

Abstract
Microglia have been shown to be of critical importance to the progression of temporal lobe epilepsy. However, the broad transcriptional changes that these cells undergo following seizure induction is not well understood. As such, we utilized RNAseq analysis upon microglia isolated from the hippocampus to determine expression pattern alterations following kainic acid induced seizure. We determined that microglia undergo dramatic changes to their expression patterns, particularly with regard to mitochondrial activity and metabolism. We also observed that microglia initiate immunological activity, specifically increasing interferon beta responsiveness. Our results provide novel insights into microglia transcriptional regulation following acute seizures and suggest potential therapeutic targets specifically in microglia for the treatment of seizures and epilepsy.
Electronic supplementary material
The online version of this article (10.1186/s13041-018-0376-5) contains supplementary material, which is available to authorized users.
Introduction
Temporal lobe epilepsy (TLE) represents the most common form of focal epileptic disorder. While several pharmaceutical treatments are currently available to mitigate and reduce seizure occurrence, as many as one third of patients display resistance to medication [1]. As such, an unmet need exists, requiring further investigation into the mechanisms underlying TLE. The rodent kainic acid (KA) epilepsy model can recapitulate many of the physical features of TLE including behavioral seizures and neuropathological lesions [2]. Therefore, many investigations have focused on how KA alters the activity and viability of neurons. However, comparatively little attention has been paid to glial cells, including astrocytes and microglia, in epileptogenesis [3, 4].
Comprising between 5 and 15% of total central nervous system (CNS) cells, microglia predominantly serve as the resident immune cell of the CNS. Recent evidence has also revealed that microglia have a diverse set of roles within the CNS, including directing neuronal maturation and supporting synaptic turnover [5, 6]. With regard to epilepsy, it was established relatively early that large numbers of reactive microglia can be found within the hippocampus of temporal lobe epilepsy patients [7, 8]. Our recent studies demonstrated that seizures can acutely induce microglia-neuron interaction as well as the changes in microglial landscape [9–12]. Microgliosis and inflammatory cytokine release has been observed within areas of neuronal damage implicating microglia in promotion of neuropathy [13]. However, microglia may also have neuroprotective roles such as modulating excitotoxicity.
Since microglia seem to be an important part of the epileptic response, we investigated how KA-induced seizures modulate microglial transcriptional activity and alters their phenotype. Specifically, we investigated hippocampal microglia since this brain region is one of the most affected by seizure [14]. To explore this, we performed RNAseq analysis, a powerful tool to determine wide scale phenotypic alterations, on isolated hippocampal microglia from mice that received KA. We report that KA-induced seizures resulted in significant transcriptional changes to microglia when compared to sham controls. Specifically, there are significant increases in the expression of metabolic and mitochondrial pathways. Coincidently, we observed that immune related factors were also being up-regulated, including several chemokine factors such as chemokine ligand 5 (CCL5) and C-X-C motif chemokine 10 (CXCL10). We also observed that microglia increased their responsiveness to interferon β, possibly through interferon regulatory factor 7 (Irf7). Thus, we show that KA-induced seizures significantly regulate the microglia transcriptome, providing novel directions for further investigation.
Results
Kainic acid induced seizures significantly alters microglial gene expression profile
To begin our investigation, heterozygote CX3CR1GFP/+ mice were treated with kainic acid (KA) via ICV injection to induce an acute seizure response [12]. Microglia in the mouse hippocampus show dramatic reactivity following KA-induced seizure strating at as early as 1 day and peaks at 3 days after KA treatment [15]. We therefore focused on hippocampus microglia isolated via FACS 3 days after KA-induced seizures. RNAseq libraries were constructed using the isolated cells and loaded onto an Illumina Hiseq platform. DEseq was used to determine differential gene expression. From the results, over 2300 differentially expressed genes were identified (Fig. 1a, Additional file 1: Table S1). Of these, we observed many of the suggested microglia specific genes including P2Y12, Tmem119, and Olfml3 [16]. Additionally, we detected only slight increases to myelin (e.g., PLP), neuronal (e.g., Rbfox3, Map2), and astrocytes (e.g., Gfap, Aldh1l1) markers within samples isolated from KA treated mice, with only GFAP registering as significant. These factors were not detected within the control samples. Since it has been suggested that the phagocytic capacity of microglia is substantially reduced following KA-seizure [17] and that microglial could express GFAP [18] we believe that the genes alterations that were deemed significant reflect microglia specific alterations. These results demonstrated the purity of microglia sorting. The overwhelming majority of differentially expressed genes were up-regulated in the microglia samples from KA treated mice with few genes being down-regulated when compared to sham controls (Fig. 1b-c). Table 1 lists the top 25 up-regulated and Table 2 the identified down-regulated genes. Table 3 lists the top 25 genes found only in the KA-treated animals as determined by Padj values.
Fig. 1.
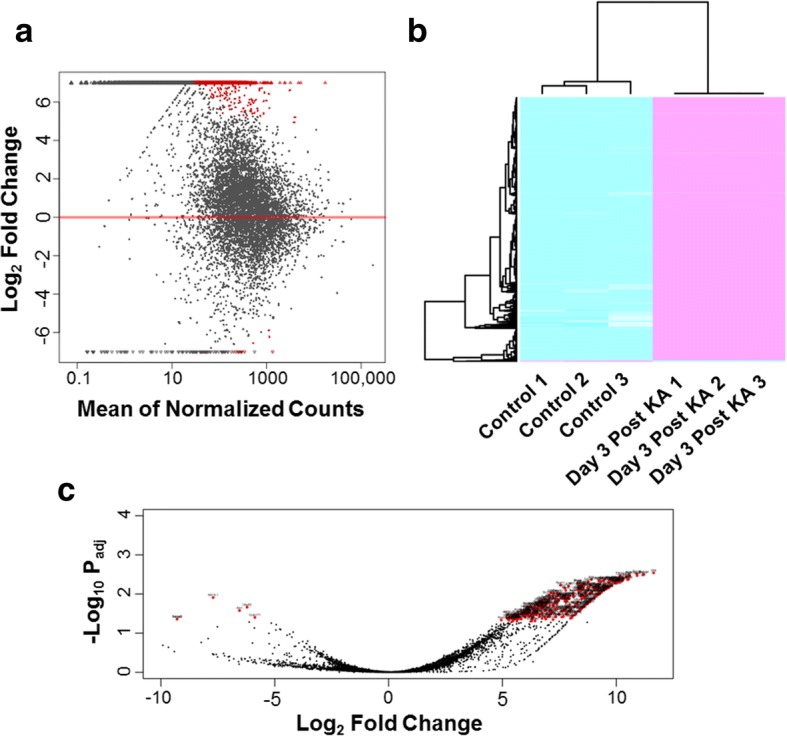
Differentially expressed genes between the sham control and KA treated groups. a MA-plot of gene expression. All significant differentially expressed genes (Padj < 0.05) and locally weighted smoothing (LOESS) line are colored in red. b Heat map and hierarchical clustering was performed based on all differentially expressed genes. Magenta indicates high relative expression, and cyan indicates low relative expression. c Volcano plot of gene expression. All significant differentially expressed genes are colored in red and labeled by gene symbols
Table 1 .
