

Abstract
Background
Complex plant-microbe interactions have been established throughout evolutionary time, many of them with beneficial effects on the host in terms of plant growth, nutrition, or health. Some of the corresponding modes of action involve a modulation of plant hormonal balance, such as the deamination of the ethylene precursor 1-aminocyclopropane-1-carboxylate (ACC). Despite its ecological importance, our understanding of ACC deamination is impaired by a lack of direct molecular tools. Here, we developed PCR primers to quantify the ACC deaminase gene acdS and its mRNA in soil communities and assessed acdS+ microorganisms colonizing maize and other Poaceae species.
Results
Effective acdS primers suitable for soil microbial communities were obtained, enabling recovery of bona fida acdS genes and transcripts of diverse genetic backgrounds. High numbers of acdS genes and transcripts were evidenced in the rhizosphere of Poaceae, and numbers fluctuated according to plant genotype. Illumina sequencing revealed taxonomic specificities of acdS+ microorganisms according to plant host. The phylogenetic distance between Poaceae genotypes correlated with acdS transcript numbers, but not with acdS gene numbers or the genetic distance between acdS functional groups.
Conclusion
The development of acdS primers enabled the first direct analysis of ACC deaminase functional group in soil and showed that plant ability to interact with soil-inhabiting acdS+ microorganisms could also involve particular plant traits unrelated to the evolutionary history of Poaceae species.
Electronic supplementary material
The online version of this article (10.1186/s40168-018-0503-7) contains supplementary material, which is available to authorized users.
Keywords: ACC deaminase functional group, Rhizosphere community, Poaceae evolution
Background
Growth, development, and health of macroorganisms are strongly influenced by the interactions they develop with their associated microbial community [1–3]. These interactions often involve nutrient exchanges between partners, the host typically providing organic carbon [4–7] while microorganisms supply amino acids or mineral nutrients, which results from various processes such as nitrogen fixation or phosphorus solubilization [8]. Many of these interactions are complex and entail also the exchange of molecular signals [2]. In the case of plants, microbial partners might influence host hormonal balance by producing molecules mimicking phytohormones (e.g., auxins, gibberellins, or jasmonate) or enzymes that modulate plant hormonal production, notably by degrading the ethylene biosynthetic precursor 1-aminocyclopropane-1-carboxylate (ACC) [2, 8, 9].
Ethylene is a plant hormone that regulates plant development and stress responses [10, 11]. Microorganisms able to produce ACC deaminase transform the ethylene precursor ACC into α-ketobutyrate and ammonia [12, 13]. By degrading ACC within roots or in exudates (thereby leading to a sink effect), root-interacting bacteria are indirectly lowering ethylene level in plant roots, thus stimulating root growth and modulating plant stress resistance [9, 14–17]. The acdS gene encoding ACC deaminase is highly conserved among microorganisms and has been used to study the phylogeny and diversity of ACC deaminase producers [18–20] in bacteria and micro-eukaryotes (i.e., fungi and stramenopiles). Although horizontal transfer of acdS between bacteria was suspected due to incongruence between acdS and 16S rRNA gene-based bacterial phylogenies [21, 22], a more exhaustive assessment suggested that acdS was mainly inherited vertically, with only occasional horizontal gene transfers [20]. Thus, acdS is a marker suitable to assess complex ACC deaminase functional communities.
In the root zone, the plant may be colonized by different types of acdS+ microorganisms [23, 24], which are likely to contribute jointly to degradation of ACC produced by roots, and the overall significance of ACC deamination for the plant is expected to result from the combined functioning of its acdS+ microbial partners [2]. However, even though various kinds of acdS+ microorganisms can be readily isolated from the rhizosphere, there is no direct PCR tool available to assess the entire functional group of root-associated acdS+ microorganisms, i.e., including non-cultured taxa and strains.
It is long established that plants shape their rhizosphere bacterial community from the telluric bacterial reservoir [25, 26]. Plant species and plant genotypes within species exhibit specific phenotypic traits, including root properties that are likely to influence rhizobacterial community composition [7]. And indeed, several studies showed the impact of plant genotype on the taxonomic composition of their associated bacterial community [27–30]. This type of effect has also been evidenced when assessing the genetic diversity of functional groups important for plant growth, such as 2,4-diacetylphloroglucinol-producing pseudomonads [31–33], nitrogen fixers [34], or microorganisms involved in other biogeochemical transformations [35], and it might well be that similar effects also take place with acdS+ microorganisms.
Therefore, this work aimed at testing the hypothesis that the functional group of acdS+ microorganisms differed according to plant genotype, which was done in the case of Poaceae. To this end, protocols for quantitative PCR (qPCR) and quantitative reverse-transcription PCR (qRT-PCR) of acdS were developed and validated for analysis of acdS+ microorganisms within the rhizosphere. These tools as well as acdS MiSeq sequencing were implemented on eight Poaceae genotypes previously used in Bouffaud et al. [36] and enabling comparisons at different Poaceae taxonomic levels, i.e., between individual inbred lines within two maize genetic groups (Corn Belt Dent and Northern Flint), between two maize genetic groups, between these maize genetic groups and a teosinte (representing maize’s pre-domestication Zea mays ancestor), and between Z. mays and another member (sorghum) from maize’s Panicoideae subfamily or a member (wheat) from the neighboring Pooideae subfamily.
Results
Validation of qPCR and qRT-PCR tools
We developed primer sets (Additional file 1: Table S1) to specifically amplify acdS gene sequences in the acdS reference database constructed. Eight primer pairs were discarded as they failed to specifically amplify acdS from pure bacterial genomic samples. Another one was discarded as it could not amplify acdS genes from a soil community, and only pair acdSF5/acdSR8 (Additional file 1: Tables S1, S2 and Additional file 2: Figure S1) was kept.
qPCR conditions were optimized to obtain an amplification efficiency of > 80% and an error below 0.1 with genomic DNA of acdS strains P. kilonenesis F113 (i.e., 81.2% and 0.06, respectively) and Burkholderia cenocepacia J2315 (i.e., 100% and 0.007, respectively). The detection limit of the qPCR method on pure cultures was 6 acdS gene copies (corresponding to 50 fg of J2315 genomic DNA template). When tested on total DNA or cDNA (reverse transcription on total RNA) obtained from rhizosphere of the different Poaceae, amplification efficiencies above 80% and errors below 0.1 were also obtained, for both soils tested. acdS could always be detected by qPCR in the two bulk soils (Additional file 3: Figure S2).
Illumina MiSeq sequencing of acdSF5/acdSR8 amplicons from bulk soils and rhizosphere soils gave 3,903,982 reads (44,287 OTUs) from cropped soil and 1,673,758 reads (28,759 OTUs) from meadow soil. Similarities of acdS sequences with known references were assessed with the in-house core-acdS database extracted from the FunGene database. The phylogenetic tree showed that none of the sequences clustered within the D-cystein-sulfydrase outgroup (Additional file 4: Figure S3). The OTUs obtained clustered in numerous clades encompassing all the known diversity of the acdS gene (Proteobacteria, Actinobacteria, and microeukaryotes; Additional file 4: Figure S3).
Size of acdS group and number of acdS transcripts in Poaceae rhizosphere
The size of the acdS group amounted to 0.5–6.2 × 106 acdS gene copies per g of rhizosphere soil in the cropped soil at 21 days, 0.5–2.0 × 107 acdS copies in the meadow soil at 21 days, and 0.2–3.0 × 106 acdS copies in the cropped soil at 42 days (Fig. 1A). Compared to bulk soil, acdS group size was higher in the presence of plant for all four maize lines and wheat at 21 days in cropped soil (all five displaying similar levels), for the two maize lines in the meadow soil at 21 days, and for all plants in the cropped soil at 42 days. A significant effect of past soil management was observed in all four treatments studied (two maize lines, tomato, and bulk soil), but an influence of sampling time was evidenced only for maize line FV4.
Fig. 1.
