

Abstract
Comparative genomics has become a central tool for evolutionary biology, and a better knowledge of understudied taxa represents the foundation for future work. In this study, we characterized the transcriptome of male and female mature gonads in the European clam Ruditapes decussatus, compared with that in the Manila clam Ruditapes philippinarum providing, for the first time in bivalves, information about transcription dynamics and sequence evolution of sex-biased genes. In both the species, we found a relatively low number of sex-biased genes (1,284, corresponding to 41.3% of the orthologous genes between the two species), probably due to the absence of sexual dimorphism, and the transcriptional bias is maintained in only 33% of the orthologs. The dN/dS is generally low, indicating purifying selection, with genes where the female-biased transcription is maintained between the two species showing a significantly higher dN/dS. Genes involved in embryo development, cell proliferation, and maintenance of genome stability show a faster sequence evolution. Finally, we report a lack of clear correlation between transcription level and evolutionary rate in these species, in contrast with studies that reported a negative correlation. We discuss such discrepancy and call into question some methodological approaches and rationales generally used in this type of comparative studies.
Keywords: RNA-Seq, transcription level, evolutionary rate, gametogenesis, embryo development, E–R correlation
Introduction
Despite the differences in terms of sexual dimorphism and behavior, males and females share almost the same genome, especially in species lacking heteromorphic sex chromosomes. Therefore, the vast majority of sex-specific characters and traits are the result of differential expression of the so-called “sex-biased genes” (Ranz et al. 2003; Ellegren and Parsch 2007; Parsch and Ellegren 2013). The study of sex-biased gene expression is crucial for understanding the mechanisms of gene regulation and evolution (Grath and Parsch 2016): several works investigated the amount of sex-biased genes among animals, showing that the proportion of these genes is extremely variable, depending on the organism, analyzed tissue, developmental, and reproductive stage. It has been reported that the number of transcribed sex-biased genes is higher in gonads, since most of them are involved in sexual dimorphism and competition (Parisi et al. 2003; Mank et al. 2010; Harrison et al. 2015). Also, genes that are more or exclusively transcribed in males (male-biased genes) show a higher rate of protein evolution—calculated as the ratio of nonsynonymous to synonymous nucleotide substitution (dN/dS)—as reported in many organisms such as insects, nematodes, birds, and mammals (Meiklejohn et al. 2003; Khaitovich et al. 2005; Pröschel et al. 2006; Zhang et al. 2007; Assis et al. 2012; Grath and Parsch 2012; Harrison et al. 2015; Wang et al. 2015). Even if female-biased genes did not receive the same attention of male-biased genes, some studies conducted in mammals, birds, fish, and insects reported evidence of high dN/dS of these transcripts compared with unbiased genes, namely genes showing no differential expression between sexes (Swanson et al. 2004; Mank et al. 2007; Yang et al. 2016). It is not clear whether the high rate of protein sequence evolution of sex-biased genes, and particularly male-biased genes, is an outcome of positive or relaxed selection. In the literature there are several studies supporting either one or the other theory. Works carried out mostly in Drosophila seem to point out that male-biased genes undergo evolution by positive selection (Swanson et al. 2004; Zhang and Parsch 2005; Pröschel et al. 2006): according to this theory, male–male competition drives a faster evolution of male reproductive proteins (Swanson and Vacquier 2002; Clark et al. 2006; Turner and Hoekstra 2008). On the other hand, several studies support the hypothesis that male-biased genes are more dispensable (Mank and Ellegren 2009) and thus under relaxed selection, while female-biased genes are under stronger constraints due to their functional pleiotropy (Duret and Mouchiroud 2000; Zhang et al. 2007; Mank et al. 2008; Wang et al. 2015; Dapper and Wade 2016). Accordingly, genes exclusively expressed in males showed a higher accumulation of deleterious mutations (Gershoni and Pietrokovski 2014). Finally, the comparison of sex-biased genes across species revealed a large variability in transcription level, suggesting that differences in regulation of sex-biased genes may have a fundamental role in speciation (Brawand et al. 2011; Romero et al. 2012). Particularly, male-biased genes seem to be the most divergent also in terms of transcription level (Torgerson et al. 2002; Meiklejohn et al. 2003; Ranz et al. 2003; Zhang et al. 2004, 2007; Khaitovich et al. 2005): this evidence inspired the hypothesis of a positive correlation between the evolution of protein sequences and transcriptional divergence (Nuzhdin et al. 2004; Khaitovich et al. 2005; Lemos et al. 2005; Liao and Zhang 2006; Sartor et al. 2006). Nevertheless, this pattern is not consistent (Jordan et al. 2004; Tirosh and Barkai 2008; Harrison et al. 2015), indicating that protein sequence evolution and transcription level divergence can be decoupled. Therefore, many questions about evolution of sex-biased genes still remain open.
What shapes the rate of protein sequence change is a central question for understanding molecular evolution. Several studies have reported different determinants that could influence dN/dS such as, for example, the functional importance of a protein, expression breadth among tissues, pleiotropy, protein–protein interaction, and secondary structure (Larracuente et al. 2008; Ridout et al. 2010). Nevertheless, according to the most recent hypotheses, transcription level has been proposed to be the main responsible for the rate of protein evolution (see Zhang and Yang 2015 for a review). In particular, a strong negative correlation, defined E–R correlation, was found between dN/dS and transcription level, across the three domains of life. One of the main hypothesis to explain the E–R correlation is that highly transcribed genes evolve more slowly thus reducing the amount of misfolded proteins, known to be cytotoxic and damaging for organism fitness (Drummond et al. 2005).
The development of High-Throughput Sequencing has significantly increased the capability to get insights into the molecular mechanisms driving evolution. Particularly, RNA-Seq produces a large amount of data about both evolution of protein sequence and transcription level, also for nonmodel organisms. The latter point is important, because our knowledge is still restricted to a very limited number of taxa; it is indeed not recommendable to formulate hypotheses and infer general evolutionary patterns based only on a small number of species. For example, although Mollusca is the second phylum of the animal kingdom for number of species, no comparative studies on the relationship between protein evolution and transcription have been performed so far in this group.
In this work, we obtained the gonadal transcriptome from males and females of the European clam Ruditapes decussatus (Linneaus 1758)—also known as grooved carpet shell—and we compared it with the available gonadal transcriptome data (Ghiselli et al. 2012) from the species Ruditapes philippinarum (Adams and Reeve 1850). Ruditapes decussatus is a bivalve species of the family Veneridae, native to the Mediterranean and European Atlantic coasts. The fishing of R. decussatus has historically had a main role in the production of seafood in Italy, Spain, and Portugal. The recent introduction in Europe of R. philippinarum—native from Philippines, Korea, and Japan—led to a replacement of R. decussatus with R. philippinarum for aquaculture purposes. Indeed, compared with R. decussatus, the Manila clam R. philippinarum, reaches sexual maturation at a smaller size, is faster growing, have a greater number of spawning events, a more extended breeding period, and a higher resistance to disease (Ghiselli et al. 2017 and references therein). All these issues probably contributed to a population decline of R. decussatus in the Southwestern Europe, as recently reported by Arias-Pérez et al. (2016). Here, we characterize the biological processes represented in male and female mature gonad in R. decussatus. The comparison with gonad transcription in R. philippinarum allows to investigate, for the first time in two bivalve species, the evolution of both protein sequence and transcription level divergence of sex-biased genes. Our analyses provided further insight into the relationship between rate of protein evolution and transcription level.
