

Abstract
In many regions of the brain, including the mammalian cortex, the magnitude and direction of activity-dependent changes in synaptic strength depend on the frequency of presynaptic stimulation (synaptic plasticity), as well as the history of activity at those synapses (metaplasticity). We present a model of a molecular mechanism of bidirectional synaptic plasticity based on the observation that long-term synaptic potentiation (LTP) and long-term synaptic depression (LTD) correlate with the phosphorylation/dephosphorylation of sites on the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor subunit protein GluR1. The primary assumption of the model, for which there is wide experimental support, is that postsynaptic calcium concentration and consequent activation of calcium-dependent protein kinases and phosphatases are the triggers for the induction of LTP/LTD. As calcium influx through the n-methyl-d-aspartate (NMDA) receptor plays a fundamental role in the induction of LTP/LTD, changes in the properties of NMDA receptor-mediated calcium influx will dramatically affect activity-dependent synaptic plasticity (metaplasticity). We demonstrate that experimentally observed metaplasticity can be accounted for by activity-dependent regulation of NMDA receptor subunit composition and function. Our model produces a frequency-dependent LTP/LTD curve with a sliding synaptic modification threshold similar to what has been proposed theoretically by Bienenstock, Cooper, and Munro and observed experimentally.
Bidirectional changes in the strength of synaptic responses are fundamental to information storage within neuronal networks. In many regions of the brain, long-term synaptic potentiation (LTP) (1), a long-lasting increase in synaptic efficacy, is produced by high-frequency stimulation (HFS) of presynaptic afferents or by pairing presynaptic stimulation with robust postsynaptic depolarization (2). Long-term synaptic depression (LTD) (3), a long-lasting decrease in the strength of synaptic transmission, is produced by low-frequency stimulation (LFS) of presynaptic afferents. HFS-induced LTP results in an increase in the amplitude of miniature excitatory postsynaptic currents (mEPSC) and an increase in the response to application of exogenous glutamate (4). In contrast, LFS-induced LTD results in a decrease in mEPSC amplitude (5, 6) and a decrease in the response to application of exogenous glutamate (7). Not only are LTP and LTD expressed in many brain regions and in many species, but the majority of synapses that express LTP also express LTD. Thus, the regulation of synaptic strength by activity is bidirectional. Such bidirectional regulation of synaptic strength has been hypothesized to depend on changes in the number and/or composition of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) subtype of glutamate receptors (AMPARs) in the postsynaptic membrane.
The AMPAR is a heteromer, composed of multiple subtypes of subunit proteins (GluR1-GluR4). AMPAR function is regulated by the composition of individual receptors and/or the phosphorylation/dephosphorylation state of individual subunit proteins. Of particular interest are serine 831 (S-831) and serine 845 (S-845) on the GluR1 subunit, which can be phosphorylated by CaMKII/protein kinase C and protein kinase A, respectively. The induction of LTP specifically increases phosphorylation of S-831 (8), which increases, by approximately 2-fold, the single channel conductance of homomeric GluR1 AMPARs (9). The induction of LTD is accompanied by a decrease in the phosphorylation of S-845, which appears to be phosphorylated at resting potential (10, 11), and phosphorylation of S-845 increases the “open time” of the AMPAR (12). Therefore, knowledge of the phosphorylation state of GluR1 may be a strong predictor of the direction of change (increase or decrease) induced by conditioning stimulation.
In many regions of the brain, including the mammalian cortex, the magnitude and sign of activity-dependent changes in synaptic strength depend on the presynaptic frequency (13) as well as the history of activity at those synapses. We refer to the curve depicting the frequency dependence of changes in synaptic strength as the LTP/LTD curve, and the crossover point between LTD and LTP, the modification threshold. Kirkwood et al. (13) have shown that the shape of the LTP/LTD curve and the value of the modification threshold depend on the history of cortical activity, which can be acutely regulated in vivo by experience or in vitro by using a stimulation paradigm (14).
