

Abstract
Cellular signal transduction is often regulated at multiple steps in order to achieve more complex logic or precise control of a pathway. For instance, some signaling mechanisms couple allosteric activation with localization to achieve high signal to noise. Here, we create a system for light activated nuclear import that incorporates two levels of control. It consists of a nuclear import photoswitch, Light Activated Nuclear Shuttle (LANS), and a protein engineered to preferentially interact with LANS in the dark, Zdk2. First, Zdk2 is tethered to a location in the cytoplasm, which sequesters LANS in the dark. Second, LANS incorporates a nuclear localization signal (NLS) that is sterically blocked from binding to the nuclear import machinery in the dark. When activated with light, LANS both dissociates from its tethered location and exposes its NLS, which leads to nuclear accumulation. We demonstrate that this coupled system improves the dynamic range of LANS in mammalian cells, yeast, and C. elegans and provides tighter control of transcription factors that have been fused to LANS.
INTRODUCTION
In cells, signal transduction is regulated by a variety of mechanisms that are commonly in use at multiple points in a single pathway. For example, Inhibitor of NF-κB (IκB) proteins apply two mechanisms to inactivate NF-κB transcription factors. First, they bind to NF-κB transcription factors and sterically occlude their nuclear localization signals (NLS) from interacting with the nuclear import machinery. Second, IκBs have nuclear export signals (NES) that direct NF-κB/IκB complexes to the cytoplasm. NF-κB pathways are activated when IκBs are phosphorylated and then degraded by the proteasome. This degradation event releases NF-κBs from the IκBs and exposes their NLSs[1]. The combined effect of losing an NES and gaining an NLS provides a strong driving force to enter the nucleus and initiate gene transcription. The spatiotemporal regulation of NF-κBs is an inspiration for developing artificial signaling pathways, and here we employ a similar strategy to an engineered nuclear import photoswitch.
A common approach for studying signaling mechanisms in biology is to perturb a pathway and then observe how the system responds. Light-activatable proteins are well suited to these types of studies because they allow pathways to be reversibly activated with high spatiotemporal resolution in living cells and organisms[2]. Recently, we developed a protein photoswitch, called LANS for Light Activatable Nuclear Shuttle, that resides in the cytoplasm and goes to the nucleus when activated with light[3]. LANS is based on the naturally occurring LOV2 domain from Avena sativa, which undergoes a large conformational change when activated with blue light[4]. To create LANS, an NLS motif was embedded at the end of the C-terminal helix (Jα-helix) of the AsLOV2 domain[5, 6]. In the dark, the Jα-helix is folded and sterically blocks binding to importins, a critical step in nuclear import. Upon light activation, the Jα-helix undocks from the AsLOV2 domain allowing the NLS to bind to importins and enter the nucleus. To maintain LANS in the cytoplasm in the dark, an NES was fused to the switch. Several NESs were tested to find a motif that would keep the switch cytoplasmic in the dark but allow it to be redirected to the nucleus when the embedded NLS is exposed[3]. We were able to achieve functional changes in nucleocytoplasmic levels using two different constructs, termed LANS1 and LANS4 that carry two different NESs. LANS4 has the higher dynamic range (light vs. dark nuclear localization) of the two constructs, but LANS1 has tighter caging (less nuclear in the dark), allowing different levels of control depending on the demands of the experiment. Despite these results, both LANS switches still enter the nucleus to some extent in the dark. This “leakiness” is not ideal for applications where it is important to have very little activity in the dark.
LANS only uses a single point of regulation to control nuclear import, the exposure of an NLS. In contrast, NF-κB includes two control points, the exposure of an NLS and the release of an NES. In order to mimic the NF-κB pathway more closely, we decided to incorporate a second layer of control for the nuclear import mediated by LANS. Recently, we engineered a system, LOVTRAP, which preferentially interacts with the AsLOV2 domain in the dark[7]. We have shown that by anchoring one of the LOVTRAP variants, Zdk2, to mitochondrial, vacuole and plasma membrane it is possible to recruit the AsLOV2 domain to the membranes in the dark and release the domain with blue light. Since LANS is built from the AsLOV2 domain, we hypothesized that we could use Zdk2 to anchor LANS in the cytoplasm in the dark and thus reduce the amount of LANS found in the nucleus in the dark. Upon light stimulation, LANS should release from the membrane and expose its NLS for translocation to the nucleus (Figure 1A). We refer to the combined system (an anchored Zdk2 with LANS) as LANSTRAP. To test our hypothesis, we performed in vitro binding studies, localization studies in mammalian cells, and activity assays in yeast and C. elegans.
Figure 1.
