

ABSTRACT
In this study, we screened poly(3-hydroxybutyrate) (PHB) synthase PhaC1 and PHB depolymerase PhaZa1 of Ralstonia eutropha for the presence of phosphorylated residues during the PHB accumulation and PHB degradation phases. Thr373 of PHB synthase PhaC1 was phosphorylated during the stationary growth phase but was not modified during the exponential and PHB accumulation phases. Ser35 of PHB depolymerase PhaZa1 was identified in the phosphorylated form during both the exponential and stationary growth phases. Additional phosphosites were identified for both proteins in sample-dependent forms. Site-directed mutagenesis of the codon for Thr373 and other phosphosites of PhaC1 revealed a strong negative impact on PHB synthase activity. Modifications of Thr26 and Ser35 of PhaZa1 reduced the ability of R. eutropha to mobilize PHB in the stationary growth phase. Our results show that phosphorylation of PhaC1 and PhaZa1 can be important for the modulation of the activities of PHB synthase and PHB depolymerase.
IMPORTANCE Poly(3-hydroxybutyrate) (PHB) and related polyhydroxyalkanoates (PHAs) are important intracellular carbon and energy storage compounds in many prokaryotes. The accumulation of PHB or PHAs increases the fitness of cells during periods of starvation and under other stress conditions. The simultaneous presence of PHB synthase (PhaC1) and PHB depolymerase (PhaZa1) on synthesized PHB granules in Ralstonia eutropha (alternative designation, Cupriavidus necator) was previously shown in several laboratories. These findings imply that the activities of PHB synthase and PHB depolymerase should be regulated to avoid a futile cycle of simultaneous synthesis and degradation of PHB. Here, we addressed this question by identifying the phosphorylation sites on PhaC1 and PhaZa1 and by site-directed mutagenesis of the identified residues. Furthermore, we conducted in vitro and in vivo analyses of PHB synthase activity and PHB contents.
KEYWORDS: PHB accumulation, PHB synthase, PhaZa1, phosphorylation, Ralstonia eutropha, PHB metabolism
INTRODUCTION
The ability to synthesize polyhydroxyalkanoates (PHAs) is a widespread feature of (aerobic) prokaryotes. They accumulate during periods of imbalanced growth, in the presence of excess suitable carbon, in the absence of nitrogen or phosphorous sources, or under oxygen-limiting conditions (1–3). PHAs are synthesized as water-insoluble granules approximately 200 to 500 nm in diameter. A thin surface layer (3- to 4-nm thickness) consisting of up to 14 species-specific proteins covers the polymer core of the poly(3-hydroxybutyrate) (PHB) granules (4–6). For an excellent overview on PHA granule structure and biotechnological applications see reference 7.
The proteins of the PHB surface layer in Ralstonia eutropha are divided into several functional groups and include (i) the PHB synthases (PhaCs) (8, 9), (ii) the PHB depolymerases (PhaZs) (10–12), (iii) the phasins (PhaPs) (13–17), proteins with (iv) regulatory (PhaR) (18, 19) and (v) activating (PhaM) (20–22) roles, and (vi) proteins with unknown functions (5). The PHB synthase (PhaC1) and PHB depolymerase (PhaZa1) proteins are the two key enzymes in R. eutropha responsible for the biosynthesis and degradation (mobilization) of PHB, respectively. Remarkably, both proteins are constitutively present during all stages of growth (5, 23, 24). Their presence implies that the activities of PHB synthase and PHB depolymerase should be regulated to avoid a futile cycle of simultaneous PHB synthesis and PHB degradation. The covalent modification of proteins, such as the phosphorylation of specific residues, is a well-known cellular tool for modifying the activity of enzymes. The phosphorylation status of PHB synthase or PHB depolymerase has not been investigated. In this contribution, we examined whether the accumulation and mobilization of PHB by PHB synthase and PHB depolymerase could be altered by phosphorylation.
RESULTS
PhaC1 is phosphorylated during the stationary growth phase in nutrient broth-grown cells.
The PHB synthase gene (phaC1) was fused with the enhanced yellow fluorescent protein (eyfp) gene in a plasmid and transformed into a ΔphaC1 background of R. eutropha. PHB synthesis in the ΔphaC1 background is restored by this plasmid (22). Pulldown experiments were performed using nutrient broth (NB)-gluconate-grown R. eutropha ΔphaC1 cells harboring the phaC1-eyfp fusion plasmid. When these cultures were harvested in the exponential growth phase (6 h after inoculation, PHB accumulation), no phosphorylation sites were detected in two independent experiments. Thus, no phosphorylation could be detected on actively PHB-synthesizing PhaC1 protein. In contrast, the residue T373 was phosphorylated in every sample at the stationary phase (24 h after inoculation). At this time point, PHB was no longer synthesized, and the cells degraded previously accumulated PHB. In addition, peptides with phosphorylated T30, T94, and T109 of PhaC1 were identified in lysates of cells harvested at the stationary phase in two of three experiments. The residues S10, T11, S16, S149, S178, T191, S196, and T198 were identified as phosphorylation sites in only one experiment (Table 1). We conclude that PhaC1 is present in a multiphosphorylated state during the stationary growth phase. Residue T373 appears to be the most essential residue because it was detected in the phosphorylated form in all repetitions. Furthermore, residue T373 is the only phosphorylated residue located in the C-terminal catalytic domain of PhaC1. Two different groups independently crystalized PhaC1 and determined the 3-dimensional structure (25) (26, 27). The four amino acids before and after T373 of PhaC1 could not be resolved (25), suggesting that the region around T373 does not have a defined structure and might be flexible. The structure solved by the other group revealed the presence of a loop around the T373 location, a finding that would also be in agreement with a high degree of flexibility of the region around T373. However, the rationale for choosing T373 for a detailed analysis will be explained later. Interestingly, cells grown on fructose-mineral medium exhibited no phosphorylation (regardless of whether the samples were taken at the exponential or the stationary growth phase) (Table 1).
TABLE 1.
Summary of all PhaC1 phosphorylation sites found in pulldown experiments
| PhaC1 expt no. | Proteome analysis sitea | Medium | Phosphorylation site during growth atb: |
||
|---|---|---|---|---|---|
| Exponential phase | Stationary phase | Undetermined growth phase | |||
| 1 | Hohenheim | NB | —d | — | T30, T373 |
| 2 | Hohenheim | NBc | — | S10, T11, S16, T30, T94, T109, T373, S178, S196, T198 | — |
| 3 | Tübingen | NBc | — | T94, T109, S149, T191, T373 | — |
| 4 | Hohenheim | MSMe | — | — | — |
Analyses were performed by the core facilities of the University Stuttgart Hohenheim (Hohenheim) or the University of Tübingen (Tübingen).
Bold type indicates phosphosites that were identified at least twice.
Exponential and stationary phases refer to the 6-h and 24-h time points, respectively.
—, no phosphorylation detected.
Exponential and stationary phases refer to the 22-h and 96-h time points, respectively.
Phosphomimetic T373D PhaC1 mutein has reduced PHB synthase activity.