Antagonist of N-methyl-d-aspartate (NMDA) receptors (NMDARs) inhibit the induction of high-frequency stimulation-induced LTP and LFS-induced LTD, suggesting that the NMDAR is the critical point of calcium entry into the postsynaptic neuron. As such, changes in NMDAR function will dramatically alter the properties of activity-dependent synaptic plasticity. NMDARs are heteromeric ion channels, composed of NR1 and NR2 subunit proteins (15, 16). Each of the four subtypes of the NR2 subunit (2A-2D) confers distinct functional properties to the receptor. As has been demonstrated both in vivo and in heterologous expression systems, NMDARs composed of NR1 and NR2B mediate long duration currents (≈250 ms), whereas inclusion of the NR2A subunit results in NMDARs with faster kinetics (≈50 ms) (17, 18). NMDARs composed of NR1 and NR2B are observed in the neocortex at birth (17–19), and over the course of development there is an increase in the ratio of NR2A/NR2B. The composition and function of synaptic NMDARs can also be acutely and bidirectionally modified by cortical activity (20–23).
Here we present a model that combines and integrates bidirectional plasticity of AMPAR by calcium-dependent phosphorylation and dephosphorylation, (LTP/LTD induction) with plasticity of NMDAR subunit composition. Our model produces a frequency-dependent LTP/LTD curve with a sliding synaptic modification threshold similar to what has been proposed on theoretical grounds (24, 25) and observed experimentally (13, 14).
Control of Synaptic Strength by Activity-Dependent Regulation of AMPAR Phosphorylation
The bidirectional regulation of phosphorylation of two sites on the GluR1 subunit of the AMPAR, based on the work of Lee, Huganir, and Bear, is schematized in Fig. 1 (10, 11, 26). The fraction of AMPARs containing GluR1 phosphorylated at S831 is denoted by Ap1, whereas Ap2 denotes the fraction of AMPARs containing GluR1s phosphorylated at S845, and A and A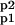 denote AMPARs phosphorylation at neither and both sites, respectively. EP1 and EP2 are used to denote the protein phosphatases that dephosphorylate these two sites, and EK1 and EK2 are used to denote the protein kinases that phosphorylate these sites.
denote AMPARs phosphorylation at neither and both sites, respectively. EP1 and EP2 are used to denote the protein phosphatases that dephosphorylate these two sites, and EK1 and EK2 are used to denote the protein kinases that phosphorylate these sites.
Figure 1.

An idealized model for the cycle of GluR1 phosphorylation/dephosphorylation at two sites. The model assumes two specific kinases (EK1, EK2) and two opposing specific phosphatases (EP1, EP2). It is assumed that high-frequency stimulation preferentially stimulates the activity of protein kinases, resulting in GluR1 phosphorylation, whereas low-frequency stimulation preferentially stimulates the activity of protein phosphatases, resulting in GluR1 dephosphorylation.
A fundamental assumption of this model, on the basis of experimental data, is that the activity of the protein kinases and protein phosphatases that target these two sites on the GluR1 protein is directly or indirectly regulated by intracellular [Ca2+] (27, 28).
Thus EP1 = EP1(Ca2+), EP2 = EP2(Ca2+), EK1 = EK1(Ca2+), EK2 = EK2(Ca2+), where we have used Ca2+, instead of [Ca2+] to simplify the notation.
The cycle shown in Fig. 1 can be quantitatively analyzed by two approaches:
(i) The Mass Action (MA) Law, where the enzymes involved are treated as calcium-dependent forward and backward rate constants of a reversible reaction:
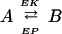 |
1 |
EK, EP ∈ {EK1, EK2, EP1, EP2} is the enzyme kinase or phosphatase; A, B ∈ {A, Ap1, Ap2, A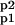 } are the reaction substrates and products.
} are the reaction substrates and products.
(ii) The Michaelis–Menten (MM) kinetics for each enzyme of the phosphorylation cycle
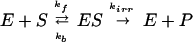 |
2 |
where E ∈ {EK1, EK2, EP1, EP2} is the protein kinase or phosphatase; S, P ∈ {A, Ap1, Ap2, A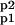 } are, respectively, the reaction substrates and products; ES are the enzyme-substrate complexes, kf, kb are the forward and backward constants, and kirr is the constant for the irreversible step.
} are, respectively, the reaction substrates and products; ES are the enzyme-substrate complexes, kf, kb are the forward and backward constants, and kirr is the constant for the irreversible step.
We can also consider the Ca2+-dependent activation of each enzyme by the following reaction:
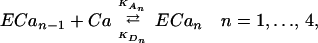 |
3 |
where KAn and KDn are, respectively, the association and the dissociation constant for each reaction, ECa0 is the bare enzyme, and ECan is the fully activated enzyme (bound with 4 Ca2+).