Materials and Methods
Library Preparation
The 12 samples of R. decussatus used for this study were collected from the Northern Adriatic Sea, in the river Po delta region (Sacca di Goro, approximate GPS coordinates: 44°50′06″N, 12°17′55″E) at the end of July 2011, during the spawning season. Six males and six females were used to obtain the RNA-Seq library. Clams were sexed by microscope inspection of gonadal liquid collected with a glass capillary tube. The samples were immediately frozen in liquid nitrogen, and preserved at −80˚C. Total RNA extraction, mRNA purification, and fragmentation, cleaning, and cDNA synthesis were carried out following the protocols of Mortazavi et al. (2008), and modifications as reported in Ghiselli et al. (2012). Samples were barcoded and sequenced over two lanes (for two technical replicates) of an Illumina Genome Analyzer IIx machine, using 76-bp paired-end reads.
The same sampling technique was used in Ghiselli et al. (2012) on R. philippinarum, with the only difference that the whole animal was flash-frozen in liquid nitrogen after being sexed, and the collection of gonadal tissue with the capillary tube was performed later, immediately before RNA extraction.
De Novo Assembly
Raw reads from Illumina sequencing were filtered with Trimmomatic version 0.36 (Bolger et al. 2014) with the following parameters: TruSeq3-PE.fa: 1:30:10 LEADING: 3 TRAILING: 3 SLIDINGWINDOW: 4:20 MINLEN: 50. We removed low abundant k-mers (k-mers < 5) with the trim-low-abund.py script, implemented in khmer version 2.1.1 (Crusoe et al. 2015). Filtered reads were assembled with Trinity-v2.4.0 (Grabherr et al. 2011) with default parameters. We used the filter_longest_trinity_subcomponents.py trinity script to select the longest isoforms among different alternative transcripts.
Assembly completeness assessment was performed using BUSCO (Simão et al. 2015) on the Metazoa ortholog set, as implemented in gVolante (Nishimura et al. 2017), and cut-off length for sequence statistics and base composition = 1.
Differential Transcription Analysis
In both the species, Bowtie 2 (Langmead and Salzberg 2012) was used to align reads of each sample to the reference transcriptome. We used SAMtools (Li et al. 2009) to retain only reads mapping exactly one time, with concordant pairs match and a mapping quality ≥ 10.
We used NOISeq R package (Tarazona et al. 2015; Costa-Silva et al. 2017) to assess quality of count data obtained from read mapping, and to perform differential transcription analysis. Correlation between technical replicates was calculated with Spearman’s and Pearson’s rank correlation coefficient, then technical replicates were summed up, as suggested by NOISeq developers. In order to remove low count genes, we used the counts per million (CPM) method, as implemented in NOISeq. A sensitivity plot was performed to investigate the percentage of genes at different CPM. Because of the high proportion of genes with CPM ≤ 1, we decided to filter genes with CPM ≤ 1 in all samples. In order to determinate whether the sequencing depth was sufficient to investigate differential transcription analysis, we performed a saturation plot, showing the number of genes detected at different simulated depth. Differential transcription between males and females was calculated with the NOISeqBio method, a nonparametric approach optimized for the use of biological replicates. Among different methods included in the NOISeq package to normalized data, we chose to use FPKM, in order to later investigate the presence of E–R correlation, as performed in other works (see Zhang and Yang 2015 for a review). We considered as female-biased genes, those with log2 of fold change between sexes (FC) > 1 and false discovery rate (FDR) < 0.05. Similarly, we considered as male-biased genes, those with log2FC < −1 and FDR < 0.05.
Orthology Detection
Orthologs between the two species were found with Proteinortho v5.16b (Lechner et al. 2011), using a TBLASTX search to compare nucleotide sequences. Only orthologs with a single sequence for each species (1:1 orthology) were considered.
Annotation
Orthologous genes were used as input for our annotation pipeline, that consists of three main steps: contaminants removal, ORF prediction, and annotation.
To identify contaminants, we first blasted our transcripts against the nr database using the BLASTX algorithm with default parameters. Then we used BLAST taxid to select only transcripts belonging to the Metazoa taxon, since most of the expected contaminats in bivalves come from fungi, plantae, and bacteria. ORF prediction was then carried out with the software findorf (Krasileva et al. 2013; https://github.com/vsbuffalo/findorf; last accessed May 28, 2018). Findorf uses BLASTX against multiple, user defined, databases, and HMMER hmmscan (e-value <1E-3) to predict ORFs. In our analyses, we used 19 databases (supplementary table 1, Supplementary Material online) for BLASTX, and the Pfam database for hmmscan. In the last step of our pipeline, we used Argot2 (Falda et al. 2012), a tool that combines the results of BLAST and HMMER searches to annotate large sequence data sets with GO terms. For this purpose, BLASTP was used to search against the Swiss-Prot database with default parameters and HMMER to search against Pfam 30.0 database (Finn et al. 2016). Outputs from BLASTP and HMMER were then used by the Argot2 tool to obtain GO annotation.
Evolution of Orthologous Genes
For each orthologous pairs, nucleotide sequences of ORFs were recovered from findorf outputs. Then, we used TranslatorX (Abascal et al. 2010) to translate nucleotide sequences, compute protein alignment with MUSCLE (Edgar 2004), and back-translate amino acid alignments into nucleotide alignments. KaKs_Calculator 2.0 (Wang et al. 2010) with default settings was used to obtain the ratio of nonsynonymous to synonymous nucleotide substitution (dN/dS). We used the EMBOSS package “distmat” (Rice et al. 2000), with uncorrected multiple substitution method for proteins, to calculate amino acid p-distance from alignments obtained from TranslatorX. We plotted the dN/dS distribution of orthologs with unbiased transcription in both species (unbiased/unbiased). Additionally, we plotted the dN/dS distribution of orthologs with sex-biased transcription in either species (unbiased/male-biased; unbiased/female-biased) and orthologs with sex-biased transcription in both species (male-biased/male-biased; female-biased/female-biased). The Dunn test with the Bonferroni correction was carried out with the R package “dunn.test” to compare the dN/dS distribution among these groups. The same groups were also adopted to analyze the correlation between dN/dS and amino acid p-distance. GO term annotation of groups of orthologs with statistically different distribution of dN/dS was obtained and visualized with REViGO (Supek et al. 2011), while a BLASTP search with default parameters, against the nr database, was used to investigate the function of genes with female-biased transcription in both the species and dN/dS > 1. Coefficient variation of gene expression (CVE; Zhou et al. 2008) was calculated as the ratio of SD and mean of transcription in genes with statistically different distribution of dN/dS and in unbiased orthologs. Finally, in order to investigate the relationship between rate of protein sequence evolution and transcription level, we first performed Spearman’s correlation between transcription level (expressed in FPKM) of orthologous genes in R. decussatus and R. philippinarum, then we plotted log2 (dN/dS) versus log2 (FPKM) of orthologous genes separately for R. decussatus and R. philippinarum.
Results
De Novo Assembly
Almost 80 million paired-end reads were generated from the Illumina sequencing from R. decussatus. A total of 63 million reads was retained after Trimmomatic and Khmer in R. decussatus, while 24 million reads were retained in R. philippinarum (raw reads= 45 million, see supplementary table 2, Supplementary Material online).
The de novo assembly of R. decussatus yielded 131,059 contigs, with a median length of 399 bp and N50 of 1,283. Since many of these contigs represent isoforms, we retained the longest isoform among alternative transcripts, for a total of 88,936 representative loci, with median length of 373 and N50 of 1,123 (see supplementary table 2, Supplementary Material online).
Similarly, we obtained 64,082 contigs for R. philippinarum (median length = 325, N50 = 602) and 45,602 representative loci (median length = 313, N50 = 595; supplementary table 2, Supplementary Material online).