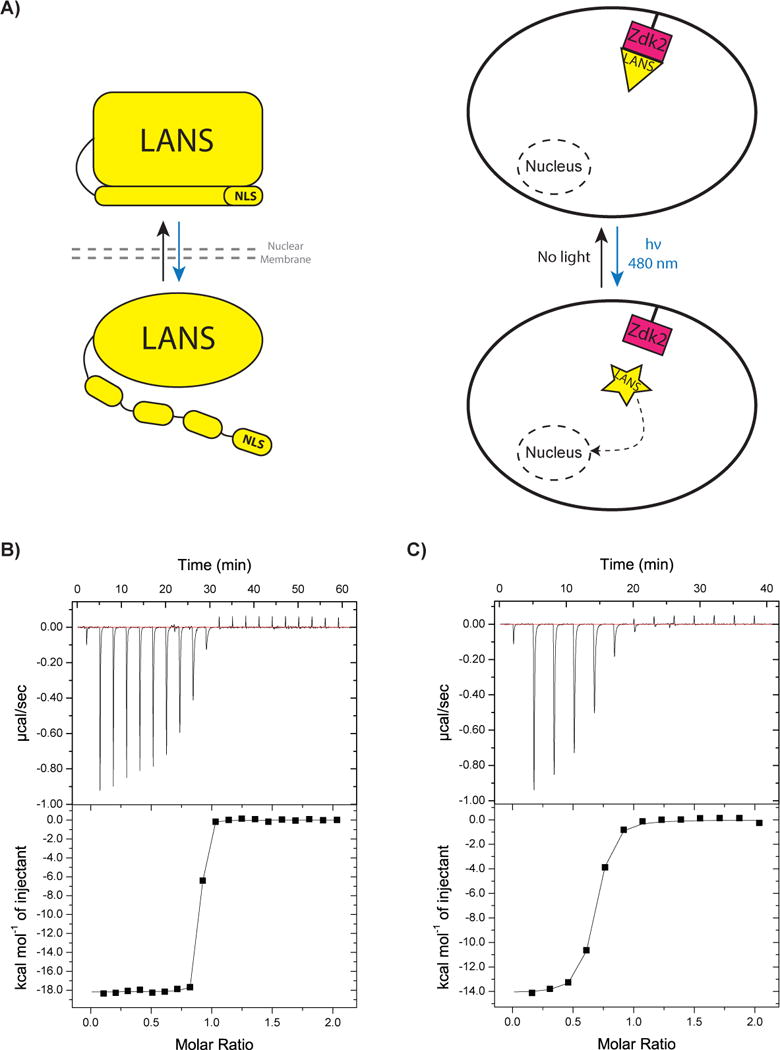
LANSTRAP system schematic and in vitro characterization. A) A schematic of LANS alone (on the left) and LANSTRAP (on the right) where LANS binds Zdk2 from LOVTRAP in the dark and sequesters it in the cytoplasm (above) until blue light illumination triggers its dissociation and translocation to the nucleus (below). The blue arrow indicates light illumination and activation and the black arrow its reversion when the stimulus is absent in the dark. B) ITC binding measurement for LANS wild-type with Zdk2 and C) LANS lit mimic (I539E) to Zdk2.
RESULTS AND DISCUSSION
While LANS preserves all of the residues from the AsLOV2 domain that interact with Zdk2, it also has additional residues at its C-terminus whose effect on this interaction have not been established. Therefore, we first tested whether LANS would preferentially bind to Zdk2 in the dark by using isothermal titration calorimetry (ITC) to measure the binding affinity between LANS and Zdk2. Measurements were made with wild-type LANS (without an NES) and a mutant form (I539E) that mimics the lit state of the switch[8]. Binding with wild type LANS represents dark state binding as the ITC experiments are performed in an enclosed cell without any light source. Wild type LANS bound to Zdk2 with an affinity of 3.9 nM, while the lit-state mimic bound 36-fold weaker with an affinity of 138 nM (Figure 1B–C). The change in binding affinity may be even larger with actual blue-light stimulation, as there are contacts between Zdk2 and LANS that are unlikely to be directly impacted by the I539E mutation but may be disrupted by light activation. Nevertheless, these results indicated that Zdk2 binds tighter to the dark state of LANS and that this may be useful for cytoplasmic sequestering of LANS in the dark.
We performed two sets of microscopy experiments to characterize the behavior of the switches in living cells. Spinning disk confocal microscopy was performed in HeLa cells with LANS4 and its lit mimetic (I539E) fused to mCherry fluorescent protein. Nuclear/cytoplasmic levels of LANS were measured with and without cotransfection of a TOM20-Zdk2 fusion. TOM20 anchors Zdk2 at the cytoplasmic face of the mitochondrial membrane. While the dark state nuclear/cytoplasmic fluorescence intensity for the LANS4 switch alone was 0.45[3], when cotransfected with TOM20-Zdk2 it was 0.21. The lit state nuclear/cytoplasmic fluorescence was similar with (2.7) and without (2.8) the coexpression of TOM20-Zdk2. Thus, an enhanced dark/light dynamic range of 12.5 was observed via confocal microscopy for LANSTRAP (LANS4 coexpressed with TOM20-Zdk2) compared to 6.2 for LANS alone (Fig. 2B). The latter is due to a decrease of the dark state nuclear/cytoplasmic fluorescence of about 2-fold for LANSTRAP (p<0.0001, unpaired two-tailed t-student test).