These approaches lead to time-evolution equations that can be written in a similar matrix form:
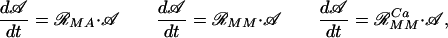 |
4 |
where 𝒜 = (A Ap1 Ap2 A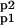 ); ℛMA, ℛMM, and ℛ
); ℛMA, ℛMM, and ℛ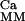 are the matrices containing the velocities of conversion between the different phosphorylation states of the AMPA receptor calculated by the MA, the MM, and the MM with calcium as activator (MMCa) approaches (see Appendix A).
are the matrices containing the velocities of conversion between the different phosphorylation states of the AMPA receptor calculated by the MA, the MM, and the MM with calcium as activator (MMCa) approaches (see Appendix A).
If we assume that the time course of Ca2+-dependent activation of each enzyme is faster than the time course of AMPA receptor phosphorylation, we can make the approximation that the enzymatic dynamics are instantaneous. Under such conditions, the MA case is exactly solvable (Appendix A), and the stable solutions have the form:
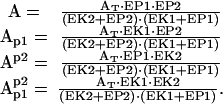 |
5 |
Although the MM cases are formally similar, the differential equations that describe their dynamics are not linear. Therefore, we do not have an explicit analytical solution for those equations. Nevertheless, they can be solved numerically. In addition, we must make assumptions about the calcium dependence of the enzymatic activity. We find two conditions necessary for obtaining realistic LTP/LTD curves: (i) The activity level of the phosphatases rises at lower calcium concentration than the activity level of the kinases. (ii) At high calcium concentrations, the activity level of the protein kinases is higher than the activity level of the protein phosphatases. We show that AMPAR conductance depends on the level of intracellular Ca2+, regardless of the form of Ca2+-dependent enzymatic activation we apply (sigmoidal, hyperbolic, or Hill function).
In Fig. 2, we show two different sets of assumptions that lead to LTD for a small elevation in calcium levels and LTP for a large elevation. The results in Fig. 2 are based on the MA model. The AMPAR conductance was calculated by conductance = A + 2 ∗ (Ap1 + Ap2) + 4 ∗ A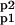 , which is roughly consistent with experimental results. Using the MM approaches leads to qualitatively similar results (data not shown).
, which is roughly consistent with experimental results. Using the MM approaches leads to qualitatively similar results (data not shown).
Figure 2.

Robustness of results of the mass-action approach to Ca2+-dependent enzymatic reactions, which regulate GluR1 phosphorylation and AMPAR conductance. (a) The kinase-phosphatase activity is assumed to be a Hill function of Ca, with exponent 2. EP1(Ca) = EP2(Ca) = 1 + 30(Ca)2/(1 + (Ca)2) and EK1(Ca) = EK2(Ca) = 1 + 100(Ca)2/(82 + (Ca)2). (b) Phosphorylation of GluR1 as a function of Ca2+, using the enzymatic activity assumed in a. (c) Conductance of AMPAR (in arbitrary units) as a function of calcium. At moderate calcium levels, LTD is attained, whereas at higher calcium levels, LTP is induced. (d) The kinase phosphatase activity of each enzyme in which a sigmoidal dependence on Ca is assumed: EP1 = (10⋅30)/(10 + 20⋅e−(2⋅Ca)), EP2 = (10⋅20)/(10 + 10⋅e−(2.5⋅Ca)), EK1 = 10·100/(10 + 90⋅e−(0.2⋅Ca)), EK2 = 10·80/(10 + 70⋅e−(0.25⋅Ca)). (e) Phosphorylation of GluR1 as a function of Ca2+, using the enzymatic activity assumed in d. (f) Conductance of AMPAR (in arbitrary units) as a function of calcium. Calcium and AMPA conductance are in arbitrary units.
NMDA Receptor Calcium Influx and Plasticity
Calcium influx through NMDA receptors, denoted by INMDA, can be broken down into two components, one fast and one slow, with time constants τf, τs (20). We assume that calcium decays passively with a time constant of τCa and a voltage dependence as described by Jahr and Stevens (29) (Appendix B). We can then obtain the average calcium concentration at steady state as:
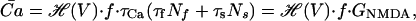 |
6 |
where GNMDA is the gain of calcium influx through NMDA receptors, V is the postsynaptic potential, f is the presynaptic frequency, and ℋ describes the voltage dependence of the calcium influx (Appendix B). This implies that the average level of calcium at steady state is linearly related to presynaptic frequency and the magnitude of the fast and slow components (Nf and Ns).
