


Abstract
The peptidyl transfer reaction on the large ribosomal subunit depends on the protonation state of the amine nucleophile and exhibits a large kinetic solvent isotope effect (KSIE ∼8). In contrast, the related peptidyl-tRNA hydrolysis reaction involved in termination shows a KSIE of ∼4 and a pH-rate profile indicative of base catalysis. It is, however, unclear why these reactions should proceed with different mechanisms, as the experimental data suggests. One explanation is that two competing mechanisms may be operational in the peptidyl transferase center (PTC). Herein, we explored this possibility by re-examining the previously proposed proton shuttle mechanism and testing the feasibility of general base catalysis also for peptide bond formation. We employed a large cluster model of the active site and different reaction mechanisms were evaluated by density functional theory calculations. In these calculations, the proton shuttle and general base mechanisms both yield activation energies comparable to the experimental values. However, only the proton shuttle mechanism is found to be consistent with the experimentally observed pH-rate profile and the KSIE. This suggests that the PTC promotes the proton shuttle mechanism for peptide bond formation, while prohibiting general base catalysis, although the detailed mechanism by which general base catalysis is excluded remains unclear.
INTRODUCTION
During the elongation phase of protein synthesis, new amino acids are added to the C-terminus of the growing peptide chain in the peptidyl transferase center (PTC) on the large ribosomal subunit. Here, the nascent peptide chain is linked to the A76 3′-oxygen of the P-site tRNA via an ester bond. When a new aminoacyl-tRNA is delivered to the ribosome, it is tested for codon-anticodon matching at the decoding center on the small ribosomal subunit. If the tRNA is correct (cognate) its CCA arm, which carries the new amino acid, will accommodate into the A-site of the PTC. The growing peptide chain will then be transferred from the P-site tRNA to the newly delivered one in the A-site. For the peptidyl transfer reaction to take place, the ester bond between the P-site A76-O3′ and the carbonyl carbon of the peptide must be broken and replaced by a peptide bond with the A-site aminoacyl-tRNA. This is achieved by nucleophilic attack of the α-amino group of the aminoacyl-tRNA on the ester carbonyl of the peptidyl-tRNA. Kinetics measurements of this reaction on the 70S bacterial ribosome both with puromycin (Pm)—a substrate analog—and aminoacyl-tRNA in the A-site show that ribosome accelerates the peptidyl transfer reaction by eliminating the entropic contribution (−TΔS‡) to the activation barrier and that the activation free energy is dominated by the enthalpic term, ΔH‡ ∼ 17 kcal/mol at 25°C (1,2).
Early mechanistic models for the peptide bond formation mechanism were based on the observation that the P-site A76 2′-OH group is crucial for this reaction (3–5), which suggests that the hydroxyl could act as a proton shuttle to the leaving 3′-oxygen during nucleophilic attack of the α-amino group. Further evidence supporting the proton shuttle mechanism was obtained from crystal structures of the PTC (6) and empirical valence bond (EVB) computer simulations (7–9), which suggested that the reaction proceeds via a transient zwitterionic tetrahedral intermediate (TI±). These simulations further indicated that the rate-limiting step corresponds to decomposition of TI±, i.e. a late transition state. Such a mechanism does not involve acid-base catalysis and, thus, is consistent with the observed pH-rate profile of peptide bond formation with aminoacyl-tRNA as the A-site substrate (10,11). Subsequent quantum mechanical density functional theory (DFT) calculations (12) predicted a late rate-limiting eight-membered ring transition state (TS) that was found to be compatible with the observed KSIE of 8.2 (13,14).
On the other hand, heavy atom kinetic isotope effect (KIE) measurements (15,16) for the considerably slower reaction with small substrate analogs on the 50S ribosomal subunit, suggest that the reaction proceeds via a negatively charged TI (TI−) and that formation of TI− is the rate-limiting step (Figure 1). That is, the transition state would then be early and correspond to the nucleophilic attack of the α-amino group on the ester carbon, with simultaneous proton transfer from the nitrogen to an unknown base. Such a mechanism does, however, not agree with the observed pH-dependence of peptide bond formation, using aminoacyl-tRNA as A-site substrate. That is, peptide bond formation depends only on the ionization state of α-amino group (11). Furthermore, crystallographic (6,17,18) and mutagenesis (19) data offer no suitable candidate for such a base. To reconcile the proposed early TS with the observed pH-rate profile and KSIE, a possibility could be that the carbonyl oxygen of the TI accepts the α-amino group proton and that this proton transfer is mediated by the P-site 2′-OH (13,16). This would result in a neutral TI and has been suggested in some computational studies of small model systems (20,21). Although such a mechanism appears to be compatible with the biochemical results, the 2′-OH and carbonyl oxygen are too far apart to participate in a concerted proton transfer without a conformational change in the active site. Crystal structures of the PTC trapped at various stages of catalysis also does not support such a conformational change (6,18). It should further be mentioned that the Brønsted coefficient for a series of Pm derivatives in the peptidyl transfer reaction has been determined and found to be near zero (22). This was interpreted as evidence that the nucleophile is uncharged in the rate-limiting transition state in line with an early TS suggested by heavy atom KIEs for the 50S subunit reaction. However, a near-zero Brønsted coefficient could also be considered as evidence for the late transition state predicted by the proton shuttle mechanism, since cleavage of the ester bond is not sensitive to the pKa of the nucleophile once the C−N bond has been formed.
Figure 1.

Schematic representation of the peptide bond formation mechanism via negatively charged (TI−) or zwitterionic (TI±) transient tetrahedral intermediates.