We exchanged the threonine codon of residue 373 in PhaC1 to aspartate; the introduction of a negative charge mimics phosphorylation at this position. The purified T373D protein had a similar circular dichroism (CD) spectrum to that of the wild-type (WT) protein, suggesting no changes in the structure of the T373D protein (see Fig. S1 in the supplemental material). Upon the testing of the T373D mutein, we could not detect PHB synthase activity in the first 8 min of the assay (Fig. 1A). In comparison, the wild-type PhaC1 protein had almost wholly polymerized the available substrate during the same period. Only after a more extended incubation with 3-hydroxybutyryl-coenzyme A (3-HB-CoA), the T373D mutein showed detectable (low) PHB synthase activity (data not shown). When the concentration of the PHB synthase was increased 10-fold to 1.3 μM, the lag phase of the wild-type (WT) protein was absent. In contrast, the PHB synthase activity of the PhaC1 T373D mutein was clearly detectable at 1.3 μM but only after a lag phase of at least 1 min and with a lower specific activity compared to that of the wild-type protein at the same concentration (Fig. 1B). A negative charge at position 373 strongly diminished the activity under the applied assay conditions. To determine whether the reduced activity was caused by the negative charge, we changed the phaC1 codon at position 373 to an asparagine codon and purified the resulting T373N mutein. Upon testing the T373N mutein, we detected low PHB synthase activity. The activity was higher than with the T373D mutein but was strongly reduced in comparison to that of the wild type (Fig. 2). Replacing the 373 threonine residue with alanine resulted in a strongly reduced state with detectable PHB synthase activity. The behavior is similar to that of the T373D mutant (Fig. 2). Lastly, we exchanged the T373 codon for glutamate and determined the activity of the purified T373E mutein. The activity was considerably reduced compared to that of the wild type but was higher than that for the T373D/N/A muteins (Fig. 2). In summary, the phosphomimetic T373D mutein resulted in the loss of most of its PHB synthase activity, but changing the residue at 373 to other residues also altered PHB synthase activity.
FIG 1.

In vitro PHB synthesis by purified His6-PhaC1. A discontinuous PHB synthase assay was performed using purified PHB synthase muteins His6-PhaC1WT or His6-PhaC1T373D in the absence of PhaM. (A) With 130 nM His6-PhaC1WT or His6-PhaC1T373D, the lag phase of PhaCT373D is approximately 7 min longer than the lag phase of PhaC1WT. (B) With 1.3 μM His6-PhaC1WT or His6-PhaC1T373D, the lag phase of PhaCT373D is approximately 1 min longer than the lag phase of PhaC1WT. Results were obtained from triplicates; error bars show the standard deviations.
FIG 2.

In vitro PHB synthesis by purified His6-PhaC1. A discontinuous PHB synthase assay was performed using purified PHB synthase mutants His6-PhaC1WT, His6-PhaC1T373A, His6-PhaC1T373D, His6-PhaC1T373E, or His6-PhaC1T373N (each 150 nM) in the presence or absence of 150 nM His6-PhaM. Results were obtained from triplicates; error bars show the standard deviations.
T373D and other PhaC1 muteins are still activated by PhaM.
It is well known that PHB synthase of R. eutropha in vitro has a lag phase of several minutes before the reaction begins and proceeds with maximal activity (28). However, in the presence of the multifunctional protein PhaM (20, 21, 29), which is attached to PHB granules in vivo, the lag phase of the PhaC1-catalyzed reaction is abolished, and the specific activity of PhaC1 is increased approximately 3-fold. To test whether the PhaC1 T373 muteins still respond to the presence of PhaM, we repeated the activity determinations of PhaC1 T373 muteins in the presence of purified PhaM. As shown in Fig. 2, the presence of PhaM reduced the lag phase of all PhaC1 muteins. Even the PhaC1 mutein with the phosphomimetic substitution (T373E) showed higher activity in the presence of PhaM than wild-type PhaC1 in the absence of PhaM. However, the activities of the PhaC1 muteins with PhaM differed among each other in the same sequential order as in the absence of PhaM. We conclude that residue 373 is not involved in the interaction of PhaC1 with PhaM or in the activation of PhaC1. However, the nature of residue 373 seems to be necessary for the general activity of the enzyme and determines the extent of the lag phase in vitro (Fig. 2).
Single mutations of PhaC1 phosphosites have no detectable effect on PHB accumulation or PHB mobilization in vivo.
The results described above clearly demonstrate that mutating the phosphosite T373 to other residues reduced the PHB synthase activity of the purified protein in vitro. To evaluate whether these mutations also influence the accumulation of PHB in vivo, we investigated the formation of PHB in R. eutropha ΔphaC1 harboring a phaC1-containing plasmid with or without mutations. The following phaC1 variant genes were tested: phaC1 (WT), phaC1 (T373D), phaC1 (T373N), and phaC1 (T373A). All plasmids had a pBBR1MCS2 backbone, and the phaC1 variant genes were cloned as fusions with eyfp under the control of the endogenous phaC1 promoter.
No substantial differences in PHB granule number, size, or eYFP-PhaC1 colocalization occurred at 0, 2, 4, 6, 8, 10, and 24 h of growth (data not shown). The growth was monitored photospectroscopically, but no difference between the various strains was observed (see Fig. S2). We conclude that the constructed single phaC1 T373 mutation caused no significant differences in the PHB contents during growth on NB-gluconate medium. However, a microscopic inspection of cells for PHB content is imprecise; therefore, we repeated the experiments with several selected strains. This time, the PHB content in samples taken during accumulation and mobilization was measured via gas chromatography; however, no significant differences were detected in PHB contents between the wild-type and the cells with T373 mutations (T373A, T373D, and T373N) (Fig. 3).
FIG 3.
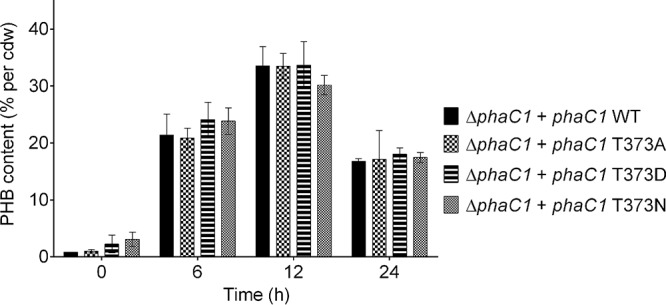
PHB contents of R. eutropha strains. R. eutropha ΔphaC1 (pBBR1MCS2_PphaC_phaC1_eyfp), R. eutropha ΔphaC1 (pBBR1MCS2_PphaC_phaC1_T373A_eyfp), R. eutropha ΔphaC1 (pBBR1MCS2_PphaC_phaC1_T373D_eyfp), and R. eutropha ΔphaC1 (pBBR1MCS2_PphaC_phaC1_T373N_eyfp) were grown on NB with 0.2% Na gluconate. Results were obtained from biological triplicates; error bars show the standard deviations.
Combined PhaC1 phosphosite mutations decreased PHB accumulation in vivo.
