, G., and Nasi, S. (1998) Design and properties of a Myc derivative that efficiently homodimerizes, Oncogene 17, 2463–2472 10.1038/sj.onc.1202199 [DOI] [PubMed] [Google Scholar]
- 16. Soucek, L., Whitfield, J., Martins, C. P., Finch, A. J., Murphy, D. J., Sodir, N. M., Karnezis, A. N., Swigart, L. B., Nasi, S., and Evan, G. I. (2008) Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683 10.1038/nature07260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henriksson, M., Bakardjiev, A., Klein, G., and Lüscher, B. (1993) Phosphorylation sites mapping in the N-terminal domain of c-myc modulate its transforming potential. Oncogene 8, 3199–3209 [PubMed] [Google Scholar]
- 18. Lutterbach, B., and Hann, S. R. (1994) Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol. Cell Biol. 14, 5510–5522 10.1128/MCB.14.8.5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salghetti, S. E., Kim, S. Y., and Tansey, W. P. (1999) Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 18, 717–726 10.1093/emboj/18.3.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gregory, M. A., and Hann, S. R. (2000) c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt's lymphoma cells. Mol. Cell Biol. 20, 2423–2435 10.1128/MCB.20.7.2423-2435.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Welcker, M., Orian, A., Jin, J., Grim, J. E., Harper, J. W., Eisenman, R. N., and Clurman, B. E. (2004) The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. U.S.A. 101, 9085–9090 10.1073/pnas.0402770101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vervoorts, J., Lüscher-Firzlaff, J. M., Rottmann, S., Lilischkis, R., Walsemann, G., Dohmann, K., Austen, M., and Lüscher, B. (2003) Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 4, 484–490 10.1038/sj.embor.embor821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patel, J. H., Du, Y., Ard, P. G., Phillips, C., Carella, B., Chen, C.-J., Rakowski, C., Chatterjee, C., Lieberman, P. M., Lane, W. S., Blobel, G. A., and McMahon, S. B. (2004) The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell Biol. 24, 10826–10834 10.1128/MCB.24.24.10826-10834.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bahram, F., von der Lehr, N., Cetinkaya, C., and Larsson, L. G. (2000) c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 95, 2104–2110 [PubMed] [Google Scholar]
- 25. Yada, M., Hatakeyama, S., Kamura, T., Nishiyama, M., Tsunematsu, R., Imaki, H., Ishida, N., Okumura, F., Nakayama, K., and Nakayama, K. I. (2004) Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23, 2116–2125 10.1038/sj.emboj.7600217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lüscher, B., Kuenzel, E. A., Krebs, E. G., and Eisenman, R. N. (1989) Myc oncoproteins are phosphorylated by casein kinase II. EMBO J. 8, 1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Noguchi, K., Kitanaka, C., Yamana, H., Kokubu, A., Mochizuki, T., and Kuchino, Y. (1999) Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J. Biol. Chem. 274, 32580–32587 10.1074/jbc.274.46.32580 [DOI] [PubMed] [Google Scholar]
- 28. Wasylishen, A. R., Chan-Seng-Yue, M., Bros, C., Dingar, D., Tu, W. B., Kalkat, M., Chan, P.-K., Mullen, P. J., Huang, L., Meyer, N., Raught, B., Boutros, P. C., and Penn, L. Z. (2013) MYC phosphorylation at novel regulatory regions suppresses transforming activity. Cancer Res. 73, 6504–6515 10.1158/0008-5472.CAN-12-4063 [DOI] [PubMed] [Google Scholar]
- 29. Huang, Z., Traugh, J. A., and Bishop, J. M. (2004) Negative control of the Myc protein by the stress-responsive kinase Pak2. Mol. Cell Biol. 24, 1582–1594 10.1128/MCB.24.4.1582-1594.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Uribesalgo, I., Buschbeck, M., Gutiérrez, A., Teichmann, S., Demajo, S., Kuebler, B., Nomdedéu, J. F., Martín-Caballero, J., Roma, G., Benitah, S. A., and Di Croce, L. (2011) E-box-independent regulation of transcription and differentiation by MYC. Nat. Cell Biol. 13, 1443–1449 10.1038/ncb2355 [DOI] [PubMed] [Google Scholar]
- 31. Kohler, J. J., Metallo, S. J., Schneider, T. L., and Schepartz, A. (1999) DNA specificity enhanced by sequential binding of protein monomers. Proc. Natl. Acad. Sci. U.S.A. 96, 11735–11739 10.1073/pnas.96.21.11735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Banerjee, A., Hu, J., and Goss, D. J. (2006) Thermodynamics of protein-protein interactions of cMyc, Max, and Mad: effect of polyions on protein dimerization. Biochemistry 45, 2333–2338 10.1021/bi0522551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ecevit, O., Khan, M. A., and Goss, D. J. (2010) Kinetic analysis of the interaction of b/HLH/Z transcription factors Myc, Max, and Mad with cognate DNA. Biochemistry 49, 2627–2635 10.1021/bi901913a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jung, K. C., Rhee, H. S., Park, C. H., and Yang, C.-H. (2005) Determination of the dissociation constants for recombinant c-Myc, Max, and DNA complexes: the inhibitory effect of linoleic acid on the DNA-binding step. Biochem. Biophys. Res. Commun. 334, 269–275 10.1016/j.bbrc.2005.06.088 [DOI] [PubMed] [Google Scholar]