Moreover, in a recent crystal structure of the PTC, an extra water molecule was observed in between the A2451 2′-hydroxyl, the A-site A76 5′-phosphate and the N-terminus of the L27 protein (18). Based on this observation, a new ‘proton wire’ mechanism was proposed in which the extra water molecule acts as the missing general base and the proton from the nucleophile is transferred to this water molecule via the P-site O2′ and A2451 O2′. This water molecule (W3 in Figure 2) is in contact with both the A76 5′-phosphate group and the L27 N-terminal amine group and the idea was that the negative charge of the former could possibly stabilize the hydronium-like form of acceptor water (18). However, given that the normal (unperturbed) pKa’s of hydronium ion, the phosphate group and the L27 amine are −1.74, 1.5 and ∼8, respectively, it is somewhat difficult to see how the water molecule could be the ultimate proton acceptor. That is, the free energies of net proton transfer from the A76 2′-OH group with a normal pKa of 13.7 (23) to these acceptors would then be 21, 17 and 8 kcal/mol uphill, respectively (ΔGPT = 1.36[pKdonor – pKacceptor]), assuming no significant pKa shifts. Hence, in order for the proton not to go the group with highest pKa, the L27 amine, a stabilization (pKa shift) of the hydronium ion by more than 13 ( = 21-8) kcal/mol would be required. The situation is further complicated by the presence of a Mg2+ ion 3.8 Å away from the nearest A76 phosphate oxygen and directly coordinated (2.6 Å) to the C2452 phosphate (Figure 2). It is thus difficult to say anything about possible pKa shifts from the crystal structure but, if the wire is operational in proton transfer, then the L27 amine would seem to be the most logical acceptor. Recent experiments with fMetPhe-tRNAPhe in the P-site and Phe-tRNAPhe or Pm as A-site substrate, however, showed that the presence of L27 does not have any effect on the chemical steps of peptide bond formation for these reactions (24), which speaks against involvement of the L27 amine in the mechanism. Moreover, whether the proton wire mechanism would be consistent with the observed pH-rate profile for peptide bond formation would, in principle, depend on whether the proton at its acceptor site (whichever one) could be effectively shielded by L27 from bulk solvent (18). However, removal of L27 was found not to change the pH dependence of peptidyl transfer (24).
Figure 2.

Optimized structure of the reactant state of the proton shuttle mechanism (R) and the deprotonated reference state for the general base mechanism (R’). A crystallographically observed Mg2+ ion (18) is shown a green sphere and constrained atoms in the model are indicated by asterisks.
Numerous quantum mechanical calculations on ribosomal peptidyl transfer have been reported over the last decade, particularly using DFT. A recurring problem with such studies is, firstly, that in order to elucidate the actual process on the ribosome, a sufficiently large model system (including key ribosomal groups in addition to the reacting fragments) must be used and, secondly, spurious conformational rearrangements must be judged cautiously if unconstrained optimization of the reacting fragments is employed. That is, optimized structures along the reaction path that deviate significantly from available crystallographic information cannot really be taken seriously. Hence, it is perhaps not so surprising that no mechanistic consensus has yet emerged from this type of computational studies. However, what is clear is that all published computational investigations of reaction paths do predict a late TS on the ribosome, dominated by C−O bond breaking (7–9,12,20,25–31). There is, however, some confusion here due to the fact that some works (20,27,28) equate the rate-limiting TS with the elementary chemical step that has the highest energy barrier, and not with the TS that has the highest energy relative to the reactant complex, which is the correct kinetic definition. Besides the stepwise type of mechanism involving a transient tetrahedral intermediate, a fully concerted mechanism has also been proposed in several studies (26,29–31).
One approach to tackle the problem of different possible mechanistic alternatives on the ribosome is thus to construct a single sufficiently large DFT model, with suitable constraints on boundary atoms in order not to compromise the active site geometry, and compare the energetics of different proposed mechanisms. This was recently done by us for the peptidyl-tRNA hydrolysis reaction in the PTC catalyzed by release factors in termination of translation (32). Among different possible mechanisms we specifically considered the case where the P-site 2′-OH releases one proton to bulk and subsequently acts as a general base in hydrolysis. The free energy cost of the deprotonation step (at pH 7.5) could then be estimated from the experimental pKa of the 2′-hydroxyl, assuming that it is not perturbed from its solution value of 13.7 (23). The calculations predicted that this is the favored reaction path for peptidyl-tRNA hydrolysis on the ribosome in accordance with the interpretation of the experimental pH-rate profile and KSIE (13,33,34). However, since peptide bond formation and peptidyl-tRNA hydrolysis are very similar reactions—ester bond cleavage—that involve different nucleophiles, it is not clear why the PTC should catalyze these reactions with different mechanisms, as indicated by the distinctly different pH-rate profiles and KSIE for peptidyl transfer (10,11,13). It can be mentioned that the earliest EVB simulations of the termination reaction showed pronounced catalysis also of a neutral hydrolysis mechanism (35,36). However, those calculations were based on a neutral description of the uncatalyzed hydrolysis reaction in water, which rather appears to occur via base catalysis (32,37), and the more recent DFT results clearly show that the base catalyzed mechanism has a lower reaction barrier (32).
In view of the above results it seems possible that two competing reaction pathways may be operational in the PTC, one that involves ionization of the P-site A76 2′-hydroxyl group and one that does not. To provide a unified picture for the reactions that catalyzed by the PTC, we therefore examine here also peptide bond formation from the perspective of two such competing mechanisms. The reaction path that involves TI− in peptidyl transfer can thus be studied using the same approach as in the termination reaction, by ionizing the A76 2′-OH group, without making any explicit decision of whether the proton goes to bulk water (at pH 7.5) or to a possible unknown base with pKa ∼7.5. The consideration of different possible reaction pathways appears important since a change of mechanism could have major implications for the interpretation of experimental results. Thus, herein we reexamine the previously proposed peptidyl transfer mechanisms and also test the possibility of general base catalysis, using a large quantum mechanical cluster model of the PTC. However, the present study does not address the proton wire mechanism explicitly, since a detailed analysis of this mechanism would require several additional phosphate groups and Mg2+ ions to be included to describe the long-range electrostatic interactions. Besides the size of such a DFT model system, another difficulty at present is that sufficiently accurate positions of Mg2+ ions can hardly be identified in the crystal structures.
