

Abstract
The plasma level of NOx, i.e., the sum of NO2− and NO3−, is frequently used to assess NO bioavailability in vivo. However, little is known about the kinetics of NO conversion to these metabolites under physiological conditions. Moreover, plasma nitrite recently has been proposed to represent a delivery source for intravascular NO. We therefore sought to investigate in humans whether changes in NOx concentration are a reliable marker for endothelial NO production and whether physiological concentrations of nitrite are vasoactive. NO2− and NO3− concentrations were measured in blood sampled from the antecubital vein and brachial artery of 24 healthy volunteers. No significant arterial-venous gradient was observed for either NO2− or NO3−. Endothelial NO synthase (eNOS) stimulation with acetylcholine (1–10 μg/min) dose-dependently augmented venous NO2− levels by maximally 71%. This effect was paralleled by an almost 4-fold increase in forearm blood flow (FBF), whereas an equieffective dose of papaverine produced no change in venous NO2−. Intraarterial infusion of NO2− had no effect on FBF. NOS inhibition (NG-monomethyl-l-arginine; 4–12 μmol/min) dose-dependently reduced basal NO2− and FBF and blunted acetylcholine-induced vasodilation and NO release by more than 80% and 90%, respectively. In contrast, venous NO3− and total NOx remained unchanged as did systemic arterial NO2− and NO3− levels during all these interventions. FBF and NO release showed a positive association (r = 0.85; P < 0.001). These results contradict the current paradigm that plasma NO3− and/or total NOx are generally useful markers of endogenous NO production and demonstrate that only NO2− reflects acute changes in regional eNOS activity. Our results further demonstrate that physiological levels of nitrite are vasodilator-inactive.
Keywords: endothelium‖blood flow‖red blood cells‖endothelial dysfunction
Previous experimental studies have demonstrated that the endothelium plays a crucial role in the maintenance of vascular homeostasis (1–4), including control of thrombosis, the interaction of platelets and leukocytes with the vessel wall, and regulation of vascular tone and growth. Among the various mediators released by the endothelium, nitric oxide (NO) is of major importance (5). NO is synthesized from the amino acid l-arginine by the constitutive calcium- and calmodulin-dependent endothelial isoform of endothelial NO synthase (eNOS; ref. 6). A disturbance in either production or availability of NO is thought to be responsible for the functional alterations that are associated with endothelial dysfunction and plays a key role in the development of atherosclerotic lesions. Because endothelial dysfunction is a crucial step in the inflammatory cascade leading to atherosclerosis (7), which precedes overt clinical manifestations, the assessment of an altered NO availability is of potentially important diagnostic and prognostic significance. Identification of such alterations may help targeting asymptomatic individuals who are at risk for cardiovascular diseases and would likely benefit from preventive measures. Traditionally, a pathological vasoconstriction in response to the administration of acetylcholine (ACh) in a regional vascular bed is taken as an indication for the impairment of endothelial NO production in the human circulation (8). The specificity of this test has been improved by simultaneous infusion of a NOS inhibitor, which unmasks the eNOS-independent part of the ACh response. Although the ACh test has received substantial critique because of its indirect nature, direct measurements of regional endothelial NO production in humans are lacking.
The rapid metabolism and short half-life of NO poses a considerable obstacle for its analytical assessment in humans. Although NO can be sensitively determined with different sophisticated assays (9–12), most of these techniques are either not suitable for the in vivo setting with complex matrices, such as blood, or require sophisticated techniques neither available nor applicable in the clinical routine (11). This is the reason why still to date eNOS activity is commonly assessed by determining the plasma concentration of nitrate or that of total NOx, i.e., the sum of nitrite and nitrate (6). The rationale for this approach is based on the experimental findings that NO is converted to nitrite and nitrate when inhaled or added to blood, and that nitrite is further oxidized to nitrate by the hemoglobin (Hb) contained in red blood cells (RBC; ref. 12). The reliability of this approach, however, requires critical reassessment because plasma nitrate levels are known to be influenced by a variety of NOS-independent factors (10, 12). Furthermore, the high background concentration of nitrate and its relatively long half-life in comparison to nitrite raises questions as to the sensitivity of NOx for detection of eNOS activity. Recently, two independent groups reported an arterial-venous gradient of nitrite (13) and total NOx (14) in humans, and the authors of one of these articles suggested that nitrite may have bioactivity in its own right.
In the present study, we sought to critically evaluate the diagnostic accuracy of nitrite and nitrate measurements for the assessment of regional NOS activity in vivo. We specifically intended to test the hypothesis that nitrate and total NOx accurately reflect acute changes in eNOS activity in humans. A second objective of our study was to test the hypothesis that circulating nitrite plays a role in the regulation of vascular tone. To achieve these goals, forearm arterial and venous plasma levels of nitrite and nitrate were measured at rest, during regional inhibition and stimulation of NO synthesis, and during application of nitrite. Blood flow was measured at the same time and related to the different N-oxide concentrations determined in arterial and venous blood. Our results demonstrate that nitrite, although vasodilator-inactive at physiological concentration, is the only N-oxide that accurately reflects acute changes in regional NOS activity. These results challenge the current paradigm on the metabolism of NO in human blood.