If we assume that synaptic strength depends on the sustained average level of calcium, we can use Eq. 6 to express AMPAR conductance as a function of presynaptic frequency (f) and postsynaptic depolarization (V). In Fig. 3a, we show an example of how AMPAR conductance depends on these two variables. This figure is based on the MA model described above. It assumes that f and V are controlled independently, such as during protocols in which presynaptic stimulation is paired with postsynaptic depolarization (30, 31). However, in most experimental paradigms, postsynaptic depolarization depends on presynaptic frequency. The diagonal line in Fig. 3a represents a possible linear dependence of V on f. In Fig. 3b, we display the AMPAR conductance along this line. This LTP/LTD curve is qualitatively similar to frequency response curves obtained experimentally (3). This qualitative result is conserved so long as V monotonically increases with f.
Figure 3.

Synaptic strength, measured as AMPAR conductance depicted as a function of presynaptic stimulation frequency (f) and postsynaptic membrane voltage (V). (a) A two-dimensional plot depicting postsynaptic membrane potential as a function of presynaptic stimulation frequency. The grey scale indicates the conductance level of the AMPAR. At low stimulation frequencies and postsynaptic voltages, the conductance is below baseline, defined as f = 0, V = −100. The diagonal line indicates a linear f − V relation, which we assume to extract the results in b. (b) AMPAR conductance as a function of presynaptic stimulation frequency, where a linear dependence of V on f is assumed (as shown in a). Low-frequency stimulation induces LTD, whereas high-frequency stimulation induces LTP.
The composition and function of NMDA receptors change over the course of development and depend on the history of cortical activity. Changing the kinetics of NMDAR-mediated synaptic currents will affect GNMDA and will therefore alter the form of the LTP/LTD curve. To account for the activity-dependent changes in NMDAR composition and function, we develop a phenomenological model for the plasticity of NMDA receptor kinetics. We assume that the total number of NMDA receptors in the membrane is fixed, and that the relative concentration of each type reflects the relative intracellular concentration. This can be expressed by the following set of equations:
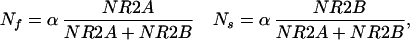 |
7 |
where α is a proportionality constant. The data indicate that NR2A levels are activity dependent, but levels of NR2B seem fixed (23). We therefore assume that NR2B is constant, and that NR2A = 〈f(V)〉τ2A, where 〈〉τ2A represents a temporal average, with a temporal window of τ2A. For illustration purposes, let us assume that NR2A = 〈(V/V0)2〉τ2A, where V0 is an arbitrary proportionality constant. We will then have:
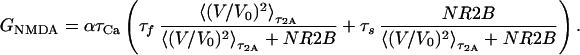 |
8 |
The rate of change depends on τ2A,** which depends on the time scale for synthesis of new NR2A subunits. The change in GNMDA is approximately inversely proportional to the modification threshold.
In Fig. 4a, we illustrate how altering the value of GNMDA changes the form of the LTP/LTD curve. This change is qualitatively consistent with experimental results that indicate that the form of the LTP/LTD curve can be altered by changing the level of cortical activity (13, 14). For comparison, we replotted the results of Kirkwood et al. (13) in Fig. 4b. The activity dependence of the LTP/LTD curve as described by Eq. 8 and illustrated in Fig. 4 is consistent with a key postulate of the BCM theory: the sliding modification threshold.
Figure 4.

The effect of changing NMDAR subunit composition on the shape of the LTP/LTD curve. (a) We use here the same approach as in Fig. 3 to produce an LTP/LTD curve as a function of frequency. The two curves reflect two different conductance levels GNMDA = 0.01 and GNMDA = 0.03. NMDAR conductance level could change as a function of NMDAR subunit composition. We used a semi-log plot to facilitate comparison to experimental results. (b) Results reproduced from Kirkwood et al. (13), in which LTP/LTD curves of light- and dark-reared animals are compared. Notice that these two sets of results are qualitatively consistent.