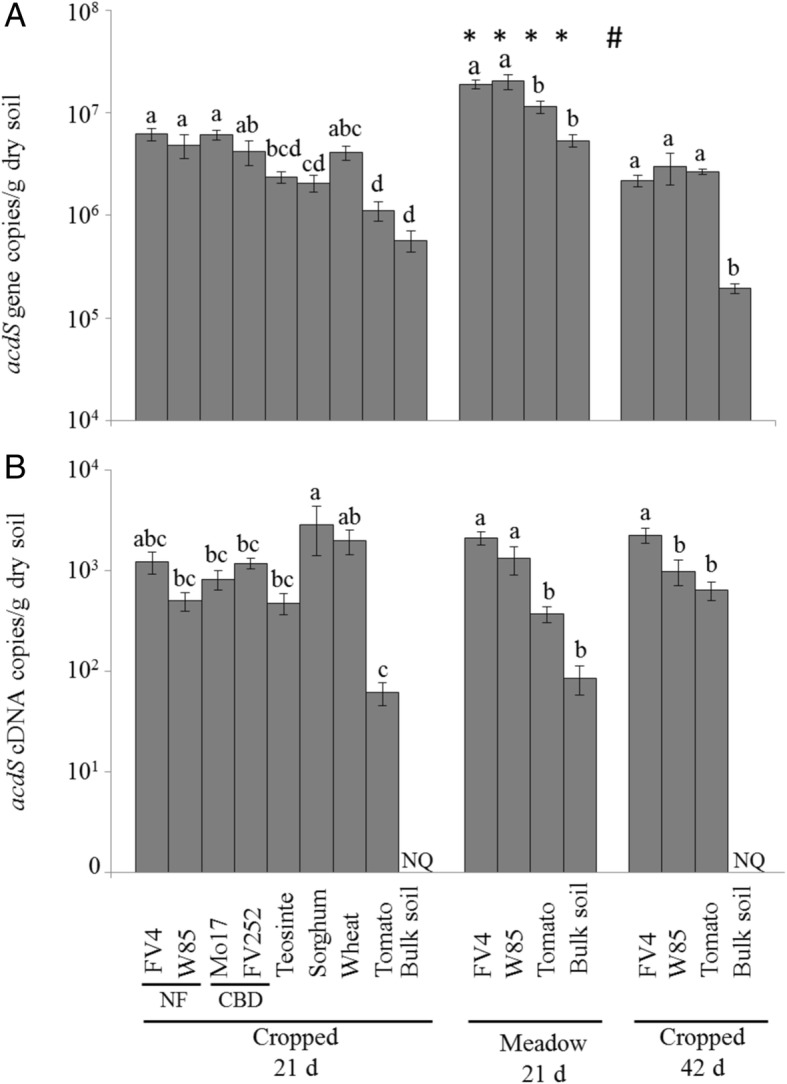
Quantification of acdS genes (A) and acdS transcripts (B) in bulk soil and rhizosphere of Poaceae genotypes and tomato by qPCR and qRT-PCR, respectively. NF means Northern Flint and CBD, Corn Belt Dent. NQ: not quantifiable. Statistical analyses were performed independently at 21 days in cropped soil, at 21 days in meadow soil, and at 42 days in cropped soil, using ANOVA and Fisher LSD tests (P < 0.05; results shown with letters a to d). For FV4, W85, tomato, and bulk soil, two-way ANOVA and Fisher LSD tests (P < 0.05) were also performed to compare treatments according to past soil management or sampling time, and differences with the same genotype at 21 days in cropped soil are indicated by symbols * and #, respectively
The number of acdS transcripts reached 0.06–2.9 × 103 acdS cDNA copies per g of rhizosphere soil in the cropped soil at 21 days, 0.08–2.1 × 103 acdS copies in the meadow soil at 21 days, and 0.6–2.2 × 103 acdS copies in the cropped soil at 42 days (Fig. 1B). The acdS transcripts in the bulk soil samples from cropped soil at 21 and 42 days were below the quantification limit. A strong positive influence of Poaceae on the number of acdS transcripts was observed in both soils at both sampling times. In addition, acdS transcript number in cropped soil was higher for sorghum than maize (except FV4) and teosinte (and tomato) at 21 days, and for FV4 maize than W85 (and tomato) at 42 days, while it was higher for both maize lines studied than for tomato and bulk soil in meadow soil at 21 days. Overall, no significant effect of past soil management or sampling time was found for acdS transcript number.
Diversity of acdS+ microorganisms colonizing Poaceae
Rarefaction analyses resulted in different saturation profiles, suggesting that acdS allele diversities differed across the different Poaceae rhizospheres (Additional file 5: Figure S4). To estimate acdS richness, a subsampling was done with 38,524 sequences per cropped soil sample and with 25,423 sequences per meadow soil sample.
Shannon, Simpson, and Chao diversity indices were calculated for each sample (Tables 1 and 2). In the cropped soil, no significant difference in the diversity of the acdS functional group was observed between treatments. In the meadow soil, Shannon (P = 0.038) and Simpson (P = 0.044) indices differed between FV4 maize and tomato (but showed opposite trends), whereas the difference was not significant for the Chao index.
Table 1.
Diversity indices of acdS functional group. Shannon, Simpson, and Chao indices in bulk soil and rhizosphere were determined at 21 days of plant growth in cropped soil
| Bulk soil | NF maize FV4 | NF maize W85 | CBD maize Mo17 | CBD maize FV252 | Teosinte | Sorghum | Wheat | |
|---|---|---|---|---|---|---|---|---|
| Shannon | 7.12 ± 0.33 | 6.26 ± 1.63 | 7.54 ± 0.19 | 7.18 ± 0.70 | 6.93 ± 0.14 | 7.21 ± 0.41 | 7.23 ± 0.11 | 7.28 ± 0.16 |
| Simpson | 0.0051 ± 0.0039 | 0.0613 ± 0.1174 | 0.0028 ± 0.0009 | 0.0075 ± 0.0102 | 0.0076 ± 0.0044 | 0.0076 ± 0.0044 | 0.0030 ± 0.0006 | 0.0054 ± 0.0041 |
| Chao | 14,326 ± 2006 | 12,578 ± 2984 | 15,996 ± 604 | 14,156 ± 3100 | 13,783 ± 579 | 14,030 ± 3095 | 14,049 ± 439 | 15,585 ± 1237 |
NF Northern Flint genetic group of maize, CBD Corn Belt Dent genetic group of maize. Data are means ± standard deviations (n = 5)
Table 2.
Diversity indices of acdS functional group. Shannon, Simpson, and Chao indices in bulk soil and rhizosphere were determined at 21 days of plant growth in meadow soil
| Bulk soil | NF maize FV4 | NF maize W85 | Tomato | |
|---|---|---|---|---|
| Shannon | 6.09 ± 1.26 ab | 6.71 ± 0.16 a | 6.68 ± 0.13 ab | 6.19 ± 0.25 b |
| Simpson | 0.054 ± 0.089 ab | 0.009 ± 0.001 a | 0.010 ± 0.002 ab | 0.020 ± 0.007 b |
| Chao | 12,870 ± 1882 | 12,353 ± 730 | 12,814 ± 834 | 11,957 ± 1316 |
NF Northern Flint genetic group of maize, CBD Corn Belt Dent genetic group of maize. Data are means ± standard deviations (n = 5). Statistical differences (when any) are indicated using letters a and b (Kruskal-Wallis tests; P < 0.05)
When considering taxa corresponding to the acdS alleles obtained, the 20 most abundant genera thus identified (all bacterial) were common to all samples and represented over 90% of the sequences (Fig. 2). Compared to bulk cropped soil, the relative abundance of the 20 most abundant taxa in the acdS community was largely similar in the rhizosphere of the different Poaceae. However, the genus Saccharotrix (1.44 ± 0.18% in bulk soil) reached as much as from 2.09 ± 0.09% (maize W85) to 2.64 ± 0.37% (maize FV252) in the rhizosphere, Amycolatopsis (2.49 ± 0.15% in bulk soil) from 2.90 ± 0.20% (wheat) to 6.14 ± 1.78% (maize W85), and Acidovorax (7.59 ± 0.67% in bulk soil) from 8.71 ± 0.27% (wheat) to 17.09 ± 0.15% (maize W85). In parallel, the genus Saccharopolyspora (3.77 ± 0.76% in bulk soil) represented only from 2.56 ± 0.54% (wheat) to 1.21 ± 0.30% (maize FV252) of all acdS sequences in the rhizosphere, and Phycicoccus (3.99 ± 0.70% in bulk soil) from 2.76 ± 0.56% (wheat) to 2.20 ± 0.22% (sorghum). Similar dynamics were observed in meadow soil when comparing bulk soil with two maize lines (FV4 and W85 from the same maize genetic group) and the non-Poaceae reference tomato (Fig. 2).