MATERIALS AND METHODS
A cluster model was constructed from the crystal structure of the T. thermophilus 70S ribosome preattack complex (18) (PDB code 1VY4). The core nucleotides C2063, A2451, U2506, U2585, and A2602 of the PTC and the N-terminal alanine residue of ribosomal protein L27 were added to the model. The A- and P-site substrate analogs Phe-NH-A76 and fMet-NH-A76, respectively, were converted to Ala-O-A76 at both positions and the terminal amine of the P-site substrate was acylated. Ala-tRNA was chosen as the A- and P-site substrates to reduce the size of active site model, since adequate modeling of larger substrates would require more ribosomal residues to be included in the model. Waters W1 to W4 were included from the crystal structure, while W5 was taken from previous MD studies (8,9). To account for the steric effect of surrounding parts of the ribosome some atoms in the periphery of the model were constrained during optimizations, which is important for maintaining the integrity of the active site model (Figure 2). The final model consists of 286 atoms and was subjected to density functional theory (DFT) calculations. The M06-2X functional (38) was used in this study and all calculations were performed with the Gaussian 09 package (39). Geometry optimizations were performed using the 6–31G(d,p) basis set and electronic energies were evaluated by single-point energy calculations at the 6–311+G(2d,2p) basis set level. Zero-point energies (ZPEs) and solvation effects were calculated at the same level of theory as the geometry optimizations. Similar to our previous study (32), the solvation effect was calculated by the SMD continuum model (40) with a dielectric constant of 24. The final energies are reported as the electronic energies corrected for the ZPE and solvation free energy. Additional single point energy calculations on the optimized structures using the B3LYP functional with the large basis set were also carried out. The KSIEs were calculated by replacing the polar hydrogens with deuterium and recalculating the ZPE. Since the fraction of exchangeable hydrogens is not known, lower and higher bounds of the KSIE were computed. For the lower bound, only the reactive fragments (the 2′-OH and α-amino groups) and waters were deuterated, while the higher bound was obtained by replacing all polar hydrogens with deuterium. The magnitudes of heavy atom kinetic isotope effects (KIEs) are generally much smaller than the KSIEs and to obtain a better estimate they were calculated according to the Bigeleisen-Wolfsberg equation (41) at 300 K.
RESULTS AND DISCUSSION
The proton shuttle mechanism yields a late TS
We initially re-examined the previously proposed proton shuttle mechanism using the large DFT cluster model of the peptidyl transferase center. The first step in this mechanism is the nucleophilic attack of the A-site α-amino group on the P-site ester carbon. In our calculations, this nucleophilic attack results in formation of the transient intermediate TI±, that lies 10.4 kcal mol−1 above the reactant state (R), and the activation energy for this step is found to be 12.1 kcal mol−1 relative to R (Figure 3). This is in agreement with previous EVB (8) and QM/MM (25) calculations and the existence of the TI± state speaks against a fully concerted mechanism (26,29–31). The optimized structure of TI± is extraordinarily similar to the crystal structure of the PTC in complex with transition state analogs (6), and the negative charge on TI± is stabilized by hydrogen bonding to W2 (Figure 4). This interaction was also predicted by the early EVB/MD simulations of the PTC (8). Furthermore, inspection of the optimized structure also suggests that the proposed neutral TI (20,21,26–28) is very improbable. That is, neutral TI formation requires a proton transfer from the α-amino group to the carbonyl oxygen, and it was suggested that this proton transfer is shuttled by the P-site A76-O2′. As mentioned earlier, this group, is too far away (∼4 Å) to participate in such a proton transfer, which also the crystal structures indicate (6). Additionally, it is clear from the positioning of W1 in the experimental structure and our optimized TI± that the P-site A76-O2′ is involved in H-bonding that does not support proton transfer to the carbonyl oxygen, either directly or via W2 (Figure 4). Therefore, unless the PTC residues undergo significant structural changes, it is difficult to see how the proposed neutral TI formation could be operational.
Figure 3.

Calculated energetics of the proton shuttle mechanism involving either an eight-membered (blue lines) or ten-membered transition state (red lines).
Figure 4.

Structure of the zwitterionic tetrahedral intermediate (yellow carbons) superimposed on the structure of a transition state analog with PDB code 1VQ7 (6) (orange carbons). Crystallographic waters are depicted as spheres (distances in Å).
During decomposition of the TI± state, the C-O3′ bond breaks while one proton is transferred from the α-amino group to the P-site A76-O3′. This proton transfer was suggested (6,12) to occur via an eight-membered TS and is mediated by the P-site A76-O2′ and a water molecule (Figure 5). In our calculations, the activation energy for this TS was found to be 25.4 kcal mol−1 relative to R. Similar energetic results were obtained in an earlier DFT study (12) of this TS and there an MP2 correction to the electronic energy was predicted to significantly lower the activation barrier. The M06-2X functional used here has, however, been shown to be quite accurate for thermochemical kinetics (38). Hence, in this study we choose to report all results with the M06-2X functional, as it is questionable whether an MP2 correction would actually improve these results. In a recent QM/MM study (25) of the eight-membered TS an activation free energy of ΔG‡ = 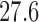 kcal mol−1 was calculated relative to the reactant complex. With an additional and somewhat arbitrarily assigned Mg2+ ion present this value was reduced to 25.4 kcal mol−1. Considering that the experimentally reported entropy contribution (1,2) is small (TΔS‡ = 0.7 kcal mol-1) the predicted activation enthalpy is thus similar to the value obtained in our model. The calculated activation energy of the eight membered TS, however, overestimates the experimental activation enthalpy by ∼8 kcal mol−1. By deuterating the reacting fragments only, the KSIE for this TS is predicted to be 5.4 and this value increases to 8.9 when all polar hydrogens are substituted. Moreover, the predicted 15N KIE is 0.977 (Table 1), similar to earlier results that also yielded an inverse effect (12).
kcal mol−1 was calculated relative to the reactant complex. With an additional and somewhat arbitrarily assigned Mg2+ ion present this value was reduced to 25.4 kcal mol−1. Considering that the experimentally reported entropy contribution (1,2) is small (TΔS‡ = 0.7 kcal mol-1) the predicted activation enthalpy is thus similar to the value obtained in our model. The calculated activation energy of the eight membered TS, however, overestimates the experimental activation enthalpy by ∼8 kcal mol−1. By deuterating the reacting fragments only, the KSIE for this TS is predicted to be 5.4 and this value increases to 8.9 when all polar hydrogens are substituted. Moreover, the predicted 15N KIE is 0.977 (Table 1), similar to earlier results that also yielded an inverse effect (12).