Three additional phosphorylated residues were detected in two independent pulldown experiments: T30, T94, and T109. Therefore, a phosphomimetic mutation and a nonphosphorylatable mutation for each of these residues were generated and introduced into R. eutropha ΔphaC1 to study the role of each phosphosite. As it was observed that the addition of PhaM could rescue the activity of PhaC1 T373 muteins in vitro, plasmids were also conjugated to a double deletion mutant of R. eutropha lacking both phaC1 and phaM. All these strains were grown on NB medium with 0.2% gluconate and analyzed for PHB content and localization. Samples were taken after 0, 2, 4, 6, 8, 10, and 24 h of growth; none showed a significant difference compared to the wild type (see Table S1). Differences in the PHB contents can also lead to differences in the optical densities. Therefore, the growth curves of these strains were documented (see Fig. S4). However, all growth curves perfectly overlaid with that of the wild type. Consequently, we hypothesized that multiple phosphorylations of PhaC1 may be necessary to regulate the activity of PhaC1 and to provoke a detectable effect on PHB accumulation in vivo.
To investigate the effect of the simultaneous mutagenesis of different phosphorylation sites, we combined the mutations for all phosphorylated residues detected at least twice in independent experiments on a single phaC1 gene. The optical density at 600 nm (OD600) was determined during growth, but no significant difference between cells harboring wild-type PhaC1 and the combined mutant strains was observed (see Fig. S3A). The PHB contents of R. eutropha strains harboring PhaC1 (T30D plus T94D plus T109D plus T373D) were slightly lower than that of the wild type. However, a significantly reduced PHB content was only seen in samples taken during the PHB accumulation phase (after 4 h of growth). In contrast, cells containing PhaC1 (T30N plus T94N plus T109N plus T373N) had the same PHB content as the wild type during all growth phases (Fig. S3B). The experiment was repeated with these two strains by harvesting the cells at more time points during PHB accumulation to ensure that the observed difference at the 4-h time point between the PhaC1 (T30D plus T94D plus T109D plus T373D) and PhaC1 wild type can be observed during the whole PHB synthesis period. Indeed, after 1, 2, 4, and 6 h of growth, the PHB contents of cells harboring PhaC1 (T30D plus T94D plus T109D plus T373D) were significantly lower than the PHB content of cells with wild-type PhaC1. The results confirm that combined phosphomimetic mutations of PhaC1 downregulate PHB synthase activity in vivo (Fig. 4).
FIG 4.
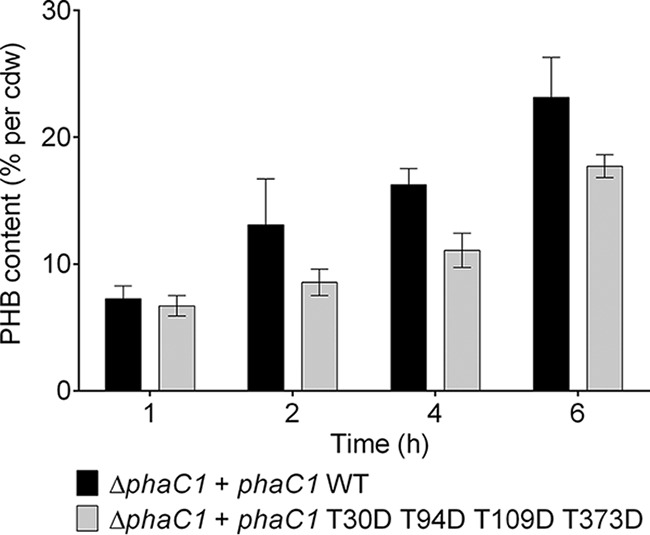
PHB accumulation of R. eutropha H16 strains during exponential growth. Data represent the PHB contents of R. eutropha ΔphaC1 (pBBR1MCS2_PphaC_phaC1_eyfp) and R. eutropha ΔphaC1 (pBBR1MCS2_PphaC_phaC1_T30D_T94D_T109D_T373D_eyfp) grown on NB medium with 0.2% gluconate. Results were obtained from biological triplicates; error bars show the standard deviations.
Residue S35 of PHB depolymerase PhaZa1 is phosphorylated.
Analog pulldown experiments as described above for eYFP-PhaC1 were performed for R. eutropha ΔphaZa1 cells that harbored a plasmid carrying an eyfp-phaZa1 fusion. One residue (S35) was identified in the phosphorylated form in the pulldown samples of all experiments, regardless of during which growth phase or from which medium the sample was taken. Residues T28 and T26 were additionally identified in phosphorylated forms in two and one of four replicates, respectively (Table 2).
TABLE 2.
Summary of all PhaZa1 phosphorylation sites found in pulldown experiments
| PhaZa1 expt no. | Proteome analysis sitea | Medium | Phosphorylation site during growth atb: |
||
|---|---|---|---|---|---|
| Exponential phase | Stationary phase | Undetermined growth phase | |||
| 1 | Hohenheim | NB | —d | — | S35 |
| 2 | Hohenheim | NBc | S35 | T26, T28, S35 | — |
| 3 | Tübingen | NBc | — | S35 | — |
| 4 | Hohenheim | MSMe | S35 | T28, S35 | — |
Analyses were performed by the core facilities of the University Stuttgart Hohenheim (Hohenheim) or the University of Tübingen (Tübingen).
Bold type indicates phosphosites that were identified at least twice.
Exponential and stationary phases refer to the 6-h and 24-h time points, respectively.
—, no phosphorylation detected.
Exponential and stationary phases refer to the 22-h and 96-h time points, respectively.
R. eutropha strains harboring a phosphomimetic T26D or S35D PhaZa1 mutein are reduced in PHB mobilization efficiency.
The identification of phosphorylated residues in the PHB depolymerase polypeptide suggested that the activity of PhaZa1 could be regulated by phosphorylation. To find evidence for this hypothesis, we exchanged the identified residues by site-directed mutagenesis and investigated the effect of these modifications on PHB metabolism. Unfortunately, the activity of PHB depolymerase PhaZa1 cannot be assayed accurately in vitro, because the enzyme requires the environment of a PHB granule to achieve the correct conformation enabling activity. The use of isolated native PHB granules as a substrate for in vitro PHB depolymerase activity determination is limited, as granules isolated from R. eutropha harbor several PHB depolymerase isoenzymes (for details, see references 5 and 12). Furthermore, the granules lack the appropriate numbers of phasins and other PHB granule-associated proteins when PHB granules are isolated from recombinant Escherichia coli strains harboring the PHB biosynthetic genes phaCAB. Therefore, the importance of the identified phosphorylation sites of PhaZa1 for the mobilization of PHB could be investigated only by an in vivo analysis. To this end, we mutagenized the identified phosphorylation sites of phaZa1 to aspartate (phosphomimetic) and to asparagine (not phosphorylatable) codons. The recombinant plasmids with the variant phaZa1 genes were then conjugatively transferred to R. eutropha ΔphaZa1. The growth on NB-gluconate medium and the PHB contents were determined for all strains (Fig. 5). A chromosomal deletion of phaZa1 led to an impairment of PHB degradation during the stationary growth phase (Fig. 5). The complementation of the chromosomal deletion of phaZa1 by a wild-type copy of phaZa1 was clearly demonstrated by the increase of the PHB content up to ∼40% of the cellular dry weight (cdw) in the first 26 h and the subsequent mobilization of three quarters of this PHB until 48 h to values of around 10% PHB (see Fig. S5). The strains with the phosphomimetic T26D and S35D mutations revealed strongly reduced abilities to mobilize accumulated PHB (residual PHB content >20% of cdw or >50% of the maximum compared to the PHB content at 26 h set to 100%). The strain with the T26N mutation showed near wild-type mobilization activity (PHB content after 48 h, <10% of cdw or <25% of the maximum). The other strains (with T28D, T28N, and S35N mutations) showed an intermediate phenotype, with PHB contents of around or slightly greater than 20% of cdw (≥50% of the maximum at 26 h) (Fig. 5; see also Fig. S6). These data show that a modification of residue T26 and/or S35 significantly decreased the efficiency of PHB mobilization by the PhaZa1 variant.