Fig. 2.

Relative abundance of the 20 most abundant acdS bacterial genera (all Proteobacteria or Actinobacteria) present in bulk soil and common to each rhizophere at 21 days in cropped (a) or meadow (b) soil. The remaining sequences (< 8% in cropped soil and < 6% in meadow soil) are not pictured. Mean values across six replicates are shown for each treatment. The same colors were used in a and b for genera common to both soils
For a more global appraisal, acdS sequence data were also processed by between-class analysis, which showed that acdS community composition in the rhizosphere differed from that in bulk soil at 21 days in cropped soil (Fig. 3a). In addition, a difference was also found between cultivated (maize, sorghum, and wheat) and spontaneous Poaceae (teosinte) (Fig. 3a). A significant rhizosphere effect also took place in the meadow soil, but it was of modest magnitude for the non-Poaceae reference tomato (Fig. 3b).
Fig. 3.

Between class analysis for comparison of acdS functional group diversity in bulk soil and the rhizosphere of different Poaceae grown in cropped-soil (a) or of maize and tomato in meadow soil (b). FV4 and W85 are Northern Flint and Mo17 and FV252 are Corn Belt Dent. For each treatment, the mean and standard error computed using all data are presented (n = 6)
Relationship between Poaceae evolution and acdS functional group
The assessment of the influence of past Poaceae evolution on recruitment of acdS microorganisms did not yield any significant correlation, regardless of whether pairwise Poaceae phylogenetic distances were crossed with (i) differences in raw numbers (or log-numbers; Additional file 1: Table S3) of acdS genes copies per g of rhizosphere soil (P = 0.64; Fig. 4b) or g of root (P = 0.24; Fig. 4c), or (ii) differences in pairwise Bray-Curtis dissimilarity indices of acdS communities (P = 0.83; Fig. 4a), at 21 days in the rhizosphere of Poaceae grown in cropped soil. When considering the functioning of the acdS community, however, a significant correlation with pairwise Poaceae phylogenetic distances was found when computing differences in raw numbers (but not log numbers; Additional file 1: Table S3) of acdS transcript copies per g of rhizosphere soil (Spearman rho = 0.65; P = 0.002; Fig. 4d) or g of root (Spearman rho = 0.53; P = 0.02; Fig. 4e).
Fig. 4.

Pairwise comparison of plant phylogenetic distance between Poaceae (X-axis) with the corresponding distance between their root-associated ACC deaminase functional group (Y-axis). The 18 maize-based comparisons are indicated using black triangles (maize-maize; n = 6), diamonds (maize-teosinte; n = 4), circles (maize-sorghum; n = 4), or squares (maize-wheat; n = 4), and the three other comparisons using a white circle (teosinte-sorghum), white triangle (teosinte-wheat), or white square (sorghum-wheat). Distances were calculated two by two, using Kimura two-parameter model for plant phylogeny, Bray-Curtis distance between rhizobacterial communities (a) and Euclidean distance for qPCR (b, c) or qRT-PCR data (d, e). The Spearman correlation coefficient is indicated
Relationship between maize/Poaceae evolution and particular rhizobacterial populations
Among the 20 most abundant genera representing over 90% of acdS functional group diversity, a significant positive correlation (P < 0.05) was found for four Actinobacteria, i.e., Agromyces (rho = 0.61; P = 0.006), Kineosphera (rho = 0.61; P = 0.005), Saccharopolyspora (rho = 0.57; P = 0.009), and Phycicoccus (rho = 0.51; P = 0.022) when comparing read abundance for these genera to the log value of the maximum likelihood phylogenetic distance between Poaceae (Additional file 6: Figure S5).
Discussion
Since the rhizosphere microbiota results mostly from the selection of soil-residing microorganisms [25, 26], root exudates and other rhizodeposits are expected to play a major role [5], which could explain why this microbial selection can differ according to host genetics [37]. Indeed, plant genetic background was shown to be an important factor shaping the root-associated microbiota, when considering both taxonomic [28, 29, 36, 38] and functional microbial groups, e.g., 2,4-diacetylphloroglucinol-producing pseudomonads [39] or nitrogen-fixing bacteria [34].
In the last two decades, much has been done to document the impact of plant genotype and/or development stage on rhizosphere microbial diversity, with an emphasis on taxonomic assessments using 16S and 18S rRNA genes as well as internal transcribed spacers [40]. However, these taxonomic assessments do not tell much about the functional potential of the microbiota, and this gap limits our capacity to understand the functioning of the plant holobiont [41]. This issue may be targeted at the scale of the entire microbial community, by metagenomics or metatranscriptomics [28, 42], as well as with a focus on particular ecological functions of prime significance, such as nitrogen fixation [34, 43, 44], nitrification [44, 45], or production of antimicrobial metabolites [46].
For certain key functional groups of soil microorganisms, primers and PCR protocols necessary for direct analysis of soil and rhizosphere populations have been used for years (e.g., [33, 46, 47]), but for others, the tools are not available yet. This was the case in particular for ACC deaminase-positive microorganisms, despite the research emphasis put on this microbial function in the last 20 years (> 10,000 papers on these microorganisms as listed in Google Scholar). The ecological importance of microbial ACC deaminase activity derives from its effect on plant metabolism of ethylene, a major phytohormone involved in various plant physiological pathways such as root elongation and immune response [16]. ACC itself acts as a phytohormone [17]. Root-colonizing microorganisms with ACC deaminase activity have the potential to indirectly lower endogenous plant levels in ACC and ethylene, and thus to alleviate environmental stress in plant and enhance root growth [16, 48].
Against this background, our first objective was to develop an acdS-based qPCR method targeting all acdS alleles, which was successfully achieved by using a set of acdS strains representative of the highest diversity possible of documented alleles (from Azospirillum lipoferum CRT1 to Ralstonia solanacearum GMI100; Additional file 4: Figure S3). This is first indicated by the ability of the method to minimize acdS quantification bias, as qPCR performance was satisfactory for two contrasted acdS strains based on the criteria of Zhang and Fang [49] (i.e., R2 > 0.95 and slope between − 3.0 and − 3.9 corresponding to PCR efficiencies of 80–115%). Second, it is also indicated by (i) primer specificity and (ii) the possibility to target the known diversity of acdS alleles in Proteobacteria, Actinobacteria, Deinococcus/Thermus, and micro-eukaryotes [19, 20, 50], as revealed by MiSeq Illumina sequencing (Additional file 4: Figure S3). Apart from the absence of sequences affiliated to the thermophilic genus Meiothermus of the Deinococcus/Thermus phylum (the unique genus of this phylum shown to harbor acdS [20]), all the other clades were represented, without over-representation of a given type of acdS sequence (Fig. 2). Therefore, this work proposes a novel tool that can be used to monitor the size, transcription, and diversity of the acdS functional group in various plant, soil, and environmental conditions.
Our second and main objective was to assess the influence of past Poaceae evolution on root interactions with acdS microorganisms, based on the rationale that bacterial taxa harboring acdS strains colonize roots of different Poaceae [27, 28, 30], acdS microorganisms are readily isolated from these plants [2, 21, 51, 52], and they can stimulate growth of cereals [53–56]. Recruitment of acdS microorganisms by all Poaceae was indeed shown by qPCR (Fig. 1), and it might be an indication of the importance of ACC deamination for the plant. Accordingly, we found a strong increase in the numbers of acdS ARNm (cDNA) when comparing the rhizosphere to bulk soil, showing for the first time the stimulation of acdS group functioning on roots. It can be expected that microbial functioning varies with different plant species [57, 58], and in this work, acdS transcript levels in the rhizosphere differed according to Poaceae genotype. Although rhizosphere effects were not the same for all Poaceae genotypes, past Poaceae evolution did not appear to have played a role on root selection of acdS microorganisms, as correlations were not significant when considering the size or genetic make-up of the acdS functional group in the rhizosphere. In fact, when considering the relationship obtained by Bouffaud et al. [36] between (i) the phylogenetic distance between Poaceae genotypes and the genetic distance between rhizobacterial communities, Proteobacteria and Actinobacteria (the main bacterial phyla containing acdS microorganisms) contributed strongly to the relationship but this concerned only 4 of the 20 principal proteobacterial and actinobacterial taxa evidenced in the current work, i.e., Pseudomonas, Acidovorax, Burkholderia, and Variovorax. Furthermore, we found that none of these four proteobacterial genera showed any correlation with Poaceae evolutionary history, whereas four actinobacterial genera not characterized in Bouffaud et al. [36] did show a significant correlation. This is reminiscent of previous findings with the functional group of diazotrophic bacteria [28, 34], and it suggests that selection for particular types of function-providing microorganisms may proceed following somewhat different ecological rules than those that apply to all members of the corresponding taxa [41].