Methods
Chemicals and Solutions.
NG-monomethyl-l-arginine (l-NMMA) and ACh were from Clinalfa (Schwalbach, Germany). Sodium chloride, sodium nitrite, and EDTA were purchased from Merck. All other chemicals were from Sigma–Aldrich (Taufkirchen, Germany). All solutions were prepared fresh in saline and kept on ice in the dark until use.
Characteristics of the Study Population.
The final study sample consisted of 24 healthy volunteers screened by clinical history, physical examination, and routine chemical analysis. None of these subjects was on a regular medication, was a smoker, or revealed present or past evidence of cardiovascular diseases known to affect endothelial function such as hypertension, hypercholesterolemia, chronic heart failure, or diabetes mellitus. Further laboratory tests also excluded elevated levels of C-reactive protein (CRP). Participants in the study were instructed to refrain from drinking alcohol and caffeine-containing beverages at least 12 h before investigation. The study protocol was approved by the ethics committee of the Heinrich-Heine-Universität, and all subjects gave written informed consent before participating in the study.
Forearm Blood Flow (FBF) Measurements.
FBF was measured simultaneously in both arms by a mercury in rubber strain-gauge plethysmography technique described in detail elsewhere (15) and expressed in ml/min/100 ml of tissue. For each determination, five FBF measurements were performed, and the results were averaged. Resting FBF was additionally determined as described (16) and amounted to 200 ± 15 ml/min. A 2F catheter was inserted into the brachial artery of the nondominant arm for local drug infusion and continuous measurement of intraarterial blood pressure. A 4F catheter was placed into the antecubital vein for blood sampling.
Measurement of Plasma Nitrite and Nitrate.
Nitrite was measured by using a flow injection analysis technique that is based on the Griess reaction and has been described in detail elsewhere (17). Blood sampled from either the brachial artery or the deep antecubital vein was immediately mixed with a precooled solution comprised of NaCl (0.9%) and citrate (3.8%). After centrifugation (10,000 × g) for 5 min at 4°C, the supernatant was subjected to ultrafiltration (cut-off 10 kDa; Centrisart, Sartorius, Göttingen, Germany). The resulting ultrafiltrate was kept on ice for up to 30–60 min until analysis. Plasma nitrate was determined by using HPLC with direct UV detection, essentially as described (18).
Protocol.
Investigations typically started at 8:00 a.m. and lasted for ≈3–4 h. Subjects were in a supine position in a quiet air-conditioned room maintained at a constant temperature of 21 ± 1°C. Twenty minutes after finishing the preparations the different infusion protocols, including saline controls, were performed in random order. Intraarterial infusion rates ranged from 0.4 to 4 ml/min. ACh was infused intraarterially at three different doses (1, 3, and 10 μg/min). After cessation of the infusion, FBF returned to baseline within ≈5 min. Subsequently, l-NMMA was infused at increasing doses (4, 8, and 12 μmol/min) to inhibit eNOS. Fifteen minutes after each dose of l-NMMA, a second dose-response curve for ACh was completed. d- and l-arginine were administered at a rate of 200 μmol/min for 15 min. Blood flow measurements and the drawing of arterial and venous blood samples for biochemical analysis of nitrite and nitrate were performed under basal conditions and during each intervention. The venous-arterial difference for nitrite across the forearm circulation was calculated by correction of venous nitrite concentrations for nitrite entering from the arterial side. NO release was estimated as the product of FBF (normalized to 100 ml of tissue) times the venous-arterial difference in nitrite concentration.
Statistical Analysis.
Data processing was performed with the different modules of the software package SPSS RELEASE 8.0 (SPSS, Chicago). Data are given as the means ± SEM, unless otherwise stated, and two-sided P values of <0.05 were considered significant. A two-way ANOVA for repeated measures with consecutive Bonferroni post hoc tests was used to test for differences from control in the dose-dependent changes in FBF, venous-arterial differences of nitrite and NO release during infusion of ACh, with and without simultaneous l-NMMA-infusion. Univariate linear regression (Spearman rank) tests were performed to evaluate the relationship between changes in FBF and NO release.
Results
The study population consisted of 16 men and 8 women, 30 ± 6 years of age, with a mean body length of 177 ± 8 cm and a body mass of 69 ± 8 kg. At the time of investigation, heart rate was determined to be 68 ± 7 min−1, and mean arterial blood pressure was 87 ± 11 mmHg. At the doses applied for local infusion, neither ACh nor l-NMMA exerted systemic vasoactive effects, as judged by the lack of effect on blood pressure, heart rate, and FBF in the control arm.
Plasma Nitrite/Nitrate Concentrations at Baseline.