FIG 5.

Growth (A) and PHB contents (B) of R. eutropha ΔphaZa1 (pBBR1MCS2_PphaC_phaZa1_eyfp), R. eutropha ΔphaZa1 (pBBR1MCS2_PphaC_phaZa1_T26D_eyfp), R. eutropha ΔphaZa1 (pBBR1MCS2_PphaC_phaZa1_T26N_eyfp), R. eutropha ΔphaZa1 (pBBR1MCS2_PphaC_phaZa1_T28D_eyfp), R. eutropha ΔphaZa1 (pBBR1MCS2_PphaC_phaZa1_T28N_eyfp), R. eutropha ΔphaZa1 (pBBR1MCS2_PphaC_phaZa1_S35D_eyfp), and R. eutropha ΔphaZa1 (pBBR1MCS2_PphaC_phaZa1_S35N_eyfp) on NB medium with 0.5% Na gluconate. The PHB content after 26 h was set to 100% for each strain (for absolute values, see Fig. S6 in the supplemental material). Results were obtained from 2 biological and 2 technical replicates; error bars show the standard deviations.
DISCUSSION
The critical enzymes of PHA synthesis (PHA synthase [PhaC1]) and PHA degradation (PHB depolymerases [PhaZs]) are constitutively expressed and present on the PHB granules in R. eutropha (23, 24, 30). This expression pattern poses the questions of how the synthesis and mobilization of carbonosomes are regulated and how a futile cycle of simultaneous synthesis and degradation is avoided. One possibility is that PHA synthesis and degradation are controlled by signaling molecules as described in a previous study (31). An additional possibility could be through allosteric regulation of PHB synthase and PHB depolymerase activities. Here, we demonstrate that the PHB synthase and PHB depolymerase can both be phosphorylated.
Remarkably, the phosphorylated peptides of PhaC1 were only detected in samples harvested during the stationary growth phase on NB medium, thus only during PHB mobilization. No phosphorylated peptides could be identified in samples harvested at the exponential growth phase or from cells grown on mineral medium with 2% fructose. As shown in previous studies, R. eutropha does not degrade PHB under this condition (24, 31). Phosphorylated peptides of the major PHB depolymerase, PhaZa1, were identified in samples at all stages of growth and on different media.
In this study, we showed that single mutations of the T373 residue of PhaC1 could inhibit PhaC1 activity in vitro, but the inhibition can be rescued (partially) by the presence of PhaM. However, the inhibitory effect of the T373D mutation was not phenotypically detectable in vivo, even in the absence of PhaM. Either undiscovered regulators of the PhaC1 activity could rescue the activity of PhaC1 T373D in vivo or the concentration of PhaC1 in vivo is higher than that in vitro. The difference in concentration thereby overcomes the inhibiting effect of the T373D mutation. Although not vital for activity, this residue plays a prominent role in PhaC1 activity.
Wittenborn and coworkers successfully crystallized the C-terminal part of PhaC1, but the region from residue 369 to 377 was disordered and could not be resolved (25). Flexible protein regions are often detected as disordered during protein crystallization (32, 33). Intrinsically disordered segments in these proteins often harbor regions that mediate molecular recognition (34). Such a flexible region could be important for substrate recognition, stabilization, and interactions with other proteins. Stubbe's group proposed a process in which the interaction between two PhaC1 monomers is stabilized by the electrostatic interactions of residue R398 with the CoA phosphate group of the incoming substrate. This stabilization could result in a conformational change in the region of the dimer interface (25). A conformational change of the region around T373 induced by phosphorylation could then affect the interaction between R398 and the substrate. Furthermore, the authors described a putative product egress channel in the C-terminal part of PhaC1 made up of a hydrophobic core that was, however, too narrow to accommodate a PHB molecule. An alternative possibility is that a conformational change induced by the phosphorylation of T373 causes the rearrangements necessary for an expansion of the channel to enable the passage of a PHB chain.
The other study describing the PHB synthase structure proposed that PhaC1 could have been proteolytically cleaved at residue R192 during the crystallization process (26, 27). This explanation aligns with results from the Stubbe lab, where only the C-terminal part could be crystallized. This group was able to provide a structure of the region around T373: they described a D-loop consisting of hydrophobic residues with T373 in the center. They proposed that this loop could mediate hydrophobic interactions between two monomers. Phosphorylation in the middle of the D-loops (T373) could abolish these hydrophobic interactions, favoring the dissociation of the active PHB synthase dimer to inactive monomers.
All phosphorylated residues detected in our study, except T373, were located in the N-terminal domain of PhaC1 (S10, T11, S16, T30, T94, T109, S149, S178, T191, S196, and T198). As previously described, the lack of a defined electron density in crystals could be caused by the high flexibility of the N-terminal part of PhaC1 due to several small conformational changes induced by phosphorylations (25, 26).
According to the PHB synthase structure solved by Lee's group, the C-terminal domains of PhaC1 are located at the center of the dimer, while the N-terminal domains are located opposite of the dimerization domain (26). It was shown that neither the N-terminal domain alone nor the C-terminal domain alone could produce PHB. A full-length protein is necessary for PHB synthesis even if the catalytic domain is located in the C-terminal part (27, 35, 36). Thus, the phosphorylation sites in the N-terminal part should not be neglected. Kim and coworkers suggested that the N-terminal domain is essential for the binding of PhaC1 to PHB granules and the interaction with PhaM (27). In accordance, our results showed that only a combination of mutated phosphorylation sites, including those of the N-terminal domain, leads to a decrease in PhaC1 activity in vivo. The polyphosphorylation of PhaC1 might have a negative effect on PHB synthase activity. The impact can be explained by a weakening of the binding to PHB granules due to repulsions arising from the negative charge of the phosphorylation sites.