Raw reads and transcriptome assemblies are available on NCBI BioProjects PRJNA170478, and PRJNA68513 (R. decussatus, and R. philippinarum, respectively).
Supplementary table 2, Supplementary Material online, shows the results of the completeness assessment performed with BUSCO: we found 97% of complete Metazoa orthologs in R. decussatus (99% complete + partial), and 69% in R. philippinarum (93% complete + partial).
Low Counts Filtering and Orthology Detection
Supplementary figures 1 and 2, Supplementary Material online, show the correlation between raw counts of technical replicates in both the species (correlation coefficients are reported in supplementary table 3, Supplementary Material online). Sensitivity plots in supplementary figures 3 and 4, Supplementary Material online, report the fraction of low expressed genes in R. decussatus and R. philippinarum, normalized for counts per million (CPM). Genes with CPM ≤ 1 in all samples were filtered in both the species, so that 29,769 genes in R. decussatus and 26,309 in R. philippinarum were retained for orthology detection and differential transcription analysis. Saturation plots in supplementary figures 5 and 6, Supplementary Material online, show that at the current sequencing depth we reached a median of 98% saturation in R. decussatus and of 87% in R. philippinarum.
Among 29,769 genes in R. decussatus and 26,309 in R. philippinarum, 3,652 were recognized as orthologs between the two species. These genes were annotated through our annotation pipeline.
Annotation
Out of 3,652 orthologous genes, about ∼10% was removed in the filtering step in both R. decussatus and R. philippinarum. A total of 3,285 ORFs was detected by findorf in R. decussatus and 3,271 in R. philippinarum. Argot2 assigned GO terms to 3,237 genes in R. decussatus and 3,236 R. philippinarum (supplementary table 4, Supplementary Material online). At the end of the annotation pipeline, a total of 3,102 orthologs was kept and used for differential transcription analysis of orthologous genes.
Differential Transcription Analysis and Sequence Evolution of Orthologous Genes
Figure 1 shows the volcano plot of the orthologous gene transcription in the two species. Out of 3,102 orthologs found between R. decussatus and R. philippinarum, 1,284 (41.3%) include at least a sex-biased gene in one of the two species. Among the 1,284 groups with at least one sex-biased gene, only in 430 the sex bias is maintained between species (33%), while in the remaining 854 (66%), the orthologs show a change in sex bias. More in detail, 435 orthologs are unbiased in one species and male-biased in the other, 402 are female-biased in one species and unbiased in the other, 330 are male-biased in both the species, 100 are female-biased in both the species, while in 17 genes the sex bias is reversed (supplementary table 5, Supplementary Material online). Figure 2 shows the distribution of dN/dS among orthologous genes between the two species, grouped according to the above-mentioned sex-bias categories. We found that the dN/dS distribution of orthologous genes with unbiased transcription in both the species (N = 1,818) is not statistically different from that of genes with an unbiased transcription in one species and a sex-biased transcription in the other, and the median dN/dS is always included between 0.043 and 0.047 (supplementary table 6, Supplementary Material online). On the contrary, when orthologs have a sex-biased transcription in both the species, the distribution of dN/dS is statistically different from that of other groups of ortholog genes (Dunn test P value = 0). In particular, orthologs with male-biased transcription in both the species (N = 330) are characterized by a lower dN/dS (median = 0.036), with GO term annotations mostly consisting in metabolic process, transport, oxidation–reduction process, and regulation of transcription (supplementary table 7 and fig. 7, Supplementary Material online). Differently, orthologs with a female-biased transcription in both the species show a higher dN/dS (median = 0.067). This group (N = 100) is characterized by a first peak of density corresponding to dN/dS = 0.05 and a second peak at 0.2 (fig. 3, red line). Supplementary table 8 and fig. 8, Supplementary Material online, reports a list of GO terms associated with orthologous genes with female-biased transcription in both the species. A BLASTP annotation of female genes with dN/dS > 0.1 (N = 25) shows that several genes are involved in cell differentiation and embryo development, and maintenance of genome stability (supplementary table 9, Supplementary Material online). Genes with a reversed sex-bias between the two species show a median dN/dS of 0.046, but given the low sample size (N = 17) we did not include this group in the statistical analysis. The analysis of the coefficient of variation of gene expression (CVE) reveals that a higher dN/dS is associated with a higher CVE in sex-biased genes (supplementary fig. 9 and table 10, Supplementary Material online), but the same pattern is not present in unbiased genes. The distribution of amino acid p-distance (supplementary fig. 10, Supplementary Material online) shows that orthologous genes between R. decussatus and R. philippinarum are characterized by a median divergence of 6.99%. In only 1.9% of genes the p-distance is ≥ 60%. We investigated the relationship between dN/dS and the amino acid p-distance in both unbiased and sex-biased orthologous genes. Among orthologs with lower amino acid divergence, approximately <40%, the relationship between dN/dS and p-distance shows a linear trend (fig. 3a, black dashed line). Instead, when the amino acid divergence is higher, the trend is better described by an exponential function (fig. 3a, red dashed line). This pattern is particularly evident in unbiased genes (fig. 3b). On the contrary, we found that among sex-biased categories, genes with a p-distance >40% are rare, a linear model fits the data best (fig. 3c–f, colored solid lines), and the trend for each sex-biased category is comparable to the linear trend of unbiased genes (fig. 3b–f, black dashed lines). Among the genes following an exponential relationship between dN/dS and amino acid p-distance (fig. 3a, red dashed line), we focused on two clusters: one includes orthologs with a p-distance included between 40% and 60% and a dN/dS < 0.2, the other includes orthologs with a p-distance > 60% and a dN/dS > 0.2. We refer to the orthologs belonging to the above-mentioned clusters as “fast-mutating” and “fast-diverging,” respectively (see Discussion).
Fig. 1.
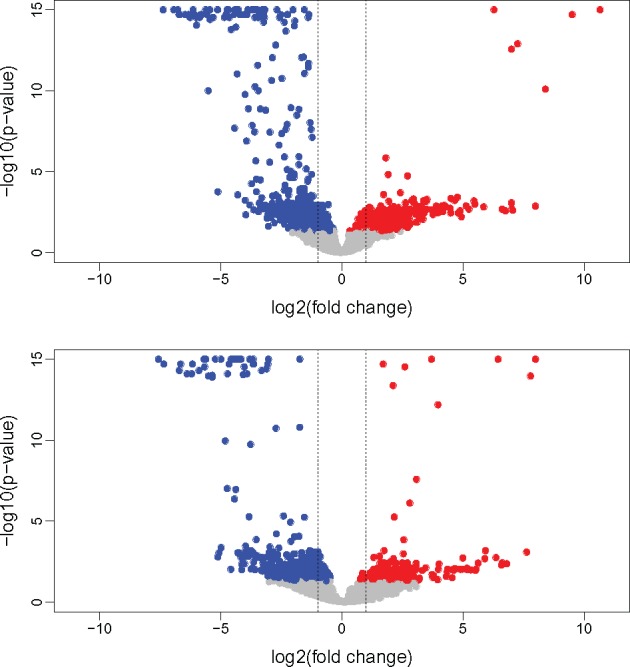
—Volcano plot of the transcription of orthologous genes in Ruditapes decussatus (top) and R. philippinarum (bottom). Male-biased transcripts are represented in blue, female-biased transcripts in red, unbiased transcripts in gray. Dashed lines: log2(fold change) = −1, 1 (fold change = −2, 2).
Fig. 2.