Discussion
We present a model of the molecular mechanism of synaptic plasticity that implies that knowing the local postsynaptic concentration of intracellular calcium is sufficient to determine the status of AMPAR subunit phosphorylation and, therefore, synaptic efficacy. The fundamental assumption of the model is that the intracellular calcium concentration is the principal trigger for the induction of LTD/LTP, an assumption that has wide experimental support. One of the principal features of this model is its relative robustness to the detailed assumptions; the same qualitative form of an LTP/LTD curve is obtained by assuming different functional forms for the calcium dependence of the enzymes and when using different mathematical formalisms.
By using a known model of NMDAR calcium conductance, and by taking a temporal average of the calcium concentration, we can convert the model to a form that depends on postsynaptic voltage and presynaptic frequency (Fig. 3a) or to a model that depends only on presynaptic frequency (Fig. 3b). We have also demonstrated that a phenomenological model for the modification of NMDAR subunit composition can account for the sliding modification threshold (13, 14, 24).
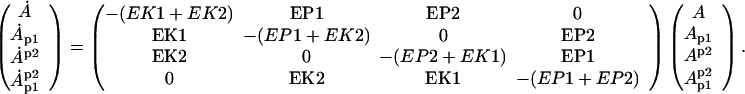 |
9 |
This mathematical model provides a link between synaptic plasticity observed in situ and biochemical/electrophysiological observations of glutamate receptor composition and function. Further, it combines plasticity of AMPA and NMDA receptors. A consequence of the proposed mechanism of the sliding modification threshold is that in low stimulation conditions, the minima of the LTP/LTD curve should occur at lower frequencies than for control conditions, and these minima should be of equivalent magnitude (Fig. 4a). Although there already is experimental evidence for a sliding modification threshold, further experiments that would measure the magnitude of these minima would test our theoretical framework.
Our biophysical model supports the postulates of the Bienenstock, Cooper, and Munro (BCM) theory (24, 25, 32) but, as expected, suggest detailed modifications. For example, the BCM theory is formulated in terms of the temporal derivative of synaptic weight. Here, we extract a rule directly for the fixed point of the synaptic weights for given presynaptic frequency and postsynaptic potential. Under certain conditions, these can be related by the addition of a weight decay term (H.Z.S., unpublished work). In addition, to attain stability, the synaptic modification threshold of the BCM theory was chosen to be a superlinear function of activity. In our model, we obtain a functional form that is more complex, and the dynamic range of the modification threshold is finite. The effect of these differences on the stability of learning, on the formation of receptive fields, and on the ability to account for various experimental results is currently being studied. Further, we are currently examining how this framework could account for more transient induction mechanisms, such a spike time-dependent plasticity (33, 34).
Acknowledgments
We thank Mark Bear, Hey-Kyoung Lee, and Irina Vayl for useful discussions. This research was supported by The Charles A. Dana Foundation. G.C.C. was supported by an exchange program between Bologna and Brown Universities and Ministero dell'Universitá e della Ricerca Scientifica e technologica (60%).
Abbreviations
- LTP
long-term synaptic potentiation
- LTD
long-term synaptic depression
- LFS
low-frequency stimulation
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPAR
AMPA receptor
- NMDA
N-methyl-d-aspartate
- NMDAR
NMDA receptor
- MA
Mass Action
- MM
Michaelis–Menten
Appendix
A. AMPAR Phosphorylation Cycle Dynamics.
A1 MA approach.
By applying the MA Law to the phosphorylation cycle showed in Fig. 1, which assumes two phosphorylation sites, we observe that it is composed of four reversible reactions:
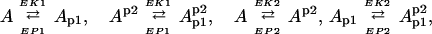 |
and it is easy to obtain the following time-evolution system:
The system (Eq. 9) can be rewritten as 𝒜̇ = ℛMA ⋅ 𝒜, where 𝒜 = (A, Ap1, Ap2, A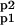 ), and ℛMA is the “coefficient matrix.” To characterize the equilibrium solution, we observe that the kernel of the matrix ℛMA is, by construction, not trivial [Det(ℛMA) = 0], and one of its bases is:
), and ℛMA is the “coefficient matrix.” To characterize the equilibrium solution, we observe that the kernel of the matrix ℛMA is, by construction, not trivial [Det(ℛMA) = 0], and one of its bases is:
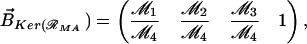 |
10 |
where
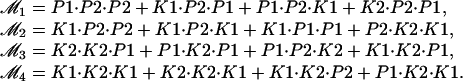 |
Now, the conservation of the total amount of AMPAR [A(t) + Ap1(t) + Ap2(t) + A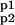 (t) = AT] leads to the following equilibrium solutions as fractions of AT:
(t) = AT] leads to the following equilibrium solutions as fractions of AT:
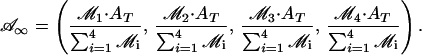 |
11 |
A2 MM approach.