In this study, we also demonstrated that the PHB depolymerase is phosphorylated during the PHB synthesis and PHB degradation phases. Mutagenesis of the phosphorylation sites leads to a downregulation of the PHB-degrading activity of PhaZa1 during the stationary growth phase. No crystal structure of PhaZa1 is available to understand in which region of the protein these phosphorylation sites are located. A previous study described a catalytic triad between the residues C183, D355, and H388 of PhaZa1 (37). The three phosphorylated residues, T26, T28, and S35, are very close to each other but far from the catalytic triad in the primary amino acid sequence. Further studies and a crystal structure of PhaZa1 are necessary to understand how the activity of PhaZa1 is affected by phosphorylation.
In a previous study, we assumed that a combination of allosteric regulation and covalent modification of the enzymes involved in PHB accumulation and mobilization is necessary to regulate the PHB content of R. eutropha. The PHB content of R. eutropha was previously shown to be dependent on the concentration of alarmones such as ppGpp (31). The presence of phosphorylations would be in line with previous data. Another study had shown that SpoT1 interacted with the nonphosphorylated form of the EIIANtr component of the phosphotransferase system (PTS) in R. eutropha (38). This finding indicates a possible phosphorylation cascade from the carbon source uptake system to SpoT1. SpoT1 can then influence the activity of the PHB synthesis and mobilization system by the phosphorylation of PhaC1 and PhaZa1.
Conclusion.
The major finding of this study is that the key enzymes of PHB synthesis and PHB mobilization, PHB synthase and the PHB depolymerase, are reproducibly phosphorylated at the identified residues. Mutagenesis of these residues indicated that the phosphorylation sites are involved in the activities of these enzymes. Further studies are necessary to understand how these phosphorylations can regulate the activities of PhaC1 and PhaZa1 in vivo. It remains unclear whether these phosphorylations are parts of a signaling cascade.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are shown in Table 3. R. eutropha H16 strains were grown aerobically on nutrient broth ([NB] 0.8% [wt/vol]) with or without sodium gluconate (0.2% or 0.5% [wt/vol] as indicated) at 30°C. Alternatively, R. eutropha was grown in mineral salts medium (MSM) (39) with fructose (2% [wt/vol]) as a carbon source at 30°C. Growth experiments for optical density at 600 nm (OD600) and PHB content determinations were routinely performed with two or three biological replicates (as indicated) and two technical replicates. Values and error bars in the figures correspond to the mean values and standard deviations, respectively. E. coli strain XL1-Blue was used for site-directed mutagenesis experiments. Recombinant PhaC1 was expressed in E. coli BL21(DE3)/pLys. E. coli strains were grown on lysogeny broth (LB) supplemented with kanamycin.
TABLE 3.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristicsa | Reference or source and strain collection no. |
|---|---|---|
| Strains | ||
| Escherichia coli BL21(DE3)/pLys | For protein expression | Novagen, 4512 |
| E. coli S17-1 (SN490) | For conjugation | 44 |
| E. coli XL1-Blue | For site-directed mutagenesis | Stratagene, 4505 |
| Ralstonia eutropha H16 (SN1342) | R. eutropha wild-type strain | DSMZ 428 |
| R. eutropha H16 ΔphaC1 (SN6525) | R. eutropha H16 with knockout of phaC1 | 16 |
| R. eutropha H16 ΔphaC1 ΔphaM (SN6506) | R. eutropha H16 with knockout of phaC1 and phaM | This study |
| R. eutropha H16 ΔphaZa1 (SN6167) | R. eutropha H16 with knockout of phaZa1 | This study |
| Plasmids | ||
| pBBR1MCS2 | Broad host range vector; Kmr | 44 |
| pBBR1MCS2-PphaC_eyfp-C1 | Universal vector for construction of fusions C-terminal to eyfp under the control of the phaCAB promoter | 29 |
| pBBR1MCS2_PphaC_phaC1_eyfp | N-terminal fusion of PhaC1 to eYFP | 16 |
| pBBR1MCS2_PphaC_phaC1_T30A_eyfp | N-terminal fusion of PhaC1 (T30A) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T30D_eyfp | N-terminal fusion of PhaC1 (T30D) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T373A_eyfp | N-terminal fusion of PhaC1 (T373A) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T373D_eyfp | N-terminal fusion of PhaC1 (T373D) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T373N_eyfp | N-terminal fusion of PhaC1 (T373N) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T373E_eyfp | N-terminal fusion of PhaC1 (T373E) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T94N_eyfp | N-terminal fusion of PhaC1 (T94N) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T94D_eyfp | N-terminal fusion of PhaC1 (T94D) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T109N_eyfp | N-terminal fusion of PhaC1 (T109N) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T109D_eyfp | N-terminal fusion of PhaC1 (T109D) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T94N_T373N_eyfp | N-terminal fusion of PhaC1 (T94N, T373N) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T94N_T109N_T373N_eyfp | N-terminal fusion of PhaC1 (T94N, T109N, T373N) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T94D_T373D_eyfp | N-terminal fusion of PhaC1 (T94D, T373D) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T94D_T109D_T373D_eyfp | N-terminal fusion of PhaC1 (T94D, T109D, T373D) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T30D_T94D_T109D_T373D_eyfp | N-terminal fusion of PhaC1 (T30D, T94D, T109D, T373D) to eYFP | This study |
| pBBR1MCS2_PphaC_phaC1_T30N_T94N_T109N_T373N_eyfp | N-terminal fusion of PhaC1 (T30N, T94N, T109N, T373N) to eYFP | This study |
| pBBR1MCS2_PphaC_phaZa1_eyfp | N-terminal fusion of PhaZa1 to eYFP | This study |
| pBBR1MCS2_PphaC_phaZa1_S35D_eyfp | N-terminal fusion of PhaZa1 (S35D) to eYFP | This study |
| pBBR1MCS2_PphaC_phaZa1_S35N_eyfp | N-terminal fusion of PhaZa1 (S35N) to eYFP | This study |
| pBBR1MCS2_PphaC_phaZa1_T26D_eyfp | N-terminal fusion of PhaZa1 (T26D) to eYFP | This study |
| pBBR1MCS2_PphaC_phaZa1_T26N_eyfp | N-terminal fusion of PhaZa1 (T26N) to eYFP | This study |
| pBBR1MCS2_PphaC_phaZa1_T28D_eyfp | N-terminal fusion of PhaZa1 (T28D) to eYFP | This study |
| pBBR1MCS2_PphaC_phaZa1_T28N_eyfp | N-terminal fusion of PhaZa1 (T28N) to eYFP | This study |
| Fusions to His6 tag | ||
| pET28a | His tag expression vector; Kmr | Novagen |
| pET28a_phaC1 | Plasmid for expression of His6-PhaC1 | 20 |
| pET28a_phaC1 (T30A) | Plasmid for expression of His6-PhaC1 (T30A) | This study |
| pET28a_phaC1 (T30D) | Plasmid for expression of His6-PhaC1 (T30D) | This study |
| pET28a_phaC1 (T373A) | Plasmid for expression of His6-PhaC1 (T373A) | This study |
| pET28a_phaC1 (T373D) | Plasmid for expression of His6-PhaC1 (T373D) | This study |
| pET28a_phaC1 (T373E) | Plasmid for expression of His6-PhaC1 (T373E) | This study |
| pET28a_phaC1 (T373N) | Plasmid for expression of His6-PhaC1 (T373N) | This study |
| pET28a_phaM | Plasmid for expression of His6-PhaM | 22 |
| Knockout | ||
| pLO3_phaZa1 | Plasmid used to generate phaZa1 knockout, Tcr | 30 |
Kmr, kanamycin resistance; Tcr, tetracycline resistance.
