

Abstract
Background & Aims
Binge alcohol causes gut leakiness, contributing to increased endotoxemia and inflammatory liver injury, although the molecular mechanisms are still elusive. This study was aimed at investigating the roles of apoptosis of enterocytes and nitration followed by degradation of intestinal tight junction (TJ) and adherent junction (AJ) proteins in binge alcohol-induced gut leakiness.
Methods
The levels of intestinal (ileum) junctional complex proteins, oxidative stress markers and apoptosis-related proteins in rodents, T84 colonic cells and autopsied human ileums were determined by immunoblot, immunoprecipitation, immunofluorescence, and mass-spectral analyses.
Results
Binge alcohol exposure caused apoptosis of gut enterocytes with elevated serum endotoxin and liver injury. The levels of intestinal CYP2E1, iNOS, nitrated proteins and apoptosis-related marker proteins were significantly elevated in binge alcohol-exposed rodents. Differential, quantitative mass-spectral analyses of the TJ-enriched fractions of intestinal epithelial layers revealed that several TJ, AJ and desmosome proteins were decreased in binge alcohol-exposed rats compared to controls. Consistently, the levels of TJ proteins (claudin-1, claudin-4, occludin and ZO-1), AJ proteins (β-catenin and E-cadherin) and desmosome plakoglobin were very low in binge-alcohol exposed rats, wild-type mice, and autopsied human ileums but not in Cyp2e1-null mice. Additionally, pretreatment with specific inhibitors of CYP2E1 and iNOS prevented disorganization and/or degradation of TJ proteins in alcohol-exposed T84 colonic cells. Furthermore, immunoprecipitation followed by immunoblot confirmed that intestinal TJ and AJ proteins were nitrated and degraded via ubiquitin-dependent proteolysis, resulting in their decreased levels.
Conclusions
These results demonstrated for the first time the critical roles of CYP2E1, apoptosis of enterocytes and nitration followed by ubiquitin-dependent proteolytic degradation of the junctional complex proteins in promoting binge alcohol-induced gut leakiness and endotoxemia, contributing to inflammatory liver disease.
Keywords: Cytochrome P450-2E1 (CYP2E1), ethanol, gut leakiness, nitroxidative stress, apoptosis, protein nitration, ubiquitin-dependent proteolysis, tight and adherent junction proteins
Graphical abstract
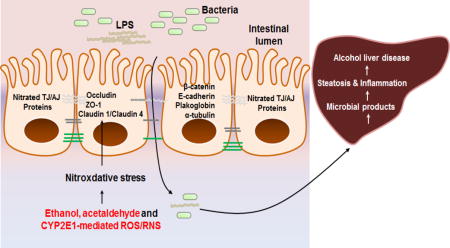
Introduction
Excessive amounts of alcohol intake can damage many organs or deaths in severe cases. Heavy alcohol intake is also known to cause gut leakiness, contributing to increased endotoxemia and inflammatory tissue damage in the liver and brain [1-4]. Various pathological conditions, such as HIV infection [5-7], obesity [8], and burn injury [9], are known to increase gut leakiness and endotoxemia. In addition, binge alcohol [10] and nonalcoholic substances such as high fat diets [11] and fructose [12] can stimulate gut leakiness, leading to elevated serum endotoxin and liver inflammation. Furthermore, alcoholic patients with cirrhosis had higher levels of endotoxin than those without cirrhosis [4, 13]. These conditions observed in humans can be replicated in experimental animals or cultured Caco-2 and T84 colonic cells. In fact, other laboratories and we have reported that binge alcohol caused gut leakiness in an ethanol-inducible cytochrome P450-2E1 (CYP2E1)-dependent manner [1, 2], since Cyp2e1-null mice were resistant to alcohol-induced gut leakiness despite extremely high doses of alcohol (3 oral doses of 6 g ethanol/kg/dose) at 12-h intervals [10]. Furthermore, pretreatment with a specific siRNA to CYP2E1 in Caco-2 human colonic cells [2] or a chemical inhibitor of CYP2E1 chlormethiazole (CMZ) or an antioxidant N-acetylcysteine [10] efficiently prevented alcohol-induced epithelial barrier dysfunction or gut leakiness, supporting the contributing role of CYP2E1-dependent oxidative stress in gut leakiness. Despites numerous reports, the detailed molecular mechanisms by which binge alcohol and CYP2E1-mediated oxidative stress stimulate gut leakiness are still poorly understood.
We have noticed that Cyp2e1-null mice and WT mice on Svj129 background do not drink the ethanol liquid diet as similar to the C57BL/6 mice. Thus, we had to use an acute alcohol exposure model by using oral gavages. To clearly determine the preventive role of CYP2E1 deletion (or suppression) in binge alcohol-induced gut leakiness and tissue injury, we chose to use a rather harsh condition of alcohol exposure (i.e., 3 consecutive oral gavages of 6 g/kg/dose at 12-h intervals) for mice, although this acute model is not likely to reflect the patterns of alcohol drinking in humans. Binge alcohol, a pattern of acute alcohol drinking leading to blood alcohol concentration (BAC) ≥0.08% (w/v) within 2 h, can increase and/or activates CYP2E1 and inducible nitric oxide synthase (iNOS) [10] while it can suppress mitochondrial electron transport chain [14], leading to elevated production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), resulting in increased oxidative/nitrative (nitroxidative) stress with production of potently toxic peroxynitrite, which can nitrate Tyr residues and/or S-nitrosylate Cys residues [15]. Based on elevated nitroxidative stress, we hypothesized that many proteins, especially in intestinal epithelial junctional complexes [16], including tight junction (TJ) and adherent junction (AJ) proteins, are altered by post-translational modifications (PTMs) (e.g., oxidation, nitration, phosphorylation, adduct formation, etc) followed by their proteasomal degradation and/or suppression, contributing to increased gut leakiness and serum endotoxin levels. In fact, Keshavarzian and colleagues reported that the decreased levels of TJ and associated proteins, such as microtubule assembly proteins [17, 18], played a causal role in alcohol-induced gut leakiness. Based on these reports [16-18], it is possible that decreased levels of AJ, desmosome and other junctional complex proteins could contribute to increased gut leakiness. We also hypothesized that increased apoptosis of enterocytes can stimulate gut permeability, resulting in elevated levels of gut bacteria in the circulating blood and other organs, including the liver, following binge alcohol exposure. Therefore, we aimed at investigating the roles of covalent protein modifications, especially nitration, of TJ and AJ proteins and apoptosis of enterocytes in binge alcohol-induced gut leakiness in experimental models of rats, mice, and cultured colonic cells, since nitrated proteins are known to be degraded by ubiquitin-dependent proteolysis [19-21]. Furthermore, we performed mass-spectral analyses of the TJ-enriched proteins from the intestinal epithelial layers to determine protein identities and demonstrate the loss of TJ, AJ and associated proteins in binge alcohol-exposed rats compared to controls. We also verified our rodent data with the ileums from eight autopsied individuals who suddenly died from heavy alcohol intoxication versus six people who perished from nonalcoholic causes (Table 1).
Table 1.
Clinical Data of Autopsied Human Subjects.
| Sample No. | Gender | Age at death | BFAC* (%) | Group | Autopsy diagnosis |
|---|---|---|---|---|---|
| 1 | Male | 53 | 0 | Control | – |
| 2 | Male | 45 | 0 | Control | Acute cardiac death |
| 3 | Male | 41 | 0 | Control | Acute cardiac death |
| 4 | Male | 59 | 0 | Control | Cerebral hemorrhage |
| 5 | Male | 62 | 0 | Control | Cerebral hemorrhage |
| 6 | Male | 67 | 0 | Control | Acute cardiac death |
| 7 | Male | 37 | 0.163 | Binge | – |
| 8 | Male | 57 | 0.22 | Binge | Stroke |
| 9 | Male | 25 | 0.231 | Binge | – |
| 10 | Male | 36 | 0.017 | Binge | Acute cardiac death |
| 11 | Male | 57 | – | Chronic Alcoholism | Chronic Alcoholism |
| 12 | Male | 30 | 0.134 | Binge | Acute cardiac death |
| 13 | Male | 59 | 0.121 | Chronic Alcoholism | Chronic Alcoholism |
| 14 | Male | 46 | 0.154 | Binge | Acute cardiac death |
BFAC, body-fluid alcohol concentration
Materials and Methods, including statistical analyses not described here are described in detail in the Supplementary data file.