Baseline levels of nitrite, measured at the same time in the brachial artery and in the antecubital vein, amounted to 322 ± 42 and 305 ± 43 nmol/liter (n = 24; range: 227–428 nmol/liter). No significant arterial-venous nitrite gradient was observed. In 16 of the 24 individuals investigated, basal arterial concentrations of nitrite were slightly higher than venous levels, whereas in the other 8 individuals nitrite levels were found to be higher in the antecubital vein than in the brachial artery (Fig. 1). Resting levels of arterial and venous plasma nitrate were not significantly different from each other and amounted to 25.3 ± 1.8 and 25.1 ± 1.9 μmol/liter, respectively.
Figure 1.

Nitrite concentrations at baseline in arterial and venous plasma samples of the 24 individuals enrolled in the study. Individual plasma level (○) and mean data (●) ± SEM are presented.
Effect of eNOS Inhibition on Plasma Nitrite/Nitrate and FBF.
In a subset of 8 volunteers, infusion of l-NMMA dose-dependently reduced FBF from 2.7 ± 0.3 down to 0.7 ± 0.3 ml/min/100 ml of tissue (P < 0.05) at the highest dose administered (Fig. 2). In parallel, venous plasma nitrite concentrations stepwise decreased from 321 ± 51 nmol/liter to a minimum of 131 ± 26 nmol/liter (P < 0.05) at 12 μmol/min l-NMMA. In contrast, venous plasma nitrate concentrations (23.5 ± 1.7 μmol/liter) remained virtually unchanged at each dose of NOS-inhibitor infused. Arterial nitrite and nitrate concentrations did not change significantly during l-NMMA infusion (data not shown), which is in agreement with the lack of systemic effects on local infusion of the NOS inhibitor. The effect of l-NMMA could be almost fully reversed by coinfusion of a 10-fold molar excess of l-arginine (n = 8).
Figure 2.

FBF and venous NO2−, NO3−, and NOx levels at baseline (C, control) and during increasing i.a. doses of l-NMMA (4, 8, and 12 μmol/min); means ± SEM; n = 8. The * indicates significant difference from control (P < 0.05). Note differences in the y-axis scale between NO2− and NO3− concentrations (nM vs. μM).
Plasma Nitrite/Nitrate and FBF Changes After Stimulation of eNOS.
Intraarterial infusion of ACh augmented FBF almost 4-fold and increased venous plasma nitrite from 322 ± 41 nmol/liter up to 550 ± 102 nmol/liter (P < 0.05; Fig. 3). As with eNOS inhibition, stimulation with ACh did not result in a significant change in venous plasma nitrate concentration. Furthermore, systemic arterial nitrite and nitrate levels remained unaffected during infusion of ACh (data not shown). Consequently, the venous-arterial difference for nitrite increased to 89 ± 40, 146 ± 34, and 245 ± 100 nmol/liter at dose levels of 1, 3, and 10 μg/min ACh, respectively. Calculated NO release during stepwise increases of intraarterial doses of ACh increased more than 10-fold to a maximum of 3,200 ± 800 pmol/min/100 ml of tissue (Fig. 4). During simultaneous infusion of l-NMMA, the ACh-induced increase in NO release was dose-dependently reduced. The level of NOS inhibition achieved was >90% (P < 0.01) at the highest dose administered as judged by comparison of net plasma nitrite increases in the absence and presence of l-NMMA. Neither inhibition of cyclooxygenase by acetylsalicylic acid (500 mg i.v.) nor of endothelium-derived hyperpolarizing factor production by ouabain (i.a. infusion of 5 μg/min) had any influence on the increase in plasma nitrite and FBF in response to ACh (n = 3 for each intervention). In 3 further experiments, l-arginine (200 μmol/min) induced a marked increase in plasma nitrite (165 ± 21%), which was accompanied by a parallel dilation of resistance arteries in the forearm circulation (61 ± 8%). This effect was stereospecific, because infusion of the same dose of d-arginine had no effect on either parameter (Δ NO2−: 1.8 ± 0.6%; Δ FBF: −1.3 ± 0.5%).
Figure 3.

FBF and venous NO2−, NO3−, and NOx levels at baseline (C, control) and during increasing i.a. doses of ACh (1, 3, and 10 μg/min); n = 24. The * indicates significant difference from control (P < 0.05). Note differences in the y-axis scale between NO2− and NO3− concentrations (nM vs. μM).
Figure 4.

Calculated NO release during infusion of different doses of ACh (1, 3, and 10 μg/min) with and without (C, control) simultaneous inhibition of eNOS (4, 8, and 12 μmol/min l-NMMA i.a.); n = 8. (*, P < 0.05; **, P < 0, 01).
Effects of Papaverine and Nitrite on Plasma Nitrite/Nitrate and FBF.
Additional control experiments with the endothelium-independent vasodilator papaverine demonstrated that increases in FBF are not necessarily accompanied by changes in plasma nitrite concentration. Local application of a 3 μmol bolus of papaverine directly into the brachial artery produced a 382 ± 21% increase in FBF, which was comparable in extent to that elicited by the highest dose of ACh. In contrast to the latter, however, plasma nitrite (−0.1 ± 0.5%) and nitrate (1.6 ± 1.2%) levels did not change (n = 3).
