

Abstract
Interest in histone deacetylase (HDAC)-based therapeutics as a potential treatment for stroke has grown dramatically. The neuroprotection of HDAC inhibition may involve multiple mechanisms, including modulation of transcription factor acetylation independent of histones. The transcription factor Nrf2 has been shown to be protective in stroke as a key regulator of antioxidant-responsive genes. Here, we hypothesized that HDAC inhibition might provide neuroprotection against mouse cerebral ischemia by activating the Nrf2 pathway. We determined that the classic HDAC inhibitor trichostatin A increased neuronal cell viability after oxygen-glucose deprivation (from an OD value of 0.10±0.01 to 0.25±0.08) and reduced infarct volume in wild-type mice with stroke (from 49.1±3.8 to 21.3±4.6%). In vitro studies showed that HDAC inhibition reduced Nrf2 suppressor Keap1 expression, induced Keap1/Nrf2 dissociation, Nrf2 nuclear translocation, and Nrf2 binding to antioxidant response elements in heme oxygenase 1 (HO1), and caused HO1 transcription. Furthermore, we demonstrated that HDAC inhibition upregulated proteins downstream of Nrf2, including HO1, NAD(P)H:quinone oxidoreductase 1, and glutamate-cysteine ligase catalytic subunit in neuron cultures and brain tissue. Finally, unlike wild-type mice, Nrf2-deficient mice were not protected by pharmacologic inhibition of HDAC after cerebral ischemia. Our studies suggest that activation of Nrf2 might be an important mechanism by which HDAC inhibition provides neuroprotection.
Keywords: HDAC inhibition, TSA, Cerebral ischemia, Nrf2
Introduction
Stroke, resulting from occlusion or hemorrhage of blood vessels, is still the leading cause of morbidity and mortality in humans. It is generally accepted that the mechanisms of stroke damage involve multiple processes, including oxidative stress, excitotoxicity, ionic imbalance, inflammation, and apoptosis [1]. Oxidative stress, the consequence of reactive oxygen species (ROS) production, in particular plays a critical role in stroke pathogenesis and contributes to brain infarction and excitotoxicity [2]. Specific antioxidative enzymes, including NAD(P)H:quinine oxidoreductase 1 (NQO1), heme oxygenase 1 (HO1), glutathione peroxidases, glutathione S-transferase, and glutamate cysteine ligase, protect cells from the deleterious effects of ROS. The genes for these enzymes each contain antioxidant response elements (AREs) in their promoter regions that enable their regulation by the transcription factor Nrf2 [3].
Nrf2 is a broadly expressed transcription factor and a key regulator of antioxidant-responsive genes and phase II detoxifying enzymes. Under basal conditions, Nrf2 is bound to its inhibitor, Kelch-like ECH-associated protein 1 (Keap1), which promotes its proteasomal degradation. Stress-induced modification of cysteine residues in Keap1 causes the release of Nrf2, which translocates from the cytosoplasm into the cell nucleus, where it binds to AREs in genes that encode antioxidant enzymes that protect the cell from the deleterious effects of ROS [3]. The Nrf2 signaling pathway has been shown to be protective in many pathological conditions, including ischemia and inflammation in the central nervous system (CNS) [4]. Many natural and synthetic compounds, such as resveratrol [5], Ginkgo biloba extract (EGb 761) [6], and 1,2-dimethoxy-4,5-dinitrobenzene (LAS0811) [7], can activate Nrf2 and increase expression of antioxidative genes. Nrf2 activity also can be regulated by a variety of other pathways. For example, protein kinase C has been suggested to phosphorylate Nrf2 at Ser40 and cause its release from Keap1, thereby increasing its activity [8]. Acetylation is probably the best understood form of transcriptional regulation: hyperacetylation leads to an increase in the expression of particular genes, and hypoacetylation has the opposite effect [9]. A recent study has also demonstrated that Nrf2 can be acetylated within its Neh1 DNA-binding domain by p300/CBP. This acetylation increases its DNA-binding capacity and downstream gene expression [10], suggesting that Nrf2 acetylation may provide cell protection.
Histone deacetylases (HDACs), which prevent acetylation of histones, have been recognized as potentially useful therapeutic targets for a number of human disorders, including CNS diseases. HDAC inhibition leads to hyperacetylation of chromatin proteins and alterations in gene expression that are known to induce growth arrest, differentiation, or apoptosis of cancer cells [11]. Over the years, many different types of HDAC inhibitors have been developed, ranging from complicated structures of bacterial or fungal origin [trichostatin A (TSA), trapoxin] to the very simple butyrate. Each is capable of inhibiting HDACs with varying efficiency [9]. Pharmacological manipulation with small-molecule HDAC inhibitors, which may restore transcriptional balance to neurons and affect immune responses, has been beneficial in several experimental models of CNS disorders. For example, valproic acid [12,13], sodium butyrate [12], and TSA [14], which have differing degrees of HDAC inhibitory activity, have been shown to possess neuroprotective properties after stroke damage.
The present study was undertaken to investigate whether postin-sult treatment with the HDAC inhibitor TSA protects against brain injury and neurological deficits after cerebral ischemia in mice and whether HDAC inhibition-induced neuroprotection is associated with Nrf2 activation.
Materials and methods
Animals
Six- to eight-week-old wild-type (WT) mice (Jackson Laboratory) and Nrf2-deficient (Nrf2−/−) C57BL/6 male mice (with permission from Dr. Yamamoto Masayuki) were used in this study. Mice were housed in a pathogen-free facility with 12-h light and dark cycles and continuous access to food and water. Mice were acclimatized for at least 4 days before any experiment. They were fasted overnight before any procedure. All experiments were conducted in accordance with the National Institutes of Health and Johns Hopkins Animal Care and Use Committee guidelines.
Drugs
For in vivo experiments, TSA (Sigma-Aldrich) was dissolved in 100% ethanol to a concentration of 1 mg/mL and stored at −20 °C as a stock solution. Before administration to mice, the working solution was made by diluting the TSA stock 10 fold with saline. For cell culture experiments, a TSA stock solution was made with 100% ethanol at a concentration of 1 mg/L and diluted directly into cell culture medium. Sodium butyrate (Sigma-Aldrich) was used at a final concentration of 5 mg/L. A stock solution of MS275 (Cayman Chemical Co.) was made in DMSO and used at a final concentration of 10 μmol/L. Ginkgo biloba leaf extract EGb 761 (IPSEN Laboratories) was dissolved in DMSO and added directly to the culture media to achieve different final concentrations (3, 10, 30, or 100 mg/L).