Figure 2.
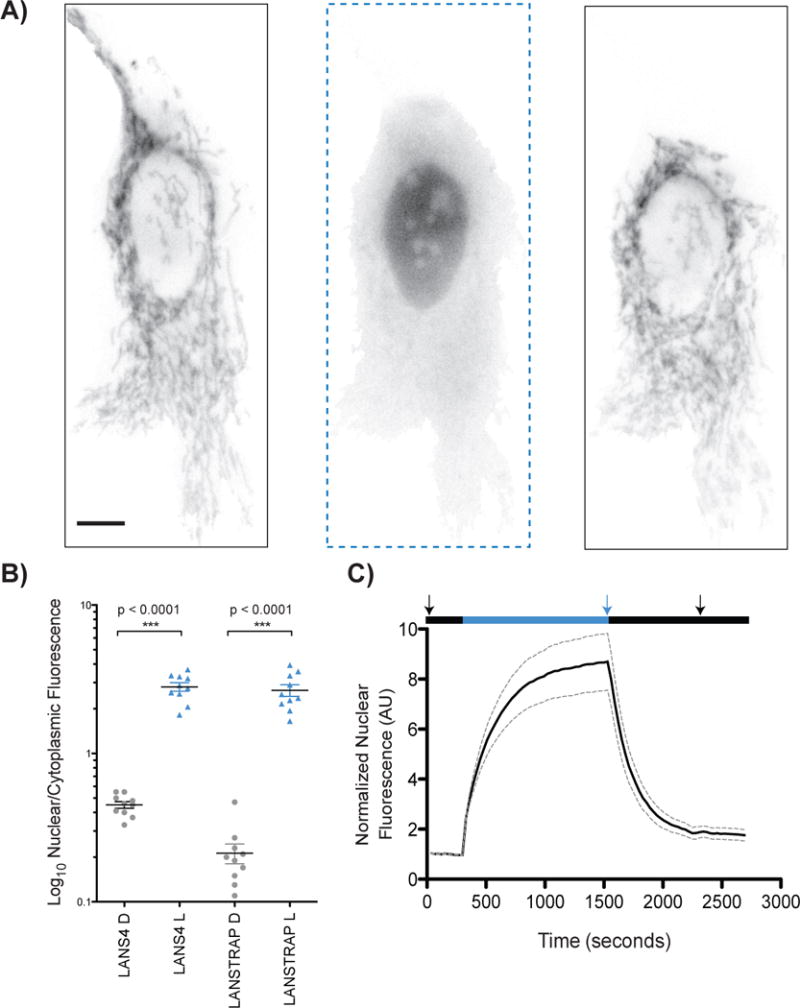
Microscopy characterization of LANSTRAP in HeLa tissue culture cells. A) Snapshots of real time activation and reversion of LANSTRAP with epifluorescence microscopy with starting dark state (left), full activation (central) and reversion (right) image. The scale bar represents 10 μm and applies to all three images. The arrows in C) correspond to the time points of these images. B) Confocal microscopy quantification of LANS4[3] and LANSTRAP. All the imaging was performed in the dark with “D” corresponding to wild-type LANS4 switches and the “L” to the LANS4 (I539E) lit mimetic. Ten cells for each state were used for the quantification. Statistical significance was measured with unpaired two-tailed student’s t-test; C) Quantification of real-time light activated nuclear import and reversion using epifluorescence microscopy (n = 10, mean reported ± SEM with dashed line). The black (dark) and blue (intermittent blue light) indicates the condition applied during the time course.
We also used epifluorescence microscopy to monitor photoinduced nuclear localization in real time with cells cotransfected with the TOM20-Zdk2 fusion. Upon blue light illumination, LANS4 released from the mitochondria and entered the nucleus. Switching the light off resulted in the protein returning to the cytoplasm and the mitochondrial membrane (Supplemental Movie 1). Nuclear fluorescence was quantified over 10 different cells. The normalized fluorescence intensity peaked at about 8.4 ± 0.54 fold higher than the starting fluorescence, and we observed a t1/2 = 2.9 ± 1.5 minutes for import and t1/2 = 2.5 ± 0.5 minutes for export (Fig. 2A and 2C). In experiments without co-expression of TOM20-Zdk2, the change in nuclear fluorescence is 2.8 ± 0.6 fold with time constants for import and export similar to those observed with Zdk2: t1/2 = 3.3 ± 0.02 minutes for import and t1/2 = 2.5 ± 0.01 minutes for export[3].