Intraarterial application of nitrite (NaNO2 in 0.9% saline) was found to be devoid of vasodilator activity at doses up to 36 μmol/min (tested range: 0.01–36 μmol/min; n = 3). Venous plasma nitrite concentrations achieved at the highest dose level exceeded 130 μM and were thus ≈200 times greater than the concentrations measured during maximal eNOS stimulation with ACh. Only minimal conversion of nitrite to nitrate was observed on passage through the forearm circulation when nitrite concentrations were kept within the physiologically relevant concentration range (200–1,000 nM), and 92–97% of the applied nitrite was typically recovered on the venous side (see Table 1). However, venous nitrate concentrations increased significantly with nitrite infusion rates >300 nmol/min (n = 2; data not shown). Nitrate (NaNO3 in 0.9% saline) was devoid of any vasodilator activity at doses up to 36 μmol/min (n = 2). These results rule out the possibility that physiological plasma concentrations of nitrite and nitrate exert regional vasodilator activity and further demonstrate that little nitrite is converted to nitrate during transit through the forearm vasculature.
Table 1.
Effect of intraarterial infusion of nitrite on venous plasma nitrite concentration and FBF in one representative individual (for details see Results)
| Nitrite infusion, nmol/min | Theoretical venous NO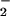 , ΔμM , ΔμM |
Measured NO , ΔμM , ΔμM |
Estimated recovery, % | ΔFBF, ml/min/100 ml of tissue |
|---|---|---|---|---|
| 10 | 0.05 | 0.046 | 92 | 0 |
| 30 | 0.15 | 0.145 | 97 | −0.1 |
| 100 | 0.50 | 0.469 | 94 | 0 |
Theoretical nitrite concentrations in venous blood were estimated based on a resting flow through the forearm circulation of ≈200 ml/min. Venous plasma nitrite concentrations were measured in samples drawn 1 min after start of nitrite infusion and were corrected for arterial nitrite inflow (basal arterial NO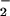 concentration: 262 nM).
concentration: 262 nM).
Relationship Between NO Release and Changes in FBF.
A plot of the venous-arterial difference of plasma nitrite and FBF data (n = 69) revealed a highly significant positive association with a correlation coefficient of r = 0.72 (r2 = 0.52; P > 0.001). The relationship between the ACh-induced NO release into the forearm circulation and the increase in FBF with and without l-NMMA coinfusion is depicted in Fig. 5. The respective correlation coefficient amounted to r = 0.85 (r2 = 0.73; P < 0.001), indicating that the difference in plasma nitrite concentration could explain, with 73% probability, the extent of FBF response to ACh. In contrast, no significant correlation was observed between venous nitrate or NOx levels and FBF (r = 0.10; P = 0.78). Furthermore, the increase in NO2− always closely preceded the increase in FBF (n = 4; Fig. 6), whereas nitrate concentrations remained unchanged.
Figure 5.

Relationship between NO release and FBF. Responses in individual subjects during increasing doses of ACh (1, 3, and 10 μg/min) with and without simultaneous inhibition of eNOS (4, 8, and 12 μmol/min l-NMMA i.a.). Experimental data from 8 individuals are depicted. Spearman rank correlation coefficient for single data points: r = 0.85, r2 = 0.73; P < 0.001.
Figure 6.

Comparison of the time course of ACh-induced changes in FBF and NO2− concentration in one representative individual. ACh was infused for 40 sec at a dose of 10 μg/min. *1 indicates the time of maximal NO2− concentration and *2 indicates the time of maximum FBF.
Discussion
The key findings of the present study are (i) plasma concentrations of nitrite sensitively reflect acute changes in regional eNOS activity; (ii) plasma nitrate and total NOx levels do not change during pharmacological modulation of the l-arginine:NO pathway; (iii) in healthy subjects, ACh-induced blood flow responses and plasma nitrite increases are almost entirely mediated by eNOS, and 12 μmol/min l-NMMA is required for complete NOS inhibition in the human forearm circulation; and (iv) physiological concentrations of plasma nitrite are vasodilator-inactive.
Critique of Methods.
The measurement of plasma nitrite in samples taken from the antecubital vein does not allow us to determine which contribution the arterial, capillary, and venous endothelial cells make to the overall signal. Immunohistological and functional studies, however, indicate that eNOS expression along the vascular tree decreases with decreasing vessel size. Major sites of eNOS, and presumably also of NO formation, seem to be the large and small conduit, resistance, and feeding arteries, whereas a considerably lower activity is present in the veins and a negligible portion in the capillaries (19, 20). Thus, the majority of nitrite detected in the venous forearm effluent is likely to originate from the arterial side.
Origin of Plasma Nitrite and Nitrate and NO Metabolism in Blood.