Figure 5.

Transition states for decomposition of the zwitterion tetrahedral intermediate (TI±) with one (A) and two (B) water molecules involved in the proton transfer chain. In this step, ester bond cleavage occurs concurrently with proton transfer. The crystallographic water molecule W2 stabilizes the negative charge of TI±.
Table 1. Calculated heavy atom kinetic isotope effects (15N, 18O, 13C) for transition states corresponding to zwitterionic and base catalyzed peptidyl transfer mechanisms.
| α-Amino N | O2′ | O3′ | Carbonyl C | |
|---|---|---|---|---|
| TI± − TS1 | 1.000 | 0.998 | 1.008 | 1.037 |
| TI± − TS2 8-membered | 0.977 | 1.017 | 1.022 | 1.035 |
| TI± − TS2 10-membered | 0.978 | 1.014 | 1.018 | 1.035 |
| Base catalyzed TS1 | 1.000 | 1.020 | 1.002 | 1.031 |
| Base catalyzed TS2 | 0.989 | 1.001 | 1.064 | 1.026 |
On the other hand, the possibility of an alternative mechanism for proton transfer cannot be readily dismissed. To investigate this, we also optimized a TS in which proton transfer is shuttled by two water molecules in addition to the P-site A76-O2′ (Figure 5). Both of these water molecules were, in fact, predicted by the early EVB/MD simulations and the second of these (W5) consistently donates a hydrogen bond to the O3′ in the simulations (8,9). Although no clear density is observed for this second water in the crystal structures of the PTC (6,18), there is clearly an empty cavity at its predicted MD position. That is, the P-site A76-O3′ would appear to be exposed to a hole on one side, with no well-defined electron density, which is likely to actually be solvated. An explanation for this situation could be that the substitution of the O3′ atom for an NH-group in the crystallized TS analogs reverts the dipole at this position and precludes the hydrogen bond donation seen in the MD simulations with a true substrate. Hence, a putative TS involving two bridging water molecules is not implausible and it is reasonable to assume that the extra water molecule can indeed be accommodated in the PTC. This is also apparent from Figure 5, where the larger TS can be seen not to perturb the active site.
The activation energy for this ten-membered TS was found to be lower and lies 22.2 kcal mol−1 above R (Figure 3). A similar decrease in the activation energy was also obtained between the six- and eight-membered ring transition states (12,25). This lowering of the activation energy, as also observed in previous studies, is partly due to relief of enthalpic strain, yielding more optimal angles for proton transfer. Furthermore, comparison of the two transition states (Figure 5) suggests that electrostatic stabilization may also play a role in reducing the barrier. In contrast to the eight-membered TS, the positive charge of the transferring proton is screened by two water molecules in the ten-membered transition state. Although addition of the extra water molecule decreased the activation energy compared to earlier studies, it is still higher than the experimental value by about 5 kcal mol−1. This may be partly due to the fact that only the ZPE correction is considered in our calculations. The contributions from thermally accessible vibrational degrees of freedom, however, may be significant for such a TS and these corrections can be expected to decrease the calculated activation energy for the ten-membered TS. Since some atoms are constrained in our model, calculation of this correction is rather inaccurate. For this TS, we further computed the magnitude of the KSIE which was found to be 5.9, when only the reacting groups are deuterated, and 8.6 if all polar hydrogens are exchanged. Hence, the predicted magnitude of the KSIE for the ten-membered TS is also in reasonable agreement with the experimental value of 8.2 (13). Since the N-terminal alanine residue of ribosomal protein L27 is not that distant from the active site, the protonation state of this residue could potentially affect the reaction energetics. To investigate the magnitude of this effect we also protonated the N-terminal alanine, yielding a system with a net charge of +1. The recalculated reaction energy for TI± and the activation energy for the ten-membered TS were then found to be 11.2 kcal mol and 22.0 kcal mol, respectively. Hence, the ionization state of the L27 N-terminal alanine does not strongly affect the energetics of this mechanism. This is also expected from an electrostatic point of view since the net charge of TI± is zero and the L27 nitrogen is ∼10 Å away from the reaction center, so that its interaction is largely screened. It has further been shown experimentally that L27 does not have any effect on the rate of peptide bond formation (24).
According to these results, the favored proton shuttle mechanism would proceed via formation of the transient TI± intermediate. The TI± formation is fast which implies that the proposed fully concerted mechanisms do not appear to be kinetically relevant for peptide bond formation catalyzed by ribosome. The structure and the H-bond network of the transient TI± intermediate is not consistent with a neutral TI involving protonation of the carbonyl oxygen either (Figure 5). Furthermore, decomposition of TI± is found to be rate-limiting here, that is, a late transition state is predicted with an inverse 15N KIE of 0.978 (Table 1). The heavy atom KIEs for the eight- and ten-membered TSs reveal that these transition states are similar, as is also evident from their geometries, and that the additional water molecule mainly facilitates proton transfer to the leaving group. Our calculations thus indicate that proton transfer in the rate-limiting step is mediated by two water molecules and the P-site A76 O2′. It can also be noted that the key hydrogen bond between A2451 2′-OH and A76 O2′ is present along the reaction and stabilizes the TS structure (Figures 4 and 5). This 10-membered TS results in a lower activation energy compared to previous calculations (12,25). However, an eight-membered TS can still not be completely dismissed since long-range interactions could potentially change the activation energy of such a transition state.