In order to test if the improved dynamic range observed in the microscopy experiments translates to tighter control over function, we used LANSTRAP to control gene transcription in yeast. For this experiment, we fused LANS4 to the LexA DNA binding domain and the Gal4 activation domain, and used a yeast strain with the β-Galactosidase reporter gene downstream of the LexA binding site. Previously, when using LANS4 alone, we had observed that we needed to also add a larger protein, MBP, as a bulky add-on to the LANS4 construct (total protein size = 98 kDa) to mitigate passive diffusion through the nuclear pore. For experiments performed here with LANS4 and LANSTRAP, we used a smaller fusion partner, the fluorescent protein Venus (total protein size = 84 kDa), as we expected that cytoplasmic anchoring with Zdk2 should counteract leakiness due to passive diffusion. With these constructs, using LANS4 alone does not provide tight control of β-Galactosidase expression. We only observed a 1.8-fold increase in expression under blue light stimulation and relatively high transcription levels were observed in the dark (Fig. 3B). To test if LANSTRAP could improve dynamic range, we generated three yeast strains using the reporter strain (NMY51) as the parent. We fused Zdk2 to the C-terminus of Vma21qq, a small protein localized to the vacuole and modulated the expression of Vma21qq-Zdk2 using three variants of the TEF1 constitutive promoter (Fig. 3A). The three promoter variants used here, Pm3, Pm7 and Pm2, exhibit decreasing levels of expression for their downstream ORF[9]. All three of the strains expressing Vma21qq-Zdk2 fusion exhibited improved light/dark photoswitching dynamic range in the β-Galactosidase assays. While the lit state expression levels of β-Galactosidase were not perturbed, dark state transcription decreased in response to increasing Vma21qq-Zdk2 expression level. With Pm2 a 4.6-fold increase in expression was observed with blue light stimulation, with Pm7 a 35-fold change, and with Pm3 a 55-fold light/dark transcriptional response (Fig. 3B). Notably, the dark state transcription levels with Pm3 decreased to 14.8 Miller Units, which constitutes a 37-fold decrease compared to the dark state transcription in the absence of the cytoplasmic anchor. In comparison, the original transcription factor alone (LexA-MBP-Gal4AD-LANS4), in the absence of Zdk2 coexpression, exhibited a 21-fold dynamic range between the dark and light level of β-galactosidase expression levels.
Figure 3.
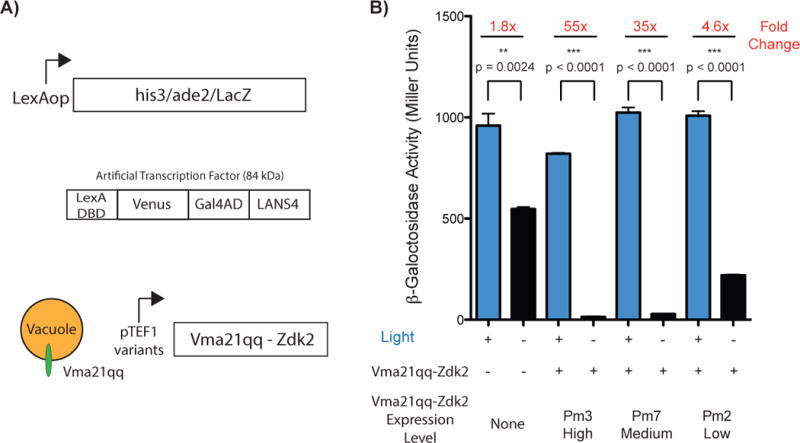
Control of transcription in yeast using LANSTRAP. A) schematic of the NMY51 reporter genes (above), LexA artificial transcription factor used in this experiments (center), Vma21qq-Zdk2 construct schematic and illustration (below); B) Assessment of transcriptional control via LANSTRAP by β-galactosidase assays in NMY51 with and without Zdk2 coexpression.
Finally, we investigated the applicability of LANSTRAP to influence C. elegans vulval development via light-mediated control of the LIN-1/ETS transcription factor. The C. elegans vulva develops from a group of six precursor cells, all of which are competent to initiate vulval development[10]. LIN-1/ETS is expressed in all six precursor cells and acts to repress the primary vulval cell fate[11]. In wild-type animals, an EGF signal from a neighboring cell, called the anchor cell, triggers MAPK-mediated phosphorylation and inactivation of LIN-1 in one of the vulval precursor cells[12]. As a result, that cell can adopt the primary vulval fate and initiate vulval development (Fig. 4A). LIN-1 remains active in the other vulval precursor cells, which do not initiate vulval development. Since LIN-1 is a repressor of the vulval fate, loss of LIN-1 activity leads to a Multivulval phenotype in which more than one vulval precursor cell initiates vulval development[11]. Conversely, gain-of-function mutations in LIN-1 result in a Vulvaless phenotype[12].
Figure 4.
