

Abstract
Among three series of 1,2,4-trioxane derivatives, five compounds showed good in vitro antimalarial activity, three compounds of which exhibited better activity against P. falciparum resistant (RKL9) strain than the sensitive (3D7) one. Two best compounds were one from aryl series and the other from heteroaryl series with IC50 values of 1.24 µM and 1.24 µM and 1.06 µM and 1.17 µM, against sensitive and resistant strains, respectively. Further, trioxane derivatives exhibited good binding affinity for the P. falciparum cysteine protease falcipain 2 receptor (PDB id: 3BPF) with well defined drug-like and pharmacokinetic properties based on Lipinski’s rule of five with additional physicochemical and ADMET parameters. In view of having antimalarial potential, 1,2,4-trioxane derivative(s) reported herein may be useful as novel antimalarial lead(s) in the discovery and development of future antimalarial drug candidates as P. falciparum falcipain 2 inhibitors against resistant malaria.
Keywords: P. falciparum, resistance, trioxane derivatives, falcipain 2 inhibitors, antimalarial
Introduction
Malaria continues to be a major lethal infectious disease of human beings, affecting around 200–300 million people and causing approximately 430,000 deaths every year globally. The most affected populations are children and infants as well as pregnant women 1 . The disease is caused by five species of the parasite Plasmodium, namely, Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malariae, and Plasmodium knowlesi. Of these, P. falciparum is the most prevalent and deadly species that causes severe malaria such as cerebral malaria, and it is also responsible for most of the malaria-related deaths in humans 2–4 . Every year over 50% of all malarial infections are caused by P. falciparum with about 20–50% of total mortality cases commonly occur in children younger than five years old and pregnant women 5 . During the past two decades, the emergence of drug resistant strains of P. falciparum has become an increasingly serious concern in malaria control and prevention worldwide. Reduced clinical efficacy of currently available drugs, especially those in the aminoquinoline group (chloroquine, amodiaquine, mefloquine, and piperaquine) including non-quinolines like antifolates (sulfadoxine, proguanil, pyrimethamine), lumefantrine, atovaquone, and antibiotics, and the novel artemisinin (ART) derivatives (dihydroartemisinin, artemether, and artesunate), to multi-drug resistant P. falciparum strains has limited their clinical usefulness in the treatment of resistant malaria 2 . Artemisinin-based combination therapies (ACTs) such as artemether/lumefantrine/amodiaquine and artesunate/mefloquine/piperaquine recommended by WHO for the treatment of multi-drug resistant P. falciparum malaria have also been reported to develop resistance in South-East Asia 2 , 6 . This increasing burden of resistant malaria has stimulated drug discovery scientists to search for new antimalarial drugs or alternative therapeutic options to combat the problem of drug resistance. In view of this, the development of new antimalarial drugs with novel mode(s) of action could be an attractive strategy to address the above challenging issue.
Though ART and related endoperoxides are the mainstay of current antimalarial therapy, high treatment cost (relative to quinoline-based drugs), unsatisfactory physicochemical/pharmacokinetic properties, toxicities and limited availability of drugs are some other notable pitfalls besides having potential resistance problem 7 , 8 . All above issues are adversely affecting clinical outcomes of ART-based drug regimens used in the treatment of malaria. The clinical significance of ART-based drugs is attributed to be due to the excellent antiparasitic action of the endoperoxide component. Pharmacodynamic study also suggests that the key pharmacophoric 1,2,4-trioxane ring system is responsible for the biological activity of ART (natural lead molecule), its semi-synthetic analogues and related endoperoxides 6 , 9 , 10 . In recent years, research into several synthetic endoperoxide scaffolds such as 1,2,4-trioxane, 1,2,4-trioxolane and 1,2,4,5-teraoxane derivatives and their hybrid analogues have gained significant interest in discovery and development of potent antimalarial drugs. Some endoperoxide-based antimalarial compounds 2 that are currently under clinical phase of development are represented in Figure 1. Endoperoxide molecules bearing 1,2,4-trioxane core have been identified to exhibit a broad spectrum of biological effects such as anticancer, antimicrobial, and anti-parasitic effects 11 . Several design strategies have been initiated by MMV (Medicines for Malaria Venture) program for the development of effective and affordable synthetic peroxides as alternative therapy to existing ART-based drugs 2 , 12 . However, considering all above facts, development of peroxide-based synthetic compounds is expected to provide effective antimalarials agents with limited toxicity issues and ready to be available at affordable cost. It would offer a better therapeutic option for the treatment of malaria with good clinical outcomes (desired potency with optimal pharmacokinetics) and resistant preventing action. It may therefore represent a new class of synthetic, new-generation and orally active peroxides, with optimal antimalarial potency against resistant P. falciparum malaria.
Figure 1.

Structures of some new endoperoxide antimalarials.
As a part of our research program towards developing potent antimalarial drugs that would be active against resistant P. falciparum malaria, novel molecular manipulation approach was adopted to design and develop newer series of 1,2,4-trioxane derivatives as single drug conjugates (single molecular entity rather than hybrid drugs) based on the basic structural scaffold of 1,2,4-trioxane ring system. After the synthesis of compounds, in vitro antimalarial screening, molecular docking and drug-likeness studies including ADMET prediction using in silico tools were also carried out. Since, ART and its semi-synthetic analogues or ART derived peroxides possess unfavourable pharmacokinetic issues and certain tissue toxicities because of their high structural complexities and some physicochemical insufficiencies 7 , 8 , it is expected that designed compounds would possess modified pharmacokinetic properties and toxicities due to less complexities in the molecular structure. Moreover, single drug conjugate-based trioxane molecule can be given as single drug therapy that would also be advantageous over hybrid antimalarial drugs and combination therapies.
Molecular docking study was performed to validate the antimalarial efficacy of synthesised 1,2,4-trioxane derivatives by investigating binding modes as well as orientation of ligands in the receptor pocket of target falcipain 2 protein (P. falciparum cysteine protease enzyme). In our study, three-dimensional (3D) structure of falcipain 2 protein molecule was used as possible antimalarial drug target for 1,2,4-trioxane derivatives. Docking rationalises finding new antimalarial lead molecules and also gives an insight into structure–activity relationships (SARs) and mode(s) of antimalarial action based on scoring function and further binding modes analysis from ligand–protein binding/interaction studies 13 . Drug-likeness study includes calculation of fundamental molecular (physicochemical) properties and Lipinski’s parameters, which assesses the acceptability of compounds as drug molecules. They represent the combined physicochemical, pharmacokinetic and pharmacodynamic properties (collectively termed as drug-like properties) of molecules to exhibit good drug-likeness behaviour in human body 14 . In fact, molecular properties are the fundamental structural parameters which determine the physicochemical (solubility and permeability) and biochemical (metabolic stability, transport property, and protein/tissue affinity) properties, which ultimately determine molecule’s in vivo pharmacokinetics (bioavailability and half life), toxicity, and pharmacodynamics (receptor affinity and efficacy) 15 . ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties have a predictable influence on pharmacokinetic and pharmacodynamic effects of drug molecules. The calculation of ADMET properties is therefore intended as the first step towards optimising the new drug molecules, whether to check the failure of lead molecules which may cause toxicity and one unable to cross the intestinal membranes or metabolised by the body in to an inactive form 16 .
Experimental
Synthesis and analysis
All of the chemicals were procured commercially from Sigma-Aldrich Corporation (St. Louis, MO), Merck Specialists Pvt. Ltd. (Darmstadt, Germany), HiMedia Lab. Pvt. Ltd. (Einhausen, Germany), or Spectrochem Pvt. Ltd. (Mumbai, India) and were used without further purification. All the reactions were performed in oven dried glass ware using synthetic grade chemicals. Melting points (MP) were measured in open capillaries on an electrically heated MP apparatus. Ultraviolet (UV)–visible spectra were recorded on Shimadzu UV-1700 UV–visible spectrophotometer and the wave lengths of maximum absorption (λ max, nm) are reported. Infrared (IR) spectra were obtained on a Bruker Alpha Fourier Transform (FT-IR) spectrophotometer (Billerica, MA) using KBR disc and are reported in terms of frequency of absorption (υ, cm−1). 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on Bruker Avance II 400 FT-NMR spectrometer (Billerica, MA) at 400 and 100 MHz, respectively, using tetramethylsilane (TMS) as an internal standard (δ 0.00 ppm) and CDCl3 as a solvent. Chemical shift (δ) values were expressed in parts per million (ppm) relative to TMS (δ 0.00 ppm). 1HNMR data are assigned in order: peak multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet), number of protons (numerical integral value), coupling constants (J value) in hertz. 13C NMR data represents different types of structural carbons with respect to their corresponding chemical shift values. Mass spectra were obtained on a LC–MS Water 4000 ZQ instrument using electrospray ionisation (ES+). The m/z values were recorded in the range of m/z between 100 and 500 and the m/z values of the most intense molecular ion [M]+ peak, with relative intensities in parentheses, are given followed by peaks corresponding to major fragment ions. Elemental analysis was performed on a ThermoFinnigan CHNS Analyzer (Somerset, NJ).
General procedure of synthesis 11 , 17 , 3a–e, 3′a–n, 3″a–e
A mixture of p-cresol, 1 (1 mmol, 0.1045 ml), distilled water (16.0 ml) and acetonitrile (CH3CN, 4.0 ml) was vigorously stirred until complete solution was achieved. A mixture of oxone (5 mmol, 1.537 g) and NaHCO3 (15 mmol, 1.261 g), previously ground into powder, was slowly added in portion, and a septum with an empty balloon was immediately placed into the flask in order to avoid overpressure and loss of generated singlet oxygen. The mixture was vigorously stirred at room temperature until total disappearance of the phenol. After about 1 h of stirring, the reaction was quenched with water and extracted several times (3–4 times) with ethyl acetate (EtOAc). Combined EtOAc extracts were dried in Na2SO4 and the solvent was removed under reduced pressure to yield the product of p-peroxyquinol, 2. The crude compound was recrystallised from EtOAc/ethanol mixture (3:1) to obtain a pure product of white crystalline solid.
The intermediate compound, p-peroxyquinol (4-methyl-4-hydroperoxycyclohexa-2,5-dienone) 2 (1 mmol) was added slowly into a mixture of aldehyde (1.5 mmol) and dichloromethane (4.0 ml), followed by the addition of an acid catalyst, p-toluenesulfonic acid (PTSA, 0.05 mmol). The reaction mixture was stirred at 45–5 °C till the completion of reaction (8–12 h) and then concentrated in vacuum. Colum chromatography (silica gel 60, 230–400 mesh) of the resulting residue was carried out using a solvent system of hexane:EtOAc (80:20) to obtain the desired pure product of target compounds, 3a–e, 3′a–n, 3″a–e as crystalline solids or semi-solids.
3-Ethyl-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3a)
UV spectrum (MeOH), λ max, nm: 212.8; IR (KBr, cm−1) υ: 2940.31, 2883.27 (C–H, CH3), 1656.48 (C=O, conj.), 1145.20 (C–O), 892.28 (O–O); 1H NMR (400 MHz, CDCl3) δ: 0.89 (t, J = 6.0 Hz, 3H, CH3), 1.21 (s, 3H, CH3), 1.43 (q, J = 6.0 Hz, 3H, CH 2–CH3), 2.46 (d, J = 3.0 Hz, 2H, CO–CH 2–CH), 4.12 (d, J = 3.0 Hz, 1H, CO–CH2–CH), 5.24 (t, J = 6.0 Hz, 1H, CH–CH2–CH3), 6.04 (d, J = 9.0, 1H, CO–CH=CH), 6.32 (d, J = 9.0, 1H, CO–CH=CH); 13C NMR (100 MHz, CDCl3) δ: 14.10, 18.24 (CH3), 22.90, 26.23 (CH2), 36.81, 42.37 (CH), 84.77, 92.40 (CH=CH), 188.27 (C=O); MS (ES+), m/z (%): 198.78 (100), [M]+, 182.12 (52), 176.12 (28), 152.38 (6); CHN anal. for C10H14O4 (198.22), calc. (%): C, 60.59, H, 7.12, O, 32.29, found (%): C, 61.09, H, 6.72, O, 31.69.