—Kernel density plots and boxplots of dN/dS in unbiased genes (green line), in genes that are unbiased in one species and male- or female-biased in the other species (respectively, blue line and pink line) and in genes where the sex bias is maintained (black line for male-biased genes, red line for female-biased genes).
Fig. 3.

—(a) Relationship between dN/dS and amino acid p-distance of all orthologous genes between Ruditapes decussatus and R. philippinarum. A linear function (black dashed line) describes the relationship between dN/dS and p-distance in genes with lower p-distance. In genes with p-distance >40%, the relationship is better explained by an exponential function (red dashed line). (b) unbiased genes in both the species (green); (c) genes that are unbiased in one species and male-biased in the other (blue); (d) genes that are unbiased in one species and female-biased in the other species (pink); (e) genes where a male-bias is maintained (black); (f) genes where a female-bias is maintained (red). Dashed lines in (b–f): regression lines calculated for all genes; solid colored lines in b–f represent the regression lines calculated for the specific subset of genes.
Finally, we show that transcription level (FPKM) of orthologous genes in the two species is correlated (Spearman’s rank correlation ρ = 0.71; P value < 2.2E-16; supplementary fig. 11, Supplementary Material online). By analyzing the relationship between dN/dS of orthologous genes and FPKM in R. decussatus (fig. 4), we found a slightly negative correlation between rate of protein evolution and transcription level (Spearman’s rank correlation ρ = −0.16; P value < 2.2E-16). On the contrary, no correlation was detected between dN/dS and FPKM in R. philippinarum (Spearman’s rank correlation ρ = −0.06; P value = 0.0001).
Fig. 4.

—Relationship between the rate of amino acid sequence evolution, indicated as log2 (dN/dS), and transcription level, indicated as log2 (FPKM), in Ruditapes decussatus (left), and R. philippinarum (right). Black lines: regression lines corresponding to the linear model.
Discussion
In this work, we obtained the transcriptome of mature gonads in male and females of R. decussatus, and performed a comparative analysis with the related species R. philippinarum. Since gonads are known to be the tissue with the higher transcription of sex-biased genes (Torgerson et al. 2002; Reinke et al. 2004; Zhang et al. 2004, 2007; Good and Nachman 2005) this experiment gave the opportunity to investigate the evolution of sex-biased genes in two bivalve species, both in terms of protein sequence and transcription level. Also, we report here an analysis of the relationship between transcription level and rate of protein evolution.
Comparative analysis of transcription level and rate of protein evolution of sex-biased orthologous genes between R. decussatus and R. philippinarum.
In R. decussatus and R. philippinarum, ∼30% and 20% of the assembled contigs show a sex-biased transcription. These values are lower compared with what reported in other taxa, even when detection of sex-biased genes was performed in somatic tissues: >81% of genes showed a sex-biased transcription in frogs (Malone et al. 2006), ∼75% in wasps (Wang et al. 2015), up to 57% in Drosophila (Ranz et al. 2003), 50% in Daphnia pulex (Eads et al. 2007), and similar patterns were found in copepods (Poley et al. 2016), Anopheles (Papa et al. 2017), fish (Small et al. 2009), birds (Mank et al. 2010), and mammals (Yang et al. 2006). Nevertheless, it should be considered that the amount of sex-biased genes increases with the number of tissues analyzed (Yang et al. 2006; Ellegren and Parsch 2007), while only gonads were investigated in this work. Furthermore, clams lack sexual dimorphism as well as mating behavior, which are responsible for the majority of differential transcription between sexes (Ellegren and Parsch 2007; Harrison et al. 2015). Therefore, in these bivalves all the genes showing differential transcription between sexes are likely involved in gametogenesis, and this analysis offers the opportunity to observe the transcriptional difference between female and male gonads and gametes. Since gonads were sampled during the same stage of gametogenesis in two related species that lack sexual dimorphism, we did not expect to detect considerable differences in transcription of sex-biased genes. Nevertheless, in 66% of the orthologous genes the sex bias is not maintained between the two species. The most frequent condition is represented by genes that are unbiased in one species and sex-biased in the other species.
Sex-biased genes are considered to evolve faster, in particular those expressed preferentially or exclusively in reproductive tissues (Parisi et al. 2003; Mank et al. 2008; Harrison et al. 2015). In order to test this condition, we investigated the rate of protein sequence evolution in both unbiased and sex-biased genes, and among the latter, we separated those where the bias was maintained from those where the bias changed between the two species.
We found that genes with a male-biased transcription in both the species are characterized by a significantly lower dN/dS compared with unbiased genes or genes with a sex-biased transcription in only one species; conversely, genes where the female-biased transcription is maintained have a significant higher dN/dS. This is an uncommon pattern of evolution compared with most taxa investigated so far, where male-biased genes have the higher rate of protein evolution, particularly those involved in sexual competition (Swanson and Vacquier 2002; Clark et al. 2006; Turner et al. 2008). Alternatively, it was reported that both expression breadth and CVE are correlated to the rate of protein evolution. In particular, genes that are broadly transcribed in different tissues are expected to evolve slower than genes with a tissue specific transcription (Meisel 2011), since they are possibly involved in more essential functions (“functional pleiotropy,” see: Zhang et al. 2007; Mank and Ellegren 2009). Similarly, genes with a higher variation in transcription across samples should be characterized by higher dN/dS (Schrader et al. 2017). Since only gonads were analyzed in this study, we could not investigate the expression breadth of sex-biased genes. We instead analyzed the CVE of unbiased and sex-biased genes (supplementary fig. 9 and table 10, Supplementary Material online): female-biased genes present higher CVE in these animals and the opposite was found in male-biased genes, nevertheless, this pattern is not always consistent in unbiased genes, where a lower dN/dS in R. philippinarum is associated with a higher CVE; therefore, a congruous association between CVE and dN/dS is not evident in these animals.
Few other works reported a faster evolution of female-biased genes (see, e.g., Whittle and Johannesson 2013; Lipinska et al. 2015; Dean and Mank 2016; Papa et al. 2017). In zebrafish, both male- and female-biased genes are characterized by higher dN/dS; this pattern was ascribed to external fertilization in these animals, that leads to a strong competition of eggs and sperm, and a consequent higher selection acting on both male- and female-biased genes (Yang et al. 2016). Bivalves are characterized by external fertilization as well, and a similar discussion could be extended to these animals to explain the higher evolution of female-biased genes; nevertheless, the same selective force should also act on male-biased genes, that show instead a very low dN/dS. In a time-series global transcription profile of birds, Mank et al. (2010) reported that sex-specific dN/dS changed throughout the life cycle, acting equally strongly on female- and male-biased genes but at different stages. For example, they found that the level of sequence divergence on female-biased genes transcribed in late embryogenesis is similar to that seen in male-biased genes transcribed in adult gonads. This suggests the possibility that in the bivalve species analyzed here, female-biased genes transcribed in mature gonads might undergo a stronger sequence divergence, and that male-biased genes might do the same in other developmental stages. A comparative longitudinal study on different stages of gonad development would be an interesting follow up study, to help claryfing this issue. By analyzing the annotation of female-biased genes with the highest dN/dS, we found that they are mainly involved in embryo development, cell proliferation and migration, and genome maintenance.