Top 25 most up-regulated genes
| ENSEMBL | Gene ID | Gene Symbol | Gene Name | Log2 Fold Change | Padj |
|---|---|---|---|---|---|
| ENSMUSG00000019505 | 22187 | Ubb | ubiquitin B(Ubb) | 11.64 | 2.89E-03 |
| ENSMUSG00000006418 | 81018 | Rnf114 | ring finger protein 114(Rnf114) | 11.21 | 3.18E-03 |
| ENSMUSG00000005881 | 66366 | Ergic3 | ERGIC and golgi 3(Ergic3) | 11.17 | 3.27E-03 |
| ENSMUSG00000090841 | 17904 | Myl6 | myosin, light polypeptide 6, alkali, smooth muscle and non- muscle(Myl6) | 10.91 | 3.39E-03 |
| ENSMUSG00000040952 | 20085 | Rps19 | ribosomal protein S19(Rps19) | 10.90 | 3.07E-03 |
| ENSMUSG00000042650 | 268420 | Alkbh5 | alkB homolog 5, RNA demethylase(Alkbh5) | 10.58 | 3.27E-03 |
| ENSMUSG00000047215 | 20005 | Rpl9 | ribosomal protein L9(Rpl9) | 10.54 | 3.27E-03 |
| ENSMUSG00000020664 | 13382 | Dld | dihydrolipoamide dehydrogenase(Dld) |
10.54 | 4.14E-03 |
| ENSMUSG00000025959 | 93691 | Klf7 | Kruppel-like factor 7 (ubiquitous)(Klf7) | 10.52 | 4.28E-03 |
| ENSMUSG00000022982 | 20655 | Sod1 | superoxide dismutase 1, soluble(Sod1) | 10.51 | 4.21E-03 |
| ENSMUSG00000026213 | 71728 | Stk11ip | serine/threonine kinase 11 interacting protein(Stk11ip) | 10.49 | 4.28E-03 |
| ENSMUSG00000031483 | 244373 | Erlin2 | ER lipid raft associated 2(Erlin2) | 10.45 | 3.66E-03 |
| ENSMUSG00000029298 | 236573 | Gbp9 | guanylate-binding protein 9(Gbp9) | 10.27 | 4.58E-03 |
| ENSMUSG00000034855 | 15945 | Cxcl 10 | chemokine (C-X-C motif) ligand 10(Cxcl10) | 10.27 | 3.78E-03 |
| ENSMUSG00000070031 | 434484 | Sp140 | Sp140 nuclear body protein(Sp140) | 10.24 | 4.59E-03 |
| ENSMUSG00000054920 | 71778 | Klhl5 | kelch-like 5(Klhl5) | 10.22 | 4.74E-03 |
| ENSMUSG00000040447 | 216892 | Spns2 | spinster homolog 2(Spns2) | 10.17 | 4.91E-03 |
| ENSMUSG00000022884 | 13682 | Eif4a2 | eukaryotic translation initiation factor 4A2(Eif4a2) | 10.16 | 3.39E-03 |
| ENSMUSG00000028962 | 20535 | Slc4a2 | solute carrier family 4 (anion exchanger), member 2(Slc4a2) | 10.15 | 4.93E-03 |
| ENSMUSG00000047153 | 219094 | Khnyn | KH and NYN domain containing(Khnyn) | 10.15 | 5.07E-03 |
| ENSMUSG00000030298 | 110379 | Sec13 | SEC13 homolog, nuclear pore and COPII coat complex component(Sec13) | 10.12 | 4.98E-03 |
| ENSMUSG00000031378 | 11666 | Abcd1 | ATP-binding cassette, sub-family D (ALD), member 1(Abcd1) | 10.11 | 4.28E-03 |
| ENSMUSG00000004568 | 102098 | Arhgef18 | rho/rac guanine nucleotide exchange factor (GEF) 18(Arhgef18) | 10.02 | 5.27E-03 |
| ENSMUSG00000030577 | 12483 | Cd22 | CD22 antigen(Cd22) | 10.02 | 5.22E-03 |
| ENSMUSG00000031858 | 74549 | Mau2 | MAU2 sister chromatid cohesion factor(Mau2) | 10.01 | 4.58E-03 |
Table 2.
Down-regulated genes
| ENSEMBL | Gene ID | Gene Symbol | Gene Name | Log2 Fold Change | Padj |
|---|---|---|---|---|---|
| ENSMUSG00000000562 | 11542 | Ccdc171 | adenosine A3 receptor(Adora3) | −5.87 | 3.98E-02 |
| ENSMUSG00000090137 | 22186 | Uba52 | ubiquitin A-52 residue ribosomal protein fusion product 1(Uba52) | −6.22 | 2.17E-02 |
| ENSMUSG00000052407 | 320226 | Atn1 | coiled-coil domain containing 171(Ccdc171) | −6.55 | 2.65E-02 |
| ENSMUSG00000092995 | 387134 | Mir16–1 | microRNA 16–1(Mir16–1) | −7.71 | 1.23E-02 |
| ENSMUSG00000004263 | 13498 | Adora3 | atrophin 1(Atn1) | −9.29 | 4.39E-02 |
| ENSMUSG00000074344 | 69296 | Tmigd3 | transmembrane and immunoglobulin domain containing 3(Tmigd3) | −9.29 | 4.39E-02 |
Table 3.