Permanent middle cerebral artery occlusion (pMCAO) and neurological deficit scoring
The pMCAO model is recognized as an alternative focal cerebral ischemic model that has high clinical relevance for testing neuroprotective agents [15,16]. Before the surgery, WT and Nrf2−/− mice were randomly placed into two groups each: vehicle-treated (n=9 per group) and TSA-treated (n=10 per group). Briefly, with the mice under halothane anesthesia, a 1.0-cm vertical skin incision was made between the right eye and the ear. The temporal muscle was moved, and the temporal bone was exposed. Under a surgical microscope, a 2.0-mm burr hole was made just over the middle cerebral artery (MCA), visible through the temporal bone. The main trunk of the distal part of the MCA was directly occluded with a bipolar coagulator, and complete interruption of blood flow at the occlusion site was confirmed by severance of the occlusion site of the MCA. Core body temperature was maintained between 36.5 and 37.5 °C during and after the procedures. A successful occlusion was confirmed by placing the laser-Doppler probe above the temporal ridge to establish that the blood flow into this region was terminated. TSA (1 mg/kg) or vehicle (saline) was administered to mice intraperitoneally immediately after and at 6 h after the surgery based on previous studies [12,17–19] and our preliminary studies in which we found that one injection immediately after pMCAO plus a second injection at 6 h thereafter protected mice against cerebral ischemic brain damage (data not shown).
Neurological deficits were assessed 48 h after pMCAO by an experimenter blinded to the groups using a robust 28-point scoring system [20]. The tests were divided into motor and sensory functions. Motor function tests included: (1) spontaneous activity, (2) symmetry of walking, (3) head/neck movement when suspended by tail, (4) symmetry of forelimbs when suspended by tail, (5) climbing at a 45° angle, and (6) balance on a rod. Sensory function was measured with the vibrissae test, in which the whiskers were stimulated with a cotton-tipped applicator. Each of the seven tests was graded from 1 to 4, establishing a maximum deficit score of 28. After completion of the behavioral tests, the mice were euthanized and the brains harvested.
Quantification of infarct volume after pMCAO
Mice were deeply anesthetized, and brains were harvested and cooled in a −20 °C freezer. Five coronal sections of 2-mm thickness were cut and then incubated in 1% 2,3,5-triphenyl-tetrazolium chloride (TTC) in saline for 15 min at 37 °C. The area of infarct, which remains white, was measured on the rostral and caudal surfaces of all five slices and numerically integrated across the thickness of the slice to obtain an estimate of infarct volume by SigmaScan software (SPSS Inc.). Infarct volume was calculated as a percentage of the contralateral cortex with correction for swelling.
Cell cultures and related materials
Cortical neuronal cells were isolated from 17-day embryos of timed pregnant mice and cultured onto poly-D-lysine-coated 24-well dishes at a density of 5×105 cells/well in serum-free, high-glucose, Neurobasal medium (Invitrogen) supplemented with B27 minus antioxidant (Invitrogen) and Hepes (Sigma-Aldrich) as described previously [21]. Cells were maintained in growth medium at 37 °C in a 95% air/5% CO2 humidified atmosphere. Half of the initial culture medium was removed at Day 4 and replaced with fresh medium. All experiments were performed after 14 days in vitro.
The mouse macrophage cell line RAW 264.7 (American Type Culture Collection TIB-71) was grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and penicillin/streptomycin, as detailed before [22]. The cells were used in experiments after 80–90% confluence had been reached.
Oxygen/glucose deprivation (OGD) and cell viability
Serum-free primary neuronal cultures were treated with different concentrations of TSA (3, 10, or 30 ng/mL) for 1 h after 8 to 10 days in vitro and then were subjected to OGD for 150 min in a balanced salt solution at pO2 < 2 mm Hg. After OGD, the cells were assessed for viability as described previously [23]. The condition of cells at various TSA concentrations was determined morphologically by phase contrast microscopy. Then, cell viability was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Furthermore, the trypan blue (TB) staining experiment was performed to support MTT assay. After each treatment, cells were stained with 0.4% TB (Sigma-Aldrich). Unstained cells were regarded as viable, and stained cells were regarded as dead. Total cell number and number of TB-positive cells were counted via a light microscope under magnification×200 by an independent, blinded investigator. Survival value was calculated by using the formula: detected unstained cells/detected whole cells shortly before treatment [24]. Experiments were repeated with at least three separate batches of cultures.
Furthermore, the trypan blue staining assay was performed to confirm MTT assay results. After each treatment, cells were stained with 0.4% TB (Sigma-Aldrich). Unstained cells were regarded as viable, and stained cells were regarded as dead. Total cell number and the number of TBA-positive cells were counted via a light microscope in a blinded way. Survival value was calculated by using the formula: number of stained cells/number of total cells×100%
Immunoprecipitation and Western blot
For immunoprecipitation [25], 15 μL of antibody (anti-Nrf2 or anti-Keap1, Santa Cruz Biotechnology) was added to 300 μg of total protein in a volume of 500 μL. The mixture was rotated overnight at 4 °C, incubated with 30 μL of glutathione Sepharose 4B beads (GE Healthcare) for 4 h, spun down at 500 g, and washed four times with lysis buffer (RIPA buffer, Sigma-Aldrich). The beads were collected and combined with 25 μL of loading buffer (LDS sample buffer 4X, Invitrogen). For Western blot, equal amounts of protein were separated by SDS-PAGE on 4–12% Bis-Tris gels, transferred to nitrocellulose membranes, and incubated in primary antibodies overnight at 4 °C. The antibodies used were anti-Keap1 (1:1000, Santa Cruz), anti-Nrf2 (1:1000, Santa Cruz), anti-HO1 (1:1000, Stressgen Biotechnologies), anti-NQO1 (1:1000, Novus Biologicals), anti-glutamate cysteine ligase catalytic (GCLC) subunit (1:1000, Santa Cruz), anti-β-actin, (1:3000, Sigma), and anti-acetylation (1:500, Cell Signaling). Subsequently, the membranes were incubated for 1 h with the corresponding secondary antibodies. Protein bands were detected with ECL reagents (GE Healthcare).