Construction of gene deletions and site-directed mutagenesis.
Precise genomic deletions of R. eutropha genes were constructed using the sacB-sucrose selection method (10% sucrose used for selection) and pLO3 as the deletion vector as described previously (40). The genotypes of all generated constructs and mutants were verified by PCR of the respective DNA regions and determinations of their DNA sequences. Two primers, each complementary to one strand of the DNA, were used to mutate the genes carried by the plasmid, with the mutation in the middle of the oligonucleotide. A PCR with PrimeSTAR HS DNA polymerase (TaKaRa Bio, Inc.) was run under conditions recommended by the supplier. The template DNA was degraded by a DpnI restriction digest after the PCR, leaving only the newly synthesized mutated DNA uncut. The DpnI-digested DNA was transformed into E. coli XL1-Blue cells. The mutated plasmids were identified by DNA sequencing of the complete mutated phaC1 gene.
Protein pulldown and screen for phosphorylations.
Genes of interest were fused to eyfp and expressed in R. eutropha. Cultures were harvested at the time points and during the growth phases of interest by centrifugation at 5,000 rpm at 4°C for 15 min. The pellets were resuspended in 3 ml pulldown buffer (25 mM sodium phosphate buffer, 5% glycerol, 50 mM NaCl, 5 mM MgSO4, 5 mM KCl, 20 mM imidazole, pH 7.5) supplemented with phosphatase inhibitors (5 mM β-glycerophosphate, 5 mM NaF, 10 mM sodium vanadate). The cells were disrupted twice by using a French press (Aminco, Silver Spring, MD, USA) with a pressure of 1,000 lb/in2. For the pulldown step, magnetic beads from ChromoTek (Germany) with a binding affinity for eYFP (GFP-Trap_MA for immunoprecipitation of GFP fusion proteins from mammalian cells) were used. For this, 500 μl of lysate was mixed with 20 μl of magnetic beads that had been equilibrated three times with 500 μl of pulldown buffer. The pulldown mixture was shaken for 1 h on ice. Subsequently, the beads were washed 5 times with pulldown buffer, and the samples were transferred to fresh low-bind tubes. The samples were eluted in 40 μl of 2.5% SDS and denatured for 5 min at 95°C.
The samples prepared by protein pulldown were allowed to migrate ≈1 cm into the separating gel of a 12% SDS-PAGE gel, and the gel was stained with 0.12% (wt/vol) colloidal Coomassie. A proteome analysis was performed as indicated either by the proteome core facility of the Life Science Center, University of Hohenheim (Stuttgart, Germany) (41), or by the Proteome Center of the University of Tübingen. In Hohenheim, the samples were digested with trypsin and extracted with acetonitrile (10 min). The supernatants were vacuum dried and dissolved in formic acid (15 μl; 0.1% [vol/vol]). Five microliters was injected into a liquid chromatograph (LC) tandem mass spectrometer (MS-MS). The data were analyzed using Scaffold proteome software (version 4.8.4). In Tübingen, tryptic in-gel digestion (above the thick blue running front) was performed before running the samples on a Proxeon Easy-nLC coupled to a QExactive HF mass spectrometer (method: 60 min, Top7, higher energy collisional dissociation [HCD]). The data were processed using MaxQuant software (version 1.5.2.8. with integrated Andromeda search engine). The spectra were searched against an R. eutropha H16 UniProt database (6,615 entries). The data were processed with a setting of 1% for the false discovery rate (FDR), i.e., with an estimation that 1% of all identifications are false positive. The posterior error probability (PEP) (42) was calculated for each peptide. Phosphorylations were identified by a mass shift of 80 Da at the position of the phosphorylated residue in the spectrum from electrospray ionization tandem mass spectrometry (ESI-MS/MS) of the fragmented peptide.
Circular dichroism.
Circular dichroism (CD) spectra were measured using a J-815 circular dichroism spectrophotometer (Jasco, Groß-Umstadt, Germany). Ten microliters of the protein solution with a concentration of 11 mg/ml was mixed with 30 μl 200 mM KC; 39 μl of this protein solution was pipetted into a 0.2-mm cuvette. The spectra were collected in a wavelength range between 185 nm and 260 nm with standard sensitivity and a bandwidth of 1 nm. The spectra recorded with buffer only were used as a baseline and subtracted from the protein spectra. For each sample, 50 scans were collected, averaged, and divided by the concentration.
Overexpression and purification of His6-PhaC1.
The expression of His6-PhaC1 from recombinant E. coli BL21(DE3)/pLys harboring pET28a-phaC1 was induced with 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at an OD600 of ∼0.6 and with 4 h of subsequent incubation at 30°C. His6-PhaC1 was purified in the presence of 0.05% (wt/vol) Hecameg via Ni-agarose chromatography according to a protocol described in detail previously (20). Imidazole and other low-molecular-weight molecules were removed by size exclusion chromatography (HiPrep 26/10, desalting column, 53-ml bed volume). All purified PhaC1 proteins were of high purity (see Fig. S7 in the supplemental material). The purification of PhaM-His6 was performed as described previously (22).
Discontinuous PHB synthase activity assay.
PHB synthase PhaC1 is the key enzyme of PHB synthesis. The precursor 3-HB-CoA is polymerized to polyhydroxybutyrate, while the release of coenzyme A can be monitored in vitro. Coenzyme A reacts with dithionitrobenzoate (DTNB) to yellow-colored nitrothiobenzoate (NTB). The formation of NTB can be measured spectroscopically at 412 nm. For the DTNB assay, the protein concentration is determined using a bicinchoninic acid (BCA) kit and adjusted for all enzymes (PhaC1 and PhaM). PhaC1 assay buffer (150 mM KH2PO4, 0.2% glycerol, pH 7.2), 0.5 mM 3-HB-CoA, and for some experiments, 150 nM PhaM-His6 were mixed in a total volume of 200 μl in an Eppendorf tube incubator at 30°C. The reaction was started by adding PHB synthase PhaC1. Aliquots of the reaction mixture (20 μl) were taken at different time intervals, and the reactions were immediately terminated by mixing with 40 μl of 10% (wt/vol) trichloroacetic acid. Samples were centrifuged to remove precipitated protein, and 145 μl of 1 mM DTNB in 0.5 M potassium phosphate (pH 7.8) was added to 55 μl of the assay mixture. The concentration of formed NTB (i.e., the absorbance at 412 nm) was determined with a microplate reader (Eon; BioTek) after a 10-min incubation at room temperature. The reaction was limited by the amount of substrate (3-HB-CoA) (20).
Microscopic methods.