It has been shown in fasted healthy volunteers that the major source of plasma nitrite is the l-arginine:NO pathway (21). The plasma concentration of nitrite in a particular vascular bed depends on the overall rate of formation of NO and related N-oxides and their subsequent interaction with reactive oxygen species, thiols, and other biomolecules such as Hb (22–24). In addition, the plasma concentration is affected by the amount of nitrite entering from the arterial side and the rate of diffusion/uptake into either blood cells or tissue or the interstitial space. As a result of the complexity of NO metabolism in the different compartments, little is known about the kinetics and fate of endothelial NO in blood. In vitro studies revealed that in the presence of oxygen, NO is rapidly oxidized to nitrite, obeying pseudo-first-order kinetics with a strict 1:1 stoichiometry (25–27). Plasma nitrite is rapidly taken up by RBCs where it is oxidized in an Hb-dependent manner to nitrate, which is subsequently released into plasma (28). An alternative metabolic pathway includes direct interaction of NO with Hb in the erythrocyte. Depending on the oxygenation state of the hemeprotein, NO can either react with oxyHb to form metHb and nitrate (29) or with deoxyHb to form nitrosylHb (NOHb; refs. 13 and 24). The latter may interconvert, by reaction with the sulfhydryl group of the Cys93 of the β-globin chains, to form S-nitrosated Hb (SNO-Hb; ref. 24) or slowly degrade to nitrite (30). Given that the reaction of NO with Hb outcompetes that with molecular oxygen by at least one order of magnitude [second-order rate constants for the reaction of NO with oxyHb and deoxyHb are in the range of 3–5 × 107 M−1s−1 (29, 31), whereas the third-order rate constant for the NO autooxidation is 6.3 × 106 M−2s−1) (32)], our finding that plasma nitrite selectively increased during eNOS stimulation is surprising. Although NO autooxidation is accelerated in hydrophobic environments (33), it may still not be fast enough to account for this reaction. Because NO is predominantly formed in the larger vessels (19, 20) where 90–95% of Hb is in the oxygenated form, the NOHb/SNO-Hb chemistry is similarly unlikely to account for nitrite formation because ≈10 times more nitrate than nitrite should have been formed under these conditions. If the reaction of NO with oxyHb occurred to any significant extent in vivo, nitrate would be expected to be the major product of NO metabolism in blood and little or no nitrite should be formed. Our results clearly demonstrate that this was not the case. If one assumes that, according to the diffusion concept of NO, 50% goes to the vascular lumen and 50% to the abluminal space, one would expect equal amounts of nitrite and nitrate to be formed, which should translate into comparable changes in nitrite and nitrate concentration. Although small, a nitrate increase of ≈4% would have been analytically detectable. However, only changes in plasma nitrite were measured in the forearm circulation after eNOS stimulation and inhibition, respectively, suggesting that the NO:oxyHb reaction plays only a minor role for the inactivation of NO in vivo, and a sizeable portion of endothelium-derived NO undergoes metabolism via other routes. Alternatively, conversion of NO to nitrite may occur at the surface of endothelial cells via a yet unknown pathway, the rate of which must be well above that of the NO:oxyHb reaction. One such pathway might be represented by the interaction with the peroxidase/H2O2 system. Recent in vitro studies suggested that mammalian peroxidases may serve as a catalytic sink for NO (34). Interestingly, one presumed intermediate formed, nitrosium cation (NO+), is known to be rapidly hydrolyzed in aqueous solution to form nitrite. It is also possible that the conversion of NO to nitrite is a result of the breakdown of circulating S-nitrosothiols or enzymatically catalyzed by vascular cells, although such an activity has not yet been described. At present, there is no way to distinguish which fraction of plasma nitrite is derived from NO directly and which from NO-related adducts and metabolites.
In contrast to nitrite, we demonstrate here that plasma nitrate levels do not change and thus do not correlate with FBF—neither at rest nor during inhibition or stimulation of NOS. This finding does not come as a complete surprise as this parameter is known to be influenced by a variety of NOS-independent factors, including dietary nitrate intake, saliva formation, bacterial nitrate synthesis within the bowels, denitrifying liver enzymes, inhalation of atmospheric gaseous nitrogen oxides, and renal function (12, 35). Furthermore, with 5–8 h its half-life in humans (35) is considerably longer than that of nitrite. Hence, although frequently used, neither nitrate nor NOx are reliable markers of acute changes in regional eNOS activity. Our results do not rule out the possibility that some nitrate was formed from NO inside RBCs. Additional analysis of changes in nitrite/nitrate concentration in this cellular compartment was beyond the scope of the present study. It is conceivable, however, that the exchange of nitrate across the erythrocyte membrane is a rather slow process which exceeds the average transit time of blood through the human forearm circulation (≈4 s at rest). Moreover, the total amounts of nitrate formed via this pathway are relatively small and therefore might not be detectable against the high background in plasma (compare nM levels of NO2− to μM levels of NO3− in Figs. 2 and 3).