Possibility of a base catalyzed mechanism
Overall, two types of mechanisms have been suggested to have an early transition state for peptide bond formation. The first involves formation of the neutral TI via proton transfer from the nucleophile to the carbonyl oxygen during the nucleophilic attack. The second is the general base mechanism that results in formation of TI− by a concerted proton transfer from the nucleophile to a base during nucleophilic attack (13,16). As discussed above, protonation of the carbonyl oxygen by the nucleophile appears very unlikely due to the active site geometrical restrictions and hydrogen bonding network. The general base mechanism, on the other hand, does not appear compatible with the observed pH-rate profile for peptide bond formation. Nevertheless, considering the experimental complexity in analyzing and interpreting the peptidyl transfer kinetics, it is of considerable interest to study also the general base catalyzed mechanism to obtain a detailed view of its characteristics.
One approach to study the general mechanism—without any prior assumption about the nature of the base—is to examine the case where the P-site A76 2′-OH is already deprotonated, as was done for the hydrolysis reaction (32). This results in a negatively charged reactant state (R’) which was optimized by DFT calculations (Figure 2). In the first step of such a mechanism (see Figure 1), the α-amino group makes a nucleophilic attack on the ester carbonyl and one proton is transferred from the nucleophile to the deprotonated 2′-oxygen, yielding the transient negative TI− tetrahedral intermediate (Figure 6). Although N−H bond breaking is not very advanced in this TS as is evident from the bond distance (Figure 6), no stationary state corresponding to TI± was found for this mechanism, suggesting that the proton transfer occurs asynchronously. The activation energy and reaction energy for this step are calculated to be 7.9 and 3.4 kcal mol−1 relative to R’, respectively (Figure 7). Subsequently, cleavage of the ester bond occurs in the second step resulting in collapse of TI−, which is found to be rate-limiting. The activation energy for this TS (Figure 6) was calculated to be 10.7 kcal mol−1 relative to R’. Similar to the proton shuttle mechanism, we also tried to recalculate the energetics of the general base mechanism in presence of the protonated N-terminal amine of L27, by protonating this group in the R’ state. The protonated L27 amine, however, turns out be unstable in the R’ state and the proton moves back to A76 O2′ during geometry optimization, thus forming the reactant state (R) of the proton shuttle mechanism. No stationary states were either found for a possible (W3) hydronium ion or protonated A76 phosphate group. By constraining the proton to stay on the L27 nitrogen the cost of proton transfer to it from the A76 2′-OH group was estimated to be 11.5 kcal/mol, which is only slightly higher than proton transfer to bulk water at pH 7.5 (see below). This result thus suggests that a possible proton transfer along the proposed proton wire (18) to L27 is uphill in energy as expected, but that it is not too different from proton transfer to the bulk. However, as said above, a larger DFT model would be required to address such longer range proton transfers with confidence.
Figure 6.

Transition states for formation (A) and decomposition (B) of the negatively charged tetrahedral intermediate (TI−). In this mechanism the P-site A76-O2′ acts as general base during nucleophilic attack.
Figure 7.

Calculated energetics of the general base catalyzed mechanism. The cost of the initial proton transfer step (R→R’) is estimated based on experimental data (see text).
In the calculations on the base catalyzed mechanism (Figure 7), we made no assumption yet regarding the group that accepts the proton from the P-site A76 2′-hydroxyl. If we, as in the termination reaction (32), assume that the pKa of the 2′-OH group is not significantly perturbed from its solution value of 13.7 (23), the free energy cost of the initial proton transfer to bulk water would be 8.4 kcal mol−1 at pH 7.5. Using this approximation, the activation energies for TI− formation and decomposition are 16.4 and 19.2 kcal mol−1, respectively (Figure 7), relative to the true reactant state with the 2′-OH protonated (R). Such a mechanism would presumably result in a pH-rate profile with two different slopes. Up to the pH where the α-amino group becomes fully deprotonated the slope would be two, and at higher pH it would be one. However, it could also be possible that the pH-rate profile reaches a plateau at high pH, if the chemical transformation is no longer rate-limiting but is masked by some conformational process. It should be noted that the same activation barriers as above would be obtained for this mechanism if an unidentified base with pKa of ∼7.5 is the final acceptor of the 2′-OH proton, similar to the interpretation of experimental results for the Pm (42) and CPm (43) reactions on the 70S and 50S ribosomes, respectively. As an additional check of the reaction energy profiles reported above, we also carried out single point energy calculations on the optimized structures using the B3LYP functional and the large basis set, which resulted in very similar energetics (Supplementary Figures S1 and S2).
These calculations suggest that peptide bond formation via the TI− state may be qualitatively different from what was previously proposed based on the heavy atom KIEs (15,16). That is, in contrast to the proposed mechanism, the rate-limiting step in our calculations corresponds to collapse of TI− and thus a late transition state (TS2 in Figure 7). The corresponding KSIE for TS2 relative to R is predicted to be 1.1 with all polar hydrogens are substituted. Hence, the small value reflects the situation that no protons are in flight. Interestingly, proton transfer from the nucleophile to the base occurs late in the first transition state (TS1 in Figure 7), even in the presence of the deprotonated P-site 2′-oxygen—a strong base. This is because the second pKa of the α-amino group is too high to donate the proton before the C−N bond is practically formed. This clearly affects the KSIE associated with the first step (TS1), which is predicted to be similarly small as for TS2 (1.6 and 1.9 with reacting fragments and all polar hydrogens deuterated, respectively). These calculated KSIEs are, in fact, completely dominated by the equilibrium isotope effect (1.6) of the initial 2′-OH deprotonation step, which was approximated from the dissociation constant in water and deuterium oxide of 2-chloroethanol (32). Hence, the small calculated solvent isotope effects for the base catalyzed mechanism are more similar to the measured KSIE of the peptide hydrolysis reaction and not consistent with the experimentally measured value of 8.2 for peptide bond formation (13).