Conclusion
In summary, we report the first acdS-based PCR method to monitor acdS alleles and transcripts of the ACC deaminase functional group in natural soil and plant systems. We then analyzed the microbial ACC deaminase functional group in the context of Poaceae evolutionary history. Correlations were significant when considering pairwise differences in raw numbers of acdS transcripts versus the genetic distance between acdS groups. This is the first evidence of a link between past Poaceae evolution and the functioning of a root-associated microbial group, such a link having not been observed for instance when considering N-fixing bacteria [34]. It also raises the possibility that microbial modulation of ethylene metabolism has evolved to different extents in various Poaceae lineages, and this issue will deserve further research attention.
Methods
Identification of acdS primers
All available (in February 2016) sequences from the acdS/D-cysteine-sulfhydrase FunGene sequence database were retrieved and aligned using MUSCLE. Key amino acid positions known to be important for ACC deaminase activity, namely Lys51, Ser78, Tyr295, Glu296, and Leu322 [59] were searched using Pseudomonas kilonensis F113 as a reference. Sequences presenting different amino acids in the key positions were discarded, as they are likely to correspond to D-cysteine desulfhydrases [59].
The acdS sequences and acdS homologs (also included in the alignment to serve as non-target sequences) from the D-cysteine desulfhydrase producer strains Pseudomonas syringae DC3000 (AE16853), Achromobacter xylosoxidans BM1 (AY604540), Achromobacter sp. CM1 (AY604541), Rhizobium sp. TAL1145 (EU183544), Serratia proteamaculans SUD165 (AY604543), Escherichia coli K12 (CP014348), Pseudomonas marginalis (AY604542), and Enterobacter aerogenes Cal3 (AY604544) were aligned using MUSCLE [60]. This alignment was used to perform a phylogenetic analysis on acdS sequences. The tree was inferred from 1000 nucleotides using the neighbor-joining (NJ) method in MEGA4 [61] with the Kimura two-parameter method for distance calculation [62]. Nodal robustness of the tree was assessed using 1000 bootstrap replicates.
Using the acdS alignment, primers (25–30 bp in length) were visually selected in regions conserved among the acdS sequences and absent from the non-target sequences. The new primers (Additional file 1: Table S1) were then assessed based on the following criteria: (i) a melting temperature (Tm) of 60 to 67 °C, (ii) an absence of predicted hairpin loops and primer-dimer formations [63], (iii) a Tm difference between primers not exceeding 1 °C, (iv) an amplification product not exceeding 300 bp, (v) a maximum of three mismatches between each primer and the 1304 acdS sequences, and (vi) the ability to specifically amplify acdS in genomic DNA samples (using 6 ng of genomic DNA from the acdS+ strains Azospirillum lipoferum TVV3, 4B and RSWT1, Burkholderia cepacia LMG 1222, B. cenocepacia LMG 16656 and J2315, Burkholderia stabilis LMG 14294, Burkholderia dolosa LMG18941, Pseudomonas thivervalensis PITR2, P. kilonensis F113, Ralstonia solanacearum GMI1000, and the non-acdS strains Pseudomonas protegens CHA0, E. coli K12, and A. lipoferum CRT1). Primer melting temperature, predicted hairpin loops, and predicted primer-dimer formations were determined using Oligo 6 (Molecular Biology Insights, West Cascade, CO) and the nearest-neighbor method [64]. Amplification specificity was determined by checking the Tm and size of the amplification product through (i) melting curve analysis followed by Tm determination (described below) and (ii) gel electrophoresis analysis and the observation of a single band of the expected size. Following this, one acdS-specific primer pair was selected for development of acdS qPCR and qRT-PCR protocols.
Development of acdS qPCR and qRT-PCR
Quantitative PCR assays were conducted using LightCycler 480 SYBR Green I Master mix in a final volume of 20 μL and a LightCycler 480 (Roche Applied Science, Meylan, France). Cycle threshold (Ct) of individual samples was calculated using the second derivative maximum method in the LightCycler Software v.1.5 (Roche Applied Science). The standard curves were obtained by plotting the mean Ct value of the three replicates (per DNA concentration) against the log-transformed DNA concentration. Amplification efficiency (E), calculated as E = 10(−1/slope) − 1, and the error of the method (mean squared error of the standard curve) were determined using the LightCycler Software v.1.5 (Roche Applied Science). Standard curves were generated using genomic DNA of P. kilonensis F113 and B. cenocepacia J2315, two bacterial genomes harboring a single acdS copy. acdS copy number was computed as [DNA (g) × Avogadro’s number (molecules mol−1)]/[number of DNA base pairs in acdS fragment × 660 (g mol−1)], based on an average of 660 g mol− 1 per base pair. Amplification specificity was assessed by melting curve analysis of PCR products, done by ramping the temperature to 95 °C for 10 s and back to 65 °C for 15 s, followed by increases of 0.1 °C s−1 up to 95 °C.
qPCR optimization was sought to improve acdS amplification efficiency (above 80%) and error (below 0.01) for the acdS strains P. kilonensis F113 and B. cenocepacia J2315. Three primer concentrations (0.5, 0.75, and 1 μM), four annealing temperatures (66, 67, 68, and 70 °C), two annealing times (30 and 15 s), and three elongation times (30, 15, and 10 s) were tested.
The final qPCR protocol used primers acdSF5/acdSR8 amplifying a fragment of 133 nt. Reaction mix contained 10 μL of LightCycler 480 SYBR Green I Master (Roche Applied Science), 1 μM of each primer, and 2 μL of DNA extract. The final cycling program included a 10-min incubation at 95 °C, 50 amplification cycles of 30 s at 94 °C, 7 s at 67 °C, and 15 s at 72 °C, and the fusion program for melting curve analysis described above. The generated standard curve from genomic DNA of B. cenocepacia J2315 was subsequently used as the external standard curve for determination of acdS copy number in uncharacterized DNA samples. Two DNA standards from genomic DNA of B. cenocepacia J2315 were included as reference in each run to detect between-run variations.
Greenhouse experiment
We used samples from a previous greenhouse experiment [36] performed with one wheat (Triticum aestivum L. cv. Fiorina; Agroscope, Changins, Switzerland), one sorghum (Sorghum bicolor L. cv. Arprim; Semences de Provence, Fourques, France), one teosinte (Zea mays ssp. parviglumis; UNAM, Cuernavaca, Mexico), four maize (Zea mays L.) inbred lines (FV252 and Mo17 from group Corn Belt Dent, and FV4 and W85 from group Northern Flint; INRA, St Martin de Hinx, France), and one tomato (Solanum lycopersicum L. cv. Marmande; Vilmorin, La Ménitré, France) considered as an arbitrary, external (non-Poaceae) reference.
The plants were cultivated by Bouffaud et al. [36] in one or two sieved (6 mm) soils collected from the topsoil of two neighboring fields (luvisols) at La Côte Saint-André (France). All were grown in soil from a maize-monocropping field (loam, organic matter 2.3%, pHH20 7.3, N 1.6 g kg−1) and three selected genotypes (see below) also in soil from a permanent meadow (loam, organic matter 5.5%, pHH20 6.0, N 3.2 g kg− 1). In short, pots of 3 dm3 holding 2.5 kg soil were sown with surface-disinfected seeds (to get one seedling per pot) or kept as non-planted pots, with five pots per treatment, and they were placed in a greenhouse (randomized blocks). For all treatments, samples were taken at 21 days in cropped soil, whereas maize lines FV4 and W85 and tomato were also studied at 42 days in the same soil and at 21 days in meadow soil. Root systems were individually unearthed and shaken to discard loosely adhering soil. Roots and tightly adhering soil were frozen in liquid nitrogen and lyophilized, and rhizosphere soil was collected from roots (0.5–6 g soil per plant) and placed at − 20 °C. Root, rhizosphere soil, and shoot dry weights were measured (Additional file 7: Figure S6). Bulk soil was taken from each non-planted pot (5 g) at 21 (two soils) and 42 days (cropped soil), frozen, lyophilized, and stored at − 20 °C.