3-Butyl-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3c)
UV spectrum (MeOH), λ max, nm: 218.6; IR (KBr, cm−1) υ: 2923.67, 2888.36 (C–H, CH3), 1668.59 (C=O, conj.), 1177.34 (C–O), 886.56 (O–O); 1H NMR (400 MHz, CDCl3) δ: 0.92 (t, J = 6.0 Hz, 3H, CH3), 1.18 (s, 3H, CH3), 1.46 (m, 2H, CH2–CH 2–CH3), 1.52 (m, 2H, CH 2–CH2–CH3), 1.63 (m, 2H, CH2–CH2–CH2–CH3), 2.52 (d, J = 3.0 Hz, 2H, CO–CH 2–CH), 4.23 (d, J = 3.0 Hz, 1H, CO–CH2–CH), 6.06 (t, J = 6.0 Hz, 1H, CH–CH2–CH3), 6.09 (d, J = 10.0, 1H, CO–CH=CH), 6.34 (d, J = 9.0, 1H, CO–CH=CH); 13C NMR (100 MHz, CDCl3) δ: 15.53, 18.18 (CH3), 22.43, 28.10, 32.62 (CH2), 36.36, 46.21 (CH), 86.54, 93.26 (CH=CH), 187.60 (C=O); MS (ES+), m/z (%): 226.27 (100), [M]+, 182.12 (52), 176.12 (28), 152.38 (6); CHN anal. for C12H18O4 (198.22), calc. (%): C, 63.70, H, 8.02, O, 28.28, found (%): C, 64.42, H, 8.71, O, 27.78.
3-(2-Hydroxyphenyl)-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3′b)
UV spectrum (MeOH), λ max, nm: 284.2; IR (KBr, cm−1) υ: 3174.10 (O–H, bonded), 2970.21, 2879.23 (C–H, CH3), 1667.13 (C=O, conj.), 1595.12, 1514.23, 1423.14 (C=C, aryl), 1161.15 (C–O), 832.19 (O–O); 1H NMR (400 MHz, CDCl3) δ: 1.25 (s, 3H, CH3), 7.01 (1H, CO–CH=CH), 7.02 (1H, CO–CH=CH), 7.82 (Ar–H), 7.84 (Ar–H), 9.83 (Ar–OH); 13C NMR (100 MHz, CDCl3) δ: 15.42 (CH3), 29.72 (CH), 116.02, 129.69, 132.55, 161.79 (Ar–C), 191.34 (C=O); MS (ES+), m/z (%): 262.26 (100), [M]+, 230.38 (59), 192.37 (34), 176.03 (21); CHN anal. for C14H14O5 (262.26), calc. (%): C, 64.12, H, 5.38, O, 30.50, found (%): C, 64.62, H, 4.78, O, 30.00.
3-(3-Methoxyphenyl)-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3′c)
UV spectrum (MeOH), λ max, nm: 292.3; IR (KBr, cm−1) υ: 2956.33, 2880.16 (C–H, CH3), 1674.20 (C=O, conjugated), 1642.13, 1582.30, 1424.10 (C=C, aryl), 1152.82 (C–O), 878.41 (O–O); 1H NMR (400 MHz, CDCl3) δ: 1.19 (s, 3H, CH3), 2.43 (d, J = 4.0 Hz, 2H, CO–CH 2–CH), 4.02 (s, 3H, OCH3), 4.23 (d, J = 4.0 Hz, 1H, CO–CH2–CH), 6.04 (d, J = 10.0, 1H, CO–CH=CH), 6.37 (d, J = 10.0, 1H, CO–CH=CH), 7.03 (d, 1H, J = 7.0 Hz, Ar–H), 7.28 (d, J = 7.0 Hz, Ar–H), 7.44 (d, J = 8.0 Hz, Ar–H), 7.82 (d, 1H, J = 8.0 Hz, Ar–H); 13C NMR (100 MHz, CDCl3) δ: 15.42 (CH3), 29.12 (OCH3), 38.40 (CH), 84.65, 92.28 (CH=CH), 118.18, 126.12, 132.17, 140.72, 152.32, 162.25 (Ar–C), 190.20 (C=O); MS (ES+), m/z (%): 276.48 (100), [M]+, 223.01 (64), 182.98 (32), 142.04 (18); CHN anal. for C15H16O5 (276.28), calc. (%): C, 65.21, H, 5.84, O, 28.95, found (%): C, 65.72, H, 6.24, O, 28.55.
3-(4-Chlorophenyl)-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3′d)
UV spectrum (MeOH), λ max, nm: 255.0; IR (KBr, cm−1) υ: 2912.56, 2898.20 (C–H, CH3), 1672.17 (C=O, conj.), 1142.10 (C–O), 1054.36 (Ar. C–Cl), 878.12 (O–O); 1H NMR (400 MHz, CDCl3) δ: 1.16 (s, 3H, CH3), 2.41 (d, J = 4.0 Hz, 2H, CO–CH 2–CH), 4.22 (d, J = 4.0 Hz, 1H, CO–CH2–CH), 6.06 (d, J = 10.0, 1H, CO–CH=CH), 6.34 (d, J = 10.0, 1H, CO–CH=CH), 7.12 (d, 1H, J = 8.0 Hz, Ar–H), 7.46 (d, J = 8.0 Hz, Ar–H); 13C NMR (100 MHz, CDCl3) δ: 15.47 (CH3), 36.38 (CH), 89.42, 94.17 (CH=CH), 117.20, 128.13, 134.60, 140.44, 152.02, 160.12 (Ar–C), 188.00 (C=O); MS (ES+), m/z (%): 262.71 (100), [M]+, 242.12 (62), 222.13 (18), 154.72 (6); CHN anal. for C14H13O4Cl (280.70), calc. (%): C, 59.90, H, 4.67, O, 22.80, found (%): C, 60.50, H, 4.07, O, 20.41.
3-(4-Nitrorophenyl)-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3′e)
UV spectrum (MeOH), λ max, nm: 265.0; IR (KBr, cm−1) υ: 2932.21, 2887.12 (C–H, CH3), 1664.20 (C=O, conj.), 1543.69 (N–O, NO2), 1346 (N–O, NO2), 1142.78 (C–O), 873.11 (O–O); 1H NMR (400 MHz, CDCl3) δ: 1.20 (s, 3H, CH3), 2.44 (d, J = 3.0 Hz, 2H, CO–CH 2–CH), 4.62 (d, J = 3.0 Hz, 1H, CO–CH2–CH), 6.13 (d, J = 10.0, 1H, CO–CH=CH), 6.38 (d, J = 10.0, 1H, CO–CH=CH), 7.24 (d, 1H, J = 8.0 Hz, Ar–H), 7.74 (d, J = 8.0 Hz, Ar–H); 13C NMR (100 MHz, CDCl3) δ: 14.13 (CH3), 39.11 (CH), 86.70, 90.12 (CH=CH), 116.12, 124.34, 132.26, 138.25, 147.10, 158.33 (Ar–C), 192.12 (C=O); MS (ES+), m/z (%): 291.45 (100), [M]+, 272.11 (52), 237.18 (27), 192.16 (12); CHN anal. for C14H13NO6 (291.26), calc. (%): C, 57.73, H, 4.50, N, 4.81, O, 32.96, found (%): C, 58.32, H, 4.10, N, 4.21, O, 32.16.
3-(4-(Dimethylamino)phenyl)-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3′j)
UV spectrum (MeOH), λ max, nm: 242.0, 344.0; IR (KBr, cm−1) υ: 2928.15, 2876.42 (C–H, CH3), 1664.74 (C=O, conj.), 1623.16, 1590.12, 1482.49 (C=C, aryl), 1278.32 (C–N), 1164.23 (C–O), 846.45 (O–O); 1H NMR (400 MHz, CDCl3) δ: 1.22 (s, 3H, CH3), 2.18 (d, J = 3.0 Hz, 2H, CO–CH 2–CH), 2.82 (d, J = 12.0 Hz, 3H, N–(CH3)2), 2.96 (d, J = 12.0 Hz, 3H, N–(CH3)2), 4.24 (d, J = 3.0 Hz, 1H, CO–CH2–CH), 4.88 (t, J = 7.0 Hz, 1H, CH–CH2–CH3), 6.23 (d, J = 9.0, 1H, CO–CH=CH), 6.36 (d, J = 9.0, 1H, CO–CH=CH), 7.22 (d, 1H, J = 8.0 Hz, Ar–H), 7.68 (d, J = 8.0 Hz, Ar–H); 13C NMR (100 MHz, CDCl3) δ: 14.25, 15.60, 18.13 (CH3), 38.03 (CH), 86.40, 89.34 (CH=CH), 116.45, 128.02, 132.57, 136.44, 150.13, 160.11 (Ar–C), 190.34 (C=O); MS (ES+), m/z (%): 289.56 (100), [M]+, 262.22 (60), 232.17 (28), 182.03 (11); CHN anal. for C16H19NO4 (289.33), calc. (%): C, 66.42, H, 6.62, N, 4.84, O, 22.12, found (%): C, 66.82, H, 6.12, N, 4.98, O, 21.52.
3-(4-Hydroxy-3-methoxyphenyl)-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3′l)
UV spectrum (MeOH), λ max, nm: 277.6, 305.4; IR (KBr, cm−1) υ: 3269.19 (O–H, bonded), 3089.21 (C–H, aryl), 2972.24, 2944.25 (C–H, CH3), 1679.11 (C=O, conj.), 1642.14, 1593.10, 1510.14 (C=C, aryl), 1043.31 (C–O), 820.17 (O–O); 1H NMR (400 MHz, CDCl3) δ: 1.18 (s, 3H, CH3), 3.94 (s, 3H, OCH 3), 7.03 (1H, CO–CH=CH), 7.05 (1H, CO–CH=CH), 7.42 (Ar–H), 7.43 (Ar–H), 9.81 (Ar–OH); 13C NMR (100 MHz, CDCl3) δ: 56.08 (OCH3), 108.84, 114.47, 127.61, 129.73, 147.23, 151.84 (Ar–C), 191.11 (C=O); MS (ES+), m/z (%): 292.80 (100), [M]+, 266.34 (62), 218.15 (36), 178.36 (12); CHN anal. for C15H16O6 (292.28), calc. (%): C, 61.64, H, 5.52, O, 32.84, found (%): C, 62.04, H, 5.93, O, 32.26.