Results
Increased gut leakiness and nitroxidative stress in alcohol-exposed rats
Similar to our previous study with mice [10], we used the same dosage of binge alcohol (3 oral doses of 6 g/kg/dose at 12-h intervals) in rats. Histologically, epithelial detachment and abnormal shapes of intestines (ileums) were frequently observed in alcohol-exposed rats compared to dextrose-controls (Fig. 1A). BAC at 1 h after the last oral ethanol administration was determined to be approximately 0.2% (w/v) (Fig. 1B). Plasma endotoxin levels (Fig. 1C) and hepatic levels of E. coli mRNA transcripts determined by real-time RT-PCR (Fig. 1D) were significantly increased in binge alcohol-exposed rats compared to controls. Plasma ROS and intestinal oxidized protein levels were also significantly elevated in binge alcohol-exposed rats (Fig. 1E, F, respectively). Additionally, immunoblot showed increased the levels of intestinal nitroxidative marker proteins such as CYP2E1, iNOS and nitrated proteins detected with anti-3-NT antibody, in binge-alcohol exposed rats compared to controls (Fig. 1G).
Fig. 1. Binge alcohol induced the elevation of gut damage, endotoxin, and various nitroxidative stress markers in small intestines of rats.
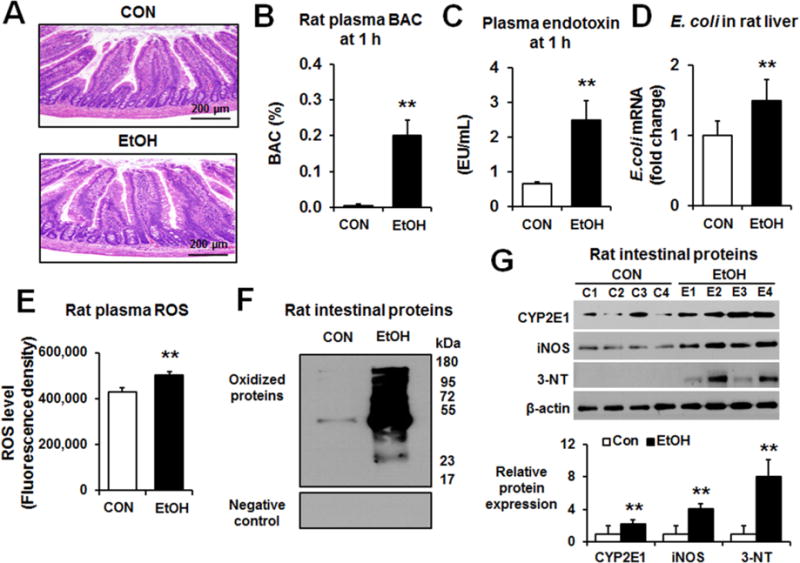
(A) Representative H&E staining of formalin-fixed rat intestines for the indicated groups. The levels of (B) plasma BAC (C) plasma endotoxin, (D) E. coli mRNA transcripts in the indicated liver extracts determined by real-time RT-PCR, (E) plasma ROS, (F) oxidized gut proteins for the different groups determined by using Oxyblot analysis (upper panel) or negative control (lower panel), and (G) CYP2E1, iNOS, or nitrated proteins in the intestines of control or alcohol-exposed rats are presented. Each lane indicates the mixture proteins combined from 2 different rats/group. Densitometric quantitation of the immunoblots for each indicated protein relative to β-actin, used as a loading control, is shown. Data represent means ± SD where statistical significance was determined using two-tailed t-test: **P<0.01.
Increased fatty liver, hepatic CYP2E1, hepatocyte apoptosis, and hepatic inflammation following binge alcohol exposure in rats
Increased intestinal permeability promotes translocation of bacterial products like lipopolysaccharide (LPS) to reach the liver, interacts with hepatic toll-like receptors (TLRs) and stimulate inflammatory steatohepatitis [8, 22, 23]. Histological analysis showed multiple lipid droplets in hepatocytes after binge alcohol-exposure (Supplementary Fig. 1A). Plasma ALT and hepatic total TG levels were elevated in binge alcohol-exposed rats compared to controls (Supplementary Fig. 1B and C).
The hepatic levels of nitroxidative stress markers CYP2E1, iNOS, nitrated proteins, and TLR-4 were significantly increased by binge alcohol exposure (Supplementary Fig. 1D). In addition, hepatic apoptosis-related proteins, phospho-c-Jun-N-terminal protein kinase (p-JNK), p-p38K, cleaved caspase-3, and cleaved caspase-9, were markedly elevated in binge alcohol-exposed rats compared to controls (Supplementary Fig. 1E). These results suggest that increased gut permeability and endotoxin likely promoted fat accumulation and hepatocyte apoptosis in alcohol-exposed rats.
Moreover, we observed significantly elevated amounts of hepatic cytokine (TNF-α) and chemokine (MCP-1) in alcohol-exposed rats compared to those of controls (Supplementary Fig. 2A-E). The hepatic amounts of inflammation related proteins and mRNAs were significantly elevated in binge alcohol-exposed rats (Supplementary Fig. 2D and E). These results showed that binge alcohol stimulated inflammatory hepatic injury.
Mass-spectral analyses and decreased gut junctional proteins and increased apoptosis after alcohol exposure in rats
Mass spectrometry analyses of the purified, TJ-enriched fractions from epithelial layers of control or alcohol-exposed rats identified numerous proteins, similar to the previous report [24]. However, unlike the previous results obtained with T84 cells where only two TJ proteins (i.e., claudin-16 and ZO-2) were identified [24], our quantitative, differential mass-spectral analyses not only identified the additional TJ proteins and other epithelial junctional proteins [16], but also found down-regulation of many TJ and AJ proteins, desmosome proteins, and α/β-tubulins in alcohol-exposed rats compared to controls (Fig. 2A).
Fig. 2. Binge alcohol decreased intestinal TJ and AP proteins with increased intestinal apoptosis and inflammatory marker proteins in rats.