Chromatin immunoprecipitation (ChIP)
RAW 264.7 cells were plated in 6-well plates. One day later, formaldehyde solution was added to the cells (1% final concentration), and the culture was incubated at 37 °C for 10 min to crosslink the protein and DNA. Then 100 μL of 10X glycine was added, and the plate was incubated at room temperature for 5 min. The cells were washed twice with 1X cold phosphate-buffered saline (PBS), resuspended in 200 μL of SDS lysis buffer [1% SDS, 10 mM EDTA, 50 mM Tris-HCl (pH 8.0)] containing phenylmethylsulfonyl fluoride (PMSF) and complete protease inhibitor cocktail (Roche), and sonicated on ice. ChIP was then carried out with the lysates by using a ChIP assay kit from Millipore according to the manufacturer’s instructions. The crosslinks were reversed by adding 20 μL of 5 M NaCl and heating at 65 °C for 5 h; DNA was recovered by phenol-chloroform extraction and ethanol precipitation. Relative amounts of DNA in the complex were quantified by PCR [26]. The PCR primers for 4 K and 10 K HO1 enhancers and GAPDH control were as follows: 4 K enhancer, 5′-GCGATATCTGCCTGCCTGTCAGAGGG-3′ (forward) and 5′-GCGATATCTGCAGAGCCCCACTGGAG-3′ (reverse); 10 K enhancer, 5′-GGCAGGGTCCACCAGATCAC-3′ (forward) and 5′-CCTGCCTTTAAGGGCTGTATCTGAACC-3′ (reverse); GAPDH (control), 5′-AGGCCCGGTGCTGAGTATGTC-3′ (forward) and 5′-TGCCTGCTTCACCACCTTCT-3′ (reverse). The products were separated by electrophoresis on a 1 to 1.5% agarose gel.
Nuclear extraction and electrophoresis mobility shift assay (EMSA)
RAW 264.7 cells (approximately 5×107) were washed with ice-cold PBS and incubated on ice in PBS containing 5 mM diisopropyl-fluorophosphate (Sigma-Aldrich) for 15 min. The cells were then resuspended in buffer containing (in mM) 60 KCl, 15 NaCl, 5 MgCl2, 10 Tris (pH 7.4), 300 sucrose, 1 dithiothreitol (DTT), 2 benzamidine, 0.5 spermidine, and 0.4 PMSF, as well as 1% nonfat dried milk and complete protease inhibitor cocktail. After 5 min, Nonidet P-40 (NP-40) was added to 0.05% (v/v). After an additional 5 min, the released nuclei were pelleted at 850 g for 5 min and resuspended in 1 mL of the buffer without NP-40. The nuclei were centrifuged at 16,000 g for 5 min and collected into 0.6 mL of buffer solution containing 1 mM DTT, 2 mM benzamidine, 0.5 mM spermidine, 0.4 mM PMSF, and protease inhibitors. The extracts were rocked at 4 °C for 30 min, centrifuged at 16,000 g for 15 min, separated into aliquots, snap-frozen in liquid nitrogen, and stored at −80 °C. Protein concentrations were assessed by the Bradford assay (Bio-Rad). EMSA was carried out with a radiolabeled oligonucleotide containing the murine HO1 ARE sites as described previously [22]. For oligonucleotide competitions, unlabeled or mutated competitors were added 5 min before the radiolabeled probes.
Reverse transcription-PCR (RT-PCR)
Total RNA was isolated from cultured cells with Trizol reagent (Invitrogen). Reverse transcription was performed with an iScript cDNA synthesis kit (170-8890, BioRad) according to the manufacturer’s instructions, and PCR were carried out with a PCR kit (Advantage 2 PCR Kit, 639201, Clontech) based on previously established protocols. The PCR parameters were 95 °C for 5 min, 94 °C for 30 s, and 68 °C extension for 1 min for a total of 30 cycles. PCR primers used were as follows: HO1, 5′-GTAACAGACTTGCCACACTCATA-3′ (forward) and 5′-GCGTCGACGACGCTCCTTCACCGGACTG-3′ (reverse); GAPDH (control), 5′-AGGCCCGGTGCTGAGTATGTC-3′ (forward) and 5′-TGCCTGCTTCACCACCTTCT-3′ (reverse).
Statistical analysis
All values are expressed as means±SEM. Statistical analysis among groups was performed with one-way analysis of variance followed by the Student-Newman-Keuls multiple-range test. Differences were considered significant at P<0.05.
Results
TSA protects neurons from OGD and mice from cerebral ischemic damage
To test whether HDAC inhibition induced by TSA protects neurons from OGD through activation of Nrf2, we first performed the MTT assay and trypan blue staining on cultured mouse neurons that had been treated with different concentrations of TSA and were subjected to OGD. In MTT assay, TSA reduced OGD-induced cell damage in a dose-dependent manner, but only 30 ng/mL of TSA significantly enhanced cell viability compared with the control group (Fig. 1A). Meanwhile, the TB staining showed that 30 or 60 ng/mL of TSA significantly increased cell viability after OGD exposure, although there was no significant difference between these two doses (Fig. 1B), which was consistent with the MTT assay results. To further confirm the results in vivo, we examined whether TSA provided neuroprotection in WT mice after pMCAO. At 48 h after pMCAO, vehicle-treated mice had an average cortical infarct volume of 49.1±3.8%, but TSA-treated mice had an average cortical infarct volume of only 21.3± 4.6% (P<0.01; Fig. 1C). Mice administered TSA also had lower neurological deficit scores than mice in the control group (6.87±0.21 vs 8.46±0.45, P<0.05; Fig. 1D). Therefore, our data confirmed that the HDAC inhibitor TSA is neuroprotective against cerebral ischemic damage.
Fig. 1.