Extraction of DNA and RNA from rhizosphere and bulk soil
We used total nucleic acids that were extracted by Bouffaud et al. [36], as follows. Briefly, 0.5 g bulk or rhizosphere soil, 0.5 ml extraction buffer (5% hexadecyltrimethylammonium bromide, 1 mM 1,4-dithio-DL-threitol, in a 0.12 M phosphate buffer at pH 8), and 0.5 g zirconium beads (VWR, Fontenay-sous-Bois, France) were used in a bead beater (TissueLyser II Retsch; Qiagen, Courtaboeuf, France) for 90 s at 30 m s−1. After 10 min centrifugation at 16,000g, supernatants were extracted twice in phenol-chloroform-isoamyl alcohol (24:24:1 v/v/v) and once in chloroform-isoamyl alcohol (24:1 v/v). Nucleic acids were precipitated overnight using absolute ethanol and potassium acetate (3 M, pH 4.8) at − 20 °C. After centrifugation for 30 min at 16,000g, pellets were washed in 70% ethanol and dissolved in 100 μL RNase-free DNase-free water (giving 50–100 ng nucleic acids μL−1).
Reverse-transcription synthesis of cDNA
DNA-free RNA was obtained by treating 20 μL of nucleic acid solution with 4 U of DNase I (Invitrogen, Cergy Pontoise, France) in 1× DNase I reaction buffer at room temperature. DNA digestion repeated to remove remaining DNA traces, the reaction was stopped in presence of 1 μL of 25 mM EDTA (10 min at 65 °C), and RNA was purified with RNeasy Mini kit (Qiagen) according to manufacturer’s protocol. DNA contamination after the DNase I treatment of RNA samples was indicated by lack of qPCR amplification (performed as described below), and in the few cases where amplification did take place, an additional DNase I treatment was performed and no qPCR amplification took place then.
Total cDNA synthesis was carried out with 8 μL of resulting purified RNA extract, using random hexanucleotide primers (Invitrogen) and Omniscript reverse transcription kit (Qiagen) following the manufacturer’s instructions (90 min at 37 °C). The reverse transcriptase was inactivated 10 min at 95 °C, and cDNA was stored at − 20 °C.
qPCR and qRT-PCR analyses of acdS
The quantities of acdS genes (qPCR) and mRNA (qRT-PCR) were estimated using 20-μL containing 4 μL of PCR grade water, 1 μL of each primer (final concentration 1 μM), 10 μL of LightCycler-DNA Master SYBR Green I master mix (Roche Applied Science), and 2 μL of sample DNA (10 ng). The cycling program entailed 10 min incubation at 95 °C, followed by 50 cycles of 94 °C for 15 s, 67 °C for 15 s, and 72 °C for 10 s. The fusion program for melting curve analysis is described above. Amplification specificity was assessed by melting curve analysis of PCR and RT-PCR products, done by ramping the temperature to 95 °C for 10 s and back to 65 °C for 15 s, followed by increases of 0.1 °C s−1 up to 95 °C. Melting curve calculation and determination of Tm values were performed using the polynomial algorithm function of LightCycler Software v.1 (Roche Applied Science).
Standard curves were obtained using DNA from B. cenocepacia J2315, whose genome contains one acdS copy, after diluting in triplicate from 5 × 10−9 to 5 × 10−15 g DNA μL−1. PCR efficiency was derived from standard curves as E = 10(−1/slope). All five samples for rhizosphere or bulk soil treatment were assessed, and data expressed in g μL−1 were converted into numbers of acdS copies computed as [DNA (g) × Avogadro’s number (molecules mol− 1)]/[number of DNA base pairs in PCR template × 660 (g mol−1)], based on an average of 660 g mol−1 per base pair.
acdS sequencing from rhizosphere DNA
Primer specificity was assessed by Illumina MiSeq sequencing of acdSF5/acdSR8 amplicons (size ~ 133 nt), using bulk soils and rhizosphere soils from tomato and six Poaceae cultivated in cropped or meadow soil. DNA extracts were sent to MR DNA laboratory (www.mrdnalab.com; Shallowater, TX) for acdS sequencing. The qPCR primers acdSF5/acdSR8 were used for the sequencing library, the forward primer carrying a barcode. The 30-cycle PCR (done five times, hence 150 cycles in total) used the HotStarTaq Plus Master Mix Kit (Qiagen, Valencia, CA) with 94 °C for 3 min, followed by 28 cycles of 94 °C for 30 s, 53 °C for 40 s, and 72 °C for 1 min, and a final elongation at 72 °C for 5 min. Samples were pooled together in same proportions, purified with calibrated Ampure XP beads and the DNA library was obtained with Illumina TruSeq DNA library protocol. Sequencing was carried out on a MiSeq following the manufacturer’s instructions.
Sequence data were treated with MR DNA pipeline. Briefly, sequences were depleted of barcodes, sequences < 120 bp or > 160 bp or with ambiguous base calls removed, the remaining sequences denoised, operational taxonomic units (OTUs; defined at 3% divergence threshold) generated, and chimeras removed. OTUs were then classified using BLASTn and a curated acdS database (described in acdS database supplemental material). Briefly, the acdS in-house database (see the “Identification of acdS primers” section) developed to define the qPCR primers was adapted to exhibit only the 133 nt corresponding to the amplified PCR fragments. Moreover, when different accessions presented 100% identity in nucleotide sequences and were affiliated to a same bacterial species, only one sequence was kept, reducing the database entries to 1304 different sequences (named core-acdS database; see acdS database supplemental material) representing the phylogenetic diversity of ACC deaminase producers defined based on published data [18–20, 50]. Dataset without singletons was used to generate rarefaction curves and Shannon, Simpson, and Chao diversity indices (calculated using sequencing subsample data for which each sample had the same number of sequences).
Alignment of selected acdS sequences (i.e., ten randomly chosen OTUs per genus) of the core-acdS database and eight related D-cystein desulfhydrase genes (see the “Identification of acdS primers” section) used as outgroup was carried out using MAFFT v7.123b (2013/10/15) [65, 66]. An acdS phylogenetic tree was constructed based on maximum likelihood method using RAxML 8.2.8 software [67]. Trees were annotated using iTOL V3 [68].
Statistical analyses
Experimental treatments were compared based on log-numbers of acdS genes and mRNA, using ANOVA and Fisher’s LSD tests, in each of the soils and at each sampling time. Two-factor ANOVA and Fisher’s LSD tests were also done, to consider sampling time effects in cropped soil (9 treatment × 2 samplings) as well as past soil management at 21 days (4 treatments × 2 past soil managements). Comparisons for bacterial composition data were carried out by between-class analysis (BCA) (ADE4 R and ggplot2 packages) and Kruskal-Wallis rank sum test associated to Tukey’s HSD test.
To evaluate the influence of past Poaceae evolution on root interactions with acdS microorganisms, Spearman correlation analysis was carried out between the phylogenetic distance between Poaceae genotypes and the corresponding pairwise differences in (i) acdS gene or transcript raw numbers (based on Euclidean distances) or (ii) acdS microbial community composition (based on Bray-Curtis dissimilarity indices). The former was computed by Bouffaud et al. [36] from concatenated chloroplastic sequences of gene rps16 and intergenic regions rps16-trnK and atpI-atpH, using the maximum likelihood method and Kimura two-parameter model.
All analyses were done at P < 0.05, using R 2.10.1 software (https://www.r-project.org).
Nucleotide sequence accession numbers
Reads have been deposited in the European Bioinformatics Institute (EBI) database under accession number PRJEB24637.