Top 25 differentially expressed genes only observed in KA treated group
| ENSEMBL | Gene ID | Gene Symbol | Gene Name | Padj |
|---|---|---|---|---|
| ENSMUSG00000069516 | 17105 | Lyz2 | lysozyme 2 | 6.62E-04 |
| ENSMUSG00000060938 | 19941 | Rpl26 | ribosomal protein L26 | 1.15E-03 |
| ENSMUSG00000002602 | 26362 | Axl | AXL receptor tyrosine kinase | 2.25E-03 |
| ENSMUSG00000031320 | 20102 | Rps4x | ribosomal protein S4, X-linked | 2.25E-03 |
| ENSMUSG00000049313 | 20660 | Sorl 1 | sortilin-related receptor, LDLR class A repeats-containing | 2.25E-03 |
| ENSMUSG00000062006 | 68436 | Rpl34 | ribosomal protein L34 | 2.25E-03 |
| ENSMUSG00000063524 | 619547 | Rpl34-ps1 | ribosomal protein L34, pseudogene 1 | 2.25E-03 |
| ENSMUSG00000069516 | 100043876 | Gm4705 | predicted gene 4705 | 2.25E-03 |
| ENSMUSG00000063524 | 13806 | Eno1 | enolase 1, alpha non-neuron | 2.25E-03 |
| ENSMUSG00000069892 | 245240 | 9,930,111 J21 Rik2 | RIKEN cDNA 9,930,111 J21 gene 2 | 2.25E-03 |
| ENSMUSG00000089809 | 319818 | A930011G23Rik | RIKEN cDNA A930011G23 gene | 2.25E-03 |
| ENSMUSG00000090733 | 57294 | Rps27 | ribosomal protein S27 | 2.25E-03 |
| ENSMUSG00000073418 | 12268 | C4b | complement component 4B | 2.83E-03 |
| ENSMUSG00000001794 | 12336 | Capns1 | calpain, small subunit 1 | 2.94E-03 |
| ENSMUSG00000003518 | 72349 | Dusp3 | dual specificity phosphatase 3 | 2.94E-03 |
| ENSMUSG00000005566 | 21849 | Trim28 | tripartite motif-containing 28 | 2.94E-03 |
| ENSMUSG00000009687 | 18301 | Fxyd5 | FXYD domain-containing ion transport regulator 5 | 2.94E-03 |
| ENSMUSG00000022415 | 20972 | Syngr1 | synaptogyrin 1 | 2.94E-03 |
| ENSMUSG00000022477 | 11429 | Aco2 | aconitase 2, mitochondrial | 2.94E-03 |
| ENSMUSG00000022565 | 18810 | Plec | plectin | 2.94E-03 |
| ENSMUSG00000024679 | 68774 | Ms4a6d | membrane-spanning 4-domains, subfamily A, member 6D | 2.94E-03 |
| ENSMUSG00000025498 | 54123 | Irf7 | interferon regulatory factor 7 | 2.94E-03 |
| ENSMUSG00000026222 | 20684 | Sp100 | nuclear antigen Sp100 | 2.94E-03 |
| ENSMUSG00000026430 | 54354 | Rassf5 | Ras association (RalGDS/AF-6) domain family member 5 | 2.94E-03 |
| ENSMUSG00000034854 | 73822 | Mfsd12 | major facilitator superfamily domain containing 12 | 2.94E-03 |
We next determined whether KA-induced seizures affected microglial specific markers. Using the list determined by Hickman et al. [16], we found that seven of the listed microglial markers were differentially expressed (Fig. 2a, Additional file 2: Figure S1). These were adenosine A3 receptor (Adora 3), crystallin beta A4 (Cryba4), galactose-3-O-sulfotransferase 4 (Gal3st4), lipase member H (Liph), membrane-spanning 4-domains, subfamily A, member 6B (Ms4a6b), serine peptidase inhibitor Kunitz type 1 (Spint1), and toll-like receptor 12 (Tlr12). Since KA treatment has also been shown to induce inflammatory responses [15], we also investigated our list of differentially expressed genes for potential inflammatory markers. Indeed, we found a number of inflammatory factors are increased within microglia isolated from KA treated mice, including C-C motif chemokine ligand 5 (Ccl5), Ccl7, and C-X-C motif chemokine ligand 10 (Cxcl10) (Fig. 2b, Additional file 2: Figure S2). We determined that expression of several inflammatory and immunological response receptors are also increased (Fig. 2c). These receptors included C-C motif chemokine receptor 2 (Ccr2), C-X-C motif chemokine receptor 4 (Cxcr4), and Tlr1. Finally, a significant number cluster of differentiation (CD) markers were significantly increased (Fig. 2d). The majority of identified CD markers are related to immunological responses including CD40, CD69, and CD80 [19, 20]. These results suggest that microglia are undergoing immunological activation in response to KA-induced seizures.
Fig. 2.

Selected differential expressed genes. Expression results were investigated for genes relating to microglial specificity and inflammatory and immunological regulation. a Microglial markers. b Secreted factors. c Related receptors. d CD markers. Values are expressed and mean ± standard error. **Padj < 0.05. All gene listed in panel (c and d) had a Padj < 0.05
Gene ontology analysis indicates significant increases to metabolic processes
Our next step was to identify if any unifying features existed within our differential expression data set. As such, we utilized clusterProfiler to perform gene ontology (GO) analysis [21]. We investigated our data set using the three major classifications, cellular component, biological process, and molecular function (Additional file 3: Table S2, Additional file 4: Table S3 and Additional file 5: Table S4). To further visualize our results, identified GO terms were input into REViGO [22]. This web-based application allows for long lists of GO terms to be summarized and grouped based on semantic similarities. REViGO analysis was run using the associated Padj for each identified GO term, with medium allowed similarity (0.7), and SimRel similarity measurement. TreeMaps were then generated for each ontology classification. Each box represents GO terms that are then grouped and colored based on keyword similarities. Box size indicates each terms level of significance as determined by input Padj values. Added labels highlight overarching grouping terms. As Fig. 3a illustrates there are significant alterations to intracellular factor expression, especially within the mitochondria. Moreover, Biological process GO analysis showed that there seems to be significant alterations to microglial metabolism, with catabolism being at the forefront (Fig. 3b). It also identified that microglia were activating viral defense mechanisms following seizure. Finally, we observed that a number of transferase activities were being undertaken following seizure (Fig. 3c).
Fig. 3.

Functional classification of the differentially expressed genes. a Cellular component. b Biological process. c Molecular function. Visualization of identified Gene Ontology terms was completed using REViGO [22]. Analysis was run using the Padj for each identified term, medium allowed similarity (0.7), and SimRel similarity measurement. Individual term size weight within each TreeMap was determined by associated Padj.
Kainic acid treatment may sensitize microglia to interferon beta
Delving deeper into the identified GO terms it was observed that a number of related terms were pertinent to type I interferons, specifically interferon β (IFN-β). Table 4 summarizes these identified GO terms. IFN-β is a type-I interferon that binds interferon-α/β receptor (IFNAR) to regulate a multitude of signaling cascades particularly the JAK/STAT pathway [23]. IFN-β has also been suggested to modulate microglial activity in multiple sclerosis and pathological neovascularization [24, 25]. Since IFN-β signaling was well represented within our GO analysis, we believe that IFN-β is important to the microglial modulation that occurs following KA-induced seizures.
Table 4.
Type I interferon related GO terms
| GO Term ID | Term Name | Padj |
|---|---|---|
| GO:0032480 | negative regulation of type I interferon production | 0.0012 |
| GO:0032479 | regulation of type I interferon production | 0.0019 |
| GO:0034340 | response to type I interferon | 0.0046 |
| GO:0032606 | type I interferon production | 0.0055 |
| GO:0032648 | regulation of interferon-beta production | 0.0109 |
| GO:0032608 | interferon-beta production | 0.0166 |
| GO:0035456 | response to interferon-beta | 0.0173 |
| GO:0060337 | type I interferon signaling pathway | 0.0204 |
| GO:0071357 | cellular response to type I interferon | 0.0204 |
| GO:0032688 | negative regulation of interferon-beta production | 0.0375 |
| GO:0060340 | positive regulation of type I interferon-mediated signaling pathway | 0.0375 |
| GO:0035458 | cellular response to interferon-beta | 0.0429 |
| GO:0060338 | regulation of type I interferon-mediated signaling pathway | 0.0497 |
Pathway analysis reveals both metabolic and immune response processes are altered
Finally, we performed pathway analysis on the differential expression data set using the clusterProfiler enrichKEGG function (Fig. 4). Unsurprisingly, this analysis corroborated our GO analysis results in that metabolism was significantly enriched in our data set. We also identified several pathways relating to neurological diseases (i.e., Parkinson’s, Alzheimer’s, and Huntington’s disease) (Fig. 4). Using KEGGmapper we were able to further investigate which specific metabolic pathway were being affected. We found that Glycan, fatty acid and lipid, and nucleotide metabolism are all up-regulated within the KA treated samples. Moreover, we observed several pathways involving glutamate utilization and isoprenoid biosynthesis were also affected (Fig. 5a).