The HDAC inhibitor TSA protects neurons from OGD and mice from cerebral ischemic damage. (A, B) Primary cortical neurons were treated with 3, 10, 30, or 60 ng/mL TSA solution or DMSO (control) for 1 h and then subjected to 150 min of oxygen/glucose deprivation (OGD). The MTT assay and TB staining were used to evaluate cell viability. The number of viable neurons significantly decreased after OGD compared to control group. TSA treatment (30 or 60 ng/mL) significantly improved cell viability after OGD but did not affect the viability of control cells that were not exposed to OGD. (C, D) Mice were subjected to pMCAO and then administered either vehicle (Veh) or TSA (1 mg/kg) immediately after the procedure and at 6 h. Infarct volume and neurological deficits were evaluated after 2 days. (B) Infarct volume was significantly larger in the vehicle-treated group than in the TSA-treated group. (C) Mice that received TSA had lower neurological deficit scores than did mice in the vehicle-treated group. n=9 per group; *P<0.05, **P<0.01.
TSA and other HDAC inhibitors suppress Nrf2 inhibitor Keap1 expression
To evaluate how HDAC inhibition influences Nrf2 activation, we first investigated whether TSA affects intracellular Nrf2 inhibitor Keap1 in RAW 264.7 cells. EGb 761 was used as a relevant control because it is known to induce Nrf2 activation and enhance HO1 expression [27]. We found that TSA (10, 30, and 100 ng/mL) but not EGb 761 (3, 10, 30, and 100 μg/mL) reduced Keap1 protein level (Fig. 2A). The inhibition of Keap1 protein expression caused by 30 ng/mL of TSA was greatest at 16 h (Fig. 2B). Based on these results and those shown in Fig. 1A, we chose 30 ng/mL TSA, 100 μg/mL EGb 761, and 16 h of exposure for the remaining in vitro experiments.
Fig. 2.

TSA and other HDAC inhibitors suppress Nrf2 inhibitor Keap1 expression. Different concentrations of TSA (10, 30, and 100 ng/mL), EGb 761 (3, 10, 30, and 100 μg/mL), or both were applied to mouse RAW 264.7 cells for 16 h. Then Keap1 protein levels were measured by Western blot analysis. (A) TSA and TSA+EGb 761, but not EGb 761 alone, decreased Keap1 expression at all doses tested. (B) TSA (30 ng/mL) significantly reduced Keap1 protein level at 16 h. (C) TSA (30 ng/mL) and TSA+EGb 761 (100 μg/mL) reduced Keap1 mRNA levels as measured by RT-PCR. (D, E) Commonly used HDAC inhibitors sodium butyrate (NaB, 5 mg/L) and MS275 (10 μmol/L) decreased Keap1 expression in the presence or absence of EGb 761 after 16 h of treatment. (F) Treatment with TSA (30 ng/mL) or TSA+EGb 761 (100 μg/mL) produced a trend toward Nrf2 acetylation. Ac-Nrf2, acetylated Nrf2; T-Nrf2, total Nrf2.
Next, using RT-PCR, we determined that TSA also reduced Keap1 mRNA levels (Fig. 2C), suggesting that TSA might affect Keap1 transcription or stabilization of its mRNA. Additional investigation will be needed to verify the mechanism. To determine whether TSA affects Keap1 expression via inhibition of HDAC, we tested two other commonly used HDAC inhibitors, sodium butyrate and MS275. Both compounds inhibited Keap1 protein expression in a manner similar to that of TSA after 16 h treatment (Figs. 2D and E), suggesting that the ability of TSA to suppress Keap1 expression might result from its inhibition of HDAC. We subsequently treated cells with TSA, EGb 761, or both and immuno-precipitated cell lysates with antibodies to Keap1 or Nrf2, and then immunoblotted the lysates with anti-acetylation antibody. We could not detect Keap1 acetylation after TSA/EGb 761 treatment (data not shown); however, the data showed a trend toward enhancement of Nrf2 acetylation by TSA and TSA plus EGb 761 compared to vehicle control (Fig. 2F), suggesting that TSA might induce acetylation of Nrf2.
TSA activates Nrf2 and enhances Nrf2-ARE binding
Based on the ability of TSA to inhibit expression of Keap1, we hypothesized that TSA might activate Nrf2, enhance Nrf2-ARE binding, and consequently increase downstream gene expression. Through this mechanism, TSA might provide neuroprotection after permanent ischemia. To test our hypothesis, we first examined the effect of TSA on Nrf2 activation. Using coimmunoprecipitation of RAW 264.7 cell lysates that had been treated with TSA, EGb 761, or both for 4 h, we found that TSA and cotreatment of TSA and EGb 761 reduced the association of Keap1 and Nrf2 (Fig. 3A). Examination of Nrf2 nuclear translocation by immunoblotting revealed that TSA and TSA plus EGb 761 increased Nrf2 translocation from the cytoplasm to the nucleus (Fig. 3B). EMSA results showed that TSA and EGb 761 each enhanced nuclear protein Nrf2 binding to AREs in the HO1 enhancer and that cotreatment promoted more binding than treatment with either individual agent. This binding specificity was confirmed by the competition of radiolabeled probes with excess cold probes or with mutant probes that had mutated Nrf2-binding sites (Fig. 3C). Additionally, to test whether nuclear Nrf2 actually bound to the HO1 enhancer region, we employed the ChIP assay. We used anti-Nrf2 antibody to immunoprecipitate DNA fragments from cells that had been treated with TSA, EGb 761, or both. Then we amplified these DNA fragments by PCR using specific primers for HO1 enhancers at −4 K and −10 K. Our results suggested that TSA and EGb 761, applied individually or together, induced Nrf2 association with HO1 enhancers located at both −4 K and −10 K and that the association was specific, as Nrf2 did not bind to HO1 proximal promoters with non-Nrf2-binding sites (Fig. 3D). Finally, to determine whether nuclear Nrf2 could actually transactivate the downstream genes, we constructed a reporter plasmid that contained a fragment with multiple Nrf2-binding sequences from the HO1 enhancer followed by a luciferase reporter gene. The plasmid was transfected into cells that were treated with TSA, EGb 761, or both for 16 h. The luciferase assay showed that individual or cotreatment increased luciferase activity and that cotreatment resulted in more activity than either drug individually (Fig. 3E). Taken together, the results suggest that TSA might activate transcription factor Nrf2 and its downstream reactions and that these effects might be enhanced by EGb 761.