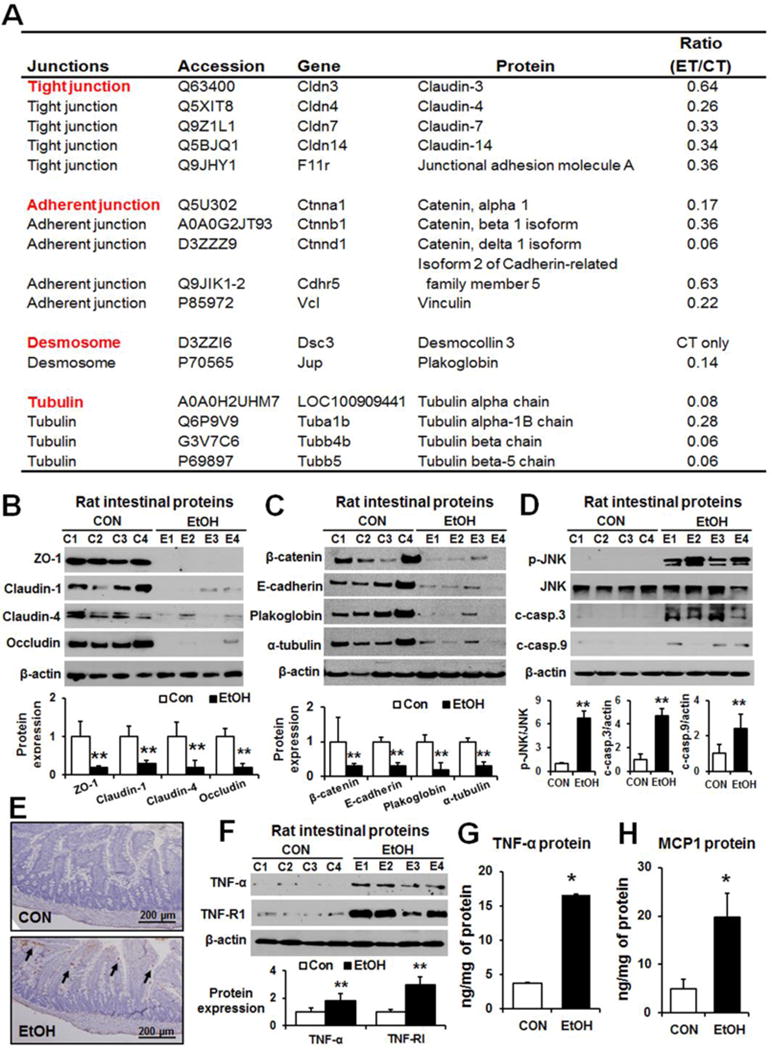
(A) Intestinal junction proteins changed upon ethanol exposure. The ratio of ET/CT represents the relative quantity of each intestinal protein detected in ethanol-exposed rats versus control rats. Representative levels of (B) gut TJ proteins ZO-1, claudin-1, claudin-4, and occludin, (C) AJ proteins (β-catenin and E-cadherin), plakoglobin and α-tubulin, and (D) p-JNK, JNK, cleaved caspase-3 and -9 in the indicated groups are shown. Each lane indicates the mixture proteins combined from 2 different rats. Densitometric quantitation of the immunoblots for each protein relative to β-actin is shown. (E) Representative TUNEL staining for apoptotic enterocytes (arrows). (F-H) The expressed amounts of TNF-α (immunoblot), TNFR-I (immunoblot), TNF-α (ELISA) and MCP-1 (ELISA) in the small intestine lysates from indicated rat groups are presented. Data show means ± SD where statistical significance was determined using two-tailed t-test: *P<0.05, **P<0.01.
Consistently, immunoblot analyses confirmed the decreased levels of some junctional complex proteins that were identified from the differential mass-spectral analyses. Immunoblot analyses showed that binge alcohol exposure markedly decreased the levels of TJ proteins ZO-1, claudin-1, claudin-4, and occludin (Fig. 2B). In addition, the levels of gut AJ proteins (e.g., β-catenin and E-cadherin), desmosome plakoglobin and junction associated proteins such as α-tubulin [16] were markedly reduced in alcohol-exposed rats compared to controls (Fig. 2C).
We next studied the role of apoptosis of enterocytes in alcohol-mediated gut leakiness. Immunoblots showed markedly elevated levels of apoptosis-related marker proteins [15] activated p-JNK, cleaved-caspase-3 and cleaved-caspase-9 in alcohol-exposed rats (Fig. 2D). Histological TUNEL analysis clearly showed markedly elevated apoptosis of enterocytes in small intestine of alcohol-exposed rats compared to those of controls (Fig. 2E and Supplementary Fig. 3). Additionally, the gut levels of proinflammatory cytokine TNF-α, known to disrupt intestinal TJ promoting intestinal barrier dysfunction [25, 26], and its receptor protein TNFR-I were significantly increased in binge alcohol-exposed rats compared to controls (Fig. 2F). Furthermore, ELISA data showed that binge alcohol significantly up-regulated the intestinal levels of cytokine TNF-α (Fig. 2G) and chemokine MCP-1 (Fig. 2H). These results demonstrate elevated gut leakiness through decreased levels of intestinal TJ, AJ and other junctional proteins along with increased apoptosis of enterocytes in alcohol-exposed rats.
Role of CYP2E1 and oxidative stress in decreased gut junctional proteins and increased apoptosis in mice
We further evaluated whether the results with rats could be also observed in alcohol-exposed mice. Consistently, the levels of the markers of gut leakiness, such as plasma endotoxin, fecal albumin, and the E. coli mRNA transcripts present in the liver, were significantly elevated in binge alcohol-exposed WT, but not in the corresponding Cyp2e1-null mice (Fig. 3A, B and C), as indicated. Additionally, the levels of oxidized gut proteins were markedly elevated in binge-alcohol exposed WT compared to the untreated controls or the alcohol-exposed Cyp2e1-null mice (Fig. 3D).
Fig. 3. Binge alcohol exposure increased endotoxemia, oxidized proteins and apoptosis marker proteins with decreased TJ and AJ proteins in WT compared to Cyp2e1-null mice.
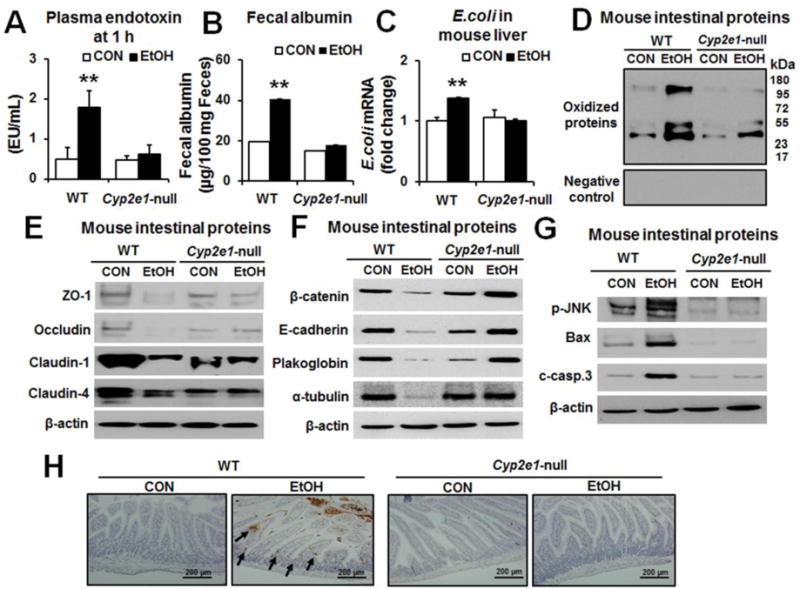
The levels of (A) plasma endotoxin, (B) fecal albumin, (C) E. coli mRNA transcripts in the liver extracts assessed by real-time RT-PCR, (D) oxidized intestinal proteins (upper panel) or negative control (lower panel), (E) gut TJ proteins, (F) AJ and associated proteins, (G) gut p-JNK, cleaved caspase-3, Bax and a loading control β-actin for the indicated mouse groups (n≥8/group) are presented. Each lane indicates the mixture proteins combined from 4 different mice randomly selected within the same group. (H) Representative TUNEL staining for apoptotic enterocytes (arrows) in the indicated mouse groups. Data represent means ± SD where statistical significance was determined using two-tailed t-test: **P<0.01
Decreased TJ, AJ and desmosome protein contents can contribute to the intestinal barrier dysfunction and leakage [17, 18, 27]. Immunoblots revealed that the amounts of intestinal TJ proteins ZO-1, occludin, claudin-1 and claudin-4 were markedly decreased in alcohol-exposed WT, while their levels were unchanged or changed little, if any, in the corresponding Cyp2e1-null mice (Fig. 3E). Similarly, the levels of gut claudin-2, -3, and -7 isoforms were decreased in alcohol-exposed WT but not in the Cyp2e1-null mice (Supplementary Fig. 4A). Significantly decreased claudin isoforms were also observed in the ileums of autopsied people died from heavy alcohol-intoxication compared to controls (Supplementary Fig. 4B). The levels of intestinal AJ proteins and desmosome plakoglobin were also markedly decreased in the alcohol-exposed WT compared to the corresponding Cyp2e1-null mice (Fig. 3F). Furthermore, the levels of apoptosis-related proteins p-JNK, pro-apoptotic Bax and cleaved (activated) caspase-3 were elevated in the alcohol-exposed WT with little changes in the corresponding Cyp2e1-null mice (Fig. 3G). Histological TUNEL analysis showed markedly elevated apoptosis of enterocytes in small intestine of the alcohol-exposed WT, but not in the corresponding Cyp2e1-null mice (Fig. 3H). These results indicate the similar mechanisms of alcohol-induced gut leakiness exist between rats and mice.