In our data, dN/dS distribution is strongly skewed toward 0 in all cases, values >0.4 are quite rare, and we did not find any sign of positive selection. Given what reported in the literature, this is surprising because proteins involved in male reproductive traits, such as sperm-eggs recognition, or sperm competition are expected to have dN/dS >1, or at least close to this value (Swanson and Vacquier 2002; Clark et al. 2006; Dapper and Wade 2016). This should be even more evident in marine invertebrates where, as mentioned before, fertilization occurs externally, and positive selection acting on proteins involved in fertilization is thought to trigger the evolution of reproductive isolation (Turner and Hoekstra 2008). We wondered if these results were due to a biological condition or to a technical artefact. On the one hand, if we obtained an accurate representation of the actual evolutionary rates, then orthologs transcribed in the gonads of these two bivalve species experience only purifying selection. So why such a slow evolutionary rate? In species with heteromorphic—thus nonrecombining—sex chromosomes, sex-biased genes are nonrandomly distributed across the genome, so there can be cooperation and conflict among different chromosomal regions depending on whether they are cotransmitted as part of male or female sex determination. Such transmission asymmetry entails a high probability of sex chromosomes being involved in genomic conflicts, leading to sexually antagonistic variation (Rice and Chippindale 2008; Rice 2013), resulting in faster evolution. In particular, the Y chromosome does not recombine and is male-limited, and males generally evolve faster, so the conflicts will result in a faster evolution of male-biased genes. One could argue that the Y chromosome contains only few genes, but—to use Rice’s words—Y is a “coding dwarf” but a “regulatory giant” (Rice 2013): in Drosophila, polymorphisms at loci on Y chromosome influence thousands of genes (Stewart et al. 2010). So given that bivalves lack heteromorphic sex chromosomes, such conflicts—thus evolutionary rates—should be different, perhaps softer. Another nonmutually exclusive hypothesis should be considered. The sex determination system in bivalves is still unknown, but there is experimental evidence that sex is determined by maternal nuclear genome (Zouros 2013), and—since triploids develop male gonads—that maleness is achieved by exceeding a threshold of some yet unknown masculinizing factor. It was proposed that the activation of sex determination genes depends on genetic elements (RNAs, proteins) stored in the oocyte, whose concentration depends on maternal genotype. In this way, F1 sex depends only on maternal genotype, while paternal genotype contributes to F2 sex (see fig. 7 in Ghiselli et al. 2012 for a scheme). Brisson and Nuzhdin (2008) showed that in a system with a strong reproductive skew between males and females (pea aphids), male-biased genes undergo mutational decay, while female-biased genes are under constant selection. Clams do not experience such a strong reproductive skew, but if the above-mentioned model is true, feminizing factors—which should be female-biased genes—and masculinizing factors—which should be male-biased genes—might experience different selective pressures, mainly because masculinizing genes do not produce their effect in the embryos receiving them, but in their progeny. Basically their transmission does not depend on their masculinization action, and this might result in the elimination of pressure on male-biased genes to evolve fast. This idea is still at the stage of speculation, but we think it deserves further investigation. On the other hand, to explain the unexpectedly low dN/dS in sex-biased genes, we also want to consider the possibility of a too strict method for orthology detection: this could lead to a considerable number of false negatives, especially among the more variable orthologous genes, and the consequent exclusion of such sequences from the analyses. This would be particularly damaging, because fast evolving genes are the most informative and interesting genes for studying molecular evolution. In order to understand whether the method, we used to identify orthologs introduced a bias toward slow evolving sequences, we investigated the relationship between dN/dS and amino acid p-distance (fig. 3). The analysis showed that the method inferred orthology in sequences that diverged even >80%. Nevertheless, orthologs with a p-distance ≥ 60% are ∼2% of the total, and they are absent among sex-biased genes, that are expected to be the most variable. Given all the published literature on this subject, the complete absence of orthologous genes showing a dN/dS ≥ 0.8 is quite surprising, especially considering that we are analyzing genes transcribed in gonads. A possible explanation is that most of the rapidly evolving genes were not recognized as orthologs. Detection of orthologs is a well-known problem: to date, basically two approaches are used: the graph-based methods and the tree-based methods (Kristensen et al. 2011). In the graph-based method, clusters of orthologs are based on sequence similarities, but, as mentioned before, the most rapidly evolving and thus informative sequences could be discarded, due to high sequence divergence. On the other hand, a tree-based method requires good a priori knowledge of both gene family trees and species trees, that is not easily obtainable especially for nonmodel taxa (such as bivalves), which are poorly represented in the construction of gene family trees. Besides, by comparing different software based on both methods on a curated database of orthologs, Trachana et al. (2011) found that all repositories predict only a fraction of these orthologs.
Going back to our data, it is worth noting that the relationship between dN/dS and amino acid p-distance has two different trends, depending on whether the p-distance is lower or higher than 40% (fig. 3a). Below 40% the relationship follow a linear trend; on the contrary, when the amino acid divergence is >40%, the correlation is better described by an exponential function. We can think the x axis on the plot (amino acid p-distance) as a measure of nonsynonymous change, while the y axis represent the amount of nonsynonymous change “normalized” by the proportion of synonymous change. Therefore, for a fixed p-distance, an increasing dN/dS value corresponds to a decreasing proportion of synonymous changes, namely a faster rate of amino acid sequence evolution. Accordingly, we can observe that between 40% and 60% of p-distance there is a cluster of genes that shows a low dN/dS, meaning that the high number of nonsynonymous changes is coupled with an even higher proportion of synonymous changes. These genes therefore seem to experience a high mutation rate, but a relatively low rate of evolution, intended as the proportion of nonsynonymous change (“fast-mutating” orthologs). Between a p-distance of 60% and 80% the dN/dS rate is much higher, meaning that for the genes included in this interval most of the change is nonsynonymous, so they undergo a faster evolution (“fast-diverging” orthologs). Unexpectedly, most of such genes did not show a sex-biased transcription in either R. decussatus or R. philippinarum.
The Controversial Relationship between Evolutionary Rate and Transcription Level
According to recent studies, the evolutionary rate of a protein would be mainly influenced by its transcription level (Zhang and Yang 2015 and references therein), and a negative correlation between transcription level and evolutionary rate, defined E–R correlation, was found across a variety of species. Despite FPKM of orthologs being highly correlated between R. decussatus and R. philippinarum (Sperman’s rank correlation = 71%, supplementary fig. 11, Supplementary Material online), the E–R correlation analysis performed reciprocally (dN/dS between species vs. Ruditapes decussatus FPKM, and dN/dS between species vs. R. philippinarum FPKM) gave different results (fig. 4). No evidence for E–R correlation was found in R. philippinarum, while a negative correlation is present in R. decussatus, albeit quite weak compared with that reported in other taxa (see Zhang and Yang 2015). All that considered, we did not find clear evidence for E–R correlation in these two bivalve species. One hypothesis to explain such result is that bivalves undergo different patterns of protein evolution and/or that protein sequence evolution is not driven by transcription level, for yet unknown reasons. In this case, bivalves—or at least the two clam species we analyzed here—would represent an exception, and further investigation would be necessary to understand how, and why. Alternatively, since we obtained contrasting results by investigating the relationship between dN/dS and FPKM from a reciprocal analysis on the same two species, a second hypothesis concerns the methodology: is our opinion that the common practice used for investigating the relationship between dN/dS and transcription level could yield inaccurate results, for three main reasons.
1) Transcription level is extremely variable. Transcription is a quite noisy process—especially in multicellular eukaryotes—and it is influenced by both genetic factors—that modify gene expression depending, for example, on the tissue, developmental stage, and phase of life cycle—and environmental factors (e.g., diet, stress). Most of the times, the variation caused by genetics, environment, and by the combination of both is unpredictable. There could also be technical issues, like different sequencing methods and RNA quality, which are known to influence the measurement of transcription level. In addition, in multicellular organism the transcription level is often calculated averaging the mRNA concentration across several tissues: such values are likely inaccurate and not representative of the physiological condition of the organism. In order to perform reliable comparative analyses, it is important to use homogeneous data, and for all the above-mentioned reasons, this is a condition which is not often achieved.