3-(3-Formylphenyl)-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3′n)
UV spectrum (MeOH), λ max, nm: 226.4, 244.6; IR (KBr, cm−1) υ: 2932.12, 2882.00 (C–H, CH3), 2812.65, 2773.26 (C–H, CHO), 1672.45 (C=O, conjugated), 1632.12, 1568.23, 1534.00, 1482.34 (C=C, aryl), 1163.67 (C–O), 879.34 (O–O); 1H NMR (400 MHz, CDCl3) δ: 1.34 (s, 3H, CH3), 2.19 (d, J = 3.0 Hz, 2H, CO–CH 2–CH), 4.24 (d, J = 3.0 Hz, 1H, CO–CH2–CH), 6.13 (d, J = 10.0, 1H, CO–CH=CH), 6.32 (d, J = 10.0, 1H, CO–CH=CH), 7.10 (t, J = 8.0 Hz, 1H, Ar–H), 7.23 (d, J = 7.0 Hz, 1H, Ar–H), 7.44 (d, J = 8.0 Hz, 1H, Ar–H), 7.53 (s, 1H, Ar–H), 9.71 (CHO); 13C NMR (100 MHz, CDCl3) δ: 14.87 (CH3), 84.60, 92.30 (CH=CH), 112.43, 116.29, 122.70, 132.67, 148.70, 156.13 (Ar–C), 189.76 (C=O); MS (ES+), m/z (%): 274.27 (100), [M]+, 243.37 (54), 210.00 (32), 189.56 (14); CHN anal. for C15H14O5 (274.27), calc. (%): C, 65.69, H, 5.15, O, 29.17, found (%): C, 66.49, H, 5.34, O, 28.56.
3-(1H-pyrrol-2-yl)-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3″c)
UV spectrum (MeOH), λ max, nm: 242.6, 259.2; IR (KBr, cm−1) υ: 3247.01 (NH, 1H-pyrrol-3-yl), 2920.12, 2889.12 (C–H, CH3), 1657.26 (C=O, conj.), 1123.05 (C–O), 874.11 (O–O); 1H NMR (400 MHz, CDCl3) δ: 1.32 (s, 3H, CH3), 2.17 (d, J = 3.0 Hz, 2H, CO–CH 2–CH), 4.26 (d, J = 3.0 Hz, 1H, CO–CH2–CH), 6.17 (d, J = 10.0, 1H, CO–CH=CH), 6.38 (d, J = 10.0, 1H, CO–CH=CH), 7.01 (d, J = 8.0 Hz, 1H, 1H-pyrrole-H3), 7.12 (t, 1H, J = 8.0 Hz, 1H-pyrrole-H4), 7.42 (t, 1H, J = 8.0 Hz, pyrrole-H5), 7.67 (s, 1H, NH, 1H-pyrrole-H1); 13C NMR (100 MHz, CDCl3) δ: 15.32 (CH3), 86.52, 88.46 (CH=CH), 107.23, 114.26, 118.40, 122.35 (Ar–C, 1H-pyrrole-2-yl), 186.38 (C=O); MS (ES+), m/z (%): 235.44 (100), [M]+, 182.48 (39), 136.59 (24), 84.72 (7); CHN anal. for C12H13NO4 (235.24), calc. (%): C, 61.27; H, 5.57; N, 5.95; O, 27.21, found (%): C, 62.06, H, 5.22, N, 5.22, O, 26.68.
3-(1H-indol-3-yl)-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3″d)
UV spectrum (MeOH), λ max, nm: 288.4, 295.8; IR (KBr, cm−1) υ: 3358.34 (NH, 1H-indol-3-yl), 3066.28, 3040.28 (C–H, aryl), 2872.17 (C–H, CH3), 1694.10 (C=O, conj.), 1481.36, 1450.25 (C=C, conj., aryl), 1144.17 (C–O), 865.45 (O–O); 1H NMR (400 MHz, CDCl3) δ: 1.24 (s, 3H, CH3), 7.23 (1H, CO–CH=CH), 7.21 (1H, CO–CH=CH), 7.46 (d, J = 7.0 Hz, 1H, 1H-indole-H4), 7.48 (1H, J = 7.0 Hz, 1H-indole-H5), 8.08 (1H, J = 8.0 Hz, 1H-indole-H6), 8.15 (s, J = 9.0 Hz, 1H, 1H-indole-H7), 8.17 (s, 1H, 1H-indole-H2), 9.87 (s, 1H, NH, 1H-indole-H1); 13C NMR (100 MHz, CDCl3) δ: 113.15, 120.08, 122.39, 123.63, 125.02, 125.68, 139.77, (Ar–C, 1H-indole-3-yl), 187.44 (C=O); MS (ES+), m/z (%): 285.47 (100), [M]+, 253.12 (43), 226.10 (25), 187.19 (14); CHN anal. for C16H15NO4 (285.29), calc. (%): C, 67.36, H, 5.30, N, 4.91, O, 22.43, found (%): C, 67.96, H, 4.70, N, 5.41, O, 17.95.
3-(Pyridin-4-yl)-8a-methyl-4a,5-dihydrobenzo-1,2,4-trioxin-6(8aH)-one (3″e)
UV spectrum (MeOH), λ max, nm: 296.8; IR (KBr, cm−1) υ: 3023.22, 3010.23 (C–H, aryl), 2932, 2822.11 (C–H, CH3), 1667.34 (C=O, conj.), 1481.36, 1450.25 (C=C, conj., aryl), 1154.20 (C–O), 872.13 (O–O); 1H NMR (400 MHz, CDCl3) δ: 1.26 (s, 3H, CH3), 2.63 (d, J = 4.0 Hz, 2H, CO–CH 2–CH), 4.32 (d, J = 4.0 Hz, 1H, CO–CH2–CH), 6.21 (d, J = 8.0, 1H, CO–CH=CH), 6.40 (d, J = 8.0, 1H, CO–CH=CH), 7.48 (d, J = 8.0 Hz, 1H, 4-pyridyl-H2/6), 7.68 (1H, J = 8.0 Hz, 4-pyridyl-H3/5); 13C NMR (100 MHz, CDCl3) δ: 15.65 (CH3), 84.72, 92.68 (CH=CH), 116.20, 122.58, 128.34, 140.67 (Ar. C, 4-pyridyl), 188.42 (C=O); MS (ES+), m/z (%): 247.47 (100), [M]+, 204.78 (67), 172.09 (38), 156.20 (24); CHN anal. for C13H13NO4 (247.25), calc. (%): C, 63.15; H, 5.30; N, 5.67; O, 25.88, found (%):C, 63.87; H, 5.06; N, 6.12; O, 25.06.
Evaluation of antimalarial activity
Chemicals and parasite strains
All the synthesised compounds, 3a–e, 3′a–n, 3″a–e were evaluated for in vitro antimalarial activity at multiple doses against CQ-sensitive (3D7) and CQ-resistant (RKL9) strains of P. falciparum using blood (erythrocytic) stage of parasite. The solvents and reagents used in the antimalarial study were of analytical grade and were procured from Sigma-Aldrich Corporation (St. Louis, MO) and HiMedia Lab. Pvt. Ltd. (Einhausen, Germany).
Assay procedure
The in vitro antimalarial activity screening was carried out by microculture Giemsa Stained slide method based on light microscopy. Schizont Maturation Inhibition assay technique was used for the quantitative assessment of parasitaemia and evaluation of drug sensitivity. The assay was conducted using the blood parasite of in vitro P. falciparum culture according to the method described by Trager and Jensen 18 , 19 . Briefly, a continuous culture of P. falciparum strain was maintained in vitro in A+ human red blood cells (RBCs) diluted to 5% haematocrit (HCT) in RPMI (Roswell Park Memorial Institute)-1640 medium supplemented with 25 mM HEPES [(N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid)] buffer, 1% d-glucose, 5.0% sodium bicarbonate, gentamycin (40 μg/ml), and 10% human AB+ serum. Incubations were done at 37 °C and 5% CO2 level in a modular incubator. The culture was passaged with fresh mixture of erythrocytes and complete RPMI medium for every day to maintain cell growth. d-Sorbitol (5%) synchronised 1% ring stage parasitaemia in 5% HCT was used for the antimalarial assay using 96 well microtitre plate. A stock solution of 1.0 mg/ml of the test compound was prepared in DMSO and subsequent dilutions were made with incomplete RPMI to obtain different concentrations (50, 25, 12.5, 6.25, 3.13, 1.56, 0.78, 0.39, and 0.19 µg/ml) of samples (test/standard) in duplicate. In addition, drug free negative control to assess the parasite growth and chloroquine diphosphate as positive control to assess the integrity of the assay were also maintained in duplicate in the microtitre plate. After 40 h of incubation at 37 °C, the smears were prepared from each well (depending on the maturation of schizonts in negative control, ≥10%), stained with 3% Giemsa and examined under light microscope to ascertain drug sensitivity by assessing the level of parasitaemia, i.e. the inhibition of parasite growth in terms of percent inhibition of schizonts’ maturation 20 , 21 .
Observation and assessment of activity
Each test compound was assayed in duplicate and number of schizonts was counted against 200 asexual parasites per replica under light microscope. The number of schizonts count was also assessed in negative control maintained in duplicate in the microtitre plate. Mean number of schizonts for duplicate observations was calculated at each concentration of test/standard samples compounds. Test values were compared with the control values. The percentage inhibition of parasite growth for each concentration was calculated as inhibition =100 – A, where A is the percentage inhibition of schizonts in test wells, which was determined using the following formula:
Further, the IC50 (concentration at which the inhibition of parasite growth represents 50%) values in µg/ml were also calculated using the NonLin v1.1 software 22 . Test results were compared with the standard results of CQ.
In silico studies
Molecular docking study
Molecular modelling studies were carried out using Dell Precision work station T3400 running Intel Core2 Duo Processor, 4 GB RAM, 250 GB hard disk and NVidia Quodro FX 4500 graphics card. Two-dimensional (2D) structures of all compounds were built on Chemdraw Ultra 10.0 (Cambridge Soft Co., Cambridge, MA, 2010) and Marvin Sketch (ChemAxon LLC, Cambridge, MA, 2015) software. The 2D structures were later transformed into 3D structures using the converter module of Biovia Discovery Studio (DS) v 4.5 (2015) software (San Diego, CA). The compounds were prepared using the prepare ligand module of the DS which includes assigning bond orders, generating various tautomers, ring conformations and stereochemistries. All the conformations generated were then energetically minimised by a single step Steepest Descent method using CHARMM (Chemistry at HARvard Macromolecular Mechanics, Cambridge, MA) Force Field with 5000 iterations and a minimum root mean square (RMS) gradient of 0.01 kcal/mol/Å 23 .
The X-ray crystal structure of falcipain 2-E-64 (PDB id: 3BPF) was retrieved from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/) and Chain A of the protein determined at a resolution of 2.9 Å was used in the study 24 . Prior to docking, protein was prepared using protein preparation wizard tool of DS. Polar hydrogen atoms were added to the proteins and charges were assigned. All bound water molecules, other heteroatoms and ligands were excluded from the crystal structure as they were not significant for the proteins’ function. Subsequently, the 3D structure of protein was optimised by energy minimisation using CHARMM Force Field. It was done in two steps to remove the bad steric clashes using Steepest Descent and Conjugate Gradient methods for 5000 steps at RMS gradients of 0.01 and 0.05 kcal/mol/Å, respectively 23 .