The predicted 15N KIE for TS1 in the base catalyzed mechanism is only marginally normal, as for TS1 in the proton shuttle mechanism (Table 1), which indicates that the nitrogen bond order is also similar in both TSs. For the early TS1 (Figure 6) to yield a larger normal 15N isotope effect, proton transfer would have to be synchronous with the nucleophilic attack (15,16). This is, however, difficult to achieve for the base catalyzed mechanism considering that the second pKa of the α-amino group is much higher than that of the A76 2′-OH group, as reflected by the KSIE for the TI− formation. The usual qualitative interpretation in terms of valence bond structures is that if bond order to the heavy atom increases in the TS compared to the reactant complex we would have an inverse KIE (<1) and if the bonding weakens we would have a normal effect (>1). The interpretation of the normal KIE observed for small substrates in the 50S reaction was that the presumed rate-limiting formation of TI− has a TS where proton transfer is so advanced that the bond order to nitrogen significantly decreases (15,16). Our calculations indicate that this is not really the case for the optimized transition state leading to TI− (Figure 6). However, if one considers both the energetics (Figure 7) and the heavy atom KIEs, it appears that TS1 of the base catalyzed mechanism is that which is most compatible with the KIE experiments for the 50S reaction with small A- and P-site substrates (16). In view of the fact that this reaction is 300-fold slower than on 70S ribosomes (15) and also that its pH-dependence with CPm as A-site substrate is different (43,44), it appears that the transition states for peptidyl transfer may actually differ between the two reactions. With full-length tRNA as the P-site substrate, however, the 50S reactions with Pm and CPm seem to have more similar kinetics to 70S ribosomes (45). Additional KSIE experiments on the 50S fragment reaction could possibly shed further light on this issue. Furthermore, a caveat with regard to the frequency calculations underlying heavy atom KIEs in our cluster model is that they cannot be expected to be highly accurate, due to the large size of the system.
Overall, the above results show that the α-amino group is a sufficiently strong nucleophile not to require activation beyond the initial –NH3+ dissociation and, even in the presence of a strong base, proton transfer does not occur until late (after TS1) in formation of TI−. Our calculations further suggest that the rate-limiting step for the general base mechanism is the collapse of TI−. Interestingly, this is in contrast to our previous study of peptide-tRNA hydrolysis where we used the same approach and found that TI− formation is rate-limiting and that proton transfer occurs synchronously with nucleophilic attack of water. The difference stems from the fact that the α-amino group is a stronger nucleophile than water.
Possible competing reaction pathways
Although the general base catalyzed mechanism does not appear to be consistent with either pH-rate profile or KSIE measurements for peptide bond formation, the calculated activation energy for this mechanism is not high enough to be prohibitive. That is, either ionization of the A76 2′-OH or the existence of an unknown base with appropriate pKa in the PTC would seem to render such a mechanism potentially operational. As noted above, a curious and somewhat puzzling result is the pH-dependence of peptide bond formation with CPm as A-site substrate. For this substrate, peptide bond formation catalyzed by the 70S ribosome depends only on the ionization state of the nucleophile (44), while the reaction catalyzed by the 50S ribosome shows a pH-rate profile that can be assigned to two ionizing groups, the nucleophile and an unknown group (43). Although the observed change in the pH-rate profile may be caused by other processes such as a pH-dependent conformational change (44,46,47), a change in mechanism also seems possible. The fact that peptidyl-tRNA ester bond hydrolysis catalyzed by the PTC occurs via the general base mechanism would appear to further strengthen such a hypothesis.
The way by which either of the two mechanisms would be favored in different phases of protein synthesis is not clear, especially if one considers that the overall structure of the PTC is almost identical in crystal structures representative of peptide bond formation and peptidyl-tRNA ester bond hydrolysis. However, a distinct difference is, of course, the replacement of the A-site tRNA by the universally conserved release factor GGQ loop in the termination complexes. Both crystal structures (48) and our earlier DFT calculations (32) indicate that the backbone NH group of the release factor glutamine residue can hydrogen bond to the A76 2′-oxygen in the reactant state, which would provide stabilization of its anionic form. This could possibly help maintaining a lower pKa of the 2′-OH in the termination reaction than in peptidyl transfer and thereby favor the general base mechanism in the former case. Another explanation could be that the PTC is able to structure water molecules in the active site differently in the elongation and termination complexes. For the general base mechanism to be operational, one proton needs to be transferred to the bulk or an acceptor base. Blocking such a proton transfer pathway may thus prohibit general base catalysis.
CONCLUDING REMARKS
In this study, we examined various mechanisms for the peptide bond formation reaction to understand why it seems to proceed via a different pathway than the hydrolysis reaction in translation termination, as indicated by biochemical experiments. Another interesting question is whether the peptidyl transfer reaction involves an early or a late rate-limiting TS. Our calculations, in agreement with previous studies, suggest that the proton shuttle mechanism results in reasonable energetics and has a late transition state. For this reaction pathway, in addition to the previously proposed eight-membered TS, we also optimized a ten-membered TS (involving two water molecules) which is found to be in agreement with the observed activation energy. This mechanism is compatible with both pH-rate profile and KSIE measurements. Surprisingly, we found that also the general base mechanism has a calculated activation energy in agreement with the experimental values. In this case, the collapse of TI− was found to be rate-limiting rather than formation of TI−. More importantly, the α-amino group seems not to participate in a synchronous proton transfer even when P-site 2′-OH is deprotonated. This is because the α-amino group is a sufficiently good nucleophile that can readily attack the carbonyl carbon. An implication of this is that if general base catalysis would be operational, the magnitude of the KSIE would be distinctly lower than what is observed for this reaction on the 70S ribosome. Our results also suggest formation of a neutral TI is not either a viable mechanism. In this case, the rate-limiting step has been hypothesized to involve the nucleophilic attack of the α-amino group with simultaneous proton transfer from the nucleophile to the carbonyl oxygen. But according to our calculations, even in the presence of a deprotonated P-site 2′-OH, the α-amino group does not readily participate in early proton transfer. Therefore, proton transfer to the carbonyl oxygen prior to almost complete C−N bond formation appears highly improbable. Moreover, the suggested concerted proton transfer via the 2′-OH group to the carbonyl oxygen would require a conformational change not seen in any crystal structures, as these groups are too far apart in the active site.