The AMPAR cycle shown in Fig. 1 is composed of four coupled enzymatic reactions, one phosphorylation and one dephosphorylation:
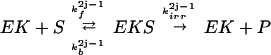 |
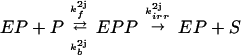 |
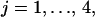 |
where EK, EP ∈ {EK1, EK2, EP1, EP2}; S, P ∈ {A, Ap1, Ap2, A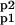 }. The constants k
}. The constants k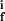 , k
, k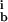 , k
, k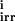 i = 1, … 8 are the rate constants for the forward, backward, and irreversible steps. According to MM analysis, we define the MM constants k
i = 1, … 8 are the rate constants for the forward, backward, and irreversible steps. According to MM analysis, we define the MM constants k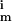 = k
= k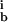 + k
+ k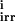 /k
/k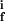 i = 1, … 8. The application of the Pseudo-Steady-State hypothesis as well the MA Law allows us to write the following time-evolution system for the concentrations of the different fractions of AMPAR for each phosphorylation state:
i = 1, … 8. The application of the Pseudo-Steady-State hypothesis as well the MA Law allows us to write the following time-evolution system for the concentrations of the different fractions of AMPAR for each phosphorylation state:
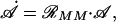 |
12 |
where 𝒜 = (A, Ap1, Ap2, A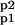 ), and ℛMM is a “coefficient matrix:”
), and ℛMM is a “coefficient matrix:”
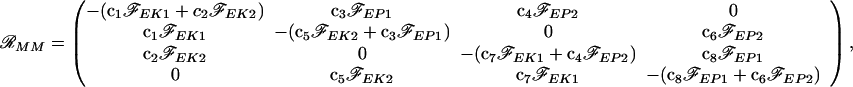 |
13 |
where:
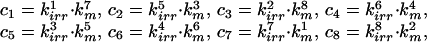 |
and the four “fluxes” have the form:
 |
where EK1T, EP1T, EK2T, EP2T are the total amounts of each enzymes. Note that Eq. 12 has a form that is similar to the one written down for the reversible reaction [we can characterize Ker(ℛMM)], however, there is a significant difference, because the “fluxes” are functions of the dynamic variables, not constants. Thus this equation is nonlinear, and a full solution of the dynamics has not been analytically obtained. Previous derivations have assumed that the levels of the enzymes' activity depend on calcium levels; we made ad hoc assumptions about this dependence. The calcium dependence of these enzymes can be calculated if we know how calcium interacts with them; for example, EK1 needs to be bound to calcium to switch an active state (Ca2+ is an activator for EK1). More generally, and according to various experimental findings, we can assume that each enzyme needs to be bound to more than one molecule of calcium. Here we will assume that activation depends on calcium binding at four sites; thus we have the following reaction sequence:
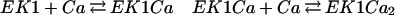 |
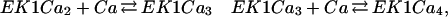 |
with the association constant KI, KII, KIII, KIV for each binding site. With these assumptions, we can write the “fluxes” for the AMPAR cycle with four enzymes:
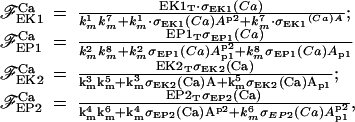 |
where the σi i ∈ (EK1, EK2, EP1, EP2) are the functions for the calcium bound of each enzyme:
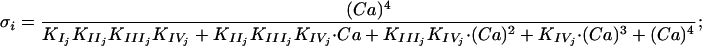 |
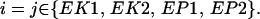 |
With this formalism, we can write the system again as 𝒜̇ = ℛ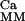 ⋅𝒜, where the matrix ℛ
⋅𝒜, where the matrix ℛ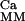 is obtained by substitution of the fluxes ℱj with the calcium-dependent fluxes ℱ
is obtained by substitution of the fluxes ℱj with the calcium-dependent fluxes ℱ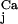 . So also in this case, we can characterize Ker(ℛ
. So also in this case, we can characterize Ker(ℛ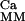 ) and numerically solve the system.