Fig. 4.

Pathway enrichment of differentially expressed genes. KEGG pathway enrichment of up-regulated genes following KA treatment with a q-value < 0.05
Fig. 5.

Functional analysis of genes from identified pathways. a KEGG mapper was utilized to determine associated metabolic pathways. Differentially expressed genes relating to the identified (b) neurodegenerative and (c) viral pathways were loaded into GeneMANIA [26] to generate putative interaction diagrams. Displayed interactions were limited to only experimentally determined relations
While metabolism was by far the most significantly altered pathway term identified, several other pathways of note were identified, specifically those relating to neurodegenerative diseases (i.e., Parkinson’s, Huntington’s, Alzheimer’s) and viral response (i.e., Herpes simplex, Epstein-Barr, viral carcinogenesis). While it was consistent with our exploration avenue to observe pathways relating to neurodegenerative diseases, we observed viral responses in both GO and pathway analysis. As such, we further explored the gene relationships underlying these identified pathway terms. Differential expressed genes identified to be part of the indicated KEGG pathway terms were analyzed with the GeneMANIA application for Cytoscape V3.5.1 [26]. GeneMANIA utilizes both published information and computational predictions to identify relationships between input genes. It will also suggest possible interaction partners not initially input into the query. Indeed, we demonstrate that the overwhelming majority of genes associated with the identified neurodegenerative pathways were related to mitochondrial function, specifically the electron transport chain (Fig. 5b). This is consistent with our GO analysis. Investigation of viral pathway term genes however revealed a more diverse set of groupings (Fig. 5c). These include genes related to RNA polymerase complexes and histones. Both of which are consistent with the high levels of transcriptional modulation observed. Additionally, several genes were associated with immunological regulation, such as complement C3, signal transducer and activator of transcription 2 (Stat2), and antigen peptide transporter 1 (Tap1).
Discussion
The majority of research into epilepsy has focused on neuronal hyperactivities and cell death. However, the role of glia, particularly microglia, in the pathogenesis of epilepsy is an important emerging area of study. Specifically, the transcriptomic alterations of microglia following KA-induced seizure have not been well studied. In this regard, we utilized RNAseq analysis on isolated hippocampal microglia to investigate microglial response during the acute phase after seizure. In total, our results clearly demonstrate that microglia undergo significant alterations following KA-induced seizures, including up-regulation of several inflammatory factors and modulation of mitochondrial activity.
Microglia may undergo oxidative stress response following KA-induced seizure
The most obvious phenotypic alteration was mitochondrial activity in microglia after seizures. While it is possible that up-regulation of mitochondrial genes is merely indicative of microglia transitioning from a resting to active state, it is also possible that microglia are increasing production of mitochondria-derived reactive oxygen species (ROS). While NADPH oxidase has often been described as the primary source of ROS, it has been well established that NADH dehydrogenase (electron transport chain complex I) can also contribute to ROS formation [27]. Indeed, several complex I subunits (e.g., Ndufs8, Ndufa5, Ndufb8) were differentially expressed in our dataset but no NADPH oxidase subunits were up-regulated. This idea is also supported by the observed up-regulation of superoxide dismutase (Sod) 1 and 2, both of which can convert the superoxide generated by the electron transport chain into hydrogen peroxide [28]. Sod 1 and 2 are critically important for the mitigation of oxidative stress and are altered during epilepsy.
When considering our other results, specifically the observed utilization of glutamate, there is further indication that microglia are responding to oxidative stressors. Our results identified two possible means by which glutamate could be utilized, conversion to either 1) proline or 2) glutathione. Of these, the generation of glutathione may be of significance. From our results we observed differential expression of glutathione peroxidase 3 (Gpx3), glutathione S-transferase omega 1 (Gsto1), glutathione transferase zeta 1 (Gstz1), and glutathione reductase (Gsr) expression, all of which are important to the mitigation of oxidative stress [29, 30]. Understanding the consequence of this response could open new avenues into attenuating oxidative damage following seizures. Moreover, it is interesting that of the four main glutathione peroxidase variants, we only observed increases in Gpx3, which is found within the extracellular space [31]. It is possible that microglia are attempting to mitigate not only their own endogenous oxidative stress but also that within the environment.
Microglia increase metabolic activity in response to KA-induced seizure
Our results also showed that many genes relating to metabolic activity are significantly up-regulated. Specifically, GO and pathway analysis determined that microglia up-regulated lipid, nucleotide, and glycan metabolism. These metabolic activities have also been observed during transcriptional analysis of total rat cortex following sarin-induced seizure [32]. We also identified that geranylgeranyl diphosphate synthase 1 (Ggps1) was up-regulated. Ggps1 is responsible for synthesis of the isoprenoid intermediate geranylgeranyl diphosphate (GGPP), which can be attached to a wide assortment of proteins via geranylgeranyltransferases (GGT) like Rab GGT, whose alpha subunit was differentially expressed in our data set [33, 34]. Within Alzheimer’s disease it was shown that GGPP may influence microglial inflammatory response via modulation of Rho GTPase [35]. Moreover, many of the positive effects of statin drugs (e.g., reducing excitotoxicity and inflammation) within Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis could be attributed to mitigation of isoprenoid intermediates, like GGPP [36–39]. The observed mitigation of KA induced seizure symptoms by statins may also involve similar mechanisms [40]. Given our results, it would be of interest to determine if statins improve seizure recovery by attenuating microglial inflammatory response.
Microglia undergo immunological activation in response to KA-induced seizure
Microglia as a principal immune cell in the brain are activated in human epileptic brain and rodent seizure models [4]. Not surprisingly, we also observed that microglia underwent immunological activation, as seen by enrichment of viral response pathways, at 3 days post KA-induced seizures in mice. However, underlying each of these pathways was a shared set of up-regulated genes, including a number of histocompatibility genes, many of which seem to correlate with non-classical major histocompatibility complexes. More specifically, we observed that H2-T23, which encodes Qa-1, and several genes that make up Qa-2 (i.e., H2-Q6, H2-Q7, and H2-Q8) were differentially expressed within our data set [41, 42]. These histocompatibility complexes have been shown to modulate the activity of natural killer (NK) cells [43, 44]. In regards to neuroinflammation, it was reported that the soluble forms of MHC-E and MHC-G might be related to inflammation protection within multiple sclerosis [45]. However, very little has been done to investigate the roles of these histocompatibility complexes within epilepsy. Given the indications that NK cells are increased following temporal lobe epilepsy, it is worth investigating whether microglia are modulating NK cell activity within the hippocampal region following seizure induction, and whether this modulation is inhibitory of stimulatory.