2) dN/dS are calculated between the species for which transcription level is measured and one related species. Since the rate of protein evolution is influenced by the phylogenetic distance between the considered species, dN/dS are variable as well, depending on the species used in the comparison. Since available transcriptome data are not equally representative of all the taxa, it is often difficult to find species with homogeneous evolutionary distances, so that the comparisons would be consistent across all the analyzed taxa.
3) For each dN/dS, we do not know whether it is the result of an even accumulation of divergence along the two branches that separate the two species under analysis (in our opinion, an unlikely pattern), or if it is due to just one species evolving much faster than the other. In the latter case, the evolutionary rate is overestimated in one species, and underestimated in the other. In addition, if transcription levels are not strongly correlated between the two species the relationship between dN/dS and transcription level would show different trends, depending on what species is used for the transcription quantification. In such case, what is the correct trend?
In the present work, we tried our best to overcome the problems mentioned in point 1 by comparing data obtained using the same experimental design (number of biological and technical replicates, sex, tissue, sampling season, and reproductive phase), the same protocols performed in the same lab, the same sequencing technology, and the same pipeline of analysis. The fact that we used two species and a single tissue eliminated the other issues raised in point 1, and those in point 2. Point 3 is more difficult to address: despite R. decussatus and R. philippinarum are morphologically and ecologically very similar, during their evolutionary history they experienced very different population dynamics (Arias-Pérez et al. 2016; Cordero et al. 2017), which could have resulted in significantly different evolutionary rates between the two species. Unfortunately, with the available data, a more accurate estimate is not possible. For what concerns the transcription level of orthologous genes, the two species show a strong correlation (Spearman’s rank correlation ρ = 0.71, P value < 2.2E-16, see supplementary fig. 11, Supplementary Material online). In this case, despite the high correlation, the final result is ambiguous (i.e., no correlation in one species, low negative correlation in the other). Even more, in cases where the transcription level of orthologs is less correlated, results might be significantly different, potentially even opposite: this would affect the E–R plots, that could yield different results depending on the transcription data used. Thus, the chance of getting ambiguous results increases with the transcriptional divergence between the analyzed species, which, in turn, increases with phylogenetic distance plus a large number of factors which—as discussed earlier—are difficult to predict or standardize.
More work is needed to establish whether the very weak/absent E–R correlation reported here is an exception—and what biological/evolutionary causes are responsible for it—or it is a more widespread feature. In the latter case, the hypothesis that transcription level is the main responsible for the rate of protein evolution should be revised (Havird and Sloan 2016). The results of this work are not compelling enough to reject the E–R correlation theory, but in our opinion, they show that caution is needed when performing comparative analyses, especially when doing it across a wide range of distantly related species. Consequently, we think that more clear-cut evidence is needed to support the hypothesis by which transcription level drives protein evolution. Alternatively, many other features are thought be involved in protein evolution, such as functional importance, subcellular localization, pleiotropy, protein–protein interaction, network property, and structural constraints (Larracuente et al. 2008; Liao et al. 2010; Ridout et al. 2010; Sojo et al. 2016).
Transcripts Involved in Embryo Development Are Stored in Gametes
Animal eggs contain the substances required for sustaining the first stages of development. Other than vitamins, minerals, fatty acids, and other nutrients provided by the yolk, egg cells contain RNAs and proteins produced by the so-called “maternal genes” during oogenesis. Such products sustain and guide embryo development, especially before the activation of zygotic gene expression (Marlow 2011). Indeed, the first cell divisions after fertilization occur in absence of new transcription, and in this phase the embryonic development relies solely on maternal gene products. Therefore, it is not surprising to find GO terms involved in embryonic organ morphogenesis, and in development and formation of primary germ layer, among female-biased genes (supplementary fig. 8 and table 8, Supplementary Material online). This is consistent with maternal gene products (in this case mRNAs) that would be used during early embryo development being stored in the eggs. Interestingly, GO terms involved in embryo development and organ morphogenesis were detected also among male-biased genes (supplementary fig. 7 and table 7, Supplementary Material online). Until few years ago it was thought that the primary function of sperm was to deliver the paternal DNA to the embryo. Recently, it was discovered in mammals, insects, and plants that sperm carry thousands of RNAs (Dadoune 2009; Hosken and Hodgson 2014). These transcripts persist in spermatozoa, where the machinery for their translation is inactive (Miller et al. 2005), so it is unlikely that the RNAs stored in spermatozoa are necessary for its survival, but it is more plausible that they contribute to embryo development, even if their function is still unknown (Hosken and Hodgson 2014). Accordingly, in the last years, evidence of sperm transcriptional contribution to the offspring development is increasing. Several experiments highlighted the importance of epigenetic inheritance acquired through sperm RNAs (Chen et al. 2016), and the role of paternal miRNAs in the first embryo division (Liu et al. 2012; Yuan et al. 2015). A role of paternal factors (RNAs, proteins) in embryo development and sex determination has been already proposed for R. philippinarum (Ghiselli et al. 2012; Milani et al. 2013; Pozzi et al. 2017), and given these additional results, in the future would be interesting to better investigate the role of sperm-transmitted transcripts in embryo development.
Conclusions
This work provides new information about transcription dynamics and sequence evolution of sex-biased genes in a group of Metazoa for which such data are missing. Compared with other taxa, both the bivalve species analyzed showed a low number of sex-biased genes, probably due to the absence of sexual dimorphism and other sex-specific features (e.g., mating behavior). Surprisingly, we found striking differences in transcription between the two species: the transcriptional bias is maintained in only 33% of the orthologs between the two species, and—contrarily to what reported in multiple studies on other animals—female-biased genes show the highest dN/dS. It is plausible that, as observed by Mank et al. (2010), sex-biased genes show different dN/dS across different developmental stages, and that male-biased genes in these bivalve species might show a higher dN/dS in other phases of the life cycle.
During the development of this work, we had to face several technical challenges typical of comparative analyses performed on nonmodel organisms. This allowed us to think about the difficulties in inferring orthology and about some downsides of the common practices used to investigate the relationship between protein sequence evolution and transcription level. The concerns we raised about such technical approaches do not have straightforward solutions, but we think a significant improvement can be achieved through more careful experimental designs—from both methodological and biological points of view—and a wider sampling across the whole “Forest of Life.” Our growing understanding of the diversity of life clearly advise against the routine of formulating hypotheses and inferring general evolutionary patterns based only on a small number of species. Therefore, since comparative genomics has a fundamental role in almost every field of biology, an improvement in comparative methods will represent one of the main challenges for the next future. Such improvement require a better knowledge of genomes and transcriptomes that, in turn, depends on our ability in annotating genes and inferring phylogenetic relationships across taxa. The problem is evidently circular, and at the moment the focus should be on getting a more uniform representation of the actual biodiversity in genomics data. This is a demanding endeavor, but in the last few years numerous international collaborative projects have been established with the goal of filling the gap of knowledge sequencing an increasing number of species (Voolstra et al. 2017 and references therein).
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Liliana Milani for providing useful comments and suggestions about the manuscript, and the Waitt Foundation. This work was supported by the Italian Ministry of Education, University, and Research (MIUR) FIR Programme no. RBFR13T97A funded to F.G., the Canziani Bequest funded to M.P., and the NIH grant no. RO1GM098741 funded to S.V.N.