) and numerically solve the system.
B. Calcium Dynamics Through NMDA Channels.
A simple differential equation that qualitatively captures the main features of the calcium dynamics in spines is
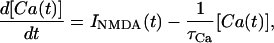 |
14 |
where [Ca(t)] is the calcium concentration at the spine at time t, and τCa is the decay time constant of calcium in the spine. This equation can be solved exactly to yield the solution
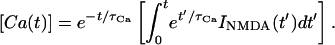 |
15 |
Given the NMDA current, we can calculate the instantaneous level of calcium. We now assume INMDA(t) has the following form:
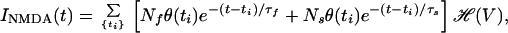 |
16 |
where ti are the times at which presynaptic spikes are delivered and θ(t) is zero for t < 0 and one for t > 0. The calcium current through NMDARs is assumed to have a fast and a slow component with time constants τf and τs, respectively, and magnitudes Nf and Ns. The voltage dependence of the calcium current through the NMDARs is summarized by ℋ, where ℋ(V) expresses the voltage dependence of the calcium current through the NMDARs. This depends on the voltage dependence of the magnesium block and on the voltage-dependent driving force. Thus we use ℋ(V) = B(V)(V − Vr), where B represents the dependence on the magnesium block, and (V − Vr) is the driving force for calcium molecules. For B, we use a model based on experimental results (29), which gives the functional form of effect of the magnesium block as:
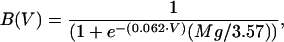 |
where Mg is the extracellular magnesium concentration. In all subsequent results, we assume that Mg = 1 mM. The linear model for the voltage dependence of the driving force is not exact for calcium channels (35); however, it is sufficient for our purposes. We also assume a calcium reversal potential of 130 mV.
By substituting Eq. 16 into Eq. 15 and carrying out the integration, we obtain:
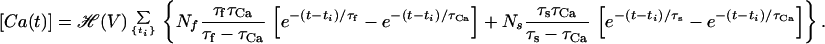 |
17 |
This result is valid on the basis of the assumption that the postsynaptic potential V is constant, which enables us to take this out of the integral. If we stimulate the synapse with a steady frequency f, then the period between two consecutive presynaptic spikes is T = 1/f and ti = i/f, where i = 0 … N and N⋅f < t. We can then redefine t = N/f + Δt, where Δt is the time since the last presynaptic spike. Thus t − ti = Δt + m/f, where m = 0, … N. We then have that
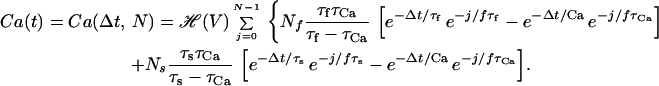 |
This result could be expected from the single spike result, because it is a solution of a linear differential equation. Now we will concentrate on the steady-state solution on the calcium current. Mathematically, this implies that N → ∞. The steady-state solution exists if the infinite series converges. In the infinite series above, we have terms of the form ∑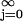 e−j/fτf = 1/1 − e−1/fτf = μf. This series converges because e−1/fτf < 1.
e−j/fτf = 1/1 − e−1/fτf = μf. This series converges because e−1/fτf < 1.
Thus
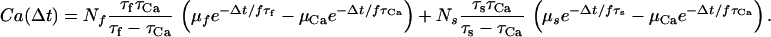 |
18 |
Notice that this steady-state solution is periodic, with periodicity T = 1/f. We will now calculate the average calcium concentration at steady state by averaging over a period, thus
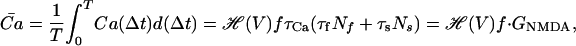 |
19 |
where GNMDA = τCa(τfNf + τsNs) is the gain of the NMDAR calcium conductance.
This result implies that the average level of calcium at steady state is linearly dependent on presynaptic frequency, and that its dependence on the magnitude of the fast and slow components (Nf and Ns) is also simply linear. Eq. 19 can now be used to rewrite the equations describing AMPAR plasticity in terms of frequency (f) and postsynaptic depolarization (V), variables that are more readily controlled than intracellular calcium concentration. To do this, we have to know how calcium concentrations depend on both presynaptic activity and postsynaptic depolarization.