Fig. 3.

TSA activates Nrf2 and enhances Nrf2–ARE binding. (A) RAW 264.7 cells were treated with TSA (30 ng/mL), EGb 761 (100 μg/mL), or TSA+EGb 761 for 16 h. Coimmuno-precipitation showed that antibodies to Keap1 and Nrf2 precipitated significantly less of the other protein after treatment with one or both drugs, indicating a reduction in interaction between Nrf2 and Keap1. Total Keap1 and total Nrf2 were used as protein loading controls. (B) Nrf2 protein expression was assessed in nuclear and cytosolic fractions. Nrf2 expression was unchanged in the cytosolic fraction, but it was significantly increased in the nuclear fraction after TSA treatment and cotreatment with TSA and EGb 761. (C) Based on EMSA, Nrf2–ARE-binding activity was enhanced after TSA treatment and substantially increased after treatment with TSA+EGb 761. Competition with excess cold (nonradioactive) probes markedly suppressed Nrf2–ARE binding, but mutant probes were unable to disrupt this binding. (D) In a chromatin immunoprecipitation assay, treatment with TSA alone or with EGb 761 induced Nrf2 association with HO1 enhancers at both −4 K and −10 K; Nrf2 did not bind to HO1 proximal promoters with non-Nrf2-binding sites. GAPDH was a loading control. (E) The luciferase assay showed that TSA or TSA+EGb 761 induced luciferase activity; TSA+EGb 761 increased transcription more than either drug individually. *P<0.05, **P<0.01 vs no drug.
TSA upregulates expression of Nrf2 downstream proteins
Because TSA was able to activate transcription factor Nrf2 and provide neuroprotection in cerebral ischemia, we hypothesized that TSA might regulate the Nrf2 downstream pathway. Therefore, we tested the effect of TSA on expression of antioxidative proteins downstream of Nrf2. We found that TSA alone induced HO1 expression in both RAW 264.7 cells (Fig. 4A) and in primary neuronal cells (Fig. 4B) and enhanced EGb 761-induced HO1 expression (Figs. 4A and B). TSA also induced the expression of two other Nrf2-regulated proteins, NQO1 and GCLC, in cultured neuronal cells (Fig. 4C) and in brain tissue (Fig. 4D). Furthermore, TSA enhanced the effect of EGb 761 on NQO1 and GCLC expression (Fig. 4C). These results indicate that the HDAC inhibitor TSA induces expression of Nrf2 downstream proteins through activation of Nrf2.
Fig. 4.

TSA upregulates the expression of proteins downstream of Nrf2. TSA (30 ng/mL) and EGb 761 (100 μg/mL) increased HO1 expression in mouse RAW 264.7 cells (A) and in primary neuronal cells (B). TSA further enhanced EGb 761-induced HO1 expression (A, B). Additionally, immunoblotting showed that the expression of two other Nrf2-regulated proteins, NQO1 and GCLC, was elevated by TSA treatment in cultured neuronal cells (C) and in brain tissue (D). TSA further increased NQO1 and GCLC expression induced by EGb 761 in neurons (C). Actin was used as a loading control.
TSA protection against cerebral ischemia is abolished in Nrf2-deficient mice
To determine whether Nrf2 activation by TSA contributes to the neuroprotection after cerebral ischemia, we evaluated infarct volume and neurological deficit scores in Nrf2-deficient mice subjected to pMCAO. The mice were treated with vehicle or TSA immediately after and at 6 h after pMCAO. As reported previously, at 48 h post-pMCAO, Nrf2-deficient mice had significantly larger infarct volume (61.3±7.2% vs 49.1±3.8%, P<0.05) and more neurological deficits (9.55±1.50 vs 8.46±0.45, P>0.05) than WT mice in the absence of TSA (Figs. 5A and C). However, TSA treatment had no protective effect in Nrf2-deficient mice on both infarct volume (61.3±7.2% vs 54.8± 5.1%, P>0.05) and neurological deficits (9.55±1.50 vs 9.10±2.30, P>0.05) (Figs. 5B and D), suggesting that Nrf2 is needed for HDAC inhibition-mediated neuroprotection.
Fig. 5.
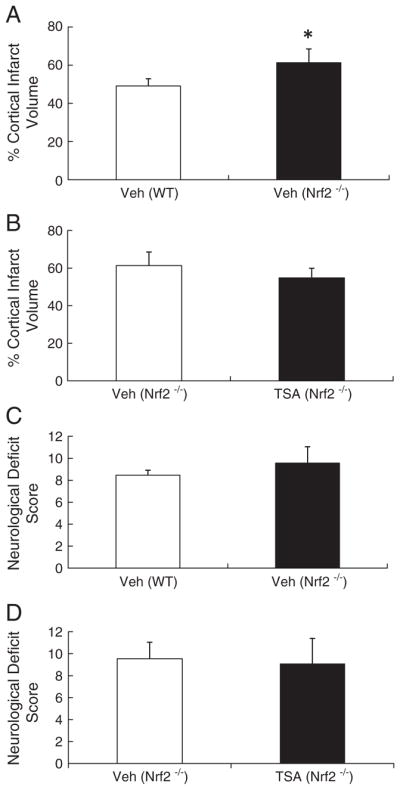
TSA protection against cerebral ischemia is absent in Nrf2-deficient mice. Nrf2−/− mice had increased infarct volume compared to WT counterparts in the absence of TSA (A), and also had more neurological deficits than WT mice, but no statistical significance (C). TSA treatment (1 mg/kg) had no effect on cortical infarct volume or neurologic deficit scores in Nrf2−/− mice compared to vehicle-treated controls (B and D); n=10 per group.