Additional files
Table S1. Universal primers designed to target acdS alleles. Table S2. Universal primer pairs tested to amplify specifically acdS alleles, with selected primer pair indicated in bold. Table S3. Spearman correlation analysis of the relation of pairwise plant phylogenetic distance between Poaceae with various Euclidean distances between log-transformed qPCR data describing the corresponding acdS communities. (DOCX 74 kb)
Figure S1. Examples of PCR amplification with the different primer pairs tested. (A) acdsF5/acdsR7; (B) acdsF5/acdsR8; (C) acdsF6/acdsR7; (D) acdsF6/acdsR8; (E) acdsF8/acdsR10. The different strains tested were 1. Azospirillum lipoferum 4B; 2. A. lipoferum TVV3; 3. A lipoferum CRT1 (acdS-); 4. A. lipoferum RSWT1; 5. Burkholderia cepacia LMG1222; 6. B. cenocepacia LMG16656; 7. B. stabilis LMG14294; 8. B. dolosa LMG18941; 9. Pseudomonas thivervalensis PITR2; 10. P. kilonensis F113; 11. P. protegens CHA0 (acdS-); and 12. Ralstonia solanacearum GMI1000. (TIFF 13453 kb)
Figure S2. Example of the acdS amplification curves (A), standard curve (B), melting peaks (C) obtained using DNA from Burkholderia cenocepacia J2315, and (E) and (F) obtained using rhizospheric metagenomics DNA serially diluted. (TIF 602 kb)
Figure S3. RAxML bipartition tree of 3322 sequenced acdS alleles from Poaceae rhizosphere. The tree was visualized using iTOL software. Branches colored in violet represent the out-group of D-cystein desulfhydrase genes, whereas acdS alleles affiliated to Betaproteobacteria are shown in khaki, to Gammaproteobacteria in blue, to Actinobacteria in green, to Alphaproteobacteria in red, and to microeukaryotes in orange. Branches are indicated in bold when corresponding to at least two genera (http://itol.embl.de/shared/acdStree). (PDF 892 kb)
Figure S4. Rarefaction curves showing the number of microbial OTUs according to the number of acdS reads, based on observed data obtained from bulk soil or rhizosphere. Data for cropped soil are in full-lines and for meadow soil in dash-lines. (PDF 473 kb)
Figure S5. Pairwise comparison of plant phylogenetic distance between Poaceae (X-axis) with the corresponding Euclidean distance between acdS reads for each of the 20 most abundant microbial genera representing over 90% of acdS functional group diversity (Y-axis). The 18 maize-based comparisons are indicated using black triangles (maize-maize; n = 6), diamonds (maize-teosinte; n = 4), circles (maize-sorghum; n = 4), or squares (maize-wheat; n = 4), and the three other comparisons using a white circle (teosinte-sorghum), white triangle (teosinte-wheat) or white square (sorghum-wheat). Distances were calculated two by two, using Kimura two-parameter model for plant phylogeny and Euclidean distance for taxa. (TIF 155 kb)
Figure S6. Root (A), rhizosphere soil (B), and shoot (C) dry weights. Statistical analyses were performed independently at 21 days in cropped soil, at 21 days in meadow soil, and at 42 days in cropped soil, using ANOVA and Fisher LSD tests (P < 0.05; differences shown with letters a to d). For maize lines FV4, W85, tomato, and bulk soil, two-way ANOVA and Fisher LSD tests (P < 0.05) were also performed to compare treatments according to past soil management or sampling time, and differences with the same genotype at 21 days in cropped soil are indicated by symbols * and #, respectively. (PDF 115 kb)
Acknowledgements
We are grateful to D. Abrouk (iBio platform, UMR CNRS 5557 Ecologie Microbienne) for helpful discussion, J.P. Magnière (Ferme expérimentale, La Côte Saint André, France) for the choice of soil, and Florence Gérin for technical help. This work made use of Serre and DTAMB plateforms of FR3728 BioEnviS at Université Lyon 1.
Funding
This work was supported in part by the Ministère Français de la Recherche and the ANR project Azodure (ANR-12-AGRO-0008) coordinated by L. Legendre (UMR CNRS 5557 Ecologie Microbienne).
Authors’ contributions
MLB, YML, and DM conceived and designed the project. MLB performed the greenhouse experiment and qPCR. SR and AD analyzed the acdS sequences. MLB, SR, YML, and DM analyzed the findings and wrote the manuscript. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40168-018-0503-7) contains supplementary material, which is available to authorized users.
References
- 1.Pieterse CMJ, Zamioudis C, Berendsen RL, Weller DM, Van Wees SCM, Bakker PAHM. Induced systemic resistance by beneficial microbes. Annu Rev Phytopathol. 2014;52:347–375. doi: 10.1146/annurev-phyto-082712-102340. [DOI] [PubMed] [Google Scholar]
- 2.Vacheron J, Desbrosses G, Bouffaud M-L, Touraine B, Moënne-Loccoz Y, Muller D, Legendre L, Wisniewski-Dyé F, Prigent-Combaret C. Plant growth-promoting rhizobacteria and root system functioning. Front Plant Sci. 2013;4:356. doi: 10.3389/fpls.2013.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Werner GDA, Strassmann JE, Ivens ABF, Engelmoer DJP, Verbruggen E, Queller DC, Noë R, Johnson NC, Hammerstein P, Kiers ET. Evolution of microbial markets. Proc Natl Acad Sci U S A. 2014;111:1237–1244. doi: 10.1073/pnas.1315980111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bais HP, Weir TL, Perry LG, Gilroy S, Vivanco JM. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu Rev Plant Biol. 2006;57:233–266. doi: 10.1146/annurev.arplant.57.032905.105159. [DOI] [PubMed] [Google Scholar]
- 5.Haichar FZ, Marol C, Berge O, Rangel-Castro JI, Prosser JI, Balesdent J, Heulin T, Achouak W. Plant host habitat and root exudates shape soil bacterial community structure. ISME J. 2008;2:1221–1230. doi: 10.1038/ismej.2008.80. [DOI] [PubMed] [Google Scholar]
- 6.Dakora FD, Phillips DA. Root exudates as mediators of mineral acquisition in low-nutrient environments. Plant Soil. 2002;245:35–47. doi: 10.1023/A:1020809400075. [DOI] [Google Scholar]
- 7.Chaparro JM, Badri DV, Bakker MG, Sugiyama A, Manter DK, Vivanco JM. Root exudation of phytochemicals in arabidopsis follows specific patterns that are developmentally programmed and correlate with soil microbial functions. PLoS One. 2013;8:e55731. doi: 10.1371/journal.pone.0055731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richardson AE, Barea J-M, McNeill AM, Prigent-Combaret C. Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms. Plant Soil. 2009;321:305–339. doi: 10.1007/s11104-009-9895-2. [DOI] [Google Scholar]
- 9.Glick BR, Penrose DM, Li J. A model for the lowering of plant ethylene concentrations by plant growth-promoting bacteria. J Theor Biol. 1998;190:63–68. doi: 10.1006/jtbi.1997.0532. [DOI] [PubMed] [Google Scholar]
- 10.Bleecker AB, Kende H. Ethylene: a gaseous signal molecule in plants. Annu Rev Cell Dev Biol. 2000;16:1–18. doi: 10.1146/annurev.cellbio.16.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Johnson PR, Ecker JR. The ethylene gas signal transduction pathway: a molecular perspective. Annu Rev Genet. 1998;32:227–254. doi: 10.1146/annurev.genet.32.1.227. [DOI] [PubMed] [Google Scholar]
- 12.Honma M, Shimomura T. Metabolism of 1-aminocyclopropane-1-carboxylic acid. Agric Biol Chem. 1978;42:1825–1831. [Google Scholar]
- 13.Glick BR. Modulation of plant ethylene levels by the bacterial enzyme ACC deaminase. FEMS Microbiol Lett. 2005;251:1–7. doi: 10.1016/j.femsle.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 14.Broekaert WF, Delauré SL, De Bolle MFC, Cammue BPA. The role of ethylene in host-pathogen interactions. Annu Rev Phytopathol. 2006;44:393–416. doi: 10.1146/annurev.phyto.44.070505.143440. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Ovakim DH, Charles TC, Glick BR. An ACC deaminase minus mutant of Enterobacter cloacae UW4 no longer promotes root elongation. Curr Microbiol. 2000;41:101–105. doi: 10.1007/s002840010101. [DOI] [PubMed] [Google Scholar]