Interferon beta may modulate microglial activity following KA-induced seizure
Another sign of microglial immunomodulation was the identification of a number of IFN-β responsive terms during GO analysis. IFN-β is typically seen as being anti-inflammatory and has become a common treatment option for relapsing-remitting multiple sclerosis patients [46]. However, there are also indications that type-I interferons may negatively regulate brain activity during aging [47]. Thus, IFN-β may have differential roles depending on disease context. In relation to microglia it has been shown that IFN-β can induce chemokine CCL5 expression, which was highly up-regulated in our data set and in our recent cytokine array [15] after KA-induced seizures. It has also been shown that interferon regulatory factor 7 (Irf7), which is suggested to be the master regulator of type-I interferon-dependent immune response, can modulate CCL5 expression [48]. We observed that Irf7 was differently expressed following KA treatment, indicating a possible means by which IFN-β could modulate microglia inflammatory responses following seizures. As a final note, it has been shown that some Irf7 activity may be tightly regulated by non-degenerative ubiquitination [49, 50]. One of the most up-regulated genes in our data set following seizure was ubiquitin (Ubb). Consequently, our data set indicates that several facets of gene regulation are at play within microglia following KA treatment.
Identified differentially expressed genes warranting further investigation
Finally, while over 2300 differentially expressed genes were identified, we believe that the following selection may be of interest for further investigation. First is osteopontin (secreted phosphoprotein 1; Spp1), which has been observed within neuronal injuries, particularly ischemic stroke [51, 52]. However, little is known about how Spp1 is involved in epileptic seizures even though other less targeted profiling analyses have also noted its up-regulation following seizures [53, 54]. What is known is that its expression seems to be localized to certain areas of the brain, including the CA1 and CA3 regions of the hippocampus [55, 56]. Moreover, it has been suggested that only a sub-set of microglia actively express Spp1, with a possible role in phagocytosis [55]. However, the exact role of Spp1 following epilepsy requires further evaluation.
Next is the adenosine A3 receptor (Adora3/A3ar). This gene is of interest as it was the only receptor to be down-regulated within our data set. Adenosine has long been viewed as an endogenous anticonvulsive and will increase dramatically during epileptic seizures [57]. As for Adora3, it was reported that its specific agonist, IB-MECA, could protect against seizures [58]. It was found that Adora3 is highly expressed in microglia and that LPS treatment down-regulates its expression [59]. Moreover, externally induced activation of Adora3 could reduce LPS-induced tumor necrosis factor alpha (TNFα) in both RAW 264.7 macrophages and BV2 microglia [60, 61]. Yet, little else is known about how Adora3 can modulate microglial activity, let alone why we observed a significant down-regulation in expression following KA-induced seizure.
Lastly, while several purinergic receptors have been shown to modulate microglial function during epilepsy, including P2ry12 and P2rx7, we only observed significantly increased expression of P2rx4 [12]. This receptor has been observed to be important to the pathogenesis of several neurological conditions including neuropathic pain and epilepsy [62, 63]. In regards to microglia, P2rx4 expression can be up-regulated via fibronectin, which was differentially expressed in our data set [64]. Within models of neuropathic pain, it has been suggested that activation of P2rx4 induced microglia to release brain-derived neurotropic factors (BDNF), which then affected neuronal activity by modulating GABAergic activity [65, 66]. Since the hippocampus has a significant population of GABAergic interneurons, particularly in the CA1 and CA3 regions, it may be of interest to determine to what extent this crosstalk exists and whether or not blockage of this communication could alleviate seizure symptoms [67].
In conclusion, our results demonstrate that KA-induced seizure acutely affects the phenotypic character of microglia within the hippocampus. Specifically, microglia seem to be undergoing a variety of activations, which could potentially regulate neuronal hyperactivities and seizure behaviors. We have identified a number of mechanisms and gene targets that could provide future directions for therapeutic intervention.
Methods
Mice
The described In vivo procedures were approved by Institutional Animal Care and Use Committee (IACUC) in both Rutgers University and Mayo Clinic. We followed the guidelines set forth by the Guide of the Care and Use of Laboratory Animals 8th Edition. Both male and female adult heterozygous microglia GFP reporter mice at two months of age were used. The mice express GFP under control of the fractalkine receptor promoter(CX3CR1GFP/+) that selectively label microglia in the CNS [68].
KA administration
An injection of kainic acid (KA) (Tocris Biosciences, Bristol, UK) via direct intracerebroventricular (ICV) injection to induce seizure was performed as previously described [12, 15]. Briefly, a guide tube (24 gauge) was implanted into CX3CR1+/GFP mice prior to KA injection. After a 24 h recovery period, a 30 gauge needle was inserted through the cannula to deliver the KA solution (0.2 μg in 5 μl). Mice were then observed for induction of seizure response using the method described previously [12]. Briefly, seizure behavior was monitored under a modified Racine scale as follows [12, 15, 69]: (1) freezing behavior; (2) rigid posture with raised tail; (3) continuous head bobbing and forepaws shaking; (4) rearing, falling, and jumping; (5) continuous level 4; and (6) loss of posture and generalized convulsion activity. Mice progressed at least to stage 3 and were sacrificed 3d after seizure. Sham controls did not receive KA administration.
Microglia isolation
All mice were perfused with ice cold PBS (pH 7.4) 3 days post KA treatment. Hippocampi were excised, minced on ice, and suspended in a trypsin/EDTA solution for 20 mins, in a 37 °C shaker. After incubation, 3 ml DMEM and 50ul DNase was added to the cell suspension. Cell pellets where then suspended in 5 ml HEPES (4-(2- hydroxyethyl)-1- piperazineethanesulfonic acid) buffer, then centrifuged again. Pellets were finally re-suspended in 800 ml HEPES buffer and transferred into a sorting tube on ice. GFP-labeled microglia were isolated via FACS on a MoFlo XDP Cell Sorter (Beckman Coulter, CA, USA). Microglia from sham controls were isolated in the same manner after a corresponding length of time.
RNAseq analysis
RNA was isolated with the RNeasy Plus Micro Kit (Qiagen, Hilden, Germany). RNA quality was evaluated by Tapestation RNA HS Assay (Agilent Technologies, CA, USA) and Bioanalyzer 2100 Eukaryote Total RNA Nano Kit (Agilent Technologies). Libraries were constructed with the SMART-Seq v4 Ultra Low Input RNA Kit (Takara-Clontech, CA, USA) using manufacturer’s instructions. Final library quantity was determined by KAPA SYBR® FAST qPCR and library quality evaluated by Tapestation RNA HS Assay (Agilent Technologies, CA, USA). Equimolar pooling of libraries were performed based on qPCR values and loaded onto an Illumina Hiseq platform (Illumina, CA, USA).