Discussion
In this study, we have shown that HDAC inhibition contributes to enhancement of neuronal cell viability after OGD and reduction of cerebral ischemic injury in WT mice subjected to permanent ischemic stroke. Our in vitro mechanistic studies provide evidence that this HDAC inhibition-related neuroprotection may occur through activation of the transcription factor Nrf2. Three HDAC inhibitors—TSA, sodium butyrate, and MS275—reduced expression of the Nrf2 suppressor, Keap1, which might contribute to the dissociation of Nrf2 from Keap1 [28]. HDAC inhibition with TSA also activated Nrf2 by promoting Keap1/Nrf2 dissociation, Nrf2 nuclear translocation, and Nrf2–ARE binding in the HO1 enhancer. In addition, TSA increased HO1 transcription activity and upregulated the expression of Nrf2 downstream proteins HO1, NQO1, and GCLC in mouse cell cultures (macrophage RAW 264.7 cells and cortical neurons) and brain tissue. Finally, in contrast to WT mice, Nrf2-deficient mice were not protected from cerebral ischemia after TSA treatment. It has been demonstrated that Nrf2 gene function is not necessary for overall mouse development, growth, and fertility. Furthermore, no detectable developmental defects have been found in Nrf2−/− mice [29]. Taken together, our studies reveal at least a partial contribution of Nrf2 to the neuroprotection by HDAC inhibition, offer underlying mechanisms for this neuroprotection, and suggest a promising therapeutic strategy for ischemic stroke.
Therapeutic benefits for diverse CNS diseases sometimes may be achieved by interfering with a set of common targets or pathways. Beginning with cancer therapy, the superfamily of HDACs has been recognized for its therapeutic possibilities in a broad range of human disorders. Now, the potential for use of HDAC-based therapeutics in CNS disorders has grown dramatically. HDACs have been implicated as direct or indirect regulators of neuronal-specific, immune-specific, and other tissue-specific gene expression patterns in brain. Furthermore, genetic evidence suggests crucial roles for HDACs in the maintenance of CNS homeostasis and in mutations of genes of histone-binding proteins that underlie neurological disorders [30]. HDAC inhibitors have emerged as a promising therapeutic intervention in neurodegenerative diseases based on their ability to raise histone acetylation levels, alter the transcription of associated genes, and provide neuroprotection.
Small molecule HDAC inhibitors include hydroxamates, short-chain fatty acids, cyclic peptides, and benzamides. TSA, an example of a hydroxamate, was first isolated in 1976 as an antifungal antibiotic [31]. It is a highly potent, specific, and reversible HDAC inhibitor, but it has not yet been developed for clinical use. However, multiple TSA analogues are being developed, and more potent HDAC inhibitors are now in early clinical trials [32]. Interestingly, a TSA-related hydroxamic acid, suberoylanilide hydroxamic acid (SAHA, also referred to as vorinostat), was recently approved by the FDA for the treatment of cutaneous T cell lymphoma. Although we had no way to measure TSA levels in plasma and CNS tissue, we observed no evidence of intoxication, and animal behavior after TSA treatment appeared normal.
In recent years it has been demonstrated that HDAC inhibitors attenuate brain damage from neurodegenerative diseases, including Huntington’s disease [33], amyotrophic lateral sclerosis [34], and experimental autoimmune encephalitis [35]. Importantly, some studies have shown that HDAC inhibitors provide neuroprotection in animal models of stroke [12,14] and intracerebral hemorrhage [13]. In a model of transient cerebral ischemia, treatment with SAHA reduced infarct volume by 30–40%, indicating that it efficiently counteracted postischemic brain damage [36]. In agreement with those findings, we demonstrated that the HDAC inhibitor TSA ameliorated mouse cerebral ischemic damage in vitro and in vivo. The protective effects of TSA were evidenced by an increase in neuronal viability after OGD in vitro and by a decrease in infarct volume and neurological deficits in mice with permanent ischemia. Yildirim et al. [14] previously reported that treatment with TSA does not influence physiological parameters such as systemic blood pressure and blood gases; therefore, these parameters do not contribute to the protective effects. We did not investigate whether the neuroprotection of TSA extends over several days or weeks after injury and plan to address this question in future studies.
The neuroprotective effects of HDAC inhibitors may involve multiple mechanisms. HDAC inhibitors not only regulate gene expression directly via promoter hyperacetylation but also selectively modulate transcription factors via hyperacetylation that is independent of histones [37]. For example, HDAC inhibitors acetylate the neuroprotective transcription factor Sp-1 [38], as well as glucose-regulated protein 78 [39]. Additionally, these drugs inhibit the excitotoxicity-induced nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase [40], which has been shown to have a proapoptotic role in neurons. HDAC inhibitors also induce brain-derived neurotrophic factor in rat brain neurons [41] and glial cell-derived neurotrophic factor in cultured astrocytes from rat midbrain, causing robust neurite outgrowth [42].
Some studies have suggested that the neuroprotection against cerebral ischemia provided by HDAC inhibitors is due to their antiapoptotic effects. Indeed, HDAC inhibitors have shown anti-inflammatory effects in vitro and in vivo that result from inhibition of cytokines and key transcription factors (NF-kB and STAT) and from proliferation or differentiation of normal cells (synoviocytes and colonic epithelial cells) [43]. In addition, it has been demonstrated that HDAC inhibitor-induced neuro-protection in the ischemic brain involves prevention of apoptotic processes. In the rodent MCAO model, HDAC inhibitors suppressed ischemia-induced p53 expression as well as neuronal caspase-3 activation and increased the expression of HSP70 and Bcl-2, both of which have been shown to protect against ischemic neuronal death [12,36]. Furthermore, the pMCAO-induced reduction in Akt phosphorylation was blocked by TSA and sodium butyrate [12]; this finding is relevant because Akt is considered to have antiapoptotic activity [44]. Moreover, HDAC inhibition can activate the ERK pathway [45] to reduce proapoptotic molecules and increase antioxidant enzymes [35] and HSP70 levels [36].
ARE is a nucleotide sequence located on the enhancer region of genes that encode phase II detoxifying enzymes, such as NQO1, HO1, and GCLC [46]. When cells are subjected to oxidative or xenobiotic stress, Nrf2 activates the expression of several dozen such cytoprotective genes [47] by binding to the ARE. Thus Nrf2 increases the production of downstream proteins that protect cells from endogenous and exogenous injuries. Our new findings suggest that HDAC inhibition with TSA might significantly activate Nrf2 and induce expression of its downstream genes. Since HDAC inhibitors may modulate the transcription factor via acetylation independent of histone, and some researchers have reported that acetylation of Nrf2 augments promoter-specific DNA binding, thereby inducing Nrf2-mediated antioxidant genes [10], we suspect that HDAC inhibition by TSA might partially contribute to Nrf2 pathway regulation.