Role of nitration for ubiquitin-dependent proteolysis in decreased levels of intestinal TJ and AJ proteins
MLCK [25] and JNK2 [28] were reported to phosphorylate TJ proteins, leading to their decreased levels with increased intestinal permeability. Thus, the levels of MLCK targets (containing p-Ser/p-Thr residues) and p-JNK target proteins (containing p-Ser-Pro and p-Thr-Pro residues) were determined. Immunoblot results showed that the levels of intestinal phospho-MLCK were increased in alcohol-exposed WT, but not in the corresponding Cyp2e1-null mice (Supplementary Fig. 5A) and in the ileums of alcohol-intoxicated people compared to controls (Supplementary Fig. 5B). However, alcohol exposure did not change the gut levels of the various phosphorylation target proteins in mice (Supplementary Fig. 5C) and in the ileums of alcohol-intoxicated people compared to controls who died from nonalcoholic causes (Supplementary Fig. 5D), although the amounts of ileum proteins containing phospho-Thr-Pro residues were elevated in people who died from alcohol-intoxication.
Mass-spectral analysis did not recognize a specific amino acid for nitration or phosphorylation in each TJ, AJ protein, or desmosome, although their amounts were markedly decreased in binge alcohol-exposed rats compared to those in controls (Fig. 2A). Since the effect of protein nitration is unknown, we further studied the role of nitration of TJ, AJ and desmosome plakoglobin in decreasing their levels with increased gut leakiness following binge alcohol exposure. Immunoprecipitation (IP) followed by immunoblot analysis revealed that nitration of claudin-1 and claudin-4 was significantly increased in alcohol-exposed WT compared to dextrose-control mice (Fig. 4A, B). Additionally, increased amounts of ubiquitin-conjugated proteins were detected in immunoprecipitated claudin-1 or claudin-4 in binge alcohol-exposed WT mice. In contrast, little changes in the amounts of nitrated and ubiquitinated proteins were observed in alcohol-exposed Cyp2e1-null mice (Fig. 4A, B), suggesting ubiquitin-dependent degradation of the nitrated TJ proteins, as reported [19-21]. Consequently, the intestinal levels of claudin-1 and claudin-4 were decreased in alcohol-exposed WT but not in the corresponding Cyp2e1-null mice. Furthermore, nitration and ubiquitin-conjugation of AJ protein β-catenin, desmosome plakoglobin and junctional complex associated proteins, including α-tubulin [16] were increased in binge alcohol-exposed WT (Fig. 4C-E), resulting in decreased levels of these AJ and α-tubulin. However, these alterations were not observed in the alcohol-exposed Cyp2e1-null mice, demonstrating a critical role of CYP2E1 in binge alcohol-mediated gut leakiness through nitration and ubiquitin-dependent degradation of junctional proteins. Additional results of similar patterns of nitration and ubiquitin-conjugation and degradation of the junctional complex proteins with longer film exposures are also shown to support our conclusion (Supplementary Fig. 6).
Fig. 4. Binge alcohol increased nitration and ubiquitin-conjugation, leading to decreased levels of intestinal TJ and AJ proteins in WT compared to Cyp2e1-null mice.
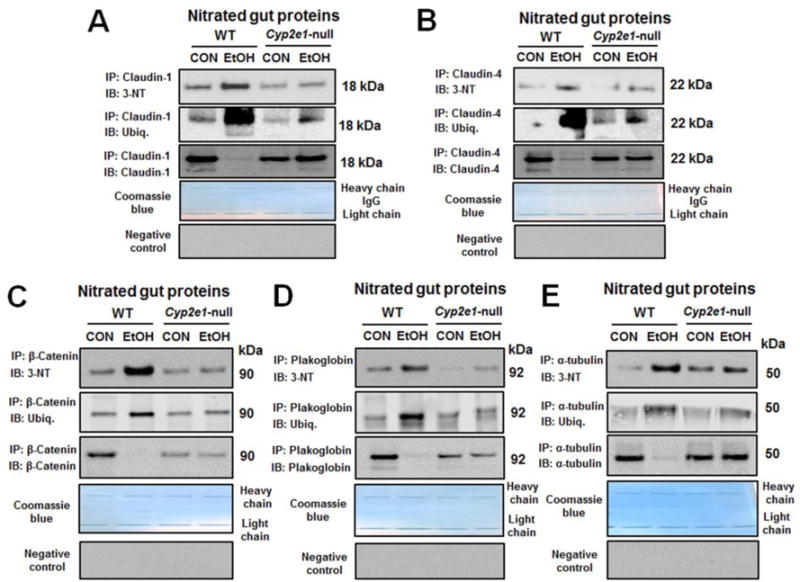
(A-E) The same amounts of intestinal proteins from the control or alcohol-exposed WT or Cyp2e1-null mice were immunoprecipitated with the specific antibody to each indicated target protein and then subjected to immunoblot analysis with the specific antibody to 3-NT (top panel), ubiquitin (middle), or each TJ or AJ protein of interest (bottom). The remaining gel with IgG heavy and light chain proteins was stained with Coomassie blue to show similar amounts of the specific antibody for each target protein used for immunoprecipitation. Normal mouse or rabbit IgG was also used as a negative control for immunoprecipitation (bottom panels).
Increased hepatic inflammation
The increased disruption of intestinal barrier function results in leaky gut and gut bacteria can easily migrate into the blood and the liver. Consequently, the bacterial translocation is likely to trigger liver inflammation and homeostasis. Histological analysis showed multiple lipid droplets in the liver after binge alcohol-exposure (Fig. 5A). Plasma ALT and hepatic total TG levels were significantly elevated in binge alcohol-exposed WT mice compared to controls (Fig. 5B and C, respectively). The amounts of hepatic cytokine (TNF-α) and chemokine (MCP-1) proteins were significantly increased in alcohol-exposed WT but not in the corresponding Cyp2e1-null mice (Fig. 5D and E, respectively). Furthermore, alcohol exposure increased the protein amounts of hepatic TLR4, TNF-α, NLRP3 and cleaved caspase-1 in WT, but not in Cyp2e1-null mice (Fig. 5F). Furthermore, significantly increased mRNA levels of hepatic NLRP3, caspase-1, IL-1β and TNF-α were observed in alcohol-exposed WT, but not in Cyp2e1-null mice (Fig. 5G). These results indicate that binge alcohol could have promoted inflammatory liver injury at least partly through elevated gut leakiness.
Fig. 5. Binge alcohol significantly increased the levels of inflammatory marker proteins in WT compared to Cyp2e1-null mice.