To the best of our knowledge, no experimental data are available on the half-life of NO in whole blood. From in vitro data it has been estimated to amount to 1.8 ms (36). However, this calculation is based on the assumption that the reaction with oxyHb is the major determinant of NO metabolism in vivo, which may be incorrect (see above). Furthermore, these calculations did not take into account that—presumably because of the formation of an RBC-free zone near the vessel wall—intravascular flow decreases erythrocyte consumption of NO by about 3 orders of magnitude (37, 38). Our finding that nitrite levels drop dramatically after regional NOS inhibition suggests that the half-lives of both NO and nitrite in the human vasculature are indeed very short. The >50% reduction of resting nitrite levels seen at the highest dose of l-NMMA used further indicates that more than half of the nitrite measured in the venous samples originates from the forearm vasculature. The relatively large negative arterial-venous gradient observed during maximal NOS inhibition suggests that nitrite is either rapidly converted to nitrate (which may have escaped our attention as a result of the reasons outlined above) or taken up by the vasculature or circulating blood cells. Little is known about the uptake and metabolism of nitrite by vascular cells. However, RBCs have a high capacity for nitrite uptake (22) and can accumulate this anion against a concentration gradient (39). It is conceivable, therefore, that the effect of l-NMMA on nitrite/nitrate plasma levels is the result of an unmasking of a continuous flux of nitrite from the endothelial surface to the RBC-rich core layer of the vascular lumen. Our data with l-NMMA are consistent with the idea that when NOS synthesis is blocked, but nitrite uptake by the RBCs continues, a negative arterial-venous gradient for nitrite ensues.
Because only individuals with normal CRP levels were included in the present study, a confounding influence on nitrite levels caused by the expression of inducible NOS (iNOS) in the forearm vasculature is unlikely. If this iNOS expression would have occurred, its activity would likely have resulted in micromolar rather than nanomolar plasma nitrite levels, and these would have changed little on stimulation with ACh. Because neuronal NOS is predominantly expressed in neurons and skeletal muscle, this NOS isoform is not expected to release relevant amounts of NO into the vascular lumen (31). Hence, our results also demonstrate that the majority of plasma nitrite under basal and stimulated conditions is of eNOS origin.
Nitrite as Delivery Source of Intravascular NO.
Recently, two groups independently reported that an arterial-venous gradient exists for nitrite (13) and NOx (14) in humans. The former was interpreted to mean that circulating nitrite represents a novel delivery source for intravascular NO as its consumption increased with handgrip exercise during regional NOS inhibition (13). Our data confirm that there is indeed a difference between mean arterial and venous nitrite concentration under resting conditions, even though it did not reach statistical significance. A closer look at the data set, however, revealed that this difference was the result of the relative higher frequency of positive compared with negative arterial-venous gradients. In fact, in 8 of 24 individuals this ratio was exactly opposite, casting doubt as to the assumption that resting nitrite levels may be bioactive. The complete lack of vasodilator activity of intraarterial infusion of nitrite clearly rules out any role for this metabolite in NO delivery. Furthermore, infused nitrite was recovered almost quantitatively on the venous side when applied at physiological concentrations. Nitrite did not dilate the forearm vasculature when applied at concentrations achieved during maximal eNOS stimulation with ACh (see Fig. 3 and Table 1), even when infused at >100-fold higher doses.
Diagnostic Accuracy of Nitrite, Nitrate, and Total NOx.
The specificity of plasma nitrite as an indicator of regional NO formation was shown under basal and stimulated conditions. We have demonstrated that plasma nitrite levels change with eNOS stimulation and inhibition in a dose-dependent manner. Moreover, the increase in plasma nitrite directly precedes the augmentation of FBF. The measured concentration range of plasma nitrite levels (228–428 nmol/liter under baseline conditions) is in good agreement with in vitro data on flow-induced release of NO from cultured endothelial cells (26). Application of the natural substrate of eNOS, l-arginine, stereospecifically increased resting plasma nitrite levels and led to a dilation of resistance arteries. In contrast, competitive inhibition of eNOS had the opposite effect with a more than 60% decrease of plasma nitrite concentration and a 70% decrease in FBF compared with resting conditions. Despite a 4-fold increase in FBF during maximal ACh stimulation (and the resultant 4-fold higher dilution of NO in the blood), plasma nitrite concentrations increased by 71% and the net venous-arterial difference of nitrite across this vascular bed increased by 245 nmol/liter, indicating an active eNOS-mediated process. In contrast, incubation of human whole blood with ACh in vitro did not affect basal plasma nitrite concentration (n = 3; data not shown).
During l-NMMA infusion, ACh-induced vasodilation decreased in a concentration-dependent manner and was almost blunted at the highest dose. A close relationship was observed between the “biochemical index” for NO release (plasma nitrite) and the “biological signal” (FBF). The strongest evidence for the specificity of nitrite as a marker for regional NO production comes from our studies with the endothelium-independent vasodilator papaverine. This compound increased FBF without affecting plasma nitrite and nitrate concentrations. As judged from the increase in FBF elicited by papaverine, NO production must have increased at least 4-fold under these conditions because venous nitrite concentrations did not change significantly. These data not only demonstrate the specificity of the response seen with ACh, but furthermore suggest that rates of endothelial NO production can be rapidly adjusted to changes in flow to keep local NO concentrations at the same level. The lack of effect of papaverine on plasma nitrite levels further rules out the possibility that the changes in nitrite seen with ACh or l-NMMA are caused by a washout phenomenon, i.e., the redistribution of nitrite from the interstitial space or RBCs secondary to changes in flow.