Interestingly, our data showed that TSA treatment with or without EGb761 increased Nrf2 acetylation in vitro, activated Nrf2, and upregulated protein expression downstream of Nrf2, including. Correa et al. demonstrated that valproic acid and TSA elevated histone acetylation levels and restored Nrf2 protein expression and Nrf2-inducible antioxidant defense [48]. This study supports the premise that HDAC inhibition increases Nrf2 activity and is consistent with our results. Two previous reports showed that HO1 gene transcription, compared with that of NQO1 or GCLM, was less or not affected byNrf2 acetylation [10], or TSA even reduced HO1 expression in a bronchial epithelia cell line [49]. These discrepancies might be caused by the fact that acetylation of Nrf2 contributes to its ability to discriminate among different ARE sequences, as ARE sequences have considerable flexibility and variability among different genes and species/cells. Also we used EGb761 to stimulate Nrf2 pathway that is a different stimulator than others. The HO1 gene, unlike other Nrf2 target genes, is highly activated by Nrf2 independent of Nrf2 acetylation; modulation of Keap1 inhibition or Nrf2 phosphorylation can also selectively regulate the binding of Nrf2 to AREs.
A variety of studies have shown that Ginkgo biloba extract induces HO1 expression through Nrf2 activation in vitro and in vivo [6,50,51]. Although Ginkgo biloba is not an HDAC inhibitor, we used it here as a relevant control to investigate the associated mechanisms by which TSA might provide neuroprotection against cerebral ischemia. We found that EGb 761 enhanced the ability of TSA to activate Nrf2 and induce HO1 in vitro without affecting the expression of Keap1. The results suggest that EGb 761 and TSA activate Nrf2 and EGb 761 might amplify the Nrf2 activation produced by HDAC inhibition. In the future, we will test the possibility that the two agents might be applied together to produce additional protection after cerebral ischemia.
Our results suggest that HDAC inhibition induced by TSA may promote neuroprotection by suppressing Keap1 expression and thereby activating the transcription factor Nrf2. Although the various effects of HDAC inhibitors and proof of their clinical efficacy for the treatment of neurodegenerative diseases remain to be verified, our findings indicate that HDAC inhibition might represent a promising new possibility for pharmacologic intervention after ischemic stroke.
Acknowledgments
This work was supported by a research grant from AHA GBIA (086576E) of USA, a grant from National Nature Science Foundation of China (81070940), and support from “High Level Innovative Talent Introduction Plan of Jiangsu Province 2011, PR China” to W. Cao. We thank Claire Levine for editorial assistance. The authors state no conflict of interest.
Abbreviations
- ARE
antioxidant response element
- ChIP
chromatin immunoprecipitation
- CNS
central nervous system
- DMEM
modified Eagle’s medium
- DTT
dithiothreitol
- EMSA
electrophoresis mobility shift assay
- GCLC
glutamate cysteine ligase catalytic
- HDAC
histone deacetylase
- HO1
heme oxygenase 1
- Keap1
Kelch-like ECH-associated protein 1
- LAS0811
1,2-dimethoxy-4,5-dinitrobenzene
- MCA
middle cerebral artery
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- NaB
sodium butyrate
- NP-40
Nonidet P-40
- NQO1
NAD(P)H:quinine oxidoreductase 1
- Nrf2−/−
Nrf2-deficient
- OGD
oxygen/glucose deprivation
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate-buffered saline
- pMCAO
permanent middle cerebral artery occlusion
- PMSF
phenylmethylsulfonyl fluoride
- ROS
reactive oxygen species
- RT-PCR
reverse transcription polymerase chain reaction
- SAHA
suberoylanilide hydroxamic acid
- SDS
sodium dodecyl sulfate
- TSA
trichostatin A
- TTC
2,3,5-triphenyl-tetra-zolium chloride
- WT
wild-type
References
- 1.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 2.Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4:461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 3.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47: 89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 4.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated anti-oxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ungvari Z, Bagi Z, Feher A, Recchia FA, Sonntag WE, Pearson K, de Cabo R, Csiszar A. Resveratrol confers endothelial protection via activation of the an-tioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol. 2010;299: H18–H24. doi: 10.1152/ajpheart.00260.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhuang H, Pin S, Christen Y, Doré S. Induction of heme oxygenase 1 by Ginkgo biloba in neuronal cultures and potential implications in ischemia. Cell Mol Biol (Noisyle-Grand) 2002;48:647–653. [PubMed] [Google Scholar]
- 7.Zhu M, Baek H, Liu R, Song A, Lam K, Lau D. LAS0811: from combinatorial chemistry to activation of antioxidant response element. J Biomed Biotechnol. 2009;420194:2009. doi: 10.1155/2009/420194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–44682. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- 9.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol. 2009;29:2658–2672. doi: 10.1128/MCB.01639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 12.Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- 13.Sinn DI, Kim SJ, Chu K, Jung KH, Lee ST, Song EC, Kim JM, Park DK, Kun Lee S, Kim M, Roh JK. Valproic acid-mediated neuroprotection in intrace-rebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis. 2007;26:464–472. doi: 10.1016/j.nbd.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Yildirim F, Gertz K, Kronenberg G, Harms C, Fink KB, Meisel A, Endres M. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp Neurol. 2008;210:531–542. doi: 10.1016/j.expneurol.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 15.Taguchi A, Kasahara Y, Nakagomi T, Stern DM, Fukunaga M, Ishikawa M, Matsuyama T. A Reproducible and simple model of permanent cerebral ischemia in CB-17 and SCID mice. J Exp Stroke Transl Med. 2010;3:28–33. doi: 10.6030/1939-067x-3.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saleem S, Shah ZA, Urade Y, Doré S. Lipocalin-prostaglandin D synthase is a critical beneficial factor in transient and permanent focal cerebral ischemia. Neuroscience. 2009;160:248–254. doi: 10.1016/j.neuroscience.2009.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, Wells AD, Hancock WW. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 18.Nervi C, Borello U, Fazi F, Buffa V, Pelicci PG, Cossu G. Inhibition of histone deacetylase activity by trichostatin A modulates gene expression during mouse embryogenesis without apparent toxicity. Cancer Res. 2001;61:1247–1249. [PubMed] [Google Scholar]