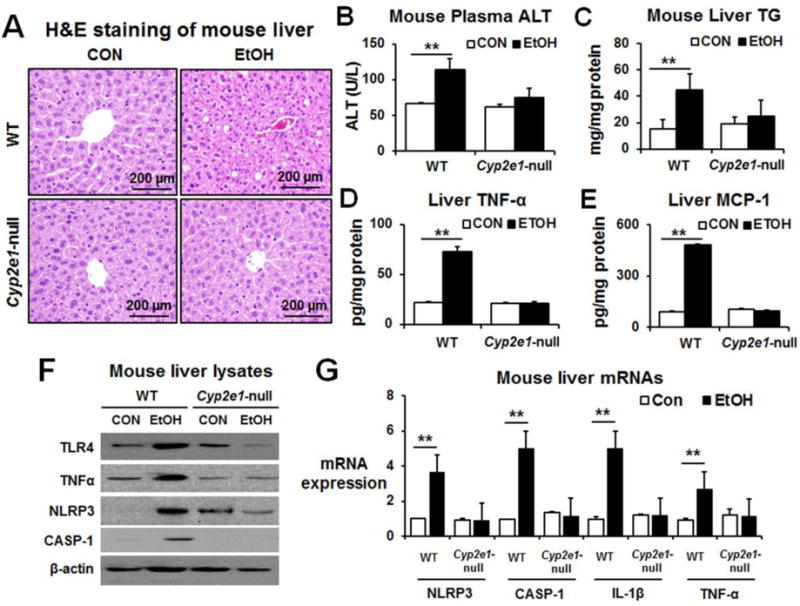
(A) Representative H&E staining of formalin-fixed mouse liver for the indicated groups (n≥8/group). The levels of (B) plasma ALT, (C) hepatic triglyceride, (D) hepatic TNF-α protein, and (E) hepatic MCP-1 protein for the indicated groups. (F) Relative levels of inflammatory marker protein TLR-4, TNF-α, NLRP3, or cleaved-caspase-1 in the control or ethanol-exposed WT or Cyp2e1-null mice are shown. Each lane indicates the mixture proteins combined from 4 different mice randomly selected within the same group. (G) Relative amounts of the mRNA transcripts of hepatic inflammatory markers in the indicated mouse groups, determined by real time RT-PCR analyses, are presented (n≥4/sample). Data show means ± SD where statistical significance was determined using two-tailed t-test: **P < 0.01.
Disruption of TJ proteins with elevated apoptosis signals in alcohol-exposed T84 colon cells
To confirm the rodent results and conduct a mechanistic study on alcohol-mediated intestinal barrier dysfunction, T84 colonic cells were treated with 40 mM ethanol and protein lysates evaluated by immunoblot or immunofluorescence analyses. Immunoblot results exhibited significantly decreased levels of TJ proteins, ZO-1, claudin-1, claudin-4 and occludin in alcohol-exposed T84 cells compared to controls (Supplementary Fig. 7A). Confocal immunofluorescence microscopy also showed that alcohol exposure disrupted the normal distribution of ZO-1 (Fig. 6A) and claudin-1 (Fig. 6B). Consistently, binge alcohol significantly reduced trans-epithelial electrical resistance (TEER) and increased permeation of FITC-labeled 4-kDa dextran (FITC-D4) (Fig. 6C and D, respectively). However, co-treatment with the CYP2E1 inhibitor CMZ or a specific iNOS inhibitor 1400W efficiently prevented disruption of tight junction proteins and epithelial cell permeability in alcohol-exposed T84 cells (Fig. 6).
Fig. 6. Confocal microscopy showed that alcohol disrupted the TJ proteins in T84 colonic cells.
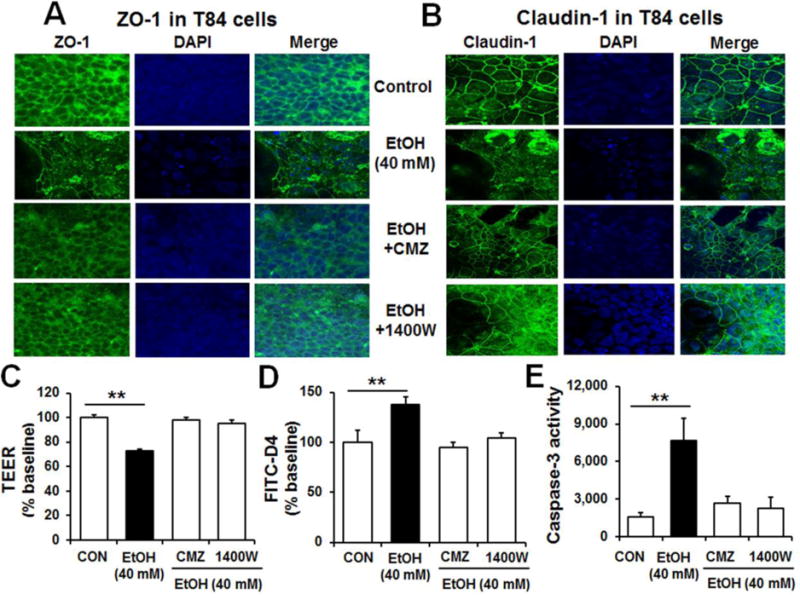
(A and B) Confocal image showing the disruption or disorganization of ZO-1 and claudin-1 in T84 cells treated with 40 mM ethanol with or without CMZ or 1400W. Cell nuclei were counterstained with DAPI. (C-D) Representative levels of TEER and permeability to FITC-D4 after 3 h pretreatment with the specific inhibitor of CYP2E1 or iNOS. Data indicate means ± SD of triplicate wells from two separate experiments. **P < 0.01. (E) Caspase-3 activity in T84 cells treated with ethanol in the absence or presence of CMZ or 1400W pretreatment (n=6/sample). **P < 0.01
Furthermore, alcohol exposure damaged T84 cells with increased apoptosis-related marker proteins p-JNK, pro-apoptotic Bax and cleaved caspase-3 (Supplementary Fig. 7B). Consistently, co-treatment with the CYP2E1 inhibitor CMZ or the iNOS inhibitor 1400W efficiently prevented the elevation of caspases 3 activity in alcohol -exposed T84 cells (Fig. 6E). Furthermore, co-treatment with the proteasome inhibitor MG-132, a JNK2 inhibitor SU3327, or an MLCK inhibitor peptide-18 (P-18) also prevented disruption of ZO-1 and epithelial cell permeability in alcohol-exposed T84 cells (Supplementary Fig. 8). These results demonstrate the important roles of disruption of TJ proteins and elevated apoptosis of T84 cells in alcohol-induced epithelial cell permeability change.
Decreased intestinal TJ and AJ proteins in autopsied people who died suddenly from heavy alcohol-intoxication
Increased levels of nitroxidative stress, as reflected by elevated levels of oxidized proteins (Fig. 7A), CYP2E1, iNOS and nitrated proteins (Fig. 7B), were observed in ileums of alcohol-intoxicated individuals compared to people who died from nonalcoholic causes. The gut levels of TJ (Fig. 7C), AJ proteins and plakoglobin (Fig. 7D) were markedly decreased in the alcohol-intoxicated people compared to the controls. Furthermore, apoptosis-related marker proteins p-JNK, pro-apoptotic Bax and cleaved caspase-3 were significantly elevated in the ileums from alcohol-intoxicated individuals (Fig. 7E). These results also indicate that heavy alcohol intoxication increased the levels of CYP2E1, nitroxidative stress and apoptosis of intestinal enterocytes with decreased amounts of TJ, AJ proteins and desmosome plakoglobin in humans, consistent with the results and elevated gut leakiness observed in the experimental models, as summarized (Fig. 7F).
Fig. 7. Increased nitroxidative stress marker proteins with decreased TJ and AJ proteins in autopsied intestines from people suddenly died from heavy alcohol intoxication.