Molecular docking studies were carried out using Biovia Discovery Studio (DS) v 4.5 (2015) software (San Diego, CA). After energy minimisation, the Chain A of falcipain 2 protein was defined as a receptor and the binding site sphere was selected based on the ligand binding location of E-64. A receptor grid was thereby generated around the binding cavity (active sites) of protein by specifying the key amino acid residues (Cys 42, Gly 83, and His 174) 24 . In DS, binding site sphere was set with a radius of 20 Å and x, y, z dimensions of –52.25, –4.46, –19.25, respectively. Flexible molecular docking was performed where the protein was held rigid while the ligands were allowed to be flexible during refinement. During docking, the falcipain 2-E-64 complex was imported and E-64 molecule (co-crystal ligand) was removed, and ligands were placed in the predicted binding site (grid box) and docking was performed using the Dock Ligands module of LibDock genetic algorithm program 25 of DS. The docking parameters were as follows: No. of hotspots 100, docking tolerance 0.25, No. of runs 100, No. of iterations 1000, conformation method best, RMS gradient 0.01. All other docking and consequent scoring parameters used were kept at their default settings. The LibDock scores of the docked ligands were calculated. Different dock poses were studied to know the best binding mode of receptor–ligand complex in terms of scoring function. All docked poses were scored, ranked and the best pose of each compound having the highest score was selected and it was later used for the receptor–ligand interaction analysis. Interaction of ligands with receptor was studied to know the best binding orientation of receptor–ligand complex having maximum LibDock score. Binding modes of the best pose for each compound was also analysed with the help of 3D receptor–ligand complex. Different non-bonding interactions (hydrogen bonding and hydrophobic) were also analysed with the help of 2D diagram of docked receptor–ligand complexes. Receptor–ligand interaction gives better understanding of the molecular interactions between the binding site residues of receptor molecule and complimentary groups/atoms of ligands involved in interactions.
Molecular properties calculation and drug-likeness studies
In silico calculations of the molecular properties and drug-likeness parameters for all compounds, 3a–e, 3′a–n, 3″a–e were performed based on theoretical approaches to identify the compounds which violate the optimum requirements for drug-likeness. Molecular properties (molecular weight, LogP value, number of hydrogen bond acceptor(s) (HBA), number of hydrogen bond donor(s) (HBD), total polar surface area) incorporated in Lipinski’s rule of five 25 and other physicochemical parameters like aqueous solubility (LogS), molar refractivity (MR), and molar volume (MV) were calculated using Calculation of Molecular Properties module of Biovia DS v 4.5 software (San Diego, CA). The number of rotatable bonds was predicted using Molsoft Online software (http://www.molsoft.com/, 2016) (San Diego, CA), and non-violation of drug-likeness and drug-likeness score was calculated using Molinspiration online software (http://www.molindpiration.com/, 2016) (Slovensky Grob, Slovakia).
ADMET prediction
ADME-Toxicity (ADMET) for all the target compounds was calculated in silico using ADMET descriptor module of Biovia DS v 4.5 software (San Diego, CA). Six mathematical models (aqueous solubility, blood–brain barrier (BBB) penetration, cytochrome P450 2D6 inhibition, hepatotoxicity, human intestinal absorption, and plasma protein binding) were used to quantitatively predict properties related to ADMET characteristics or pharmacokinetics of molecules 26 . These properties influence oral bioavailability, cell permeation, and metabolism of drug molecules.
Results and discussion
Design of compounds
Rationale behind the design strategy involved selection of 1,2,4-trioxane ring system as the parent peroxide scaffold considering the pharmacodynamic importance of 1,2,4-trioxane component for antimalarial activity of ART and related endoperoxides. Our study was aimed at developing 1,2,4-trioxane derivatives as newer synthetic peroxide-based antimalarial agents having comparatively simpler molecular framework than conventional antimalarial drug molecules with activity against resistant malaria parasites. Three series of target compounds (Figure 2) were designed with diverse substitution patterns (alkyl/aryl/heteroaryl groups) at C-3 position of the 1,2,4-trioxane structural scaffold, based on the molecular manipulation approach of drug design with due consideration to all structure and property parameters that are relevant to biological activity. The 1,2,4-trioxane ring system was considered as the basic requirement for the antimalarial activity and substitutions with groups/moieties having electronic property of (s) was the main motif behind the design task.
Figure 2.

Design of 1,2,4-trioxane derivatives.
Chemistry
In our study, three new series of 1,2,4-trioxane derivatives were synthesised and evaluated in vitro for their antimalarial activity. Substitutions with different aliphatic, aromatic, and heteroaromatic groups at the C-3 position of 1,2,4-trioxane ring system afforded target trioxane derivatives obtained as single drug conjugates. The standard reaction procedure 11 involving a facile synthetic route depicted in Figure 3 was employed for the preparation of 1,2,4-trioxane derivatives. The oxidative dearomatisation of p-cresol, 1 with oxone first yielded the intermediate compound, p-peroxyquinol, 2 which was later allowed to react with different aldehydes (aliphatic/aromatic/heteroaromatic) in the presence of an acid catalyst called PTSA, to obtain desired trioxane derivatives, 3a–e, 3′a–n, 3″a–e. The first step of reaction was a [4 + 2] cycloaddition between electron rich p-alkyl phenol and 1O2 (singlet oxygen) generated in situ in the reaction system from oxone, which gave p-peroxyquinol intermediate through a 1,4-endoperoxide, i.e. peroxyhemiacetal. Subsequent desymmetrisation of p-peroxyquinol after reaction with aldehydes with the help of PTSA catalyst yielded different trioxane derivatives (Figure 4) 17 . The progress of reactions was monitored by the silica gel-G thin-layer chromatography (TLC) and the spots were visualised by iodine vapours. The purity of the synthesised compounds was ascertained by MP determinations and silica gel G TLC. All the compounds were obtained in good yields with high purity. The intermediate compound, 4-hydroperoxy-4-methyl-2,5-cyclohexadien-1one was obtained as white solid in 89% yield with melting range of 102–104 °C and R f value of 7.8 (cyclohexane:EtOAc:acetic acid = 1:2:0.5). The physicochemical details of synthesised compounds are summarised in Table 1. The spectral (UV, IR, 1HNMR, 13CNMR, Mass) and analytical data (of representative compounds) depicted in the Experimental section are in close agreement with the structures of synthesised compounds. In UV spectra (in methanol), λ max values were observed at the range of 212.8–344.0 which was due to the presence of chromophoric trioxane ring system along with alkyl/aryl/heteroaryl substituents at the C-3 position of the parent scaffold. IR spectral data showed absorption frequencies characteristic to specific functional groups and/or bonds present in the structure of synthesised compounds. The appearance of a broad peak at 3174.10 and 3269.19 cm−1 confirmed the presence of phenolic –OH group in the structure of 3′b and 3′l, respectively. N–H stretching band of 1H-pyrrol-2-yl and 1H-indol-3-yl systems was observed at 3247.01 and 3358.34 cm−1, respectively. The carbonyl (C=O) group was assigned with a strong and sharp absorption band in the region of 1694.10–1656.48 cm−1 for all the synthesised compounds. The characteristic peak due to N–O stretching (NO2) was also appeared for the compound 3′e by a sharp peak of strong intensity at 1543.69 cm−1. A weak band at frequency of 1278.32 cm−1 was explicated due to the C–N stretch for compound 3′j. Distinguished C–O stretching band appeared in the range of 1043.24–1177.34 cm−1. A peak of weak intensity observed at 1054.36 cm−1 was due to the aromatic C–Cl stretch for compound 3′d. A weak absorption band observed at 892.28–820.17 cm−1 was due to O–O stretching. The presence of aliphatic C–H (CH3), aromatic C–H (CH=C), and aromatic C=C groups in the structure of synthesised compounds was also confirmed by characteristic peaks as depicted in the Experimental section.
Figure 3.

Scheme of synthesis of target compounds, 3a–e, 3′a–n, 3″a–e. Reagents and conditions: (a) Oxone, NaHCO3/CH3CN/H2O, rt; (b) RCHO, 40–50 °C, 8–12 h, CH2Cl2, PTSA.
Figure 4.

Reaction mechanism of synthesis of 1,2,4-trioxane derivative(s).
Table 1.
Physicochemical data of synthesised compounds.
| Comp. | R | Colour (state) | % Yield | MP (°C) | Rf a |
|---|---|---|---|---|---|
| 3a | Ethyl | Colourless (solid) | 78 | 112–114 | 0.70 |
| 3b | Propyl | Colourless (solid) | 74 | 118–122 | 0.72 |
| 3c | Butyl | Brown (semi-solid) | 69 | – | 0.74 |
| 3d | Pentyl | Brown (semi-solid) | 66 | – | 0.74 |
| 3e | 1-Formyl-but-4-yl | Brown (semi-solid) | 65 | – | 0.67 |
| 3′a | Phenyl | Colourless (solid) | 72 | 122–124 | 0.72 |
| 3′b | 2-Hydroxyphenyl | Off-white (solid) | 83 | 128–130 | 0.76 |
| 3′c | 3-Methoxyphenyl | Pale yellow (solid) | 74 | 145–147 | 0.72 |
| 3′d | 4-Chlorophenyl | Colourless (solid) | 84 | 136–140 | 0.74 |
| 3′e | 4-Nitrophenyl | Yellow (solid) | 80 | 156–158 | 0.72 |
| 3′f | 4-Tolyl | Colourless (solid) | 69 | 142–144 | 0.78 |
| 3′g | 4-Bromophenyl | Brown (semi-solid) | 68 | – | 0.68 |
| 3′h | 4-Fluorophenyl | Brown (semi-solid) | 66 | – | 0.68 |
| 3′i | 1-Naphthyl | Brown (semi-solid) | 70 | – | 0.70 |
| 3′j | 4-Dimethylaminophenyl | Pale yellow (solid) | 78 | 168–170 | 0.72 |
| 3′k | 3-Cinnamyl | Yellow (solid) | 65 | – | 0.66 |
| 3′l | 4-Hydroxy-3-methoxyphenyl | Colourless (solid) | 82 | 148–150 | 0.76 |
| 3′m | 3,4-Dimethoxyphenyl | Colourless (solid) | 76 | 128–130 | 0.74 |
| 3′n | Isophthalyl | Colourless (solid) | 69 | 134–136 | 0.79 |
| 3″a | Furan-2-yl | Dark brown (semi-solid) | 68 | – | 0.65 |
| 3″b | Thiophen-2-yl | Light brown (semi-solid) | 66 | – | 0.66 |
| 3″c | Pyrrole-2-yl | Light yellow (solid) | 64 | 145–147 | 0.67 |
| 3″d | Indole-3-yl | Colourless (solid) | 75 | 157–160 | 0.74 |
| 3″e | Pyridin-4-yl | Brown (semi-solid) | 68 | – | 0.68 |
Cyclohexane:EtOAc:acetic acid (1:2:0.5).
1HNMR spectra exhibited resonance signals with chemical shift (δ, ppm) values characteristic to various structural protons which concorded the anticipated structure of synthesised compounds (Supplementary material). A singlet for three protons of CH3 group of the trioxane ring system was observed in the range of 1.16–1.34 ppm. For olefinic protons (CH=CH) peaks at 2.17–2.63 and 4.02–4.32 ppm were assigned, while for other skeletal protons like CO–CH2CH were assigned at 6.04–6.23 and 6.32–6.40 ppm. Depending on the nature of the substituent at C-3 position of the trioxane ring, distinct δ values were also observed with expected splitting patterns (doublets, triplets, or multiplets) and coupling constant (J) values. A broad singlet at 9.71 and 9.81 ppm was observed due to aromatic OH proton for compounds, 3′b and 3′l, respectively. The CHO proton in compound 3′h was appeared at 9.71 ppm. Distinct singlet due to three protons of the OCH3 group appeared at 4.02 and 3.94 ppm for compounds 3′c and 3′l, respectively. Aromatic protons including heteroaryl substituents appeared in the range of 7.01–8.15 ppm. A broad singlet was also assigned to NH proton at 7.67 ppm and 9.87 ppm for compounds 3″c and 3″d, respectively. Aliphatic methyl and methylene protons were observed in multiplet pattern (doublet or triplet) at δ values in the downfield region for compounds 3a and 3c, as depicted in the Experimental section 28 .