Literature Cited
- Abascal F, Zardoya R, Telford MJ.. 2010. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38(Web Server issue):W7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Pérez A, et al. , . 2016. Assessing the geographic scale of genetic population management with microsatellites and introns in the clam Ruditapes decussatus. Ecol Evol. 610:3380–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assis R, Zhou Q, Bachtrog D.. 2012. Sex-biased transcriptome evolution in Drosophila. Genome Biol Evol. 411:1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 3015:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, et al. , . 2011. The evolution of gene expression levels in mammalian organs. Nature 4787369:343–348. [DOI] [PubMed] [Google Scholar]
- Brisson JA, Nuzhdin SV.. 2008. Rarity of males in pea aphids results in mutational decay. Science 3195859:58.. [DOI] [PubMed] [Google Scholar]
- Chen Q, Yan W, Duan E.. 2016. Epigenetic inheritance of acquired traits through sperm RNAs and sperm RNA modifications. Nat Rev Genet 17:733–743.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark NL, Aagaard JE, Swanson WJ.. 2006. Evolution of reproductive proteins from animals and plants. Reproduction 1311:11–22. [DOI] [PubMed] [Google Scholar]
- Cordero D, Delgado M, Liu B, Ruesink J, Saavedra C.. 2017. Population genetics of the Manila clam (Ruditapes philippinarum) introduced in North America and Europe. Sci Rep. 7:39745.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Silva J, Domingues D, Lopes FM.. 2017. RNA-Seq differential expression analysis: an extended review and a software tool. PLoS One 1212:e0190152.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crusoe MR, et al. , . 2015. The khmer software package: enabling efficient nucleotide sequence. F1000Research 4:900.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadoune J-P. 2009. Spermatozoal RNAs: what about their functions? Microsc Res Tech. 728:536–551. [DOI] [PubMed] [Google Scholar]
- Dapper AL, Wade MJ.. 2016. The evolution of sperm competition genes: the effect of mating system on levels of genetic variation within and between species. Evolution 702:502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean R, Mank JE. 2016. Tissue Specificity and Sex-Specific Regulatory Variation Permit the Evolution of Sex-Biased Gene Expression. Am Nat. 188:E74–84. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH.. 2005. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 10240:14338–14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D.. 2000. Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol Biol Evol. 171:68–74. [DOI] [PubMed] [Google Scholar]
- Eads BD, Colbourne JK, Bohuski E, Andrews J.. 2007. Profiling sex-biased gene expression during parthenogenetic reproduction in Daphnia pulex. BMC Genomics 8:464.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 325:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Parsch J.. 2007. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 89:689–698. [DOI] [PubMed] [Google Scholar]
- Falda M, et al. , . 2012. Argot2: a large scale function prediction tool relying on semantic similarity of weighted Gene Ontology terms. BMC Bioinformatics 13(Suppl 4):S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, et al. , . 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44(D1):D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershoni M, Pietrokovski S.. 2014. Reduced selection and accumulation of deleterious mutations in genes exclusively expressed in men. Nat Commun. 5:4438.. [DOI] [PubMed] [Google Scholar]
- Ghiselli F, et al. , . 2012. De Novo assembly of the Manila clam Ruditapes philippinarum transcriptome provides new insights into expression bias, mitochondrial doubly uniparental inheritance and sex determination. Mol Biol Evol. 292:771–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiselli F, et al. , . 2017. The complete mitochondrial genome of the grooved carpet shell, Ruditapes decussatus (Bivalvia, Veneridae). PeerJ 5:e3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good JM, Nachman MW.. 2005. Rates of protein evolution are positively correlated with developmental timing of expression during mouse spermatogenesis. Mol Biol Evol. 224:1044–1052. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, et al. , . 2011. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol. 297:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grath S, Parsch J.. 2012. Rate of amino acid substitution is influenced by the degree and conservation of male-biased transcription over 50 myr of Drosophila evolution. Genome Biol Evol. 43:346–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grath S, Parsch J.. 2016. Sex-biased gene expression. Annu Rev Genet. 50:29–44. [DOI] [PubMed] [Google Scholar]
- Harrison PW, et al. , . 2015. Sexual selection drives evolution and rapid turnover of male gene expression. Proc Natl Acad Sci U S A. 11214:4393–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havird JC, Sloan DB.. 2016. The roles of mutation, selection, and expression in determining relative rates of evolution in mitochondrial versus nuclear genomes. Mol Biol Evol. 3312:3042–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosken DJ, Hodgson DJ.. 2014. Why do sperm carry RNA? Relatedness, conflict, and control. Trends Ecol Evol. 298:451–455. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Mariño-Ramírez L, Wolf YI, Koonin EV.. 2004. Conservation and coevolution in the scale-free human gene coexpression network. Mol Biol Evol. 2111:2058–2070. [DOI] [PubMed] [Google Scholar]
- Khaitovich P, et al. , . 2005. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 3095742:1850–1854. [DOI] [PubMed] [Google Scholar]
- Krasileva KV, et al. , . 2013. Separating homeologs by phasing in the tetraploid wheat transcriptome. Genome Biol. 146:R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen DM, Wolf YI, Mushegian AR, Koonin EV.. 2011. Computational methods for Gene Orthology inference. Brief Bioinformatics 125:379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg S.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 94:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM, et al. , . 2008. Evolution of protein-coding genes in Drosophila. Trends Genet. 243:114–123. [DOI] [PubMed] [Google Scholar]
- Lechner M, et al. , . 2011. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 121:124.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Bettencourt BR, Meiklejohn CD, Hartl DL.. 2005. Evolution of proteins and gene expression levels are coupled in Drosophila and are independently associated with mRNA abundance, protein length, and number of protein-protein interactions. Mol Biol Evol. 225:1345–1354. [DOI] [PubMed] [Google Scholar]
- Li H, et al. , . 2009. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2516:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B-Y, Weng M-P, Zhang J.. 2010. Impact of extracellularity on the evolutionary rate of mammalian proteins. Genome Biol Evol. 2:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B-Y, Zhang J.. 2006. Evolutionary conservation of expression profiles between human and mouse orthologous genes. Mol Biol Evol. 233:530–540. [DOI] [PubMed] [Google Scholar]
- Lipinska A, et al. 2015. Sexual dimorphism and the evolution of sex-biased gene expression in the brown alga ectocarpus. Mol Biol Evol. 32:1581–1597. [DOI] [PubMed] [Google Scholar]
- Liu W-M, et al. , . 2012. Sperm-borne microRNA-34c is required for the first cleavage division in mouse. Proc Natl Acad Sci U S A. 1092:490–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone JH, Hawkins DL Jr, Michalak P.. 2006. Sex-biased gene expression in a ZW sex determination system. J Mol Evol. 634:427–436. [DOI] [PubMed] [Google Scholar]
- Mank JE, Ellegren H.. 2009. Are sex-biased genes more dispensable? Biol Lett. 53:409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE, Hultin-Rosenberg L, Axelsson E, Ellegren H.. 2007. Rapid evolution of female-biased, but not male-biased, genes expressed in the avian brain. Mol Biol Evol. 2412:2698–2706. [DOI] [PubMed] [Google Scholar]
- Mank JE, Hultin-Rosenberg L, Zwahlen M, Ellegren H.. 2008. Pleiotropic constraint hampers the resolution of sexual antagonism in vertebrate gene expression. Am Nat. 1711:35–43. [DOI] [PubMed] [Google Scholar]
- Mank JE, Nam K, Brunström B, Ellegren H.. 2010. Ontogenetic complexity of sexual dimorphism and sex-specific selection. Mol Biol Evol. 277:1570–1578. [DOI] [PubMed] [Google Scholar]