Fluorescence microscopy experiments were performed with a Nikon Ti-E microscope (MEA53100) equipped with F41-007 Cy3 and F41-54 Cy2 filters for the analysis of the fluorescent proteins (eYFP). Pictures were taken with a digital camera (Hamamatsu Orca Flash 4.0 sCMOS camera) and processed with Nikon imaging software. The fluorophore eYFP was detected with the aid of a standard filter set (excitation, 500/24 nm; emission, 542/27 nm) as previously described (6). PHB granules were visualized by fluorescence microscopy of Nile red-stained cells using standard filters (excitation, 562/40 nm; emission, 594 nm [longpass filter]) as described previously (6, 31). Insoluble inclusions could also be visualized by bright-field microscopy and appeared as dark spheres.
Other methods.
Quantitative analysis of PHA content was performed using gas chromatography (GC) after acid methanolysis of lyophilized cells according to reference 43. DNA manipulation and the construction of plasmids were completed by using standard molecular biological methods and according to suppliers' instructions. The amplification of DNA by PCR was performed using commercial oligonucleotides of at least 16 bp in length that were identical in sequence to the corresponding region of the R. eutropha genome.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft (GRK1708) and by a grant from the Ontario-Baden-Württemberg student exchange program to C.P.
We thank Stephanie Bresan, Simone Reinhardt, Anna Sznajder, Anna Schweter, Mirita Franz-Wachtel, Berit Würtz, and Jens Pfannstiel for their support with some experiments. We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00604-18.
REFERENCES
- 1.Anderson AJ, Dawes EA. 1990. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol Rev 54:450–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meng D-C, Chen G-Q. 2018. Synthetic biology of polyhydroxyalkanoates (PHA). Adv Biochem Eng Biotechnol 162:147–174. doi: 10.1007/10_2017_3. [DOI] [PubMed] [Google Scholar]
- 3.Rehm BHA. 2010. Bacterial polymers: biosynthesis, modifications and applications. Nat Rev Microbiol 8:578–592. doi: 10.1038/nrmicro2354. [DOI] [PubMed] [Google Scholar]
- 4.Jendrossek D, Pfeiffer D. 2014. New insights in the formation of polyhydroxyalkanoate granules (carbonosomes) and novel functions of poly(3-hydroxybutyrate). Environ Microbiol 16:2357–2373. doi: 10.1111/1462-2920.12356. [DOI] [PubMed] [Google Scholar]
- 5.Sznajder A, Pfeiffer D, Jendrossek D. 2015. Comparative proteome analysis reveals four novel polyhydroxybutyrate (PHB) granule-associated proteins in Ralstonia eutropha H16. Appl Environ Microbiol 81:1847–1858. doi: 10.1128/AEM.03791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bresan S, Sznajder A, Hauf W, Forchhammer K, Pfeiffer D, Jendrossek D. 2016. Polyhydroxyalkanoate (PHA) granules have no phospholipids. Sci Rep 6:26612. doi: 10.1038/srep26612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parlane NA, Gupta SK, Rubio-Reyes P, Chen S, Gonzalez-Miro M, Wedlock DN, Rehm BHA. 2017. Self-assembled protein-coated polyhydroxyalkanoate beads: properties and biomedical applications. ACS Biomater Sci Eng 3:3043–3057. doi: 10.1021/acsbiomaterials.6b00355. [DOI] [PubMed] [Google Scholar]
- 8.Rehm BHA. 2003. Polyester synthases: natural catalysts for plastics. Biochem J 376:15–33. doi: 10.1042/BJ20031254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rehm BHA. 2006. Genetics and biochemistry of polyhydroxyalkanoate granule self-assembly: the key role of polyester synthases. Biotechnol Lett 28:207–213. doi: 10.1007/s10529-005-5521-4. [DOI] [PubMed] [Google Scholar]
- 10.Uchino K, Saito T, Jendrossek D. 2008. Poly(3-hydroxybutyrate) (PHB) depolymerase PhaZa1 is involved in mobilization of accumulated PHB in Ralstonia eutropha H16. Appl Environ Microbiol 74:1058–1063. doi: 10.1128/AEM.02342-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eggers J, Steinbüchel A. 2013. Poly(3-hydroxybutyrate) degradation in Ralstonia eutropha H16 is mediated stereoselectively to (S)-3-hydroxybutyryl coenzyme A (CoA) via crotonyl-CoA. J Bacteriol 195:3213–3223. doi: 10.1128/JB.00358-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sznajder A, Jendrossek D. 2014. To be or not to be a poly(3-hydroxybutyrate) (PHB) depolymerase: PhaZd1 (PhaZ6) and PhaZd2 (PhaZ7) of Ralstonia eutropha, highly active PHB depolymerases with no detectable role in mobilization of accumulated PHB. Appl Environ Microbiol 80:4936–4946. doi: 10.1128/AEM.01056-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wieczorek R, Pries A, Steinbüchel A, Mayer F. 1995. Analysis of a 24-kilodalton protein associated with the polyhydroxyalkanoic acid granules in Alcaligenes eutrophus. J Bacteriol 177:2425–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neumann L, Spinozzi F, Sinibaldi R, Rustichelli F, Pötter M, Steinbüchel A. 2008. Binding of the major phasin, PhaP1, from Ralstonia eutropha H16 to poly(3-hydroxybutyrate) granules. J Bacteriol 190:2911–2919. doi: 10.1128/JB.01486-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfeiffer D, Jendrossek D. 2011. Interaction between poly(3-hydroxybutyrate) granule-associated proteins as revealed by two-hybrid analysis and identification of a new phasin in Ralstonia eutropha H16. Microbiology 157:2795–2807. doi: 10.1099/mic.0.051508-0. [DOI] [PubMed] [Google Scholar]
- 16.Pfeiffer D, Jendrossek D. 2012. Localization of poly(3-hydroxybutyrate) (PHB) granule-associated proteins during PHB granule formation and identification of two new phasins, PhaP6 and PhaP7, in Ralstonia eutropha H16. J Bacteriol 194:5909–5921. doi: 10.1128/JB.00779-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mezzina MP, Pettinari MJ. 2016. Phasins, multifaceted polyhydroxyalkanoate granule-associated proteins. Appl Environ Microbiol 82:5060–5067. doi: 10.1128/AEM.01161-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pötter M, Madkour MH, Mayer F, Steinbüchel A. 2002. Regulation of phasin expression and polyhydroxyalkanoate (PHA) granule formation in Ralstonia eutropha H16. Microbiology 148:2413–2426. doi: 10.1099/00221287-148-8-2413. [DOI] [PubMed] [Google Scholar]