In 13C NMR spectra, distinguished resonance signal due to the carbonyl (>C=O) carbon appeared in the range of 188.27–192.12 ppm. Aromatic/heteroaromatic carbons appeared at 107.23–162.25 ppm. Characteristic downfield signals for aliphatic carbons were observed for CH3 (14.10–15.65 ppm), CH2 (22.43–32.62 ppm), OCH3 (56.08 ppm, 3′l; 29.12 ppm, 3′c), and CH (36.36–92.68 ppm) groups. Olefinic (CH=CH) carbons were also assigned and depicted in the Experimental section. The mass spectra of 3a–e, 3′a–n, 3″a–e exhibited prominent molecular ion peaks, [M]+ which finally confirmed the structure of compounds with the anticipated mass corresponding to their respective molecular formula. The results of elemental analyses were within the acceptable limits (±0.5%) of the calculated values for all the synthesised compounds.
Antimalarial activity
The in vitro antimalarial activity data are summarised in Table 2. Results of in vitro antimalarial activity revealed that all the synthesised compounds (3a–e, 3′a–n, 3″a–e) showed activity against both CQ-sensitive (3D7) and CQ-resistant (RKL9) strains of P. falciparum in a dose dependant manner at screening doses. The IC50 values of tested compounds range from 1.24 µM to >1000 µM and 1.06 µM to >1000 µM against CQ-sensitive and CQ-resistant strains of P. falciparum, respectively. Out of 24 tested compounds, five compounds (3d, 3e, 3′k, 3″a, and 3″b) showed poor activity with IC50 values of >1000 µM against both the strains. Compounds 3a–3c, 3′a, 3′d–3′j, 3′m, 3′n, and 3″c exhibited moderate activity with IC50 values ranging from 18.37 to 220.20 µM and 14.72 to 220.68 µM against sensitive and resistant strains, respectively. Compounds, 3b, 3c, 3′a, 3′e, 3′h, and 3′j showed better activity against sensitive strain than the resistant one. Among three series of compounds, five compounds (3′b, 3′c, 3′l, 3″d, and 3″e) showed good activity, three compounds (3′b, 3′l, and 3″d) of which exhibited better activity against resistant strain than the sensitive one. These five compounds were found comparatively more potent than rest of the synthesised analogues. The IC50 values of two best compounds (3′l and 3″d), one (3′l) from aryl series and the other (3″d) from heteroaryl series were found to be 1.24 µM and 1.24 µM, respectively, against sensitive strain of P. falciparum. The IC50 values of the two compounds against resistant strain were found to be 1.06 µM and 1.17 µM, respectively. Results were comparable with that of the standard drug, CQ (IC50=1.23 µM and 47.16 µM against sensitive and resistant strain of P. falciparum, respectively). Figure 5 shows photographs of the effect of the most active compound, 3′l against both CQ-sensitive (3D7) and CQ-resistant (RKL9) strains of P. falciparum.
Table 2.
In vitro antimalarial activity data.
| IC50 in µg/ml (µM)
a
|
||
|---|---|---|
| Comp. | Pf 3D7 | Pf RKL9 |
| 3a | 0.026 (26.20) | 0.028 (28.46) |
| 3b | 0.020 (20.44) | 0.022 (22.26) |
| 3c | 0.16 (160.47) | 0.12 (120.13) |
| 3d | 3.48 (>1000) | 4.09 (>1000) |
| 3e | 3.46 (>1000) | 4.07 (>1000) |
| 3′a | 0.068 (68.46) | 0.066 (66.19) |
| 3′b | 0.0054 (5.43) | 0.0056 (5.68) |
| 3′c | 0.0074 (7.62) | 0.0078 (7.83) |
| 3′d | 0.032 (32.49) | 0.032 (32.54) |
| 3′e | 0.040 (40.78) | 0.036 (36.42) |
| 3′f | 0.052 (52.06) | 0.054 (54.17) |
| 3′g | 0.058 (58.20) | 0.058 (58.34) |
| 3′h | 0.040 (40.21) | 0.036 (36.67) |
| 3′i | 0.074 (74.22) | 0.078 (78.28) |
| 3′j | 0.018 (18.37) | 0.014 (14.72) |
| 3′k | 1.20 (>1000) | 1.48 (>1000) |
| 3′l | 0.0012 (1.24) | 0.0010 (1.06) |
| 3′m | 0.056 (56.09) | 0.064 (64.53) |
| 3′n | 0.045 (45.29) | 0.048 (48.20) |
| 3″a | 2.07 (>1000) | 5.02 (>1000) |
| 3″b | 3.50 (>1000) | 3.61 (>1000) |
| 3″c | 0.22 (220.20) | 0.22 (220.68) |
| 3″d | 0.0012 (1.24) | 0.0011 (1.17) |
| 3″e | 0.0072 (8.27) | 0.0065 (6.52) |
| CQ | 0.0012 (1.23) | 0.147 (47.16) |
Calculated using NonLin v1.1 software, mean of two replicate observations (counted against 200 cells per replicate), values in parentheses indicate activity in micromolar (µM) concentration.
Figure 5.

Photographs showing in vitro antimalarial activity of the most active compound, 3′l against (a) CQ-sensitive 3D7 and (b) CQ-resistant RKL9 strains of P. falciparum.
Results reveal that the degree of antimalarial (inhibitory) activity of 1,2,4-trioxane derivatives is dependent on the nature and degree of substitution pattern. Different structural substitutions such as non-bulky alkyl and bulky aryl/heteroaryl groups at C-3 position of the ring system modulate the antimalarial efficacy of prepared 1,2,4-trioxane derivatives. Some degree of variations in activity among synthesised analogues might be due to diverse structural substitutions. A brief SAR study can be depicted as follows: small alkyl substituents like ethyl, propyl, or butyl moieties are of considerable importance for the activity, while substitution with larger alkyl like pentyl and 1-formyl-but-4-yl moieties is comparatively less important for the activity. Substitution with bulky aryl moieties except 3-cinnamyl group also contributes good antimalarial potency to same extent or little less or even more compared to alkyl substituted analogues. Compounds with phenyl, 4-chlorophenyl, 4-nitrophenyl, 4-tolyl, 3,4-dimethoxyphenyl, and isophthalyl substitutions sufficiently retain the activity, whereas, in case of 2-hydroxyphenyl, 3-methoxyphenyl, 4-hydroxy3-methoxyphenyl substituted analogues the activity significantly increases. With bulky heteroaryl moieties like furan-2-yl and thiophen-2-yl substitutions, the activity declines, while, pyrrole-2-yl and pyridine-4-yl improve activity, and the indole-3-yl substitution still potentiates the activity. It is also clear that compounds with five membered heterocyclic rings are less important for the activity than six membered or fused heteroaryl systems. Upon critical analysis of SAR, the most notable point found is that substitutions with electron withdrawing moieties are likely less important for the activity, while electron releasing groups substituents are considerably more important for activity. More illustratively, phenyl substituent with electron releasing (OH, OCH3, etc.) groups significantly increases the activity, whereas, electron-withdrawing group (Cl, NO2, etc.) substituted phenyl ring reduces the activity (Figure 6). Substitutions of electron releasing groups at ortho and para or both have greater contributing effects than meta substitutions. Di-substitution (4-hydroxy-3-methoxyphenyl, compound 3′l) is more important for the activity than mono-substitution (2-hydroxyphenyl, compound 3′b). The findings of our present study are also consistent with earlier report 28 on trioxane-based compounds which show excellent antiparasitic activity upon incorporation of an aryl group. It is seen that bulkiness of substituents is important for the activity. However, aryl substitution seems to be more important than alkyl or heteroaryl substitution. It is important to note here that besides bulkiness of substituents, lipophilicity (hydrophobicity) and ionisation potential (basicity) of the molecule are also considered to play a crucial role for the activity. The lipophilicity (LogP) is inevitable for permeation of parasitic membrane and basicity (pKa) is required for accumulation of trioxane molecule in the acidic food vacuole of parasites.
Figure 6.

Structural requirements and effect of substitutions on antimalarial activity of 1,2,4-trioxane derivatives.
Literature report that 1,2,4-trioxane structure lacking the lactone ring plays an important role for the antimalarial activity, but 1,2,4-trioxane ring alone is insufficient for optimal antimalarial potency as compared to ART and its semi-synthetic analogues 29 . The reductive cleavage of the peroxide bond in vivo by heme [Fe(II)] (released during haemoglobin degradation process in parasitised RBC) and subsequent production of cytotoxic carbon-centred radicals target cell proteins/enzymes and destroy parasites 30–32 . From the SAR study, possible mode(s) of antimalarial action (Figure 7) of 1,2,4-trioxane derivatives can therefore be explained as: target compounds may act either as direct inhibitor of enzyme (receptor antagonist) or as prodrug by interfering the haemoglobin degradation mechanism during erythrocytic stages of P. falciparum. Electronic property of the substituent may play a significant role for the bio-activation (through O–O cleavage) of trioxane ring system to generate highly reactive oxyl radical which causes death of parasite by lipid peroxidation of parasitic cell membrane/protein.
Figure 7.

Hypothetical mode(s) of antimalarial action of 1,2,4-trioxane derivatives. (1) Inhibition of haemoglobin degrading enzyme in food vacuole of parasite, (2) direct lethal action to parasite by LPO process. Pf HDE: P. falciparum haemoglobin degrading enzyme; LPO: lipid peroxidation.
Docking simulations
Plasmodium cysteine protease falcipain 2 enzyme plays an essential role in the degradation of host haemoglobin into smaller peptides which takes place in the acidic food vacuole of P. falciparum. Recent literature 33 claims that endoperoxide antimalarials have potential to inhibit falcipain 2 enzyme and therefore, drug targeting of this particular enzyme would be an attractive avenue in the design and development of new antimalarial drugs. Accordingly, a molecular docking study was performed for all newly designed compounds 3a–e, 3′a–n, 3″a–e using the falcipain 2 enzyme as receptor molecule.
The protein model used for docking study was validated as follows: the 3D crystal structure of falcipain 2 co-crystallised with the inhibitor trans-epoxysuccinyl-L-leucylamido-(4-guanidino) butane (E-64) with active site (receptor grid model) as defined by Cys 42, Gln 36, and His 174 residues was optimised and used for the study. The co-crystal structure of falcipain 2-E-64 (PDB: 3BPF) and the receptor grid model used for docking study are represented in Figure 8. Co-crystallised ligand, E-64 was re-docked using flexible docking simulations (LibDock program of DS) into the original structure of the target protein, falcipain-2 by non-covalent docking procedure. For this study, docking parameters were set to the software’s default values. E-64 was successfully re-docked to the predicted active sites of falcipain 2 with an acceptable RMSD value of 1.124 Å. Further, in order to reproduce an experimentally observed ligand-binding mode, the co-crystallised ligand, E-64 (a selective falcipain 2 inhibitor) was used as reference ligand. Results confirmed experimental binding conformations of E-64 in the binding pocket of receptor molecule. The binding modes and ligand–protein interaction diagrams obtained in re-docking of E-64 are shown in Figure 9(a).
Figure 8.

(a) Optimised co-crystal structure of falcipain 2 (chain A)-E-64 and (b) receptor grid for docking.
Figure 9.

(a) Redocked conformer (pose) of E-64 in the active site of the protein falcipain 2 (left) and 2D representation of the binding interaction (right); (b, c) binding mode (left) and 2D receptor–ligand interaction diagram (right) of compounds, 3′l and 3″d at binding pocket of falcipain 2 (left), respectively.