Differential gene expression analysis
RNA-seq data were aligned to the mouse reference genome using STAR mapping tool [70]. Read counts were then quantified using HTSeq-count [71]. DESeq, an R Bioconductor package, was used for differential gene expression analysis [72]. It estimates variance-mean dependence in RNA-seq count data and tests for differential expression using a negative binomial distribution model. Heat map and hierarchical clustering of differentially expressed genes was performed using the heatmap function in stats package in R.
Functional classification of differentially expressed genes
Gene Ontology (GO) analysis is a commonly used approach for functional studies of RNA-seq data. To functional classify the differentially expressed genes between the control and KA treated groups, GO enrichment analysis using clusterProfiler was performed. Additionally, significant KEGG pathways were identified using the enrichKEGG function in clusterProfiler package with FDR < 0.05.
Statistics
Both KA-treated and control were collected with n = 3 mice. The R package DESeq was used on our RNAseq counts to estimate the variance-mean dependence and to test for differential expression. Differentially expressed proteins with adjusted p-values < 0.05 using the Benjamini-Hochberg procedure. These proteins were then subjected to pathway enrichment/gene ontology analysis. A Benjamini-Hochberg adjusted p-value of < 0.05 was used to identify significantly enriched pathways.
Additional files
Table S1. List of all identified differentially expressed genes. (XLSX 336 kb)
Figure S1. Expression profiles of microglia specific markers. The expression of microglia specific markers, as determined by Hickman et al. [16] was investigated. The Log2 base mean expression of each condition is presented for each gene. Presented error bars are standard error using the Log2 standard deviation of each mean. **Padj < 0.05. Figure S2. Expression profiles of cytokine markers. The expression of a variety of cytokines was investigated. The Log2 base mean expression of each condition is presented for each gene. Presented error bars are standard error using the Log2standard deviation of each mean. **Padj < 0.05. (PDF 114 kb)
Table S2. List of all identified biological process GO terms. (XLSX 44 kb)
Table S3. List of all identifed molecular function GO terms. (XLSX 23 kb)
Table S4. List of all identifed cellular compartment GO terms. (XLSX 24 kb)
Acknowledgements
The authors would like to thank all members of Wu lab at Mayo for the insightful discussions and Dr. Paul A. Stewart (The Moffitt Cancer Center, Tampa, FL) for the technical assistance.
Funding
This work was supported by National Institute of Health (R01NS088627, R21DE025689, K22NS104392), and National Natural Science Foundation of China (No. 31500845).
Availability of data and materials
The datasets supporting the conclusion of this article are included within article.
Authors’ contributions
DB, JRR, WH and WLJ designed the study and wrote the manuscript; DB, JZ, JP, and LF generated mouse seizure model; JZ, ZX, JP, UBE, LF, GW performed the cell sorting experiments; KT, CY and JH performed RNAseq and data analysis; DB and WLJ analyzed the data and revised the manuscript. All authors read and approved the final manuscript.
Ethics approval
All the animal-related procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of in both Rutgers University and Mayo Clinic and were consistent with the ethical guidelines of the National Institutes of Health. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13041-018-0376-5) contains supplementary material, which is available to authorized users.
Contributor Information
Dale B. Bosco, Email: bosco.dale@mayo.edu
Jiaying Zheng, Email: zheng.jiaying@mayo.edu.
Zhiyan Xu, Email: 15170026@yjs.ntu.edu.cn.
Jiyun Peng, Email: peng.jiyun@mayo.edu.
Ukpong B. Eyo, Email: eyo.ukpong@mayo.edu
Ke Tang, Email: ke.tang@admerahealth.com.
Cheng Yan, Email: cheng.yan@admerahealth.com.
Jun Huang, Email: jun.tt.huang@gmail.com.
Lijie Feng, Email: fenglijie1128@sina.com.
Gongxiong Wu, Email: wugongxiong@gmail.com.
Jason R. Richardson, Email: jrichardson@neomed.edu
Hui Wang, Phone: 86-513-85051859, Email: huiwangph@ntu.edu.cn.
Long-Jun Wu, Phone: 507-422-5135, Email: wu.longjun@mayo.edu.
References
- 1.Kwan P, Brodie MJ. Clinical trials of antiepileptic medications in newly diagnosed patients with epilepsy. Neurology. 2003;60(11 Suppl 4):S2–12. doi: 10.1212/WNL.60.11_suppl_4.S2. [DOI] [PubMed] [Google Scholar]
- 2.Levesque M, Avoli M. The kainic acid model of temporal lobe epilepsy. Neurosci Biobehav Rev. 2013;37(10 Pt 2):2887–2899. doi: 10.1016/j.neubiorev.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilcox KS, Gee JM, Gibbons MB, Tvrdik P, White JA. Altered structure and function of astrocytes following status epilepticus. Epilepsy Behav. 2015;49:17–19. doi: 10.1016/j.yebeh.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eyo UB, Murugan M, Wu LJ. Microglia-neuron communication in epilepsy. Glia. 2017;65(1):5–18. doi: 10.1002/glia.23006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eyo UB, Wu LJ. Bi-directional microglia-neuron communication in the healthy brain. Neural Plast. 2013;2013:456857. doi: 10.1155/2013/456857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155(7):1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beach TG, Woodhurst WB, MacDonald DB, Jones MW. Reactive microglia in hippocampal sclerosis associated with human temporal lobe epilepsy. Neurosci Lett. 1995;191(1–2):27–30. doi: 10.1016/0304-3940(94)11548-1. [DOI] [PubMed] [Google Scholar]
- 8.Wyatt-Johnson SK, Herr SA, Brewster AL. Status epilepticus triggers time-dependent alterations in microglia abundance and morphological phenotypes in the Hippocampus. Front Neurol. 2017;8:700. doi: 10.3389/fneur.2017.00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eyo UB, Gu N, De S, Dong H, Richardson JR, Wu LJ. Modulation of microglial process convergence toward neuronal dendrites by extracellular calcium. J Neurosci. 2015;35(6):2417–2422. doi: 10.1523/JNEUROSCI.3279-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eyo UB, Mo M, Yi MH, Murugan M, Liu J, Yarlagadda R, Margolis DJ, Xu P, Wu LJ. P2Y12R-dependent translocation mechanisms gate the changing microglial landscape. Cell Rep. 2018;23(4):959–966. doi: 10.1016/j.celrep.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eyo UB, Peng J, Murugan M, Mo M, Lalani A, Xie P, Xu P, Margolis DJ, Wu LJ. Regulation of physical microglia-neuron interactions by Fractalkine signaling after status epilepticus. eNeuro. 2017;3(6) [DOI] [PMC free article] [PubMed]
- 12.Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci. 2014;34(32):10528–10540. doi: 10.1523/JNEUROSCI.0416-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mika J. Modulation of microglia can attenuate neuropathic pain symptoms and enhance morphine effectiveness. Pharmacol Rep. 2008;60(3):297–307. [PubMed] [Google Scholar]