NO and ACh Response.
l-NMMA is a selective and stereospecific NOS inhibitor without intrinsic vasoconstrictor properties (40, 41). Considering that the NO release is a product of the net venous-arterial concentration difference of nitrite (luminal release) times FBF (abluminal release), the data of the present investigation are in keeping with previous studies, all of which were limited to measure only vasoconstriction as parameter for the abluminal release. This general vasoconstrictor response was shown in clinical and experimental settings as diverse as human forearm and coronary circulation, isolated organs, rat, or porcine vessels (40, 41). In human forearm circulation, the commonly used concentration of l-NMMA ranges from 1 to 8 μmol/min, which is lower than the maximal dose applied in the present study. In response to endothelium-dependent stimulation, a 20–60% reduction in FBF was reported. We show here that higher doses of l-NMMA result in a further decrease in blood flow, which is paralleled by an almost entirely blunted NO release at 12 μmol/min l-NMMA. Thus, at least in healthy humans, ACh responses are almost exclusively eNOS-mediated, and the appropriate dose for sufficient eNOS inhibition in the human forearm amounts to 12 μmol/min l-NMMA.
In conclusion, the measurement of plasma NO2− accurately reflects acute changes in eNOS activity in the human forearm circulation, and thus may help to further elucidate the pathophysiological significance of an altered eNOS activity in disease states known to be associated with endothelial dysfunction.
Acknowledgments
The skillful technical assistance of Mrs. S. Matern and C. Ferfers are gratefully acknowledged. This work was supported by Deutsche Forschungsgemeinschaft Grants Ke 405/4-1 and -4-3, and by the BMFZ (to the Biomedizinisches Forschungszentrum Düsseldorf). T.R. is a research fellow funded by Deutsche Forschungsgemeinschaft Grant RA 969/1-1.
Abbreviations
- ACh
acetylcholine
- FBF
forearm blood flow
- l-NMMA
NG-monomethyl-l-arginine
- eNOS
endothelial NO synthase
- RBC
red blood cell
- Hb
hemoglobin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Busse R, Fleming I. Ann Med. 1995;27:331–340. doi: 10.3109/07853899509002586. [DOI] [PubMed] [Google Scholar]
- 2.Furchgott R F, Vanhoutte P M. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- 3.Ignarro L J. Annu Rev Pharmacol Toxicol. 1990;30:535–560. doi: 10.1146/annurev.pa.30.040190.002535. [DOI] [PubMed] [Google Scholar]
- 4.Rubanyi G M. J Cardiovasc Pharmacol. 1993;22, Suppl 4:S1–S14. doi: 10.1097/00005344-199322004-00002. [DOI] [PubMed] [Google Scholar]
- 5.Moncada S, Higgs E A. FASEB J. 1995;9:1319–1330. [PubMed] [Google Scholar]
- 6.Moncada S, Palmer R M J, Higgs E A. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 7.Ross R. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 8.Ludmer P L, Selwyn A P, Shook T L, Wayne R R, Mudge G H, Alexander R W, Ganz P. N Engl J Med. 1986;315:1046–1051. doi: 10.1056/NEJM198610233151702. [DOI] [PubMed] [Google Scholar]
- 9.Kelm M, Dahmann R, Wink D, Feelisch M. J Biol Chem. 1997;272:9922–9932. doi: 10.1074/jbc.272.15.9922. [DOI] [PubMed] [Google Scholar]
- 10.Hampl V, Walters C L, Archer S L. In: Methods in Nitric Oxide Research. Feelisch M, Stamler J S, editors. New York: Wiley; 1996. pp. 309–318. [Google Scholar]
- 11.Vallance P, Patton S, Bhagat K, MacAllister R, Radomski M, Moncada S, Malinski T. Lancet. 1995;345:153–154. doi: 10.1016/s0140-6736(95)91211-8. [DOI] [PubMed] [Google Scholar]
- 12.Kelm M. Biochim Biophys Acta. 1999;1411:273–289. doi: 10.1016/s0005-2728(99)00020-1. [DOI] [PubMed] [Google Scholar]
- 13.Gladwin M T, Shelhamer J H, Schechter A N, Waclawiw M A, Panza J A, Ognibene F P, Cannon R O., III Proc Natl Acad Sci USA. 2000;97:11482–11487. doi: 10.1073/pnas.97.21.11482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cicinelli E, Ignarro L J, Schonauer L M, Matteo M G, Galantino P, Falco N. Clin Physiol. 1999;19:440–442. doi: 10.1046/j.1365-2281.1999.00200.x. [DOI] [PubMed] [Google Scholar]