Docking results revealed that LibDock program successfully docked all 1,2,4-trioxane derivatives into the binding pocket of falcipain 2 enzyme. All compounds which were active in in vitro test could bind with the active site of falcipain-2 with high docking score (LibDock) and binding affinities in the range of 96.077–126.390. The LibDock scores are summarised in Table 3. All trioxane derivatives except 3a and 3b exhibited more binding affinity (higher dock scores) than the co-crystal ligand, E-64 (LibDock score =100.364). Five compounds which showed most potency in in vitro study also ranked top in docking study with LibDock scores of 116.533, 126.390, 122.609, 110.646, and 124.737 for 3′a, 3′b, 3′l, 3′d, and 3′e, respectively. It is important to note here that docking results are generally analysed by a statistical scoring function which converts interacting energy (binding energy) into numerical values called as the docking score, and therefore docking score is nothing but an expression of binding energy 34 . Binding energy (ΔG) includes the sum of all non-bonded interactions including hydrogen bonding, hydrophobic and Vander Walls between protein residues and bound ligand 35 . LibDock score is useful to assess the antimalarial activity of ligands as P. falciparum falcipain 2 inhibitors. Docking scores of compounds supported the results of in vitro antimalarial activity of synthesised 1,2,4-trioxane derivatives.
Table 3.
LibDock scores, no. of H-bonds and H-bond energies.
| Comp. | LibDock score | No. of H-bond (s) | H-Bond energy |
|---|---|---|---|
| 3a | 93.077 | 3 | −2.056 |
| 3b | 96.153 | 3 | −1.648 |
| 3c | 101.69 | 3 | −2.174 |
| 3d | 111.343 | 3 | −2.056 |
| 3e | 108.143 | 5 | −5.457 |
| 3′a | 107.807 | 4 | 0.804 |
| 3′b | 116.533 | 6 | −3.727 |
| 3′c | 126.390 | 4 | −2.265 |
| 3′d | 110.908 | 3 | 0.179 |
| 3′e | 127.665 | 4 | −2.246 |
| 3′f | 118.391 | 5 | 0.173 |
| 3′g | 117.436 | 2 | 0.182 |
| 3′h | 110.983 | 3 | 0.174 |
| 3′i | 120.229 | 3 | 0 |
| 3′j | 118.985 | 4 | −2.5 |
| 3′k | 120.370 | 1 | −2.526 |
| 3′l | 122.609 | 3 | 0.976 |
| 3′m | 100.364 | 1 | −2.526 |
| 3′n | 122.864 | 4 | 0.780 |
| 3″a | 101.911 | 3 | −2.5 |
| 3″b | 105.097 | 5 | −2.5 |
| 3″c | 110.952 | 6 | −2.466 |
| 3″d | 110.646 | 5 | −2.478 |
| 3″e | 124.747 | 6 | 0.031 |
| E-64 | 100.364 | 7 | 0.086 |
Protein–ligand docking was performed to generate the bioactive binding poses of designed inhibitors in the active site of falcipain 2 enzyme. LibDock is a high throughput docking algorithm that finds various conformations of the ligands in the protein active site based on polar interaction sites (hotspots) 36 . The 3D poses of bound ligands were visualised which revealed that the best orientation of the ligand relative to the receptor as well as the conformation of the ligand and receptor (best fit of ligand in the receptor molecule). Analysis of 2D diagram indicated that various non-bonded interactions mainly polar hydrogen bonding interactions were involved between binding site residues (active site amino acids) and ligand moieties/atoms. Table 3 also depicts the number of hydrogen bonds (H-bonds) and H-bond energies for all docked compounds. Higher the number of hydrogen bonds, higher is the binding affinity. Apart from hydrogen bonding interactions, other non-bonded interactions like hydrophobic bonding were also observed.
Table 4 reveals the hydrogen bonding interactions of five most potent compounds (3′b, 3′c, 3′l, 3″d, and 3″e) with active site residues such as Asp 154, Arg 12, Gln 36, Glu 14, Glu 15, Glu 138, Gly 13, Val 152, and His 19. Compounds 3′l and 3″d which showed the highest antimalarial activity among three series, could bind the active sites of falcipain-2 mainly by hydrogen bonding interactions. The 3D binding modes and 2D interaction diagrams of two most potent compounds, 3′l and 3″d are shown in Figure 9(b,c), respectively. The first compound formed three strong hydrogen bonds with residues like Gln 36 (O··H··O), Asp 154 (O··H··N), and Arg 12 (O··H··O) with bonding distances of 2.737, 2.985, and 1.834 Å, respectively. In later case, five bonds were observed with residues like Arg 12 (O··H··O), His 19 (O··H··H), Gly 13 (O··H··O), Glu 14 (O··H··O), and Glu 15 (O··H··O) with bonding distances of 1.868, 2.938, 2.308, 2.795, and 2.489 Å, respectively. Analysis of best docking poses of 3′l and 3″d revealed that the trioxane scaffold was oriented in the binding cavity (active site residues) of falcipain 2 receptor molecule. In 2D diagram, the trioxane moiety could occupy the binding sites of falcipain-2 through strong H-bonding interactions along with hydrophobic interactions. Such interactions afforded good stability between receptor molecule and ligand. Trioxane moiety interacted with multiple amino acid residues such as Arg 12, Asp 154, Val 12 and Gly 13, and Gln 36 and Glu 14 residues interacted with the 4-hydroxy-3-methoxyphenyl and idole-3-yl substituents in compounds 3′l and 3″d, respectively. Strong H-bonding interactions between C=O group of the trioxane and amino acid residues were also observed. The OH and NH groups were involved for H-bonding interactions from the substituent moieties for compounds 3′l and 3″d, respectively. On further analysis of docking interactions, it was found that trioxane ring played a crucial role in protein–ligand binding. Substituents increased binding strength by forming additional H-bonds that facilitated much stronger interaction of ligands with the receptor (falcipain-2 protein) molecule with optimum binding affinity to achieve desired antimalarial activity.
Table 4.
Details of hydrogen bonding between five most active ligands and receptor molecule.
| H-binding ligand |
H-binding receptor |
||||||
|---|---|---|---|---|---|---|---|
| Comp. | H-bond (s) | Element | Type | Residue | Element | Type | H-bond distance (Å) |
| 3′b | 6 | O | A | Asp 154 | N | D | 2.749 |
| O | A | His 19 | H | D | 2.811 | ||
| O | A | Arg 12 | H | D | 1.720 | ||
| O | A | Arg 12 | H | D | 2.393 | ||
| H | D | Gly 13 | O | A | 2.488 | ||
| H | D | Val 152 | O | A | 2.262 | ||
| 3′c | 4 | O | A | Asp 154 | N | D | 2.234 |
| O | A | His 19 | H | D | 2.534 | ||
| H | D | Asp 154 | O | A | 2.874 | ||
| H | D | Glu 138 | O | A | 3.016 | ||
| 3′l | 3 | O | A | Gln 36 | H | D | 2.737 |
| O | A | Asp 154 | N | D | 2.985 | ||
| O | A | Arg 12 | H | D | 1.834 | ||
| 3″d | 5 | O | A | Arg 12 | H | D | 1.868 |
| O | A | His 19 | H | D | 2.938 | ||
| H | D | Gly 13 | O | A | 2.308 | ||
| H | D | Glu 15 | O | A | 2.795 | ||
| H | D | Glu 14 | O | A | 2.489 | ||
| 3″e | 6 | O | A | Arg 12 | H | D | 2.976 |
| O | A | Arg 12 | H | D | 2.107 | ||
| H | D | Glu 14 | O | A | 2.804 | ||
| O | A | Asp 154 | H | D | 2.515 | ||
| H | D | Gly 13 | O | A | 2.247 | ||
| H | D | Glu 14 | O | A | 2.735 | ||
Docking is generally used to find the best binding orientation of small molecules bound to their target protein molecules in order to predict the binding affinity and thereby biological activity of the small molecules. Hence, docking plays an important role in the rational drug design. To evaluate the ligand–receptor interactions, the best pose of receptor–ligand complex should possess the lowest free energy (binding energy) which differs from experimentally observed other structure and estimate the binding affinity. Search algorithm is used to generate different poses of the ligand within the active site of molecules, orientations of particular conformations of the molecule in the binding site. Scoring function is used to identify the most likely pose for an individual ligand to assign a priority order to a set of diverse ligands docked to the same protein and hence estimate binding affinity. Using scoring function, one can predict the binding conformation of a ligand in its receptor and the affinity between the ligand and the receptor with the correct poses of ligands in the binding pocket of a protein 34–38 .
Furthermore, a correlation study between docking scores and logIC50 (pIC50) values of top five compounds (3′b, 3′c, 3′l, 3″d, and 3″e) was carried out. Graphs presented in Figure 10 revealed that a good correlation existed between in vitro antimalarial activity (against both CQ-sensitive and CQ-resistant P. falciparum strains) of compounds and their LibDock scores. The in vitro activity was found consistent (R 2 = 0.745 or 0.782) with the binding affinity of compounds for the P. falciparum cysteine protease falcipain 2 receptor.
Figure 10.
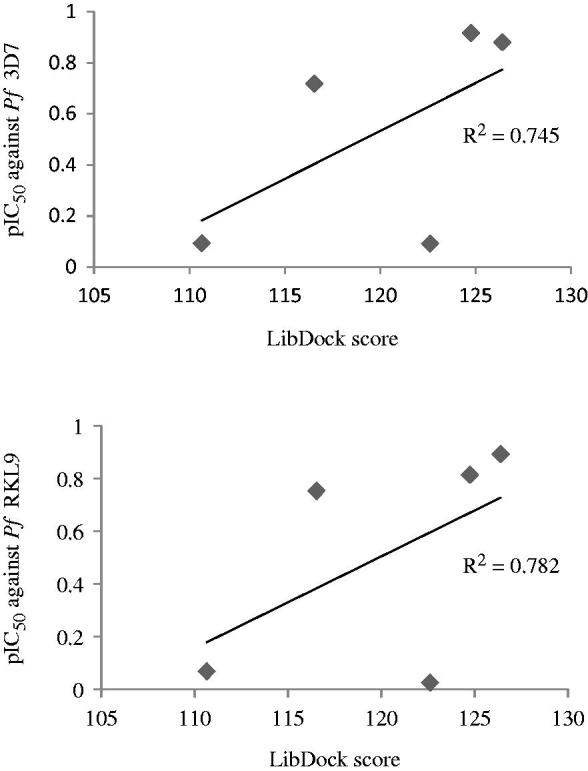
Graphs showing correlation between in vitro antimalarial activity (pIC50) and LibDock scores for five most potent compounds.