- Marlow FL. 2011. Maternal control of development in vertebrates: my mother made me do it! San Rafael (CA): Morgan and Claypool Life Sciences. [PubMed]
- Meiklejohn CD, Parsch J, Ranz JM, Hartl DL.. 2003. Rapid evolution of male-biased gene expression in Drosophila. Proc Natl Acad Sci U S A. 10017:9894–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP. 2011. Towards a More Nuanced Understanding of the Relationship between Sex-Biased Gene Expression and Rates of Protein-Coding Sequence Evolution. Mol Biol Evol. 28:1893–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani L, Ghiselli F, Nuzhdin SV, Passamonti M.. 2013. Nuclear genes with sex bias in Ruditapes philippinarum (Bivalvia, veneridae): mitochondrial inheritance and sex determination in DUI species. J Exp Zool B Mol Dev Evol. 3207:442–454. [DOI] [PubMed] [Google Scholar]
- Miller D, Ostermeier GC, Krawetz SA.. 2005. The controversy, potential and roles of spermatozoal RNA. Trends Mol Med. 114:156–163. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B.. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 57:621–628. [DOI] [PubMed] [Google Scholar]
- Nishimura O, Hara Y, Kuraku S.. 2017. gVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics. 33:3635–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzhdin SV, Wayne ML, Harmon KL, McIntyre LM.. 2004. Common pattern of evolution of gene expression level and protein sequence in Drosophila. Mol Biol Evol. 217:1308–1317. [DOI] [PubMed] [Google Scholar]
- Papa F, et al. , . 2017. Rapid evolution of female-biased genes among four species of Anopheles malaria mosquitoes. Genome Res. 27:1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, et al. , . 2003. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 2995607:697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsch J, Ellegren H.. 2013. The evolutionary causes and consequences of sex-biased gene expression. Nat Rev Genet. 142:83–87. [DOI] [PubMed] [Google Scholar]
- Poley JD, Sutherland BJG, Jones SRM, Koop BF, Fast MD.. 2016. Sex-biased gene expression and sequence conservation in Atlantic and Pacific salmon lice (Lepeophtheirus salmonis). BMC Genomics 17: 483. [DOI] [PMC free article] [PubMed]
- Pozzi A, Plazzi F, Milani L, Ghiselli F, Passamonti M. 2017. SmithRNAs: could mitochondria “bend” nuclear regulation? Mol Biol Evol. 34:1960–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pröschel M, Zhang Z, Parsch J.. 2006. Widespread adaptive evolution of Drosophila genes with sex-biased expression. Genetics 1742:893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL.. 2003. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 3005626:1742–1745. [DOI] [PubMed] [Google Scholar]
- Reinke V, Gil IS, Ward S, Kazmer K.. 2004. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development 1312:311–323. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 166:276–277. [DOI] [PubMed] [Google Scholar]
- Rice WR. 2013. Nothing in genetics makes sense except in light of genomic conflict. Annu Rev Ecol Evol Syst. 441:217–237. [Google Scholar]
- Rice WR, Chippindale AK.. 2008. Intersexual ontogenetic conflict. J Evol Biol. 145:685–693. [Google Scholar]
- Ridout KE, Dixon CJ, Filatov DA.. 2010. Positive selection differs between protein secondary structure elements in Drosophila. Genome Biol Evol. 2:166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IG, Ruvinsky I, Gilad Y.. 2012. Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet. 137:505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor MA, et al. , . 2006. A new method to remove hybridization bias for interspecies comparison of global gene expression profiles uncovers an association between mRNA sequence divergence and differential gene expression in Xenopus. Nucleic Acids Res. 341:185–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader L, Helanterä H, Oettler J.. 2017. Accelerated evolution of developmentally biased genes in the tetraphenic ant cardiocondyla obscurior. Mol Biol Evol. 343:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 3119:3210–3212. [DOI] [PubMed] [Google Scholar]
- Small CM, Carney GE, Mo Q, Vannucci M, Jones AG.. 2009. A microarray analysis of sex- and gonad-biased gene expression in the zebrafish: evidence for masculinization of the transcriptome. BMC Genomics 10:579.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sojo V, Dessimoz C, Pomiankowski A, Lane N.. 2016. Membrane proteins are dramatically less conserved than water-soluble proteins across the tree of life. Mol Biol Evol. 3311:2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AD, Pischedda A, Rice WR.. 2010. Resolving intralocus sexual conflict: genetic mechanisms and time frame. J Hered. 101(Suppl 1):S94–S99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T.. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 67:e21800.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson WJ, Vacquier VD.. 2002. The rapid evolution of reproductive proteins. Nat Rev Genet. 32:137–144. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Wong A, Wolfner MF, Aquadro CF.. 2004. Evolutionary expressed sequence tag analysis of Drosophila female reproductive tracts identifies genes subjected to positive selection. Genetics 1683:1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazona S, Furio-Tari P, Turra D, Pietro AD, Nueda MJ, Ferrer A, Conesa A.. 2015. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 4321:e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Barkai N.. 2008. Evolution of gene sequence and gene expression are not correlated in yeast. Trends Genet. 243:109–113. [DOI] [PubMed] [Google Scholar]
- Torgerson DG, Kulathinal RJ, Singh RS.. 2002. Mammalian sperm proteins are rapidly evolving: evidence of positive selection in functionally diverse genes. Mol Biol Evol. 1911:1973–1980. [DOI] [PubMed] [Google Scholar]
- Trachana K, et al. , . 2011. Orthology prediction methods: a quality assessment using curated protein families. Bioessays 3310:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner LM, Chuong EB, Hoekstra HE.. 2008. Comparative analysis of testis protein evolution in rodents. Genetics 1794:2075–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner LM, Hoekstra HE.. 2008. Causes and consequences of the evolution of reproductive proteins. Int J Dev Biol. 52(5-6):769–780. [DOI] [PubMed] [Google Scholar]
- Voolstra CR GIGA Community of Scientists (COS) Wörheide G, Lopez JV.. 2017. Advancing genomics through the Global Invertebrate Genomics Alliance (GIGA). Invertebr Syst. 311:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zhang Y, Zhang Z, Zhu J, Yu J.. 2010. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 81:77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Werren JH, Clark AG.. 2015. Genetic and epigenetic architecture of sex-biased expression in the jewel wasps Nasonia vitripennis and giraulti. Proc Natl Acad Sci U S A. 11227:E3545–E3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle CA, Johannesson H. 2013. Evolutionary dynamics of sex-biased genes in a hermaphrodite fungus. Mol. Biol. Evol. 30:2435–2446. [DOI] [PubMed] [Google Scholar]
- Yang L, Zhang Z, He S.. 2016. Both male-biased and female-biased genes evolve faster in fish genomes. Genome Biol Evol. 811:3433–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, et al. , . 2006. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 168:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, et al. , . 2015. mir-34b/c and mir-449a/b/c are required for spermatogenesis, but not for the first cleavage division in mice. Biol Open 42:212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang J-R.. 2015. Determinants of the rate of protein sequence evolution. Nat Rev Genet. 167:409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B.. 2007. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature 4507167:233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Hambuch TM, Parsch J.. 2004. Molecular evolution of sex-biased genes in Drosophila. Mol Biol Evol. 2111:2130–2139. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Parsch J.. 2005. Positive correlation between evolutionary rate and recombination rate in Drosophila genes with male-biased expression. Mol Biol Evol. 22:1945–1947. [DOI] [PubMed] [Google Scholar]
- Zhou L, Ma X, Sun F.. 2008. The effects of protein interactions, gene essentiality and regulatory regions on expression variation. BMC Syst Biol. 2:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouros E. 2013. Biparental inheritance through uniparental transmission: the doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol Biol. 401:1–31. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.