- 14.Bronen RA. The status of status: seizures are bad for your brain's health. AJNR Am J Neuroradiol. 2000;21(10):1782–1783. [PMC free article] [PubMed] [Google Scholar]
- 15.Tian DS, Peng J, Murugan M, Feng LJ, Liu JL, Eyo UB, Zhou LJ, Mogilevsky R, Wang W, Wu LJ. Chemokine CCL2-CCR2 signaling induces neuronal cell death via STAT3 activation and IL-1beta production after status epilepticus. J Neurosci. 2017;37(33):7878–7892. doi: 10.1523/JNEUROSCI.0315-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16(12):1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abiega O, Beccari S, Diaz-Aparicio I, Nadjar A, Laye S, Leyrolle Q, Gomez-Nicola D, Domercq M, Perez-Samartin A, Sanchez-Zafra V, et al. Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. 2016;14(5):e1002466. doi: 10.1371/journal.pbio.1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trias E, Díaz-Amarilla P, Olivera-Bravo S, Isasi E, Drechsel DA, Lopez N, Bradford CS, Ireton KE, Beckman JS, Barbeito L. Phenotypic transition of microglia into astrocyte-like cells associated with disease onset in a model of inherited ALS. Front Cell Neurosci. 2013;7:274. doi: 10.3389/fncel.2013.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown A. Understanding the MIND phenotype: macrophage/microglia inflammation in neurocognitive disorders related to human immunodeficiency virus infection. Clin Translat Med. 2015;4:7. doi: 10.1186/s40169-015-0049-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louveau A, Nerriere-Daguin V, Vanhove B, Naveilhan P, Neunlist M, Nicot A, Boudin H. Targeting the CD80/CD86 costimulatory pathway with CTLA4-Ig directs microglia toward a repair phenotype and promotes axonal outgrowth. Glia. 2015;63(12):2298–2312. doi: 10.1002/glia.22894. [DOI] [PubMed] [Google Scholar]
- 21.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6(7):e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreiber G, Piehler J. The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol. 2015;36(3):139–149. doi: 10.1016/j.it.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Kawanokuchi J, Mizuno T, Kato H, Mitsuma N, Suzumura A. Effects of interferon-beta on microglial functions as inflammatory and antigen presenting cells in the central nervous system. Neuropharmacology. 2004;46(5):734–742. doi: 10.1016/j.neuropharm.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 25.Luckoff A, Caramoy A, Scholz R, Prinz M, Kalinke U, Langmann T. Interferon-beta signaling in retinal mononuclear phagocytes attenuates pathological neovascularization. EMBO Mol Med. 2016;8(6):670–678. doi: 10.15252/emmm.201505994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montojo J, Zuberi K, Rodriguez H, Kazi F, Wright G, Donaldson SL, Morris Q, Bader GD. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics (Oxford, England) 2010;26(22):2927–2928. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–485. doi: 10.1016/j.redox.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen M, Zou F, Chai HS, Younkin CS, Miles R, Nair AA, Crook JE, Pankratz VS, Carrasquillo MM, Rowley CN, et al. Glutathione S-transferase omega genes in Alzheimer and Parkinson disease risk, age-at-diagnosis and brain gene expression: an association study with mechanistic implications. Mol Neurodegener. 2012;7:13. doi: 10.1186/1750-1326-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Couto N, Wood J, Barber J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic Biol Med. 2016;95:27–42. doi: 10.1016/j.freeradbiomed.2016.02.028. [DOI] [PubMed] [Google Scholar]
- 31.Olson GE, Whitin JC, Hill KE, Winfrey VP, Motley AK, Austin LM, Deal J, Cohen HJ, Burk RF. Extracellular glutathione peroxidase (Gpx3) binds specifically to basement membranes of mouse renal cortex tubule cells. Am J Physiol Renal Physiol. 2010;298(5):F1244–F1253. doi: 10.1152/ajprenal.00662.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spradling KD, Lumley LA, Robison CL, Meyerhoff JL, Dillman JF., 3rd Transcriptional analysis of rat piriform cortex following exposure to the organophosphonate anticholinesterase sarin and induction of seizures. J Neuroinflammation. 2011;8:83. doi: 10.1186/1742-2094-8-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maurer-Stroh S, Washietl S, Eisenhaber F. Protein prenyltransferases. Genome Biol. 2003;4(4):212. doi: 10.1186/gb-2003-4-4-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiemer AJ, Hohl RJ, Wiemer DF. The intermediate enzymes of isoprenoid metabolism as anticancer targets. Anti Cancer Agents Med Chem. 2009;9(5):526–542. doi: 10.2174/187152009788451860. [DOI] [PubMed] [Google Scholar]
- 35.Cordle A, Landreth G. 3-Hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors attenuate beta-amyloid-induced microglial inflammatory responses. J Neurosci. 2005;25(2):299–307. doi: 10.1523/JNEUROSCI.2544-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuipers HF, van den Elsen PJ. Immunomodulation by statins: inhibition of cholesterol vs. isoprenoid biosynthesis. Biomed Pharmacother. 2007;61(7):400–407. doi: 10.1016/j.biopha.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 37.Li L, Zhang W, Cheng S, Cao D, Parent M. Isoprenoids and related pharmacological interventions: potential application in Alzheimer's disease. Mol Neurobiol. 2012;46(1):64–77. doi: 10.1007/s12035-012-8253-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wahner AD, Bronstein JM, Bordelon YM, Ritz B. Statin use and the risk of Parkinson disease. Neurology. 2008;70(16 Pt 2):1418–1422. doi: 10.1212/01.wnl.0000286942.14552.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roy A, Pahan K. Prospects of statins in Parkinson disease. Neuroscientist. 2011;17(3):244–255. doi: 10.1177/1073858410385006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JK, Won JS, Singh AK, Singh I. Statin inhibits kainic acid-induced seizure and associated inflammation and hippocampal cell death. Neurosci Lett. 2008;440(3):260–264. doi: 10.1016/j.neulet.2008.05.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lo WF, Woods AS, DeCloux A, Cotter RJ, Metcalf ES, Soloski MJ. Molecular mimicry mediated by MHC class Ib molecules after infection with gram-negative pathogens. Nat Med. 2000;6(2):215–218. doi: 10.1038/72329. [DOI] [PubMed] [Google Scholar]
- 42.Cai W, Cao W, Wu L, Exley GE, Waneck GL, Karger BL, Warner CM. Sequence and transcription of Qa-2-encoding genes in mouse lymphocytes and blastocysts. Immunogenetics. 1996;45(2):97–107. doi: 10.1007/s002510050177. [DOI] [PubMed] [Google Scholar]
- 43.Chiang EY, Henson M, Stroynowski I. The nonclassical major histocompatibility complex molecule Qa-2 protects tumor cells from NK cell- and lymphokine-activated killer cell-mediated cytolysis. J Immunol. 2002;168(5):2200–2211. doi: 10.4049/jimmunol.168.5.2200. [DOI] [PubMed] [Google Scholar]