Molecular properties and drug-likeness
The results of predicted Lipinski’s parameters and other drug-likeness properties of the synthesised compounds, 3a–e, 3′a–n, 3″a–e are depicted in Table 5. Results revealed that all the compounds possessed good drug-like properties based on Lipinski’s rule of five with additional parameters such as molar solubility (MS), MV, MR, and number of rotatable bonds (nRotB). All compounds obeyed Lipinski’s rule of five and Veber rule. Lipinski rule of five is a rule to evaluate drug likeness to determine if a chemical compound has a certain pharmacological or biological activity to make it an orally active drug 39 . According to Lipinski’s rule, compounds are more likely to be drug-like and orally bioavailable if they obey the following criteria: LogP o/w (octanol/water partition coefficient) ≤ 5, MW (molecular weight) ≤ 500, HBA ≤ 10, and HBD ≤5 15 . To further substantiate, Veber et al. stated that compounds with ≤10 rotatable bonds and TPSA (total polar surface area) of ≤140 A2 are more likely to show membrane permeability and good bioavailability 39 , 40 . Lipinski rule of five is considered predictive for oral bioavailability; however, 16% of oral drugs violate at least one of the criteria and 6% fail in two or more 39 . In our study, compounds did not violate Lipinski rule of five parameters. Poor absorption or permeation of a ligand is more likely if a drug-like molecule have more than one of five rule violations. Values of LogP, MW, and TPSA indicated that compounds possessed good membrane permeability and oral bioavailability, whereas, nRotb bonds suggested that compounds had good intestinal availability. MS data indicated good bioavailability of compounds if given by oral route. MV and MR values were also found in permissible range which indicated good oral bioavailability for all the compounds. Hydrophobicity, membrane permeability, and bioavailability are dependent on molecule’s MW, LogP, MS, HBA, and HBD. Molecules violating more than one of these rules fail to exhibit optimum bioavailability. Sufficient water solubility is also important for optimal bioavailability of drugs. Number of rotatable bonds is important for molecular conformational studies (i.e. stereoselectivity of drug molecules) for optimal binding with the receptor molecule. Reduced molecular flexibility, as measured by the number of rotatable bonds, and polar surface area or total hydrogen bond count (sum of donors and acceptors) are some important predictors of good oral bioavailability, independent of molecular weight 15 . Further, TPSA, MV, and MR are also useful parameters for drug’s transport and biodistribution 15 . The drug score combines drug-likeness, lipophilicity, solubility, molecular weight, and the risk of toxicity into a single numerical value that can be used to predict a global value for each compound as a potential new drug candidate 22 . Drug-likeness score was obtained in the range of −0.18 and −0.79. Even though compounds exhibited negative drug-likeness values, scores were in acceptable range and indicated that compounds possessed potential as new drug candidates. The overall analysis of drug-likeness studies strongly suggested that newly designed 1,2,4-trioxane derivatives possessed good drug-likeness behaviour favourable for optimal membrane permeability, transport and bioavailability and eventual interaction with the receptor molecule.
Table 5.
Calculated molecular properties and drug-likeness parameters.
| Lipinski’s parameters |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | MW | LogP | nHBA | nHBD | TPSA (A2) | nViolations | MS | MR | MV (A3) | nRotB | DL score |
| 3a | 198.78 | 1.02 | 4 | 0 | 44.76 | 0 | −1.33 | 50.01 | 220.52 | 1 | −0.43 |
| 3b | 212.25 | 1.50 | 4 | 0 | 44.76 | 0 | −1.74 | 54.65 | 238.37 | 2 | −0.41 |
| 3c | 226.27 | 1.99 | 4 | 0 | 44.76 | 0 | −2.21 | 59.25 | 256.28 | 3 | −0.60 |
| 3d | 240.30 | 1.64 | 4 | 0 | 44.76 | 0 | −2.23 | 59.68 | 258.69 | 4 | −0.68 |
| 3e | 240.26 | 0.69 | 5 | 0 | 61.83 | 0 | −1.81 | 59.99 | 264.63 | 4 | −0.42 |
| 3′a | 246.26 | 2.17 | 4 | 0 | 44.76 | 0 | −2.68 | 65.27 | 255.79 | 1 | −0.82 |
| 3′b | 262.26 | 1.79 | 5 | 1 | 64.99 | 0 | −2.21 | 66.97 | 268.63 | 1 | −0.17 |
| 3′c | 276.29 | 2.26 | 5 | 0 | 53.99 | 0 | −2.81 | 71.74 | 287.64 | 2 | −0.54 |
| 3′d | 280.71 | 2.88 | 4 | 0 | 44.76 | 0 | −3.48 | 70.08 | 272.98 | 1 | −0.26 |
| 3′e | 291.26 | 1.84 | 6 | 0 | 90.58 | 0 | −3.19 | 72.60 | 280.74 | 2 | −1.06 |
| 3′f | 260.29 | 2.57 | 4 | 0 | 44.76 | 0 | −3.19 | 70.32 | 276.73 | 1 | −0.78 |
| 3′g | 325.16 | 3.02 | 4 | 0 | 44.76 | 0 | −3.85 | 72.90 | 277.64 | 1 | −0.65 |
| 3′h | 264.25 | 2.44 | 4 | 0 | 44.76 | 0 | −2.98 | 65.49 | 261.70 | 1 | −0.45 |
| 3′i | 296.32 | 3.50 | 4 | 0 | 44.76 | 0 | −4.39 | 81.73 | 306.92 | 1 | −0.76 |
| 3′j | 289.33 | 2.29 | 5 | 0 | 48.00 | 0 | −2.64 | 79.70 | 305.35 | 2 | −0.79 |
| 3′k | 272.30 | 3.01 | 4 | 0 | 44.76 | 0 | −3.66 | 75.59 | 296.66 | 2 | −0.70 |
| 3′l | 292.29 | 1.88 | 6 | 1 | 74.22 | 0 | −2.32 | 73.43 | 299.06 | 2 | −0.18 |
| 3′m | 306.32 | 2.23 | 6 | 0 | 63.22 | 0 | −2.89 | 78.21 | 319.92 | 3 | −0.10 |
| 3′n | 274.27 | 1.96 | 5 | 0 | 61.83 | 0 | −2.21 | 62.76 | 284.05 | 2 | −0.72 |
| 3″a | 236.23 | 1.32 | 4 | 0 | 57.90 | 0 | −2.08 | 57.68 | 241.85 | 1 | −0.70 |
| 3″b | 252.28 | 1.76 | 4 | 0 | 73.00 | 0 | −2.68 | 64.13 | 251.15 | 1 | −0.51 |
| 3″c | 235.24 | 0.99 | 4 | 1 | 60.55 | 0 | −1.63 | 59.93 | 243.60 | 1 | −1.22 |
| 3″d | 285.30 | 1.12 | 5 | 0 | 57.65 | 0 | −3.25 | 76.36 | 251.09 | 1 | −0.39 |
| 3″e | 247.25 | 2.39 | 4 | 1 | 60.55 | 0 | −2.21 | 71.87 | 296.39 | 1 | −1.24 |
MW: molecular weight; LogP: log of octanol/water partition coefficient; nHBA: no. of hydrogen bond acceptor(s); nHBD: no. of hydrogen bond donor(s); TPSA: total polar surface area; nViolations: no. of rule of five violations; MS: molar aqueous solubility; MR: molar refractivity; MV: molar volume; nRotB: no. of rotatable bonds; DL: drug-likeness.
Since all newly designed compounds showed favourable drug-like properties, a reasonable correlation can be drawn between their calculated drug-like properties and in vitro antimalarial activity profile. LogP o/w is a direct indicator of lipophilicity of drug substances. Higher the value of LogP, better the biological membrane permeability. It is inevitable for a molecule to have sufficient lipophilicity which is required for its optimal bioavailability and biological action. Hydrogen bond acceptor and donor groups are of paramount importance required to achieve optimal drug action. PSA is also closely related to the hydrogen bonding potential of a drug molecule. Practically, a compound with all drug-like properties in the desired range appears to exhibit high levels of therapeutic potency. Good drug-like properties and activity are complementary, and hence balanced attention to both properties and bioactivity could suitably transform a ligand to a good drug lead 41–44 . Better antimalarial activity of the top five compounds 3′b, 3′c, 3′l, 3″d, and 3″e might be probably due to their sufficient lipophilicity along with balanced polar properties likes HBD, HBA groups, rotatable bonds, and PSA. They collectively determine membrane permeability as well as transport property of drug molecule. Due to optimal lipophilicity, compound readily penetrated parasite’s cell membrane that led to attain an intracellular concentration for desired antimalarial activity.
ADMET prediction
The ADMET values of newly designed 1,2,4-trioxane derivatives presented in Table 6 were found in acceptable range with favourable ADMET properties. All the compounds were predicted to have good intestinal absorption and non-inhibitors of cytochrome P450 2D6 (CYP2D6) with medium to moderate BBB penetration. BBB penetration is mandatory for the drug to be used in the treatment of cerebral malaria. The CYP2D6 enzyme is one of the important enzymes involved in drug metabolism. The aqueous solubility prediction (defined in water at 25 °C) indicated that most of the compounds were soluble in water. The predictive hepatotoxicity was observed for a few compounds among three series. Some of the compounds were found to be highly bound with plasma protein, while some were poorly bound with plasma protein.
Table 6.
Theoretical ADMET parameters.
| Comp. | Aqueous solubility | BBB penetration | CYP P450 2D6 inhibition | Hepatotoxicity | Intestinal absorption | PP binding |
|---|---|---|---|---|---|---|
| 3a | 3 | 2 | FALSE | TRUE | 0 | FALSE |
| 3b | 3 | 2 | FALSE | FALSE | 0 | FALSE |
| 3c | 3 | 1 | FALSE | FALSE | 0 | FALSE |
| 3d | 3 | 3 | FALSE | FALSE | 0 | FALSE |
| 3e | 3 | 3 | FALSE | FALSE | 0 | FALSE |
| 3′a | 2 | 1 | FALSE | FALSE | 0 | TRUE |
| 3′b | 3 | 2 | FALSE | FALSE | 0 | TRUE |
| 3′c | 2 | 2 | FALSE | FALSE | 0 | TRUE |
| 3′d | 2 | 1 | FALSE | FALSE | 0 | TRUE |
| 3′e | 2 | 3 | FALSE | TRUE | 0 | TRUE |
| 3′f | 2 | 1 | FALSE | FALSE | 0 | TRUE |
| 3′g | 2 | 1 | FALSE | FALSE | 0 | TRUE |
| 3′h | 2 | 1 | FALSE | TRUE | 0 | TRUE |
| 3′i | 2 | 1 | FALSE | TRUE | 0 | TRUE |
| 3′j | 2 | 1 | FALSE | TRUE | 0 | TRUE |
| 3′k | 2 | 1 | FALSE | FALSE | 0 | TRUE |
| 3′l | 3 | 2 | FALSE | FALSE | 0 | FALSE |
| 3′m | 2 | 2 | FALSE | FALSE | 0 | TRUE |
| 3′n | 2 | 2 | FALSE | FALSE | 0 | TRUE |
| 3″a | 3 | 2 | FALSE | TRUE | 0 | TRUE |
| 3″b | 2 | 2 | FALSE | TRUE | 0 | TRUE |
| 3″c | 3 | 2 | FALSE | FALSE | 0 | FALSE |
| 3″d | 3 | 2 | FALSE | FALSE | 0 | FALSE |
| 3″e | 2 | 2 | FALSE | TRUE | 0 | FALSE |
Aqueous solubility: 3, Good, 2, Low; BBB (blood–brain barrier) penetration: 3, Low, 2, Medium, 1, Moderate; Cytochrome (CYP) P450 2D6 inhibition: false-non-inhibitor; hepatotoxicity: true-toxic, false-non-toxic; intestinal absorption: 0, Good; plasma protein (PP) binding: true-highly bounded, false-poorly bounded.