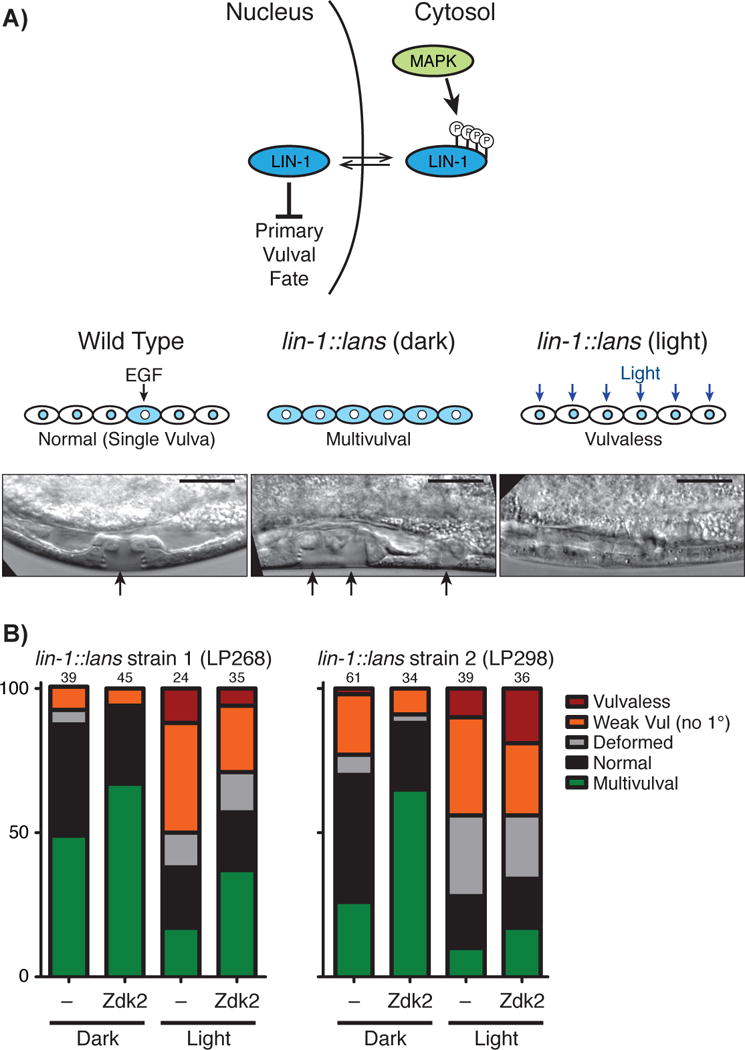
Control of vulval development in C. elegans using LANSTRAP. A) LIN-1 role in primary vulval fate determination (above), schematic of the effect on phenotype of wild type LIN-1 and LIN-1 constitutively active form controlled by LANS (central); B) Quantification of phenotypes in the indicated strains and conditions. Numbers at the top of each bar indicate the total number of animals scored in this experiment.
In previous work, we used CRISPR/Cas9-triggered homologous recombination to re-engineer the lin-1 gene, replacing the normal EGF-mediated regulation of LIN-1 with LANSbased light-dependent activity[3]. lin-1∷lans animals grown in the dark exhibited a Multivulval phenotype, consistent with LIN-1 sequestration in the cytoplasm, while lin-1∷lans animals grown in the light exhibited Vulvaless phenotypes, suggesting constitutive nuclear localization and activity of LIN-1. Although these data showed that LANS could influence developmental signaling via control of an endogenous transcription factor, the Multivulval phenotype observed for animals raised in the dark was mild compared to complete loss of lin-1 function, suggesting that LIN-1∷LANS retained some ability to enter the nucleus and activate transcription even in the dark. We therefore asked whether adding the Zdk2 anchor to this system would enhance light-mediated control of development.
To test the activity of Zdk2 in C. elegans development, we generated extrachromosomal arrays expressing Zdk2 in vulval precursor cells. Extrachromosomal arrays are semi-stable in C. elegans, so that within a population of animals, some carried the array and some did not. By following the presence of the array with a fluorescent marker, we were able to score vulval development with or without the Zdk2 anchor in worms raised under identical conditions on the same plate. We performed this experiment in parallel in two independently constructed lin-1∷lans strains. Consistent with our previous findings[3], animals lacking the array exhibited a low penetrance (<50%) of Multivulval phenotypes in the dark, while light activation suppressed the Multivulval phenotype and induced a Vulvaless phenotype (Fig. 4B). Expression of the Zdk2 anchor increased the penetrance of Multivulval phenotypes in the dark in both strains, consistent with our prediction that Zdk2 would sequester LIN-1∷LANS in the cytoplasm and reduce its activity in the dark. However, even in the presence of Zdk2, the Multivulval phenotype was weaker in both severity and penetrance than that produced by a lin-1 null allele[11], suggesting that LIN-1∷LANS retains some signaling activity even under these conditions. We next examined whether expression of the Zdk2 anchor still allowed for activation of LIN-1∷LANS by blue light. Zdk2 expression did not prevent light induction of Vulvaless phenotypes (Fig. 4B), suggesting that the anchor could be released upon light activation as expected. However, we did observe a higher penetrance of “residual” Multivulval phenotypes in light-stimulated Zdk2-expressing animals, suggesting that Zdk2 might have inhibited light activation of LIN-1∷LANS to some extent. This might be partially a consequence of expressing Zdk2 from an extrachromosomal array, since transgenes generated in this manner typically show high levels of overexpression. In summary, these results suggest that expression of Zdk2 improves the caging of LANS while still allowing for light-stimulated nuclear import in vivo.