- 19.York GM, Stubbe J, Sinskey AJ. 2002. The Ralstonia eutropha PhaR protein couples synthesis of the PhaP phasin to the presence of polyhydroxybutyrate in cells and promotes polyhydroxybutyrate production. J Bacteriol 184:59–66. doi: 10.1128/JB.184.1.59-66.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeiffer D, Jendrossek D. 2014. PhaM is the physiological activator of poly(3-hydroxybutyrate) (PHB) synthase (PhaC1) in Ralstonia eutropha. Appl Environ Microbiol 80:555–563. doi: 10.1128/AEM.02935-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ushimaru K, Tsuge T. 2016. Characterization of binding preference of polyhydroxyalkanoate biosynthesis-related multifunctional protein PhaM from Ralstonia eutropha. Appl Microbiol Biotechnol 100:4413–4421. doi: 10.1007/s00253-015-7225-6. [DOI] [PubMed] [Google Scholar]
- 22.Bresan S, Jendrossek D. 2017. New insights in PhaM-PhaC-mediated localization of PHB granules in Ralstonia eutropha H16. Appl Environ Microbiol 31:e00505-17. doi: 10.1128/AEM.00505-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence AG, Schoenheit J, He A, Tian J, Liu P, Stubbe J, Sinskey AJ. 2005. Transcriptional analysis of Ralstonia eutropha genes related to poly-(R)-3-hydroxybutyrate homeostasis during batch fermentation. Appl Microbiol Biotechnol 68:663–672. doi: 10.1007/s00253-005-1969-3. [DOI] [PubMed] [Google Scholar]
- 24.Brigham CJ, Speth DR, Rha C, Sinskey AJ. 2012. Whole-genome microarray and gene deletion studies reveal regulation of the polyhydroxyalkanoate production cycle by the stringent response in Ralstonia eutropha H16. Appl Environ Microbiol 78:8033–8044. doi: 10.1128/AEM.01693-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wittenborn EC, Jost M, Wei Y, Stubbe J, Drennan CL. 2016. Structure of the catalytic domain of the class I polyhydroxybutyrate synthase from Cupriavidus necator. J Biol Chem 291:25264–25277. doi: 10.1074/jbc.M116.756833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J, Kim Y-J, Choi SY, Lee SY, Kim K-J. 2017. Crystal structure of Ralstonia eutropha polyhydroxyalkanoate synthase C-terminal domain and reaction mechanisms. Biotechnol J 12:1600648. doi: 10.1002/biot.201600648. [DOI] [PubMed] [Google Scholar]
- 27.Kim Y-J, Choi SY, Kim J, Jin KS, Lee SY, Kim K-J. 2017. Structure and function of the N-terminal domain of Ralstonia eutropha polyhydroxyalkanoate synthase, and the proposed structure and mechanisms of the whole enzyme. Biotechnol J 12:201600649. doi: 10.1002/biot.201600649. [DOI] [PubMed] [Google Scholar]
- 28.Gerngross TU, Martin DP. 1995. Enzyme-catalyzed synthesis of poly[(R)-(−)-3-hydroxybutyratel: formation of macroscopic granules in vitro. Proc Natl Acad Sci U S A 92:6279–6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfeiffer D, Wahl A, Jendrossek D. 2011. Identification of a multifunctional protein, PhaM, that determines number, surface to volume ratio, subcellular localization and distribution to daughter cells of poly(3-hydroxybutyrate), PHB, granules in Ralstonia eutropha H16. Mol Microbiol 82:936–951. doi: 10.1111/j.1365-2958.2011.07869.x. [DOI] [PubMed] [Google Scholar]
- 30.Pötter M, Steinbüchel A. 2006. Biogenesis and structure of polyhydroxyalkanoate granules, p 109–136. In Shively JM. (ed), Inclusions in prokaryotes. Microbiology monographs, vol 1 Springer, Berlin, Germany. [Google Scholar]
- 31.Jüngert JR, Borisova M, Mayer C, Wolz C, Brigham CJ, Sinskey AJ, Jendrossek D. 2017. Absence of ppGpp leads to increased mobilization of intermediately accumulated poly(3-hydroxybutyrate) (PHB) in Ralstonia eutropha H16. Appl Environ Microbiol 83:e00755-17. doi: 10.1128/AEM.00755-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurevich VV, Gurevich EV, Uversky VN. 16 February 2018. Arrestins: structural disorder creates rich functionality. Protein Cell doi: 10.1007/s13238-017-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. 2001. Intrinsically disordered protein. J Mol Graph Model 19:26–59. doi: 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 34.Wong ETC, Na D, Gsponer J. 2013. On the importance of polar interactions for complexes containing intrinsically disordered proteins. PLoS Comput Biol 9:e1003192. doi: 10.1371/journal.pcbi.1003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng Z, Li M, Xue X-J, Tian H-L, Li Z, Chen G-Q. 2006. Mutation on N-terminus of polyhydroxybutyrate synthase of Ralstonia eutropha enhanced PHB accumulation. Appl Microbiol Biotechnol 72:896–905. doi: 10.1007/s00253-006-0371-0. [DOI] [PubMed] [Google Scholar]
- 36.Ye Z, Song G, Chen G, Chen J. 2008. Location of functional region at N-terminus of polyhydroxyalkanoate (PHA) synthase by N-terminal mutation and its effects on PHA synthesis. Biochem Eng J 41:67–73. doi: 10.1016/j.bej.2008.03.006. [DOI] [Google Scholar]
- 37.Kobayashi T, Saito T. 2003. Catalytic triad of intracellular poly(3-hydroxybutyrate) depolymerase (PhaZ1) in Ralstonia eutropha H16. J Biosci Bioeng 96:487–492. doi: 10.1016/S1389-1723(03)70136-4. [DOI] [PubMed] [Google Scholar]
- 38.Karstens K, Zschiedrich CP, Bowien B, Stülke J, Görke B. 2014. Phosphotransferase protein EIIANtr interacts with SpoT, a key enzyme of the stringent response, in Ralstonia eutropha H16. Microbiology 160:711–722. doi: 10.1099/mic.0.075226-0. [DOI] [PubMed] [Google Scholar]
- 39.Schlegel HG, Kaltwasser H, Gottschalk G. 1961. A submersion method for culture of hydrogen-oxidizing bacteria: growth physiological studies. Arch Mikrobiol 38:209–222. (In German.) [PubMed] [Google Scholar]
- 40.Lenz O, Friedrich B. 1998. A novel multicomponent regulatory system mediates H-2 sensing in Alcaligenes eutrophus. Proc Natl Acad Sci U S A 95:12474–12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poudel N, Pfannstiel J, Simon O, Walter N, Papageorgiou AC, Jendrossek D. 2015. The Pseudomonas aeruginosa isohexenyl glutaconyl coenzyme A hydratase (AtuE) is upregulated in citronellate-grown cells and belongs to the crotonase family. Appl Environ Microbiol 81:6558–6566. doi: 10.1128/AEM.01686-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Käll L, Storey JD, MacCoss MJ, Noble WS. 2008. Posterior error probabilities and false discovery rates: two sides of the same coin. J Proteome Res 7:40–44. doi: 10.1021/pr700739d. [DOI] [PubMed] [Google Scholar]
- 43.Juengert J, Bresan S, Jendrossek D. 2018. Determination of polyhydroxybutyrate (PHB) content in Ralstonia eutropha using gas chromatography and Nile red staining. Bio Protoc 8:e2748. doi: 10.21769/BioProtoc.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.