- 16.Singh RP, Shelke GM, Kumar A, Jha PN. Biochemistry and genetics of ACC deaminase: a weapon to ‘stress ethylene’ produced in plants. Front Microbiol. 2015;6:937. doi: 10.3389/fmicb.2015.00937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van de Poel B, Van Der Straeten D. 1-Aminocyclopropane-1-carboxylic acid (ACC) in plants: more than just the precursor of ethylene! Front Plant Sci. 2014;5:640. doi: 10.3389/fpls.2014.00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prigent-Combaret C, Blaha D, Pothier JF, Vial L, Poirier MA, Wisniewski-Dyé F, Moënne-Loccoz Y. Physical organization and phylogenetic analysis of acdR as leucine-responsive regulator of the 1-aminocyclopropane-1-carboxylate deaminase gene acdS in phytobeneficial Azospirillum lipoferum 4B and other Proteobacteria. FEMS Microbiol Ecol. 2008;65:202–219. doi: 10.1111/j.1574-6941.2008.00474.x. [DOI] [PubMed] [Google Scholar]
- 19.Bruto M, Prigent-Combaret C, Luis P, Moënne-Loccoz Y, Muller D. Frequent, independent transfers of a catabolic gene from bacteria to contrasted filamentous eukaryotes. Proc R Soc Lond B Biol Sci. 2014;281:20140848. doi: 10.1098/rspb.2014.0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nascimento FX, Rossi MJ, Soares CRFS, McConkey BJ, Glick BR. New insights into 1-aminocyclopropane-1-carboxylate (ACC) deaminase phylogeny, evolution and ecological significance. PLoS One. 2014;9:e99168. doi: 10.1371/journal.pone.0099168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blaha D, Prigent-Combaret C, Mirza MS, Moënne-Loccoz Y. Phylogeny of the 1-aminocyclopropane-1-carboxylic acid deaminase-encoding gene acdS in phytobeneficial and pathogenic Proteobacteria and relation with strain biogeography. FEMS Microbiol Ecol. 2006;56:455–470. doi: 10.1111/j.1574-6941.2006.00082.x. [DOI] [PubMed] [Google Scholar]
- 22.Hontzeas N, Richardson AO, Belimov A, Safronova V, Abu-Omar MM, Glick BR. Evidence for horizontal transfer of 1-aminocyclopropane-1-carboxylate deaminase genes. Appl Environ Microbiol. 2005;71:7556–7558. doi: 10.1128/AEM.71.11.7556-7558.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin Z, Di Rienzi SC, Janzon A, Werner JJ, Angenent LT, Dangl JL, Fowler DM, Ley RE. Novel rhizosphere soil alleles for the enzyme 1-aminocyclopropane-1-carboxylate deaminase queried for function with an in vivo competition assay. Appl Environ Microbiol. 2016;82:1050–1059. doi: 10.1128/AEM.03074-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruto M, Prigent-Combaret C, Muller D, Moënne-Loccoz Y. Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related Proteobacteria. Sci Rep. 2014;4:6261. doi: 10.1038/srep06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raaijmakers J, Paulitz TC, Steinberg C, Alabouvette C, Moënne-Loccoz Y. The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil. 2009;321:341–361. doi: 10.1007/s11104-008-9568-6. [DOI] [Google Scholar]
- 26.Berg G, Smalla K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol. 2009;68:1–13. doi: 10.1111/j.1574-6941.2009.00654.x. [DOI] [PubMed] [Google Scholar]
- 27.Bouffaud M-L, Kyselková M, Gouesnard B, Grundmann G, Muller D, Moënne-Loccoz Y. Is diversification history of maize influencing selection of soil bacteria by roots? Mol Ecol. 2012;21:195–206. doi: 10.1111/j.1365-294X.2011.05359.x. [DOI] [PubMed] [Google Scholar]
- 28.Bulgarelli D, Garrido-Oter R, Münch PC, Weiman A, Dröge J, Pan Y, McHardy AC, Schulze-Lefert P. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe. 2015;17:392–403. doi: 10.1016/j.chom.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coleman-Derr D, Desgarennes D, Fonseca-Garcia C, Gross S, Clingenpeel S, Woyke T, North G, Visel A, Partida-Martinez LP, Tringe SG. Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 2016;209:798–811. doi: 10.1111/nph.13697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci U S A. 2013;110:6548–6553. doi: 10.1073/pnas.1302837110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazzola M, Gu YH, Funnell DL, Cohen MF, Raaijmakers JM. Significance of host genotype in exploitation of resident disease suppressive soil microbial communities. Phytopathology. 2004;94:S125. doi: 10.1094/PHYTO.2004.94.2.125. [DOI] [Google Scholar]
- 32.Meyer JB, Lutz MP, Frapolli M, Péchy-Tarr M, Rochat L, Keel C, Défago G, Maurhofer M. Interplay between wheat cultivars, biocontrol pseudomonads, and soil. Appl Environ Microbiol. 2010;76:6196–6204. doi: 10.1128/AEM.00752-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergsma-Vlami M, Prins ME, Raaijmakers JM. Influence of plant species on population dynamics, genotypic diversity and antibiotic production in the rhizosphere by indigenous Pseudomonas spp. FEMS Microbiol Ecol. 2005;52:59–69. doi: 10.1016/j.femsec.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Bouffaud M-L, Renoud S, Moënne-Loccoz Y, Muller D. Is plant evolutionary history impacting recruitment of diazotrophs and nifH expression in the rhizosphere? Sci Rep. 2016;6:21690. doi: 10.1038/srep21690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alegria Terrazas R, Giles C, Paterson E, Robertson-Albertyn S, Cesco S, Mimmo T, Pii Y, Bulgarelli D. Plant-microbiota interactions as a driver of the mineral turnover in the rhizosphere. Adv Appl Microbiol. 2016;95:1–67. doi: 10.1016/bs.aambs.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Bouffaud M-L, Poirier M-A, Muller D, Moënne-Loccoz Y. Root microbiome relates to plant host evolution in maize and other Poaceae. Environ Microbiol. 2014;16:2804–2814. doi: 10.1111/1462-2920.12442. [DOI] [PubMed] [Google Scholar]
- 37.Micallef SA, Shiaris MP, Colón-Carmona A. Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. J Exp Bot. 2009;60:1729–1742. doi: 10.1093/jxb/erp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hacquard S, Garrido-Oter R, González A, Spaepen S, Ackermann G, Lebeis S, McHardy AC, Dangl JL, Knight R, Ley R, Schulze-Lefert P. Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe. 2015;17:603–616. doi: 10.1016/j.chom.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 39.Mazzola M, Funnell DL, Raaijmakers JM. Wheat cultivar-specific selection of 2,4-diacetylphloroglucinol-producing fluorescent Pseudomonas species from resident soil populations. Microb Ecol. 2004;48:338–348. doi: 10.1007/s00248-003-1067-y. [DOI] [PubMed] [Google Scholar]
- 40.Vandenkoornhuyse P, Mahé S, Ineson P, Staddon P, Ostle N, Cliquet J-B, Francez A-J, Fitter AH, Young JPW. Active root-inhabiting microbes identified by rapid incorporation of plant-derived carbon into RNA. Proc Natl Acad Sci U S A. 2007;104:16970–16975. doi: 10.1073/pnas.0705902104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemanceau P, Blouin M, Muller D, Moënne-Loccoz Y. Let the core microbiota be functional. Trends Plant Sci. 2017;22:583–595. doi: 10.1016/j.tplants.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Louca S, Jacques SMS, Pires APF, Leal JS, Srivastava DS, Parfrey LW, Farjalla VF, Doebeli M. High taxonomic variability despite stable functional structure across microbial communities. Nat Ecol Evol. 2016;1:0015. doi: 10.1038/s41559-016-0015. [DOI] [PubMed] [Google Scholar]
- 43.Köberl M, Erlacher A, Ramadan EM, El-Arabi TF, Müller H, Bragina A, Berg G. Comparisons of diazotrophic communities in native and agricultural desert ecosystems reveal plants as important drivers in diversity. FEMS Microbiol Ecol. 2016;92:fiv166. doi: 10.1093/femsec/fiv166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwob G, Roy M, Manzi S, Pommier T, Fernandez MP. Green alder (Alnus viridis) encroachment shapes microbial communities in subalpine soils and impacts its bacterial or fungal symbionts differently. Environ Microbiol. 2017;19:3235–3250. doi: 10.1111/1462-2920.13818. [DOI] [PubMed] [Google Scholar]