CONCLUSION
Light mediated nuclear import and export is a generalizable approach for controlling a variety of biological functions[13]. We and others have used it to control transcription factors as well as enzymes that modify the epigenome[3, 14]. In these studies, it has been important to tune the relative strength of the NLSs and NESs embedded in the switches to maximize dynamic range, and it has been challenging to minimize dark state activity. Here, we have shown that by adding an additional mode of regulation to the pathway it is possible to lower dark state activity and increase dynamic range. Our approach is inspired by the multiple mechanisms used to regulate NF-κB activity, and should be applicable to other light-activatable switches. Redchuk et al have also recently proposed using a similar mode of control by multiplexing BphP1-Q-PAS1 with LANS, thus using near-infrared and blue light as stimuli[15]. The main distinction between our approach and that of Redchuk et al is the number of stimuli required. For instance, a we have also developed LINX — a variant of the AsLOV2 domain that cages an NES in its Jα-helix and transports from the nucleus to the cytoplasm with the application of light[16]. In this case, anchoring Zdk2 in the nucleus should provide a way to more tightly hold LINX in the nucleus in the dark.
The LANSTRAP system is compatible with a variety of organisms and is fully genetically encoded. In previous work, we have shown that lasers can be used to activate LANS in specific cells in the C. elegans embryo[3]. LANSTRAP will allow even tighter control of transcription factors or the epigenome in these types of studies, and should provide a valuable approach for studying animal development.
MATERIALS AND METHODS
DNA Cloning
All cloning PCR amplifications were performed using high-fidelity Q5 polymerase and all preliminary construct screens were carried out by colony PCR using Taq polymerase. All enzymes were purchased from New England Biolabs (NEB). All plasmids were verified by DNA sequencing.
Light Illumination Setup
The illumination setup for the fluorescence polarization assays was as described in Lungu et al, 2012[5]. Briefly, collimated blue LED with maximum emission at 455 nm (Thorlabs) provided 6 mW/cm2 illumination to a sample in a 1 cm quartz cuvette.
For the S. cerevisae and C. elegans experiments, an LED strip with maximum emission at 465 nm (Mouser Electronics, cat. #: 901-SB-0465-CT) was placed in an array 25 × 35 cm, 15 cm above the samples in an incubator set at 30 °C for yeast and 20 °C for C. elegans, thus obtaining even illumination of 1 mW/cm2.
Isothermal Titration Calorimetry
Zdk2 was cloned in pQE-80L vectors via restriction digest with BamHI and HindIII. All purifications were performed as in Yumerefendi et al, 2015[3]. Purified proteins were dialysed together against 5 L of 20 mM Na2HPO4/NaH2PO4 pH 7, 100 mM NaCl and 1 mM DTT overnight at 4°C and automatic titrations performed with MicroCal Auto-iTC200 (GE Healthcare). Each titration consisted of 20 injections, where 30 μM of Zdk2 was in the cell and 250 μM of LANS wt and lit mimetic (I539E) in the syringe. Measurements were taken at room temperature, 26 °C. The baseline of each titration was determined and subtracted from all data points. Heat change titration curve was fitted to a one-site binding model using Origin software (OriginLab).
Mammalian Cell Culture
HeLa and HEK293T (ATCC) tissue cultures were grown at 37 °C, 10 % CO2 in DMEM supplemented with 10 % (v/v) fetal bovine serum (FBS) and passaged every 2–3 days usually in Nunc T-75 culture flasks (Thermo Scientific).
Mammalian cell imaging
The constructs used in this work were produced and reported in Yumerefendi et al, 2015 and Wang et al, 2016[3,7]. Coverslips were washed with PBS (GIBCO) and coated with 10 μg/ml fibronectin at room temperature for a minimum of 1.5 hours. Cells were seeded for 3 hours to overnight, then transfected using FuGENE 6 (Promega) and imaged after about 18 hours post-transfection in Ham’s F-12K medium free of Phenol red (Caisson) and containing 10 % FBS buffered with 10 mM HEPES pH 8. Coverslips were mounted in an Attofluor live cell chamber (Invitrogen) placed in a microscope stage with a heated stage adaptor (Warner) and an objective temperature controller (Bioptechs).
An Olympus DSU-IX81 Spinning Disk Confocal coupled with Andor solid-state lasers (Andor) was used to perform the confocal microscopy quantification. Z-stacks of 12 μm at 0.5 μm steps were acquired with a PlanApo 60× objective (Oil, N.A. 1.42) using a 561 nm laser set at 20% intensity (150 EM gain and 300 ms exposure). Two confocal optical slices 0.5 μm each sectioning through the nucleus were combined and mCherry fluorescence was quantified for 10 cells of each condition.