Interestingly, however, the general base mechanism cannot be excluded solely based on the calculated activation energy, despite the fact it is not compatible with the experimentally observed pH-rate profile and KSIE. Even if the L27 amine would be the proton acceptor (rather than bulk water) in the general base mechanism, it is difficult to see why the pH-dependence should then not reflect deprotonation of this group. As noted above, deletion of L27 does not change the pH-dependence of peptidyl transfer (24) and the protein is also not present in all kingdoms of life. It also remains unclear how the PTC could promote the proton shuttle mechanism for peptide bond formation, while prohibiting general base catalysis. Besides the possibility that the pKa of the 2′-OH group might be raised with tRNA bound to the A-site compared to release factors in termination, the general base mechanism also requires one proton to be transferred to the bulk or an acceptor base. Therefore, blocking of such a proton transfer pathway could potentially be a way to prohibit this reaction pathway. Alternatively, the PTC might structure water molecules so as to favor different mechanisms at different stages of protein synthesis (elongation and termination). Nonetheless, the fact that base catalysis also appears energetically viable indicates that experimental conditions may possibly affect the reaction pathway and additional KSIE measurements for peptidyl transfer assays with different substrates etc. could be very informative in this respect.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Swedish Research Council (VR) [2014-03688, 2016-0624]; Knut and Alice Wallenberg Foundation; eSSENCE e-science initiative; Swedish National Infrastructure for Computing (SNIC). Funding for open access charge: Swedish Research Council.
Conflict of interest statement. None declared.
REFERENCES
- 1. Sievers A., Beringer M., Rodnina M.V., Wolfenden R.. The ribosome as an entropy trap. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7897–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johansson M., Bouakaz E., Lovmar M., Ehrenberg M.. The kinetics of ribosomal peptidyl transfer revisited. Mol. Cell. 2008; 30:589–598. [DOI] [PubMed] [Google Scholar]
- 3. Dorner S., Polacek N., Schulmeister U., Panuschka C., Barta A.. Molecular aspects of the ribosomal peptidyl transferase. Biochem. Soc. Trans. 2002; 30:1131–1137. [DOI] [PubMed] [Google Scholar]
- 4. Dorner S., Panuschka C., Schmid W., Barta A.. Mononucleotide derivatives as ribosomal P-site substrates reveal an important contribution of the 2′‐OH to activity. Nucleic Acids Res. 2003; 31:6536–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weinger J.S., Parnell K.M., Dorner S., Green R., Strobel S.A.. Substrate-assisted catalysis of peptide bond formation by the ribosome. Nat. Struct. Mol. Biol. 2004; 11:1101–1106. [DOI] [PubMed] [Google Scholar]
- 6. Schmeing T.M., Huang K.S., Kitchen D.E., Strobel S.A., Steitz T.A.. Structural insights into the roles of water and the 2′ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol. Cell. 2005; 20:437–448. [DOI] [PubMed] [Google Scholar]
- 7. Sharma P.K., Xiang Y., Kato M., Warshel A.. What are the roles of substrate-assisted catalysis and proximity effects in peptide bond formation by the ribosome. Biochemistry. 2005; 44:11307–11314. [DOI] [PubMed] [Google Scholar]
- 8. Trobro S., Åqvist J.. Mechanism of peptide bond synthesis on the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:12395–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trobro S., Åqvist J.. Analysis of predictions for the catalytic mechanism of ribosomal peptidyl transfer. Bichemistry. 2006; 45:7049–7056. [DOI] [PubMed] [Google Scholar]
- 10. Bieling P., Beringer M., Adio S., Rodnina M.V.. Peptide bond formation does not involve acid-base catalysis by ribosomal residues. Nat. Struct. Mol. Biol. 2006; 13:423–428. [DOI] [PubMed] [Google Scholar]
- 11. Johansson M., Ieong K.W., Trobro S., Strazewski P., Åqvist J., Pavlov M.Y., Ehrenberg M.. pH-sensitivity of the ribosomal peptidyl transfer reaction dependent on the identity of the A-site aminoacyl-tRNA. Proc. Natl. Acad. Sci. U. S. A. 2011; 108:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wallin G., Åqvist J.. The transition state for peptide bond formation reveals the ribosome as a water trap. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:1888–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuhlenkoetter S., Wintermeyer W., Rodnina M.V.. Different substrate-dependent transition states in the active site of the ribosome. Nature. 2011; 476:351–354. [DOI] [PubMed] [Google Scholar]
- 14. Åqvist J., Lind C., Sund J., Wallin G.. Bridging the gap between ribosome structure and biochemistry by mechanistic computations. Curr. Opin. Struct. Biol. 2012; 22:815–823. [DOI] [PubMed] [Google Scholar]
- 15. Seila A.C., Okuda K., Nunez S., Seila A.F., Strobel S.A.. Kinetic isotope effect analysis of the ribosomal peptidyl transferase reaction. Biochemistry. 2005; 44:4018–4027. [DOI] [PubMed] [Google Scholar]
- 16. Hiller D.A., Singh V., Zhong M., Strobel S.A.. A two-step chemical mechanism for ribosome-catalysed peptide bond formation. Nature. 2011; 476:236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hansen J.L., Schmeing T.M., Moore P.B., Steitz T.A.. Structural insights into peptide bond formation. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:11670–11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Polikanov Y.S., Steitz T.A., Innis C.A.. A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nat. Struct. Mol. Biol. 2014; 21:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Youngman E.M., Brunelle J.L., Kochaniak A.B., Green R.. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell. 2004; 117:589–599. [DOI] [PubMed] [Google Scholar]