- 19.Cao W, Bao C, Padalko E, Lowenstein CJ. Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits Toll-like receptor signaling. J Exp Med. 2008;205:1491–1503. doi: 10.1084/jem.20071728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Zhuang H, Doré S. Heme oxygenase 2 is neuroprotective against intra-cerebral hemorrhage. Neurobiol Dis. 2006;22:473–476. doi: 10.1016/j.nbd.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 21.Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Doré S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147:53–59. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao W, Bao C, Lowenstein CJ. Inducible nitric oxide synthase expression inhibition by adenovirus E1A. Proc Natl Acad Sci U S A. 2003;100:7773–7778. doi: 10.1073/pnas.1337185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li RC, Saleem S, Zhen G, Cao W, Zhuang H, Lee J, Smith A, Altruda F, Tolosano E, Dore S. Heme-hemopexin complex attenuates neuronal cell death and stroke damage. J Cereb Blood Flow Metab. 2009;29:953–964. doi: 10.1038/jcbfm.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez-Martin FJ, Fernandez-Salguero PM, Merino JM. Aryl hydrocarbon receptor-dependent induction of apoptosis by 2,3,7,8-tetrachlorodibenzo-p-dioxin in cerebellar granule cells from mouse. J Neurochem. 2011;118:153–162. doi: 10.1111/j.1471-4159.2011.07291.x. [DOI] [PubMed] [Google Scholar]
- 25.Wang B, Cao W, Biswal S, Dore S. Carbon monoxide-activated nrf2 pathway leads to protection against permanent focal cerebral ischemia. Stroke. 2011;42: 2605–2610. doi: 10.1161/STROKEAHA.110.607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27:6334–6349. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saleem S, Zhuang H, Biswal S, Christen Y, Dore S. Ginkgo biloba extract neuroprotective action is dependent on heme oxygenase 1 in ischemic reperfusion brain injury. Stroke. 2008;39:3389–3396. doi: 10.1161/STROKEAHA.108.523480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanigawa S, Fujii M, Hou DX. Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic Biol Med. 2007;42:1690–1703. doi: 10.1016/j.freeradbiomed.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 29.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsuji N, Kobayashi M, Nagashima K, Wakisaka Y, Koizumi K. A new antifungal antibiotic, trichostatin. J Antibiot (Tokyo) 1976;29:1–6. doi: 10.7164/antibiotics.29.1. [DOI] [PubMed] [Google Scholar]
- 32.Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs. 2005;14:1497–1511. doi: 10.1517/13543784.14.12.1497. [DOI] [PubMed] [Google Scholar]
- 33.Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;22:40–49. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 35.Camelo S, Iglesias AH, Hwang D, Due B, Ryu H, Smith K, Gray SG, Imitola J, Duran G, Assaf B, Langley B, Khoury SJ, Stephanopoulos G, De Girolami U, Ratan RR, Ferrante RJ, Dangond F. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164:10–21. doi: 10.1016/j.jneuroim.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 36.Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, Moroni F, Chiarugi A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol. 2006;70:1876–1884. doi: 10.1124/mol.106.027912. [DOI] [PubMed] [Google Scholar]
- 37.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 38.Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci U S A. 2003;100: 4281–4286. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bown CD, Wang JF, Young LT. Increased expression of endoplasmic reticulum stress proteins following chronic valproate treatment of rat C6 glioma cells. Neuropharmacology. 2000;39:2162–2169. doi: 10.1016/s0028-3908(00)00029-0. [DOI] [PubMed] [Google Scholar]
- 40.Kanai H, Sawa A, Chen RW, Leeds P, Chuang DM. Valproic acid inhibits histone deacetylase activity and suppresses excitotoxicity-induced GAPDH nuclear accumulation and apoptotic death in neurons. Pharmacogenomics J. 2004;4:336–344. doi: 10.1038/sj.tpj.6500269. [DOI] [PubMed] [Google Scholar]
- 41.Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology (Berl) 2001;158:100–106. doi: 10.1007/s002130100871. [DOI] [PubMed] [Google Scholar]
- 42.Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, Wilson B, Lu RB, Gean PW, Chuang DM, Hong JS. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–1125. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- 43.Blanchard F, Chipoy C. Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases? Drug Discov Today. 2005;10:197–204. doi: 10.1016/S1359-6446(04)03309-4. [DOI] [PubMed] [Google Scholar]
- 44.Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- 45.Perez M, Rojo AI, Wandosell F, Diaz-Nido J, Avila J. Prion peptide induces neuronal cell death through a pathway involving glycogen synthase kinase 3. Biochem J. 2003;372:129–136. doi: 10.1042/BJ20021596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- 47.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 48.Correa F, Mallard C, Nilsson M, Sandberg M. Activated microglia decrease histone acetylation and Nrf2-inducible anti-oxidant defence in astrocytes: restoring effects of inhibitors of HDACs, p38 MAPK and GSK3beta. Neurobiol Dis. 2011;44: 142–151. doi: 10.1016/j.nbd.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mercado N, Thimmulappa R, Thomas CM, Fenwick PS, Chana KK, Donnelly LE, Biswal S, Ito K, Barnes PJ. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem Biophys Res Commun. 2011;406:292–298. doi: 10.1016/j.bbrc.2011.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah ZA, Nada SE, Dore S. Heme oxygenase 1, benecial role in permanent ischemic stroke and in Gingko biloba (EGb 761) neuroprotection. Neuroscience. 2011;180:248–255. doi: 10.1016/j.neuroscience.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen JS, Huang PH, Wang CH, Lin FY, Tsai HY, Wu TC, Lin SJ, Chen JW. Nrf-2 mediated heme oxygenase-1 expression, an antioxidant-independent mechanism, contributes to anti-atherogenesis and vascular protective effects of Ginkgo biloba extract. Atherosclerosis. 2011;214:301–309. doi: 10.1016/j.atherosclerosis.2010.11.010. [DOI] [PubMed] [Google Scholar]