- 15.Kelm M, Preik-Steinhoff H, Preik M, Strauer B E. Cardiovasc Res. 1999;41:765–772. doi: 10.1016/s0008-6363(98)00259-4. [DOI] [PubMed] [Google Scholar]
- 16.Preik M, Lauer T, Heiss C, Tabery S, Strauer B E, Kelm M. Ultraschall Med. 2000;21:195–198. doi: 10.1055/s-2000-7989. [DOI] [PubMed] [Google Scholar]
- 17.Schulz K, Kerber S, Kelm M. Nitric Oxide. 1999;3:225–234. doi: 10.1006/niox.1999.0226. [DOI] [PubMed] [Google Scholar]
- 18.Preik-Steinhoff H, Kelm M. J Chromatogr B Biomed Appl. 1996;685:348–352. doi: 10.1016/s0378-4347(96)00264-2. [DOI] [PubMed] [Google Scholar]
- 19.Lüscher T F, Diederich D, Stebenmann R, Lehmann K, Stulz P, von Segesser L, Yang Z, Turina M, Grädel E, Weber E, et al. N Engl J Med. 1988;319:462–467. doi: 10.1056/NEJM198808253190802. [DOI] [PubMed] [Google Scholar]
- 20.Addicks K, Bloch W, Feelisch M. Microsc Res Tech. 1994;29:161–168. doi: 10.1002/jemt.1070290214. [DOI] [PubMed] [Google Scholar]
- 21.Rhodes P M, Leone A M, Francis P L, Struthers A D, Moncada S. Biochem Biophys Res Commun. 1995;209:590–596. doi: 10.1006/bbrc.1995.1541. [DOI] [PubMed] [Google Scholar]
- 22.Wink D A, Grisham M B, Mitchell J B, Ford P C. Methods Enzymol. 1996;268:12–31. doi: 10.1016/s0076-6879(96)68006-9. [DOI] [PubMed] [Google Scholar]
- 23.Beckman J S, Koppenol W H. Am J Physiol Cell Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 24.Stamler J S, Slivka A. Nutr Rev. 1996;54:1–30. doi: 10.1111/j.1753-4887.1996.tb03770.x. [DOI] [PubMed] [Google Scholar]
- 25.Ignarro L J, Fukuto J M, Griscavage J M, Rogers N E, Byrns R E. Proc Natl Acad Sci USA. 1993;90:8103–8107. doi: 10.1073/pnas.90.17.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelm M, Feelisch M, Spahr R, Piper H M, Noack E, Schrader J. Biochem Biophys Res Commun. 1988;154:236–244. doi: 10.1016/0006-291x(88)90675-4. [DOI] [PubMed] [Google Scholar]
- 27.Lancaster J R., Jr Proc Natl Acad Sci USA. 1994;91:8137–8141. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.May J M, Qu Z C, Xia L, Cobb C E. Am J Physiol Cell Physiol. 2000;279:C1946–C1954. doi: 10.1152/ajpcell.2000.279.6.C1946. [DOI] [PubMed] [Google Scholar]
- 29.Doyle M P, Hoekstra J W. J Inorg Biochem. 1981;14:351–358. doi: 10.1016/s0162-0134(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 30.Wennmalm A, Benthin G, Edlund A, Kieler-Jensen, Lundin S, Petersson A-S, Waagstein F. Ann NY Acad Sci. 1994;714:158–164. doi: 10.1111/j.1749-6632.1994.tb12040.x. [DOI] [PubMed] [Google Scholar]
- 31.Eich R F, Li T, Lemon D D, Doherty D H, Curry S R, Aitken J F, Mathews A J, Johnson K A, Smith R D, Phillips G N, Jr, Olson J S. Biochemistry. 1996;35:6976–6983. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- 32.Ford P C, Wink D A, Stanbury D M. FEBS Lett. 1993;326:1–3. doi: 10.1016/0014-5793(93)81748-o. [DOI] [PubMed] [Google Scholar]
- 33.Nedospasov A, Rafikov R, Beda N, Nudler E. Proc Natl Acad Sci USA. 2000;97:13543–13548. doi: 10.1073/pnas.250398197. . (First Published November 28, 2000; 10.1073/pnas.250398197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abu-Soud H M, Hazen S L. J Biol Chem. 2000;275:37524–37532. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 35.Tannenbaum S R, Witter J P, Gatley S J, Balish E. Science. 1979;205:1333–1337. [Google Scholar]
- 36.Liu X, Miller M J S, Joshi M S, Sadowska-Krowicka H, Clark D A, Lancaster J R., Jr J Biol Chem. 1998;273:18709–18713. doi: 10.1074/jbc.273.30.18709. [DOI] [PubMed] [Google Scholar]
- 37.Butler A R, Megson I L, Wright P G. Biochim Biophys Acta. 1998;1425:168–176. doi: 10.1016/s0304-4165(98)00065-8. [DOI] [PubMed] [Google Scholar]
- 38.Liao J C, Hein T W, Vaughn M W, Huang K T, Kuo L. Proc Natl Acad Sci USA. 1999;96:8757–8761. doi: 10.1073/pnas.96.15.8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Recchia F A, Vogel T R, Hintze T H. Am J Physiol. 2000;279:H852–H856. doi: 10.1152/ajpheart.2000.279.2.H852. [DOI] [PubMed] [Google Scholar]
- 40.Rees D D, Palmer R M J, Schulz R, Hodson H F, Moncada S. Br J Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vallance P, Collier J, Moncada S. Lancet. 1989;ii:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]