- 20. Wang Q., Gao J., Liu Y., Liu C.. Validating a new proton shuttle reaction pathway for formation of the peptide bond in ribosomes: A theoretical investigation. Chem. Phys. Lett. 2010; 50:113–117. [Google Scholar]
- 21. Rangelov M.A., Petrova G.P., Yomtova V.M., Vayssilov G.N.. Catalytic role of vicinal OH in ester aminolysis: proton shuttle versus hydrogen bond stabilization. J. Org. Chem. 2010; 75:6782–6792. [DOI] [PubMed] [Google Scholar]
- 22. Kingery D.A., Pfund E., Voorhees R.M., Okuda K., Wohlgemuth I., Kitchen D.E., Strobel S.A.. An uncharged amine in the transition state of the ribosomal peptidyl transfer reaction. Chem. Biol. 2008; 15:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Åström H., Limén E., Strömberg R.. Acidity of secondary hydroxyls in ATP and adenosine analogues and the question of a 2′,3′-hydrogen bond in ribonucleosides. J. Am. Chem. Soc. 2004; 126:14710–14711. [DOI] [PubMed] [Google Scholar]
- 24. Maracci C., Wohlgemuth I., Rodnina M.V.. Activities of the peptidyl transferase center of ribosomes lacking protein L27. RNA. 2015; 21:2047–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Świderek K., Marti S., Tuñón I., Moliner V., Bertran J.. Peptide bond formation mechanism catalyzed by ribosome. J. Am. Chem. Soc. 2015; 137:12024–12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Acosta-Silva C., Bertran J., Branchadell V., Oliva A.. Quantum-mechanical study on the mechanism of peptide bond formation in the ribosome. J. Am. Chem. Soc. 2012; 134:5817–5831. [DOI] [PubMed] [Google Scholar]
- 27. Byun B.J., Kang Y.K.. A mechanistic study supports a two-step mechanism for peptide bond formation on the ribosome. Phys. Chem. Chem. Phys. 2013; 15:14931–14935. [DOI] [PubMed] [Google Scholar]
- 28. Wang Q., Gao J., Zhang D., Liu C.. A theoretical model investigation of peptide bond formation involving two water molecules in ribosome supports the two-step and eight membered ring mechanism. Chem. Phys. 2015; 450:1–11. [Google Scholar]
- 29. Gindulyte A., Bashan A., Agmon I., Massa L., Yonath A., Karle J.. The transition state for formation of the peptide bond in the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13327–13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thirumoorthy K., Nandi N.. Role of chirality of the sugar ring in the ribosomal peptide synthesis. J. Phys. Chem. B. 2008; 112:9187–9195. [DOI] [PubMed] [Google Scholar]
- 31. Xu J., Zhang J.Z.H., Xiang Y.. Ab initio QM/MM free energy simulations of peptide bond formation in the ribosome support an eight-membered ring reaction mechanism. J. Am. Chem. Soc. 2012; 134:16424–16429. [DOI] [PubMed] [Google Scholar]
- 32. Kazemi M., Himo F., Åqvist J.. Peptide release on the ribosome involves substrate-assisted base catalysis. ACS Catal. 2016; 6:8432–8439. [Google Scholar]
- 33. Shaw J.J., Trobro S., He S.L., Åqvist J., Green R.. A role for the 2′ OH of peptidyl-tRNA substrate in peptide release on the ribosome revealed through RF-mediated rescue. Chem. Biol. 2012; 19:983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Indrisiunaite G., Pavlov M.Y., Heurgué-Hamard V., Ehrenberg M.. On the pH dependence of class-1 RF-dependent termination of mRNA translation. J. Mol. Biol. 2015; 427:1848–1860. [DOI] [PubMed] [Google Scholar]
- 35. Trobro S., Åqvist J.. A model for how ribosomal release factors induce peptidyl-tRNA cleavage in termination of protein synthesis. Mol. Cell. 2007; 27:758–766. [DOI] [PubMed] [Google Scholar]
- 36. Trobro S., Åqvist J.. Mechanism of the translation termination reaction on the ribosome. Biochemistry. 2009; 48:11296–11303. [DOI] [PubMed] [Google Scholar]
- 37. Wolfenden R. The mechanism of hydrolysis of amino acyl RNA. Biochemistry. 1963; 2:1090–1092. [DOI] [PubMed] [Google Scholar]
- 38. Zhao Y., Truhlar D.G.. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008; 120:215–241. [Google Scholar]
- 39. Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A. et al. . Gaussian 09, Revision D.01. 2009; Wallingford: Gaussian Inc. [Google Scholar]
- 40. Marenich A.V., Cramer C.J., Truhlar D.G.. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 2009; 113:6378–6396. [DOI] [PubMed] [Google Scholar]
- 41. Kohen A, Limbach HH. Isotope Effects in Chemistry and Biology. 2005; Boca Raton: CRC Press. [Google Scholar]
- 42. Katunin V.I., Muth G.W., Strobel S.A., Wintermeyer W., Rodnina M.V.. Important contribution to catalysis of peptide bond formation by a single ionizing group within the ribosome. Mol. Cell. 2002; 10:339–346. [DOI] [PubMed] [Google Scholar]
- 43. Okuda K., Seila A.C., Strobel S.A.. Uncovering the enzymatic pKa of the ribosomal peptidyl transferase reaction utilizing a fluorinated puromycin derivative. Biochemistry. 2005; 44:6675–6684. [DOI] [PubMed] [Google Scholar]
- 44. Brunelle J.L., Youngman E.M., Sharma D., Green R.. The interaction between C75 of tRNA and the A loop of the ribosome stimulates peptidyl transferase activity. RNA. 2006; 12:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wohlgemuth I., Beringer M., Rodnina M.. Rapid peptide bond formation on isolated 50S ribosomal subunits. EMBO Rep. 2006; 7:699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bayfield M.A., Dahlberg A.E., Schulmeister U., Dorner S., Barta A.. A conformational change in the ribosomal peptidyl transferase center upon active/inactive transition. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:10096–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Muth G.W., Chen L., Kosek A.B., Strobel S.A.. pH-dependent conformational flexibility within the ribosomal peptidyl transferase center. RNA. 2001; 7:1403–1415. [PMC free article] [PubMed] [Google Scholar]
- 48. Jin H., Kelley A.C., Loakes D., Ramakrishnan V.. Structure of the 70S ribosome bound to release factor 2 and a substrate analog provides insights into catalysis of peptide release. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:8593–8598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.