Footnotes
Note that τf, the decay time constant of the NMDA heteromers with the NR2A subunit, and τ2A are different variables.
References
- 1.Bliss T V P, Lømo T. J Physiol (London) 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wigstrom H, Gustafsson B. J Physiol (Paris) 1986;81:228–236. [PubMed] [Google Scholar]
- 3.Dudek S M, Bear M F. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies S N, Lester R A, Reymann K G, Collingridge G L. Nature (London) 1989;338:500–550. doi: 10.1038/338500a0. [DOI] [PubMed] [Google Scholar]
- 5.Oliet S H, Malenka R C, Nicoll R A. Science. 1996;271:1294–1297. doi: 10.1126/science.271.5253.1294. [DOI] [PubMed] [Google Scholar]
- 6.Carrol R C, Lissin D V, Von Zastrow M, Nicoll R A, Malenka R C. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- 7.Kandler K, Katz L C, Kauer J A. Nat Neurosci. 1998;1:119–123. doi: 10.1038/368. [DOI] [PubMed] [Google Scholar]
- 8.Barria A, Muller D, Derkach V, Soderling T R. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 9.Derkach V, Barria A, Soderling T R. Proc Natl Acad Sci USA. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee H K, Kameyama K, Huganir R L, Bear M F. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- 11.Lee H K, Barbarosie M, Kameyama K, Bear M F, Huganir R L. Nature (London) 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 12.Banke T G, Bowie D, Lee H K, Huganir R L, Schousboe A, Traynelis S F. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirkwood A, Rioult M G, Bear M F. Nature (London) 1996;381:526–528. doi: 10.1038/381526a0. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, Wagner J J. J Neurophysiol. 1999;82:2024–2028. doi: 10.1152/jn.1999.82.4.2024. [DOI] [PubMed] [Google Scholar]
- 15.McBain C J, Mayer M L. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- 16.Zukin R S, Bennet M V. Trends Neurosci. 1995;18:306–313. doi: 10.1016/0166-2236(95)93920-s. [DOI] [PubMed] [Google Scholar]
- 17.Stocca G, Vicini S. J Physiol. 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flint A C, Maisch U S, Weishaupt J H, Kriegstein A R, Monyer H. J Neurosci. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheng M, Cummings J, Roldan L A, Jan Y N, Jan L Y. Nature (London) 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 20.Carmignoto G, Vicini S. Science. 1992;258:1007–1011. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- 21.Quinlan E M, Philpot B D, Huganir R L, Bear M F. Nat Neurosci. 1999;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- 22.Quinlan E M, Olstein D H, Bear M F. Proc Natl Acad Sci USA. 1999;96:1287–1290. doi: 10.1073/pnas.96.22.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Philpot B D, Sekhar A K, Shouval H Z, Bear M F. Neuron. 2001;29:157–169. doi: 10.1016/s0896-6273(01)00187-8. [DOI] [PubMed] [Google Scholar]
- 24.Bienenstock E L, Cooper L N, Munro P W. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Intrator N, Cooper L N. Neural Networks. 1992;5:3–17. [Google Scholar]
- 26.Kameyama K, Lee H K, Bear M F, Huganir R L. Neuron. 1998;21:1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- 27.Lisman J A. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyer T, Hanson P I, Stryer L, Schulman H. Science. 1992;256:1199–1201. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 29.Jahr C E, Stevens C F. J Neurosci. 1990;10:3178–3182. doi: 10.1523/JNEUROSCI.10-09-03178.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crair M C, Malenka R C. Nature (London) 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- 31.Feldman D E, Nicoll R A, Malenka R C, Isaac J T. Neuron. 1998;21:347–357. doi: 10.1016/s0896-6273(00)80544-9. [DOI] [PubMed] [Google Scholar]
- 32.Blais B, Shouval H, Cooper L N. Proc Natl Acad Sci USA. 1999;96:1083–1087. doi: 10.1073/pnas.96.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Markram H, Lübke J, Frotscher M, Sakmann B. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- 34.Bi G, Poo M. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnston D, Wu S M S. Foundations of Cellular Neurophysiology. Cambridge, MA: MIT Press; 1995. [Google Scholar]