- 35. Meier-Andrejszki, L., Bjelić, S., Naud, J.-F., Lavigne, P., and Jelesarov, I. (2007) Thermodynamics of b-HLH-LZ protein binding to DNA: the energetic importance of protein-DNA contacts in site-specific E-box recognition by the complete gene product of the Max p21 transcription factor. Biochemistry 46, 12427–12440 10.1021/bi701081q [DOI] [PubMed] [Google Scholar]
- 36. Jung, L. A., Gebhardt, A., Koelmel, W., Ade, C. P., Walz, S., Kuper, J., von Eyss, B., Letschert, S., Redel, C., d' Artista, L., Biankin, A., Zender, L., Sauer, M., Wolf, E., Evan, G., et al. (2017) Omomyc blunts promoter invasion by oncogenic myc to inhibit gene expression characteristic of myc-dependent tumors, Oncogene 36, 1911–1924. 10.1038/onc.2016.354 [DOI] [PubMed] [Google Scholar]
- 37. Soucek, L., Jucker, R., Panacchia, L., Ricordy, R., Tatò, F., and Nasi, S. (2002) Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 62, 3507–3510 [PubMed] [Google Scholar]
- 38. Palmer, A. G., 3rd (2004) NMR Characterization of the dynamics of biomacromolecules. Chem. Rev. 104, 3623–3640 10.1021/cr030413t [DOI] [PubMed] [Google Scholar]
- 39. Lienin, S. F., Bremi, T., Brutscher, B., Bruschweiller, R., and Ernst, R. (1998) Anisotropic intramolecular backbone dynamics of ubiquitin characterized by NMR relaxation and MD computer simulation. J. Am. Chem. Soc. 120, 9870–9879 10.1021/ja9810179 [DOI] [Google Scholar]
- 40. Marsh, J. A., Singh, V. K., Jia, Z., and Forman-Kay, J. D. (2006) Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: implications for fibrillation. Protein Sci. 15, 2795–2804 10.1110/ps.062465306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andrew, C. D., Warwicker, J., Jones, G. R., and Doig, A. J. (2002) Effect of phosphorylation on α-helix stability as a function of position. Biochemistry 41, 1897–1905 10.1021/bi0113216 [DOI] [PubMed] [Google Scholar]
- 42. Greenfield, N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Whitmore, L., and Wallace, B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–73 10.1093/nar/gkh371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Provencher, S. W., and Glöckner, J. (1981) Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20, 33–37 10.1021/bi00504a006 [DOI] [PubMed] [Google Scholar]
- 45. Janes, R. W. (2009) Reference datasets circular dichroism and synchrotron radiation circular dichroism spectroscopy of proteins. In: Modern Techniques in Circular Dichroism and Synchrotron Radiation Circular Dichroism Spectroscopy, Advances in Biomedical Spectroscopy (Wallace, B. A., and Janes, R.W., eds), pp. 183–201, IOS Press, Clifton, VA [Google Scholar]
- 46. Savino, M., Annibali, D., Carucci, N., Favuzzi, E., Cole, M. D., Evan, G. I., Soucek, L., and Nasi, S. (2011) The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 6, e22284–16 10.1371/journal.pone.0022284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krylov, D., and Vinson, C. R. (2001) Leucine Zipper. In: Encyclopedia of Life Sciences, John Wiley & Sons, Ltd., Chichester, UK, 10.1038/npg.els.0003001 [DOI] [Google Scholar]
- 48. Fletcher, J. M., Boyle, A. L., Bruning, M., Bartlett, G. J., Vincent, T. L., Zaccai, N. R., Armstrong, C. T., Bromley, E. H., Booth, P. J., Brady, R. L., Thomson, A. R., and Woolfson, D. N. (2012) A basis set of de novo coiled-coil peptide oligomers for rational protein design and synthetic biology. ACS Synth. Biol. 1, 240–250 10.1021/sb300028q [DOI] [PubMed] [Google Scholar]
- 49. Keller, S., Vargas, C., Zhao, H., Piszczek, G., Brautigam, C. A., and Schuck, P. (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84, 5066–5073 10.1021/ac3007522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Houtman, J. C., Brown, P. H., Bowden, B., Yamaguchi, H., Appella, E., Samelson, L. E., and Schuck, P. (2007) Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 16, 30–42 10.1110/ps.062558507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao, H., Piszczek, G., and Schuck, P. (2015) SEDPHAT: a platform for global ITC analysis and global multi-method analysis of molecular interactions, Methods 76, 137–148 10.1016/j.ymeth.2014.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schanda, P., Van Melckebeke, H., and Brutscher, B. (2006) Speeding up three-dimensional protein NMR experiments to a few minutes. J. Am. Chem. Soc. 128, 9042–9043 10.1021/ja062025p [DOI] [PubMed] [Google Scholar]
- 53. Lescop, E., Schanda, P., and Brutscher, B. (2007) A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J. Magn. Reson. 187, 163–169 10.1016/j.jmr.2007.04.002 [DOI] [PubMed] [Google Scholar]
- 54. Sreerama, N., and Woody, R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN,SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260 10.1006/abio.2000.4880 [DOI] [PubMed] [Google Scholar]
- 55. Gundry, R. L., White, M. Y., Murray, C. I., Kane, L. A., Fu, Q., Stanley, B. A., and Van Eyk, J. E. (2009) Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr. Protoc. Mol. Biol. Chapter 10, Unit 10.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.