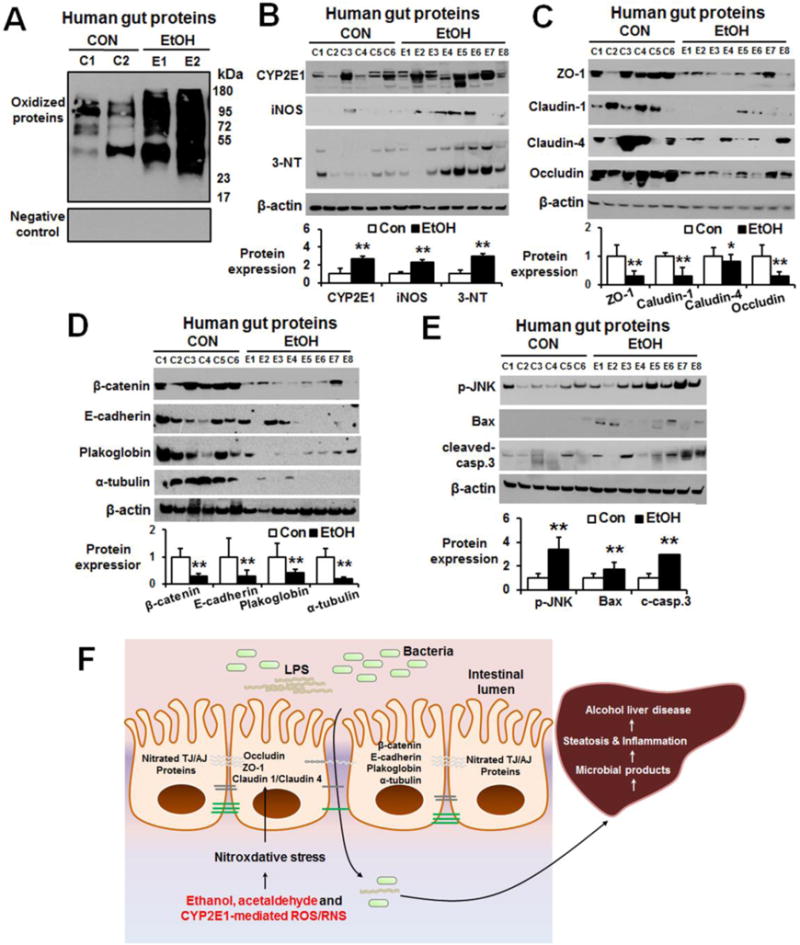
The levels of (A) oxidized intestinal proteins determined (upper panel) or negative control (lower panel) and (B) gut CYP2E1, iNOS, and nitrated proteins in control (CON, n=6) or people died from heavy alcohol intoxication (EtOH, n=8) are presented. (C, D) Immunoblots for the intestinal levels of TJ proteins (i.e., ZO-1, claudin-1, claudin-4, occludin), and AJ proteins (i.e. β-catenin, E-cadherin), plakoglobin and α-tubulin in the indicated groups of controls (CON, n=6) or heavy alcohol intoxications (EtOH, n=8). (E) The gut levels of apoptosis marker proteins p-JNK, Bax and cleaved-caspase-3 proteins are determined. Densitometric quantitation of the immunoblots for each protein relative to β-actin (n=6 or 8/group) is shown. Data represent means ± SD where statistical significance was determined using two-tailed t-test: *P<0.05, **P<0.01. (F) The proposed mechanisms of binge alcohol-induced gut leakiness and inflammatory liver injury. LPS, lipopolysaccharide.
Discussion
It is well-established that large amounts of alcohol intake can cause liver injury by directly damaging the hepatocytes. For instance, ALD including inflammatory ASH can be promoted through induction and/or activation of hepatic CYP2E1 and iNOS, producing elevated amounts of ROS and RNS, which then stimulate various PTMs of mitochondrial proteins and activate the cell death signaling pathway, contributing to mitochondrial dysfunction and apoptosis of hepatocytes [15]. Excessive amounts of alcohol can also stimulate infiltration of neutrophils into the liver with activation of hepatic Kupffer and stellate cells for inflammatory injury [29-31]. Binge drinking, which is a prevalent pattern of alcohol intake in many adults, can directly increase apoptosis of hepatocytes via hypoxic liver injury [32]. In addition, binge alcohol or chronic excessive alcohol intake can indirectly cause hepatotoxicity through changing intestinal microbiota (dysbiosis) with inflammation and gut leakiness with elevated endotoxin via induction and activation of hepatic TLR4 receptor [8, 22, 23, 31]. Various factors such as ethanol [1-4, 10], its metabolite acetaldehyde [33, 34], increased CYP2E1-mediated ROS [10, 30] and iNOS-mediated RNS [18, 35], gut dysbiosis-mediated inflammation [8, 22, 23, 26], alteration of microRNAs [35], etc, were reported to stimulate alcohol-induced gut leakiness. Co-treatment with antioxidants such as N-acetylcysteine [10], berberine [25], and glutamine [36] prevented alcohol-induced gut leakiness. Furthermore, other nonalcoholic substances such as fructose and high fat diets were reported to impair the intestinal barrier function with enhanced translocation of bacterial toxins (endotoxemia) to the liver, thereby contributing to inflammatory liver injury [8, 11, 12, 37].
The damaging effects of elevated ROS/RNS and peroxynitrite on promoting various PTMs and cell death are not restricted to the liver. The present results revealed that increased ROS/RNS through induction of intestinal CYP2E1 and iNOS also stimulated PTMs such as oxidation and nitration of junctional complex proteins with elevated apoptosis of enterocytes, leading to gut leakiness in the alcohol-exposed WT mice. In contrast, these ethanol-mediated changes were not observed in the Cyp2e1-null mice [10], supporting the critical role of increased nitroxidative stress in alcohol-induced intestinal barrier dysfunction and hepatotoxicity. However, the molecular mechanisms underlying the decreased TJ and other junctional complex proteins, resulting in increased gut leakiness following exposure to binge alcohol or nonalcoholic substances have been still unclear. To our best knowledge, the direct roles of nitration of TJ and AJ proteins and apoptosis of intestinal enterocytes have not been systematically studied. Based on the ubiquitin-dependent degradation of nitrated proteins [19-21], we hypothesized that the decreased levels of TJ and AJ proteins through nitration along with increased apoptosis of enterocytes can contribute to increase epithelial barrier dysfunction and gut permeability at least partly via CYP2E1-mediated events. Therefore, in this study, we investigated the molecular mechanisms for alcohol-induced gut leakiness by evaluating the levels of nitrated intestinal TJ and/or AJ proteins and apoptosis of enterocytes in WT versus Cyp2e1-null mice after binge alcohol exposure. Increased levels of apoptosis-related proteins and decreased intestinal junctional complex proteins led to elevated plasma endotoxin and fecal albumin levels, indicating elevated gut leakiness in alcohol-exposed WT mice in a CYP2E1-dependent manner. Similar results of increased nitroxidative stress marker proteins and decreased TJ, AJ proteins and desmosome plakoglobin with elevated endotoxin were observed in the alcohol-exposed rat intestines and autopsied human ileums compared to their respective controls. Mass-spectral and confocal immunofluorescence imaging analyses also confirmed decreased or redistributed TJ, AJ and desmosome proteins in alcohol-exposed specimens compared to the controls. Furthermore, IP followed by immunoblot analyses confirmed nitration and ubiquitin conjugation of TJ or AJ proteins and associated proteins, indicating ubiquitin-dependent proteolytic degradation of these proteins, as reported [19, 20]. Although we only studied the role of nitration of only a few selected TJ and AJ proteins out of many proteins associated with the epithelial junctional complexes [16], it is likely that other TJ or AJ proteins, not specifically identified or evaluated in this study, might be also modified by similar mechanisms. Nonetheless, out current results represent the first report to clearly demonstrate the critical roles of elevated apoptosis of enterocytes and nitration of intestinal TJ and AJ proteins and desmosome plakoglobin in decreased their levels, contributing to increased gut leakiness, endotoxemia and inflammatory liver injury following binge alcohol-exposure (Fig. 7F). Furthermore, the consistent results not only indicate conserved mechanisms of intestinal barrier dysfunction among different species but also explain the contributing roles of apoptosis of enterocytes and nitration of junctional complex proteins, resulting in decreased levels of TJ/AJ proteins with elevated gut leakiness, as previously reported by many other scientists [16, 17, 25-28]. In this regard, these two main factors may also play, at least partial, important roles in gut leakiness observed after exposure to nonalcoholic substances or under pathological conditions [5-9, 11, 12].