Conclusions
Newer series of 1,2,4-trioxane derivatives reported herein have potential in vitro antimalarial effectiveness and therefore, further studies are required for the evaluation of their toxicity, antimalarial efficacy, and pharmacokinetics in animal models. Our work is in progress to carry out in vivo toxicity and antimalarial activity of potent compounds. Structure–activity relationship study helped us to understand the importance of the core 1,2,4-trioxane ring system and also the role of substitution pattern on the overall antimalarial activity of newly designed trioxane analogues. Pharmacodynamic significance of the basic structural scaffold and unique structural simplicity of trioxane molecule highlighted the potential of 1,2,4-trioxane derivatives to be used as future antimalarial agents. In silico studies confirmed that newer 1,2,4-trioxane derivatives developed as single drug conjugates possessed target specificity for the P. falciparum cysteine protease falcipain 2 receptor with well-defined physicochemical, pharmacokinetics, and toxicity properties. All these results yielded valuable information for further optimisation of 1,2,4-trioxane-based compounds as falcipain 2 inhibitors based on the structure-based drug design approach. Further QSAR and pharmacokinetic-based optimisation studies are therefore required to develop antimalarial drug candidates as novel falcipain 2 inhibitors from our present 1,2,4-trioxane lead molecules, particularly 4-hydroxy-3-methoxysubstituted or indole substituted compounds.
Supplementary Material
Funding Statement
One of the authors is grateful to UGC, New Delhi, India, for the financial support in the form of Senior Research Fellowship (F/2015-16/NFO-17-OBC-TRI-33908).
Acknowledgements
The authors are thankful to Bioinformatics Infrastructure Facility, Centre for Biotechnology and Bioinformatics, Dibrugarh University, Dibrugarh, Assam (India) for carrying out in silico studies. The authors extend their sincere thanks to SAIF, NEHU, Shillong and SAIF, Panjab University, Chandigarh for providing spectral data of synthesised compounds. Authors gratefully thank the Director, National Institute of Malaria Research (ICMR), New Delhi for providing necessary facilities to carry out in vitro antimalarial screening of synthesized compounds.
Disclosure statement
The authors report no declarations of interest.
References
- 1. World Malaria Report. World Health Organization: Geneva, Switzerland; 2016. [Google Scholar]
- 2. Rudrapal M, Chetia D.. Endoperoxide antimalarials: development, structural diversity and pharmacodynamic aspects with reference to 1,2,4-trioxane-based structural scaffold. Drug Des Dev Ther 2016;10:3575–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pandey S, Agarwal P, Srivastava K, et al. Synthesis and bioevaluation of novel 4-aminoquinoline-tetrazole derivatives as potent antimalarial agents. Eur J Med Chem 2013;66:69–81. [DOI] [PubMed] [Google Scholar]
- 4. Gogoi J, Chetia D, Kumawat MK, et al. Synthesis and antimalarial activity evaluation of some mannich bases of tetraoxane-phenol conjugate. Indian J Pharm Educ Res 2016;50:591–7. [Google Scholar]
- 5. Geleta G, Ketema T.. Severe malaria associated with P. falciparum and P. vivax among children in Pawe Hospital, Northwest Ethiopia. Malaria Res Treat 2016;2016:1240962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kyle DE, Teja-Isavadharm P, Li Q, et al. Pharmacokinetics and pharmacodynamics of qinghaosu derivatives: how do they impact on the choice of drug and the dosage regimens? Med Trop (Mars) 1998;58:38–44. [PubMed] [Google Scholar]
- 7. Garner P, Graves PM.. The benefits of artemisinin combination therapy for malaria extend beyond the individual patient. PLoS Med 2005;2:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park VK, O’Neill PM, Maggs JL, et al. Safety assessment of peroxide antimalarials: clinical and chemical perspectives. Br J Clin Pharmacol 1998;46:521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White NJ. Clinical pharmacokinetics and pharmacodynamics of artemisinin and derivatives. Trans R Soc Trop Med Hyg 1994;88:S41–S3. [DOI] [PubMed] [Google Scholar]
- 10. White NJ. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo . Antimicrob Agents Chemother 1997;41:1413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rubush DM, Morges MA, Rose BJ, et al. An asymmetric synthesis of 1,2,4-trioxane anticancer agents via desymmetrization of peroxyquinols through a Brønsted acid catalysis cascade. J Am Chem Soc 2012;134:13554–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang Y, Dong Y, Vennerstrom JL.. Synthetic peroxides as antimalarials. Med Res Rev 2004;24:425–48. [DOI] [PubMed] [Google Scholar]
- 13. Singh SP, Konwar BK.. Molecular docking studies of quercetin and its analogues against human inducible nitric oxide synthase. SpringerPlus 2012;1:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shukla S, Srivastava RS, Shrivastava SK, et al. Synthesis, cytotoxic evaluation, docking and in silico pharmacokinetic prediction of 4-arylideneamino/cycloalkylidineamino 1, 2-naphthoquinone thiosemicarbazones. J Enzyme Inhib Med Chem 2013;28:1192–8. [DOI] [PubMed] [Google Scholar]
- 15. Sharma D, Chetia D, Rudrapal M.. Design, synthesis and antimalarial activity of some new 2-hydroxy-1,4-naphthoquinone-4-hydroxyaniline hybrid Mannich bases. Asian J Chem 2016;28:782–8. [Google Scholar]
- 16. Singh S, Srivastava P.. Molecular docking studies of myricetin and its analogues against human PDK-1 kinase as candidate drugs for cancer. Comput Mol Biosci 2015;5:20–33. [Google Scholar]
- 17. Carreno MC, Gonzalez-Lopez M, Urbano A.. Oxidative de-aromatization of para-alkyl phenols into para-peroxyquinols and para-quinols mediated by oxone as a source of singlet oxygen. Angew Chem Int Ed 2006;45:2737–41. [DOI] [PubMed] [Google Scholar]
- 18. Trager W, Jensen JB.. Human malaria parasites in continuous culture. Science 1976;193:673–5. [DOI] [PubMed] [Google Scholar]
- 19. Lambros C, Vanderberg JP.. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 1976;65:418–20. [PubMed] [Google Scholar]
- 20. Rudrapal M, Chetia D, Prakash A.. Synthesis, antimalarial- and antibacterial activity evaluation of some new 4-aminoquinoline derivatives. Med Chem Res 2013;22:3703–11. [Google Scholar]
- 21. Roy S, Chetia D, Rudrapal M, et al. Synthesis and antimalarial activity study of some new Mannich bases of 7-chloro-4-aminoquinoline. Med Chem 2013;9:379–83. [DOI] [PubMed] [Google Scholar]
- 22. Kashyap A, Chetia D, Rudrapal M.. Synthesis, antimalarial activity evaluation and drug-likeness study of some new quinoline-lawsone hybrids. Indian J Pharm Sci 2016;78:892–911. [Google Scholar]
- 23. Singh J, Kumar M, Mansuri R, et al. Inhibitor designing, virtual screening, and docking studies for methyltransferase: a potential target against dengue virus. J Pharm Bioallied Sci 2016;8:188–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sashidhara KV, Avula SR, Palnati GR, et al. Synthesis and in vitro evaluation of new chloroquine-chalcone hybrids against chloroquine-resistant strain of Plasmodium falciparum . Bioorg Med Chem 2012;22:5455–9. [DOI] [PubMed] [Google Scholar]
- 25. Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;23:3–26. [DOI] [PubMed] [Google Scholar]
- 26. Alam S, Khan F.. QSAR and docking studies on xanthone derivatives for anticancer activity targeting DNA topoisomerase IIα. Drug Des Dev Ther 2014;8:183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Silverstein RM, Webster FX.. Spectrometric identification of organic compounds. New York: Wiley Inc; 2005:205–13. [Google Scholar]
- 28. Sashidhara KV, Kumar K, Dodda RP, et al. Coumarin-trioxane hybrids: synthesis and evaluation as a new class of antimalarial scaffolds. Bioorg Med Chem Lett 2012;22:3926–30. [DOI] [PubMed] [Google Scholar]
- 29. Liu Y, Cui K, Lu W, et al. Synthesis and antimalarial activity of novel dihydro-artemisinin derivatives. Molecule 2011;16:4527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meshnick SR. Artemisinin: mechanisms of action, resistance and toxicity. Int J Parasitol 2002; 32:1655–60. [DOI] [PubMed] [Google Scholar]
- 31. Cumming JN, Ploypradith P, Posner GH.. Antimalarial activity of artemisinin (qinghaosu) and related trioxanes: mechanism(s) of action. Adv Pharmacol 1996;37:253–97. [DOI] [PubMed] [Google Scholar]
- 32. Paul M, O'Neill PM, Posner GH.. A medicinal chemistry perspective on artemisinin and related endoperoxides. J Med Chem 2004;47:2945–64. [DOI] [PubMed] [Google Scholar]
- 33. Oliveria R, Newton AS, Guedes RC, et al. An endoperoxide-based hybrid approach to deliver falcipain inhibitors inside malaria parasites. Chem Med Chem 2013;8:1528–36. [DOI] [PubMed] [Google Scholar]
- 34. Lionta E, Spyrou G, Vassilatis DK, et al. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem 2014;14:1923–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kitchen DB, Decornez H, Furr JR, et al. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev: Drug Discov 2004;3:935–49. [DOI] [PubMed] [Google Scholar]
- 36. Usha T, Middha SK, Goyal AK, et al. Molecular docking studies of anti-cancerous candidates in Hippophae rhamnoides and Hippophae salicifolia . J Biomed Res 2014;28:406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nikam MD, Mahajan PS, Damale MG, et al. Synthesis, molecular docking and biological evaluation of some novel tetrazolo[1,5-a]quinoline incorporated pyrazoline and isooxazoline derivatives. Med Chem Res 2015;24:3372–86. [Google Scholar]
- 38. Sapre NS, Gupta S, Pancholi N, et al. Molecular docking studies on tetrahydroimidazo-[4,5,1-jk][1,4]-benzodiazepinone (TIBO) derivatives as HIV-1 NNRT inhibitors. J Comput Aided Mol Des 2008;22:69–80. [DOI] [PubMed] [Google Scholar]
- 39. Khoshneviszadeh M, Shahraki O, Khoshneviszadeh M, et al. Structure-based design, synthesis, molecular docking study and biological evaluation of 1,2,4-triazine derivatives acting as COX/15-LOX inhibitors with anti-oxidant activities. Enzyme Inhib Med Chem 2016;31:1602–11. [DOI] [PubMed] [Google Scholar]
- 40. Faidallah HM, Al-Mohammadi MM, Alamry KA, et al. Synthesis and biological evaluation of fluoropyrazolesulfonylurea and thiourea derivatives as possible antidiabetic agents. J Enzyme Inhib Med Chem 2016;31:157–63. [DOI] [PubMed] [Google Scholar]
- 41. Grover J, Kumar V, Singh V, et al. Synthesis, biological evaluation, molecular docking and theoretical evaluation of ADMET properties of nepodin and chrysophanol derivatives as potential cyclooxygenase (COX-1, COX-2) inhibitors. Eur J Med Chem 2014;80:47–56. [DOI] [PubMed] [Google Scholar]
- 42. Debnath B, Ganguly S.. Synthesis, biological evaluation, in silico docking, and virtual ADME studies of 2-[2-Oxo-3-(arylimino)indolin-1-yl]-N-arylacetamides as potent anti-breast cancer agents. Monatsh Chem 2016;147:565–74. [Google Scholar]
- 43. Kerns EH, Di L.. Drug-like properties: concepts, structure design and methods from ADME to toxicity optimization. New York: Academic Press; 2008:6–14. [Google Scholar]
- 44. Kashid AM, Dube PN, Alkutkar PG, et al. Synthesis, biological screening and ADME prediction of benzylindole derivatives as novel anti-HIV-1, anti-fungal and anti-bacterial agents. Med Chem Res 2013;22:4633–40. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.