- 44.Gays F, Fraser KP, Toomey JA, Diamond AG, Millrain MM, Dyson PJ, Brooks CG. Functional analysis of the molecular factors controlling Qa1-mediated protection of target cells from NK lysis. J Immunol. 2001;166(3):1601–1610. doi: 10.4049/jimmunol.166.3.1601. [DOI] [PubMed] [Google Scholar]
- 45.Morandi F, Venturi C, Rizzo R, Castellazzi M, Baldi E, Caniatti ML, Tola MR, Granieri E, Fainardi E, Uccelli A, et al. Intrathecal soluble HLA-E correlates with disease activity in patients with multiple sclerosis and may cooperate with soluble HLA-G in the resolution of neuroinflammation. J Neuroimmune Pharmacol. 2013;8(4):944–955. doi: 10.1007/s11481-013-9459-3. [DOI] [PubMed] [Google Scholar]
- 46.Limmroth V, Putzki N, Kachuck NJ. The interferon beta therapies for treatment of relapsing-remitting multiple sclerosis: are they equally efficacious? A comparative review of open-label studies evaluating the efficacy, safety, or dosing of different interferon beta formulations alone or in combination. Ther Adv Neurol Disord. 2011;4(5):281–296. doi: 10.1177/1756285611413825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science (New York, NY) 2014;346(6205):89–93. doi: 10.1126/science.1252945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 49.Huye LE, Ning S, Kelliher M, Pagano JS. Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol Cell Biol. 2007;27(8):2910–2918. doi: 10.1128/MCB.02256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ning S, Campos AD, Darnay BG, Bentz GL, Pagano JS. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol Cell Biol. 2008;28(20):6536–6546. doi: 10.1128/MCB.00785-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gliem M, Krammes K, Liaw L, van Rooijen N, Hartung HP, Jander S. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia. 2015;63(12):2198–2207. doi: 10.1002/glia.22885. [DOI] [PubMed] [Google Scholar]
- 52.Chiu IM, Morimoto ETA, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, et al. A neurodegeneration-specific gene expression signature and immune profile of acutely isolated microglia from an ALS mouse model. Cell Rep. 2013;4(2):385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hunsberger JG, Bennett AH, Selvanayagam E, Duman RS, Newton SS. Gene profiling the response to kainic acid induced seizures. Brain Res Mol Brain Res. 2005;141(1):95–112. doi: 10.1016/j.molbrainres.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 54.Gorter JA, van Vliet EA, Aronica E, Breit T, Rauwerda H, Lopes da Silva FH, Wadman WJ. Potential new antiepileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J Neurosci. 2006;26(43):11083–11110. doi: 10.1523/JNEUROSCI.2766-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim SY, Choi YS, Choi JS, Cha JH, Kim ON, Lee SB, Chung JW, Chun MH, Lee MY. Osteopontin in kainic acid-induced microglial reactions in the rat brain. Mol Cells. 2002;13(3):429–435. [PubMed] [Google Scholar]
- 56.Borges K, Gearing M, Rittling S, Sorensen ES, Kotloski R, Denhardt DT, Dingledine R. Characterization of osteopontin expression and function after status epilepticus. Epilepsia. 2008;49(10):1675–1685. doi: 10.1111/j.1528-1167.2008.01613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pedata F, Pugliese A, Sebastião A, Ribeiro J. Adenosine A3 receptor signaling in the central nervous system. 2010. [Google Scholar]
- 58.Von Lubitz DK, Carter MF, Deutsch SI, Lin RC, Mastropaolo J, Meshulam Y, Jacobson KA. The effects of adenosine A3 receptor stimulation on seizures in mice. Eur J Pharmacol. 1995;275(1):23–29. doi: 10.1016/0014-2999(94)00734-O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113(12):E1738–E1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin L, Pingle SC, Hallam DM, Rybak LP, Ramkumar V. Activation of the adenosine A3 receptor in RAW 264.7 cells inhibits lipopolysaccharide-stimulated tumor necrosis factor-alpha release by reducing calcium-dependent activation of nuclear factor-kappaB and extracellular signal-regulated kinase 1/2. J Pharmacol Exp Ther. 2006;316(1):71–78. doi: 10.1124/jpet.105.091868. [DOI] [PubMed] [Google Scholar]
- 61.Lee JY, Jhun BS, Oh YT, Lee JH, Choe W, Baik HH, Ha J, Yoon KS, Kim SS, Kang I. Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of PI 3-kinase/Akt and NF-kappaB activation in murine BV2 microglial cells. Neurosci Lett. 2006;396(1):1–6. doi: 10.1016/j.neulet.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 62.Ulmann L, Levavasseur F, Avignone E, Peyroutou R, Hirbec H, Audinat E, Rassendren F. Involvement of P2X4 receptors in hippocampal microglial activation after status epilepticus. Glia. 2013;61(8):1306–1319. doi: 10.1002/glia.22516. [DOI] [PubMed] [Google Scholar]
- 63.Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008;28(44):11263–11268. doi: 10.1523/JNEUROSCI.2308-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsuda M, Toyomitsu E, Komatsu T, Masuda T, Kunifusa E, Nasu-Tada K, Koizumi S, Yamamoto K, Ando J, Inoue K. Fibronectin/integrin system is involved in P2X(4) receptor upregulation in the spinal cord and neuropathic pain after nerve injury. Glia. 2008;56(5):579–585. doi: 10.1002/glia.20641. [DOI] [PubMed] [Google Scholar]
- 65.Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. 2009;29(11):3518–3528. doi: 10.1523/JNEUROSCI.5714-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malcangio M. GABAB receptors and pain. Neuropharmacology. 2017. https://www.sciencedirect.com/science/article/pii/S0028390817302186?via%3Dihub. [DOI] [PubMed]
- 67.Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ. Hippocampal GABAergic inhibitory interneurons. Physiol Rev. 2017;97(4):1619–1747. doi: 10.1152/physrev.00007.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20(11):4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32(3):281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 70.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. B ioinformatics (Oxford England) 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford England) 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of all identified differentially expressed genes. (XLSX 336 kb)
Figure S1. Expression profiles of microglia specific markers. The expression of microglia specific markers, as determined by Hickman et al. [16] was investigated. The Log2 base mean expression of each condition is presented for each gene. Presented error bars are standard error using the Log2 standard deviation of each mean. **Padj < 0.05. Figure S2. Expression profiles of cytokine markers. The expression of a variety of cytokines was investigated. The Log2 base mean expression of each condition is presented for each gene. Presented error bars are standard error using the Log2standard deviation of each mean. **Padj < 0.05. (PDF 114 kb)
Table S2. List of all identified biological process GO terms. (XLSX 44 kb)
Table S3. List of all identifed molecular function GO terms. (XLSX 23 kb)
Table S4. List of all identifed cellular compartment GO terms. (XLSX 24 kb)
Data Availability Statement
The datasets supporting the conclusion of this article are included within article.