Interestingly, the basal levels of the junctional complex proteins claudin-1, occludin, ZO-1, plakoglobin are significantly decreased in control Cyp2e1-null mice compared to the WT mice, although we do not know the reason(s) for the different basal levels of some, but not all, junctional complex proteins. The changes in the expressed proteins (or genes) may be frequently observed in many knock-out (or knock-in) mice through adaptation or adjustment to the new physiological states that may develop in response to the deletion of a certain gene, depending on the physiological function of the particular gene. In our case, it is likely that the lower basal levels of the junctional complex proteins in the Cyp2e1-null mice may reflect the adjustment to the reduced levels of basal oxidative stress in the null mice than WT because of the absence of CYP2E1. The opposite case of adaptation may be also true for the WT mice. However, the most critically important fact was that the levels of these junctional proteins were not decreased in Cyp2e1-null mice in response to ethanol when compared to their control levels. In contrast, the same junctional complex proteins in WT mice were markedly decreased after exposure to alcohol. Furthermore, the lack of parallel elevation of serum endotoxin levels in ethanol-exposed Cyp2e1-null mice also support our notion that newly adjusted levels of the junctional complex proteins could be sufficient to maintain the gut epithelial barrier function, despite being moderately decreased in Cyp2e1-null mice.
Previous reports indicated that MLCK-mediated [25] or JNK2-mediated [28] phosphorylation of TJ proteins can contribute to epithelial cell barrier dysfunction. Thus, we also determined the targets of MLCK (proteins with p-Ser or p-Thr residues) and/or JNK (proteins with p-Thr-Pro or p-Ser-Pro residues) in WT and Cyp2e1-null mice without or with binge alcohol exposure. However, we did not observe significant differences in the levels of phosphorylated substrate proteins of MLCK and JNK between the alcohol -exposed mice or human ileums and their respective controls (Supplementary Fig. 5C and D), suggesting little role of MLCK or JNK-mediated protein phosphorylation in our models of binge alcohol-induced gut leakiness. However, the levels of activated p-MLCK in both alcohol-exposed mouse intestine and human ileums were significantly elevated compared to their corresponding controls (Supplementary Fig. 5A and B). Additionally, its levels were not elevated in the Cyp2e1-null mice even after binge alcohol exposure, strongly suggesting a potential role of p-MLCK in regulating gut epithelial barrier function. The latter notion was further supported by our confocal microscopic image and epithelial barrier function assay results (Supplementary Fig. 8) with the alcohol-exposed T84 colonic cells in the absence or presence of the selective inhibitor of p-MLCK or p-JNK. Although we do not know the underlying reason(s) for the different outcomes among our current results (i.e., Supplementary Fig. 5 and 8) and previous reports [25, 28], one possibility could be that the phosphorylation of a specific junctional complex protein as a target substrate of p-MPCK or p-JNK cannot be unambiguously demonstrated by grossly analyzing the whole gut lysates, as we performed (Supplementary Fig. 5C and D) with the pan-phospho-antibodies, which might not be highly specific. In addition, binge alcohol exposure in our model observed within a total span of 25 hours may not alter the gut microbiome usually observed in the chronic alcohol feeding paradigm [25, 26]. Alternatively, it is possible that phosphorylation of junctional complex proteins could be re-distributed and inactivated, thus contributing to elevated gut leakiness, although their protein levels may not be altered, as similar to the redistributed ZO-1 protein without change in its amounts in alcohol-exposed mice with gut barrier dysfunction [36]. Furthermore, under elevated nitroxidative stress, multiple types of PTM of junctional complex proteins can simultaneously take place, as discussed [15]. Consequently, it may be difficult to clearly demonstrate the role of a specific PTM (e.g., oxidation versus nitration or phosphorylation) in regulating gut leakiness or epithelial cell permeability, since oxidation, nitration, phosphorylation and other PTMs can take place simultaneously, as we reviewed [15]. In this regard, it is possible that the junctional complex proteins could be phosphorylated by activated p-JNK or p-MLCK. However, based on the well-established information about ubiquitin-dependent degradation of nitrated proteins [19-21], we specifically aimed at studying the nitration followed by ubiquitin conjugation of junctional complex proteins, leading to their decreased levels. Our results demonstrate a contributing role of nitration of junctional complex proteins in alcohol-induced gut leakiness.
In conclusion, binge alcohol exposure elevated the levels of CYP2E1, iNOS, p-JNK, nitrated and oxidized proteins in the small intestines of rats, WT mice and autopsied human specimens compared to their respective controls. Differential mass-spectral analyses revealed that the amounts of several intestinal TJ, AJ, desmosome and junctional complex associated proteins present in control rats were markedly decreased in binge alcohol-exposed rats. IP followed by immunoblot analyses revealed that selected TJ, AJ proteins and plakoglobin were nitrated and ubiquitinated, leading to their proteasomal degradation in alcohol-exposed WT mice compared to dextrose-controls or alcohol-exposed Cyp2e1-null mice. Specific inhibitors of CYP2E1 and iNOS significantly prevented disruption of TJ proteins in alcohol-exposed T84 colonic cells. In addition, elevated levels of apoptosis marker proteins in the enterocytes were observed in alcohol-exposed rats, WT mice and humans compared to their respective controls. These results from the experimental models and human specimens demonstrate for the first time that nitration followed by decreased levels of some TJ, AJ proteins and desmosomes along with elevated apoptosis of enterocytes contribute to increased gut leakiness, endotoxemia and inflammatory liver disease following alcohol exposure.
Supplementary Material
Highlights.
Binge alcohol caused apoptosis of gut enterocytes in rats and WT mice.
Binge alcohol reduced the levels of tight junction and adherent junction proteins.
Nitration of the junctional proteins led to ubiquitin-dependent degradation.
Apoptosis of enterocytes and reduced junctional proteins led to gut leakiness.
Binge alcohol-induced gut leakiness was observed in mice, rats and humans.
Layman’s Summary.
Our results demonstrated for the first time the critical roles of apoptosis of enterocytes and nitration followed by ubiquitin-dependent proteolytic degradation of the junctional complex proteins in promoting binge alcohol-induced gut leakiness and endotoxemia, contributing to inflammatory liver disease.
Acknowledgments
The authors thank Dr. Klaus Gawrisch for supporting the experiments. The information in this article is not a formal dissemination of information by NIH or FDA and does not represent agency position or policy.
Financial support
This study was supported by the Intramural Research Program of the National Institute on Alcohol Abuse and Alcoholism and in part with funds from the National Center for Toxicological Research, U.S. Food and Drug Administration (NCTR/FDA). In addition, this research was partially supported by the Korean Biomedical Scientist Fellowship Program Award (to Y.E. Cho) provided by Korean Research Institute of Bioscience and Biotechnology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
Thu authors disclose no conflicts.
Author’s contributions:
Y.E.C. and B.J.S., designed the experiment; Y.E.C., L.R.Y. and S.H.Y. performed experiments; Y.E.C. and B.J.S. wrote the paper; Y.E.C., L.R.Y., S.H.Y., M.A.A. and B.J.S. revised the manuscript. All authors contributed to the discussion of results and manuscript corrections.
Abbreviations used in this paper: AJ, adherent junction; BAC, blood alcohol concentration; CMZ, chlormethiazole; CYP2E1, cytochrome P450-2E1; DCFH-DA, 2′,7′-dichlorofluorescein diacetate; FITC-D4, FITC-labeled 4-kDa dextran; iNOS, inducible nitric oxide synthase; IP, immunoprecipitation; JNK, c-Jun N-terminal protein kinase; LPS, lipopolysaccharide; MLCK, myosin light chain kinase; ROS/RNS, reactive oxygen/nitrogen species; PTM, post-translational modification; TEER, trans-epithelial electric resistance; TLR4, toll-like receptors; TG, triglyceride; TJ, tight junction.