Live cell timelapse series were collected with an Olympus IX81 epifluorescence microscope equipped with a ZDC focus drift compensator and a Photometrics CoolSnap ES2 CCD camera (Roper Photometrics). A UPlanFLN 40× objective (Oil, N.A. 1.30) was used with an ET572/35× filter for mCherry detection and 1 % (UVND 2.0, ET430/24×) for blue light activation of LANS.
Yeast transcription
The transcription factor used in this study is derived from the pNIA-CEN-MBP plasmid constructed in Yumerefendi et al, 2015[3] substituting the MBP gene for that of Venus fluorescent protein using restriction digest cloning. The Zdk2 was cloned in yeast vectors designed to integrate at the HO gene locus. After sequence verification of the generated plasmids they were digested with Notl and the product of the reaction was transformed with high efficiency lithium acetate transformation into the NMY51 strain.
β-Galactosidase assay were performed as follows: Fresh colonies were grown overnight at 30 °C in 5 ml SC-Leucine. On the next day, the cell density was measured at OD600 and 2 mL cultures were diluted to OD600 = 0.2 in duplicates - one for a light and another for a dark condition (falcon tubes were wrapped in aluminium foil). Cultures were grown at 30 °C in a shaking incubator (250 rpm) untill they reached OD600 ± 0.8 in the presence or absence of blue light (465 nm) at 500 μW/cm2 via LED strip light wrapped around the tube rack. The resulting cultures were pelleted in triplicates and the β-Galactosidase assay using CPRG for a substrate was performed according to the manufacturer’s instructions (Clontech).
C. elegans culture and strain construction
The two independent lin-1∷lans strains used here were LP268: lin-1(cp70[lin- 1(ACT)∷mkate2∷lans1 + LoxP HygR unc-58(e665) LoxP]) IV[3] and LP298: lin-1(cp72[lin- 1(ΔCT)∷mkate2∷lans1 + LoxP HygR unc-58(e665) LoxP]) IV. LP268 and LP298 are two independent isolates of molecularly identical lin-1∷lans alleles. These strains were maintained at 20 °C on NGM medium, using E. coli strain OP50 as a food source. To express Zdk2 in vulval precursor cells, the Zdk2 sequence was codon-optimized for C. elegans expression[17] and inserted into the vector pB255[18], which contains the lin-31 promoter for expression in vulval precursor cells. To generate extrachromosomal arrays, the resulting construct was injected into young adults of strain LP268 or LP298 at a concentration of 50 ng/μL. 5 ng/μL of pCFJ104 (Pmyo-3∷mCherry;[19]) was included in the injection mix as a marker for extrachromosomal arrays.
Scoring of vulval phenotypes
Scoring of vulval phenotypes was performed exactly as previously described[3]. Briefly, animals were synchronized by bleaching and allowed to develop under blue LED illumination (see above). Dark controls were placed in the same incubator but wrapped in aluminium foil to prevent light exposure. Mid-L4 animals were imaged using DIC (for scoring of phenotypes) and mCherry fluorescence (to determine which animals carried the extrachromosomal array). To avoid bias, samples were blinded before imaging. After image acquisition, we first scored each image for phenotype without viewing the mCherry channel; then, we examined the mCherry channel to determine whether each animal was carrying the array. Finally, after scoring was complete, strain background and dark/light condition were revealed. Detailed descriptions of phenotypic categories are given in Yumerefendi et al[3]. Data were plotted using GraphPad Prism software.
Supplementary Material
Supplemental Movie S1. Timelapse of the activation and reversion of LANSTRAP in a HeLa cell where Zdk2 is anchored at the mitochondria via a fusion to TOM20. The scale bar represents 10 μm and the blue dot represents the period of time under blue light stimulation. The elapsed time is shown in min:sec format at the upper right corner.
Acknowledgments
We thank David Reiner for advice and for the pB255 plasmid. This work was supported by a Howard Hughes postdoctoral fellowship from the Helen Hay Whitney Foundation and NIH K99 GM115964 (D.J.D.); and by NIH R01 GM083071 (B.G.), NIH DA036877 (K.M.H) and DOD W911NF-15-1-0631/A16-0438-001 (K.M.H).