- 45.Philippot L, Spor A, Hénault C, Bru D, Bizouard F, Jones CM, Sarr A, Maron P-A. Loss in microbial diversity affects nitrogen cycling in soil. ISME J. 2013;7:1609. doi: 10.1038/ismej.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frapolli M, Défago G, Moënne-Loccoz Y. Denaturing gradient gel electrophoretic analysis of dominant 2,4-diacetylphloroglucinol biosynthetic phlD alleles in fluorescent Pseudomonas from soils suppressive or conducive to black root rot of tobacco. Soil Biol Biochem. 2010;42:649–656. doi: 10.1016/j.soilbio.2010.01.005. [DOI] [Google Scholar]
- 47.Almario J, Moënne-Loccoz Y, Muller D. Monitoring of the relation between 2,4-diacetylphloroglucinol-producing Pseudomonas and Thielaviopsis basicola populations by real-time PCR in tobacco black root-rot suppressive and conducive soils. Soil Biol Biochem. 2013;57:144–155. doi: 10.1016/j.soilbio.2012.09.003. [DOI] [Google Scholar]
- 48.Gamalero E, Glick BR. Bacterial modulation of plant ethylene levels. Plant Physiol. 2015;169:13–22. doi: 10.1104/pp.15.00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang T, Fang HHP. Applications of real-time polymerase chain reaction for quantification of microorganisms in environmental samples. Appl Microbiol Biotechnol. 2006;70(3):281–289. doi: 10.1007/s00253-006-0333-6. [DOI] [PubMed] [Google Scholar]
- 50.Li Z, Chang S, Ye S, Chen M, Lin L, Li Y, Li S, An Q. Differentiation of 1-aminocyclopropane-1-carboxylate (ACC) deaminase from its homologs is the key for identifying bacteria containing ACC deaminase. FEMS Microbiol Ecol. 2015;91:10. doi: 10.1093/femsec/fiv112. [DOI] [PubMed] [Google Scholar]
- 51.Subramanian P, Krishnamoorthy R, Chanratana M, Kim K, Sa T. Expression of an exogenous 1-aminocyclopropane-1-carboxylate deaminase gene in psychrotolerant bacteria modulates ethylene metabolism and cold induced genes in tomato under chilling stress. Plant Physiol Biochem. 2015;89:18–23. doi: 10.1016/j.plaphy.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 52.Liu Y, Cao L, Tan H, Zhang R. Surface display of ACC deaminase on endophytic Enterobacteriaceae strains to increase saline resistance of host rice sprouts by regulating plant ethylene synthesis. Microb Cell Factories. 2017;16:214. doi: 10.1186/s12934-017-0831-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stromberger ME, Abduelafez I, Byrne P, Canela MM, Elamari AA, Manter DK, Weir T. Genotype-specific enrichment of 1-aminocyclopropane-1-carboxylic acid deaminase-positive bacteria in winter wheat rhizospheres. Soil Sci Soc Am J. 2017;81:796–805. doi: 10.2136/sssaj2016.12.0437. [DOI] [Google Scholar]
- 54.Glick BR. Bacteria with ACC deaminase can promote plant growth and help to feed the world. Microbiol Res. 2014;169:30–39. doi: 10.1016/j.micres.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 55.Bell TH, El-Din Hassan S, Lauron-Moreau A, Al-Otaibi F, Hijri M, Yergeau E, St-Arnaud M. Linkage between bacterial and fungal rhizosphere communities in hydrocarbon-contaminated soils is related to plant phylogeny. ISME J. 2014;8:331–343. doi: 10.1038/ismej.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaharoona B, Arshad M, Zahir ZA. Effect of plant growth promoting rhizobacteria containing ACC-deaminase on maize (Zea mays L.) growth under axenic conditions and on nodulation in mung bean (Vigna radiata L.) Lett Appl Microbiol. 2006;42:155–159. doi: 10.1111/j.1472-765X.2005.01827.x. [DOI] [PubMed] [Google Scholar]
- 57.Turner TR, Ramakrishnan K, Walshaw J, Heavens D, Alston M, Swarbreck D, Osbourn A, Grant A, Poole PS. Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J. 2013;7:2248–2258. doi: 10.1038/ismej.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ofek-Lalzar M, Sela N, Goldman-Voronov M, Green SJ, Hadar Y, Minz D. Niche and host-associated functional signatures of the root surface microbiome. Nat Commun. 2014;5:4950. doi: 10.1038/ncomms5950. [DOI] [PubMed] [Google Scholar]
- 59.Todorovic B, Glick BR. The interconversion of ACC deaminase and D-cysteine desulfhydrase by directed mutagenesis. Planta. 2008;229:193–205. doi: 10.1007/s00425-008-0820-3. [DOI] [PubMed] [Google Scholar]
- 60.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 62.Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 63.Couillerot O, Bouffaud M-L, Baudoin E, Muller D, Caballero-Mellado J, Moënne-Loccoz Y. Development of a real-time PCR method to quantify the PGPR strain Azospirillum lipoferum CRT1 on maize seedlings. Soil Biol Biochem. 2010;42:2298–2305. doi: 10.1016/j.soilbio.2010.09.003. [DOI] [Google Scholar]
- 64.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 65.Katoh K, Misawa K, Ki K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Universal primers designed to target acdS alleles. Table S2. Universal primer pairs tested to amplify specifically acdS alleles, with selected primer pair indicated in bold. Table S3. Spearman correlation analysis of the relation of pairwise plant phylogenetic distance between Poaceae with various Euclidean distances between log-transformed qPCR data describing the corresponding acdS communities. (DOCX 74 kb)
Figure S1. Examples of PCR amplification with the different primer pairs tested. (A) acdsF5/acdsR7; (B) acdsF5/acdsR8; (C) acdsF6/acdsR7; (D) acdsF6/acdsR8; (E) acdsF8/acdsR10. The different strains tested were 1. Azospirillum lipoferum 4B; 2. A. lipoferum TVV3; 3. A lipoferum CRT1 (acdS-); 4. A. lipoferum RSWT1; 5. Burkholderia cepacia LMG1222; 6. B. cenocepacia LMG16656; 7. B. stabilis LMG14294; 8. B. dolosa LMG18941; 9. Pseudomonas thivervalensis PITR2; 10. P. kilonensis F113; 11. P. protegens CHA0 (acdS-); and 12. Ralstonia solanacearum GMI1000. (TIFF 13453 kb)
Figure S2. Example of the acdS amplification curves (A), standard curve (B), melting peaks (C) obtained using DNA from Burkholderia cenocepacia J2315, and (E) and (F) obtained using rhizospheric metagenomics DNA serially diluted. (TIF 602 kb)
Figure S3. RAxML bipartition tree of 3322 sequenced acdS alleles from Poaceae rhizosphere. The tree was visualized using iTOL software. Branches colored in violet represent the out-group of D-cystein desulfhydrase genes, whereas acdS alleles affiliated to Betaproteobacteria are shown in khaki, to Gammaproteobacteria in blue, to Actinobacteria in green, to Alphaproteobacteria in red, and to microeukaryotes in orange. Branches are indicated in bold when corresponding to at least two genera (http://itol.embl.de/shared/acdStree). (PDF 892 kb)
Figure S4. Rarefaction curves showing the number of microbial OTUs according to the number of acdS reads, based on observed data obtained from bulk soil or rhizosphere. Data for cropped soil are in full-lines and for meadow soil in dash-lines. (PDF 473 kb)
Figure S5. Pairwise comparison of plant phylogenetic distance between Poaceae (X-axis) with the corresponding Euclidean distance between acdS reads for each of the 20 most abundant microbial genera representing over 90% of acdS functional group diversity (Y-axis). The 18 maize-based comparisons are indicated using black triangles (maize-maize; n = 6), diamonds (maize-teosinte; n = 4), circles (maize-sorghum; n = 4), or squares (maize-wheat; n = 4), and the three other comparisons using a white circle (teosinte-sorghum), white triangle (teosinte-wheat) or white square (sorghum-wheat). Distances were calculated two by two, using Kimura two-parameter model for plant phylogeny and Euclidean distance for taxa. (TIF 155 kb)
Figure S6. Root (A), rhizosphere soil (B), and shoot (C) dry weights. Statistical analyses were performed independently at 21 days in cropped soil, at 21 days in meadow soil, and at 42 days in cropped soil, using ANOVA and Fisher LSD tests (P < 0.05; differences shown with letters a to d). For maize lines FV4, W85, tomato, and bulk soil, two-way ANOVA and Fisher LSD tests (P < 0.05) were also performed to compare treatments according to past soil management or sampling time, and differences with the same genotype at 21 days in cropped soil are indicated by symbols * and #, respectively. (PDF 115 kb)