References
- 1.Forsyth CB, Voigt RM, Keshavarzian A. Intestinal CYP2E1: A mediator of alcohol-induced gut leakiness. Redox biology. 2014;3:40–46. doi: 10.1016/j.redox.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forsyth CB, Voigt RM, Shaikh M, Tang Y, Cederbaum AI, Turek FW, et al. Role for intestinal CYP2E1 in alcohol-induced circadian gene-mediated intestinal hyperpermeability. American journal of physiology Gastrointestinal and liver physiology. 2013;305:G185–195. doi: 10.1152/ajpgi.00354.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keshavarzian A, Farhadi A, Forsyth CB, Rangan J, Jakate S, Shaikh M, et al. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. Journal of hepatology. 2009;50:538–547. doi: 10.1016/j.jhep.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. The American journal of gastroenterology. 1999;94:200–207. doi: 10.1111/j.1572-0241.1999.00797.x. [DOI] [PubMed] [Google Scholar]
- 5.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nature medicine. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 6.Nazli A, Chan O, Dobson-Belaire WN, Ouellet M, Tremblay MJ, Gray-Owen SD, et al. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS pathogens. 2010;6:e1000852. doi: 10.1371/journal.ppat.1000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nature reviews Microbiology. 2012;10:655–666. doi: 10.1038/nrmicro2848. [DOI] [PubMed] [Google Scholar]
- 8.Frazier TH, DiBaise JK, McClain CJ. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN Journal of parenteral and enteral nutrition. 2011;35:14S–20S. doi: 10.1177/0148607111413772. [DOI] [PubMed] [Google Scholar]
- 9.Carter SR, Zahs A, Palmer JL, Wang L, Ramirez L, Gamelli RL, et al. Intestinal barrier disruption as a cause of mortality in combined radiation and burn injury. Shock. 2013;40:281–289. doi: 10.1097/SHK.0b013e3182a2c5b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdelmegeed MA, Banerjee A, Jang S, Yoo SH, Yun JW, Gonzalez FJ, et al. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free radical biology & medicine. 2013;65:1238–1245. doi: 10.1016/j.freeradbiomed.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology. 2016;151:733–746.e712. doi: 10.1053/j.gastro.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology. 2009;50:1094–1104. doi: 10.1002/hep.23122. [DOI] [PubMed] [Google Scholar]
- 13.Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and nonalcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. Journal of hepatology. 1987;4:8–14. doi: 10.1016/s0168-8278(87)80003-x. [DOI] [PubMed] [Google Scholar]
- 14.Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122:2049–2063. doi: 10.1053/gast.2002.33613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song BJ, Akbar M, Abdelmegeed MA, Byun K, Lee B, Yoon SK, et al. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox biology. 2014;3:109–123. doi: 10.1016/j.redox.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neunlist M, Van Landeghem L, Mahe MM, Derkinderen P, des Varannes SB, Rolli-Derkinderen M. The digestive neuronal-glial-epithelial unit: a new actor in gut health and disease. Nature reviews Gastroenterology & hepatology. 2013;10:90–100. doi: 10.1038/nrgastro.2012.221. [DOI] [PubMed] [Google Scholar]
- 17.Banan A, Choudhary S, Zhang Y, Fields JZ, Keshavarzian A. Ethanol-induced barrier dysfunction and its prevention by growth factors in human intestinal monolayers: evidence for oxidative and cytoskeletal mechanisms. The Journal of pharmacology and experimental therapeutics. 1999;291:1075–1085. [PubMed] [Google Scholar]
- 18.Banan A, Fields JZ, Decker H, Zhang Y, Keshavarzian A. Nitric oxide and its metabolites mediate ethanol-induced microtubule disruption and intestinal barrier dysfunction. The Journal of pharmacology and experimental therapeutics. 2000;294:997–1008. [PubMed] [Google Scholar]
- 19.Abdelmegeed MA, Moon KH, Chen C, Gonzalez FJ, Song BJ. Role of cytochrome P450 2E1 in protein nitration and ubiquitin-mediated degradation during acetaminophen toxicity. Biochemical pharmacology. 2010;79:57–66. doi: 10.1016/j.bcp.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciechanover A. The unravelling of the ubiquitin system. Nature reviews Molecular cell biology. 2015;16:322–324. doi: 10.1038/nrm3982. [DOI] [PubMed] [Google Scholar]
- 21.Gow AJ, Duran D, Malcolm S, Ischiropoulos H. Effects of peroxynitrite-induced protein modifications on tyrosine phosphorylation and degradation. FEBS letters. 1996;385:63–66. doi: 10.1016/0014-5793(96)00347-x. [DOI] [PubMed] [Google Scholar]
- 22.Banerjee A, Abdelmegeed MA, Jang S, Song BJ. Increased Sensitivity to Binge Alcohol-Induced Gut Leakiness and Inflammatory Liver Disease in HIV Transgenic Rats. PloS one. 2015;10:e0140498. doi: 10.1371/journal.pone.0140498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146:1513–1524. doi: 10.1053/j.gastro.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang VW. Proteomic and bioinformatic analysis of epithelial tight junction reveals an unexpected cluster of synaptic molecules. Biology direct. 2006;1:37. doi: 10.1186/1745-6150-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao M, Wang P, Sun C, He W, Wang F. Amelioration of IFN-gamma and TNF-alpha-induced intestinal epithelial barrier dysfunction by berberine via suppression of MLCK-MLC phosphorylation signaling pathway. PloS one. 2013;8:e61944. doi: 10.1371/journal.pone.0061944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen P, Starkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology. 2015;61:883–894. doi: 10.1002/hep.27489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiological reviews. 2013;93:525–569. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samak G, Suzuki T, Bhargava A, Rao RK. c-Jun NH2-terminal kinase-2 mediates osmotic stress-induced tight junction disruption in the intestinal epithelium. American journal of physiology Gastrointestinal and liver physiology. 2010;299:G572–584. doi: 10.1152/ajpgi.00265.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. Journal of hepatology. 2013;58:395–398. doi: 10.1016/j.jhep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 31.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World journal of gastroenterology. 2010;16:1321–1329. doi: 10.3748/wjg.v16.i11.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yun JW, Son MJ, Abdelmegeed MA, Banerjee A, Morgan TR, Yoo SH, et al. Binge alcohol promotes hypoxic liver injury through a CYP2E1-HIF-1alpha-dependent apoptosis pathway in mice and humans. Free radical biology & medicine. 2014;77:183–194. doi: 10.1016/j.freeradbiomed.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao RK. Acetaldehyde-induced barrier disruption and paracellular permeability in Caco-2 cell monolayer. Methods in molecular biology. 2008;447:171–183. doi: 10.1007/978-1-59745-242-7_13. [DOI] [PubMed] [Google Scholar]
- 34.Seth A, Basuroy S, Sheth P, Rao RK. L-Glutamine ameliorates acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. American journal of physiology Gastrointestinal and liver physiology. 2004;287:G510–517. doi: 10.1152/ajpgi.00058.2004. [DOI] [PubMed] [Google Scholar]
- 35.Tang Y, Zhang L, Forsyth CB, Shaikh M, Song S, Keshavarzian A. The Role of miR-212 and iNOS in Alcohol-Induced Intestinal Barrier Dysfunction and Steatohepatitis. Alcoholism, clinical and experimental research. 2015;39:1632–1641. doi: 10.1111/acer.12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaudhry KK, Shukla PK, Mir H, Manda B, Gangwar R, Yadav N, et al. Glutamine supplementation attenuates ethanol-induced disruption of apical junctional complexes in colonic epithelium and ameliorates gut barrier dysfunction and fatty liver in mice. The Journal of nutritional biochemistry. 2016;27:16–26. doi: 10.1016/j.jnutbio.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bergheim I, Weber S, Vos M, Kramer S, Volynets V, Kaserouni S, et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. Journal of hepatology. 2008;48:983–992. doi: 10.1016/j.jhep.2008.01.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.