ABBREVIATIONS
- LOV2
Light Oxygen Voltage 2
- LANSTRAP
LANS coupled with Zdk2
- LANS
Light Activated Nuclear Shuttle
- iLID
improved Light Inducible Dimer
References
- 1.Napetschnig J, Wu H. Annu Rev Biophys. 2013;42:443–468. doi: 10.1146/annurev-biophys-083012-130338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toettcher JE, Voigt CA, Weiner OD, Lim WA. Nat Methods. 2011;8:35–38. doi: 10.1038/nmeth.f.326. [DOI] [PMC free article] [PubMed] [Google Scholar]; Toettcher JE, Weiner OD, Lim WA. Cell. 2013;155:1422–1434. doi: 10.1016/j.cell.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM. Nature. 2009;461:104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]; Levskaya A, Weiner OD, Lim WA, Voigt CA. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Nat Methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yumerefendi H, Dickinson DJ, Wang H, Zimmerman SP, Bear JE, Goldstein B, Hahn K, Kuhlman B. PLoS One. 2015;10:e0128443. doi: 10.1371/journal.pone.0128443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harper SM, Neil LC, Gardner KH. Science. 2003;301:1541–1544. doi: 10.1126/science.1086810. [DOI] [PubMed] [Google Scholar]
- 5.Lungu OI, Hallett RA, Choi EJ, Aiken MJ, Hahn KM, Kuhlman B. Chem Biol. 2012;19:507–517. doi: 10.1016/j.chembiol.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strickland D, Lin Y, Wagner E, Hope CM, Zayner J, Antoniou C, Sosnick TR, Weiss EL, Glotzer M. Nat Methods. 2012;9:379–384. doi: 10.1038/nmeth.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zimmerman SP, Kuhlman B, Yumerefendi H. Methods Enzymol. 2016;580:169–190. doi: 10.1016/bs.mie.2016.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yi JJ, Wang H, Vilela M, Danuser G, Hahn KM. ACS Synth Biol. 2014 doi: 10.1021/sb5001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Vilela M, Winkler A, Tarnawski M, Schlichting I, Yumerefendi H, Kuhlman B, Liu R, Danuser G, Hahn KM. Nat Methods. 2016;13:755–758. doi: 10.1038/nmeth.3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harper SM, Christie JM, Gardner KH. Biochemistry. 2004;43:16184–16192. doi: 10.1021/bi048092i. [DOI] [PubMed] [Google Scholar]
- 9.Nevoigt E, Kohnke J, Fischer CR, Alper H, Stahl U, Stephanopoulos G. Appl Environ Microbiol. 2006;72:5266–5273. doi: 10.1128/AEM.00530-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hill KJ, Stevens TH. Mol Biol Cell. 1994;5:1039–1050. doi: 10.1091/mbc.5.9.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sternberg PW. WormBook. 2005:1–28. doi: 10.1895/wormbook.1.6.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferguson EL, Horvitz HR. Genetics. 1985;110:17–72. doi: 10.1093/genetics/110.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ferguson EL, Sternberg PW, Horvitz HR. Nature. 1987;326:259–267. doi: 10.1038/326259a0. [DOI] [PubMed] [Google Scholar]; Sulston JE, Horvitz HR. Dev Biol. 1981;82:41–55. doi: 10.1016/0012-1606(81)90427-9. [DOI] [PubMed] [Google Scholar]
- 12.Jacobs D, Beitel GJ, Clark SG, Horvitz HR, Kornfeld K. Genetics. 1998;149:1809–1822. doi: 10.1093/genetics/149.4.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Ventura B, Kuhlman B. Curr Opin Chem Biol. 2016;34:62–71. doi: 10.1016/j.cbpa.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yumerefendi H, Lerner AM, Zimmerman SP, Hahn K, Bear JE, Strahl BD, Kuhlman B. Nat Chem Biol. 2016 doi: 10.1038/nchembio.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]; Niopek D, Benzinger D, Roensch J, Draebing T, Wehler P, Eils R, Di Ventura B. Nat Commun. 2014;5:4404. doi: 10.1038/ncomms5404. [DOI] [PMC free article] [PubMed] [Google Scholar]; Niopek D, Wehler P, Roensch J, Eils R, Di Ventura B. Nat Commun. 2016;7:10624. doi: 10.1038/ncomms10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Redchuk TA, Omelina ES, Chernov KG, Verkhusha VV. Nat Chem Biol. 2017;13:633–639. doi: 10.1038/nchembio.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yumerefendi H, Lerner AM, Zimmerman SP, Hahn K, Bear JE, Strahl BD, Kuhlman B. Nat Chem Biol. 2016;12:399–401. doi: 10.1038/nchembio.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Redemann S, Schloissnig S, Ernst S, Pozniakowsky A, Ayloo S, Hyman AA, Bringmann H. Nat Methods. 2011;8:250–252. doi: 10.1038/nmeth.1565. [DOI] [PubMed] [Google Scholar]
- 18.Tan PB, Lackner MR, Kim SK. Cell. 1998;93:569–580. doi: 10.1016/s0092-8674(00)81186-1. [DOI] [PubMed] [Google Scholar]
- 19.Frakjær-Jensen C, Davis MW, Ailion M, Jorgensen EM. Nat Methods. 2012;9:117–118. doi: 10.1038/nmeth.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie S1. Timelapse of the activation and reversion of LANSTRAP in a HeLa cell where Zdk2 is anchored at the mitochondria via a fusion to TOM20. The scale bar represents 10 μm and the blue dot represents the period of time under blue light stimulation. The elapsed time is shown in min:sec format at the upper right corner.