

Abstract
Epigenetic regulation may play an important role in mediating microbe–host interactions and adaptation of intestinal gene expression to bacterial colonization just after birth. This is particularly important after preterm birth because the immature intestine is hypersensitive to invading bacteria. We compared the intestinal DNA methylome and microbiome between conventional (CON) and antibiotics-treated (AB) preterm pigs, used as a model for preterm infants. Oral AB treatment reduced bacterial density (∼100-fold), diversity and fermentation, improved the resistance to necrotizing enterocolitis (NEC) and changed the genome-wide DNA methylation in the distal small intestine. Integration of epigenome data with previously obtained proteome data showed that intestinal immune–metabolic pathways were affected by the AB-induced delay in bacterial colonization. DNA methylation and expression of intestinal genes, related to innate immune response, phagocytosis, endothelial homeostasis and tissue metabolism (e.g. CPN1, C3, LBP, HIF1A, MicroRNA-126, PTPRE), differed between AB and CON pigs even before any evidence of NEC lesions. Our findings document that the newborn immature intestine is influenced by bacterial colonization via DNA methylation changes. Microbiota-dependent epigenetic programming of genes related to gut immunity, vascular integrity and metabolism may be critical for short- and long-term intestinal health in preterm neonates.
Keywords: epigenetics, DNA methylation, microbiota, immunity, metabolism
1. Introduction
Development of balanced host–microbe interactions is critical for adaptation of newborn infants to their external environment.1 Newborn infants are normally protected from pathogens but allow exposure to a high density of beneficial (commensal) microbes that facilitate normal development of the immune system, in accord with the “hygiene hypothesis”.2 It is not clear how bacterial colonization affects microbe–host interactions in preterm infants that have an immature innate immune system and limited access to protection from mother’s own milk just after birth.3 Up to 10% of all hospitalized preterm infants suffer from necrotizing enterocolitis (NEC), an intestinal inflammatory disease, associated with formula feeding and gut microbiota dysbiosis.4 To prevent NEC, different interventions to manipulate bacterial colonization have been used, including antibiotics (AB), probiotics and prebiotics. Delaying bacterial colonization via oral administration of AB for the first few days after preterm birth decreases NEC incidence in both infants and pigs.5–8 This NEC-preventive treatment is not used in clinical practice, mainly due to concerns of increased antimicrobial resistance. Nevertheless, it is important to investigate the molecular mechanisms responsible for the enhanced adaptation of the immature intestine when gut bacterial colonization is delayed by AB treatment just after preterm birth. This is critical to identify intestinal pathways that are affected by bacterial colonization and new ways to prevent microbe-induced intestinal dysfunction and NEC.
Recent research has documented direct connections between microbial metabolites and epigenetic modifications.9,10 For instance, microbiota-derived folate, choline and methionine are involved in the one-carbon metabolism that contributes to the methyl donor, S-adenosylmethionine, for DNA methylation. DNA methylation may regulate gene transcription, cellular differentiation and normal development.11 Therefore, DNA methylation may play an important role in mediating microbe–host interactions in early life, when rapid adaptation of the immature intestine is critical for survival, especially following preterm birth. In newborn mice, germ-free conditions alter the DNA methylation of the colonic epithelium and affect gene activation, and thereby intestinal functions.12 Correspondingly, the intestinal DNA methylome may be affected by the timing, density and diversity of bacterial colonization just after birth in preterm neonates.
Using preterm pigs as a model for preterm infants, we applied reduced representation bisulphite sequencing (RRBS) for intestinal DNA methylome profiling and 16S rRNA sequencing for microbiome profiling in animals with or without oral AB treatment for 5 days after birth. We observed marked DNA methylation differences between the two groups of pigs associated with differences in the total load and community structure of bacteria. The epigenome data were integrated with proteome data from a previous study, using the same animal model, to gain further insight into the biological pathways that shape the interface between the gut microbiota and intestinal cells.
2. Materials and methods
2.1. Animals and their treatment
All animal procedures were approved by the Danish National Committee on Animal Experimentation. Fourteen preterm pigs from three sows (Danish Landrace×Large White×Duroc) were selected from a previous larger study where the phenotypic characteristics have been described in detail.8 All pigs were caesarean-delivered at day 106 (∼90% of gestation). Preterm pigs delivered at 90% gestation show intestinal characteristics of human infants delivered at ∼70% gestation. Fourteen pigs showing limited or no severe clinical signs of NEC before tissue collection were chosen for this study on DNA methylation patterns. The newborn preterm pigs were immediately transferred to a piglet intensive care unit and reared in temperature- and oxygen-regulated incubators. The pigs were weighed and fitted with umbilical arterial and orogastric catheters and then subject to passive immunization with maternal plasma, as previously described.8 Pigs were subjected to delayed bacterial colonization, using daily oral boluses of broad-spectrum AB (AB group, n = 7), and they were compared with pigs raised conventionally (CON group, n = 7) with spontaneous microbial colonization and daily boluses of saline. For the AB group, the selection of AB and doses were adapted from the current use of AB (intravenous) to preterm infants in Denmark.7,8 Thus, a combination of ampicillin (30 mg/kg BW thrice daily), gentamicin (2.5 mg/kg BW twice daily) and metronidazole (10 mg/kg BW thrice daily) was used to target a broad range of microorganisms.
For nutrition, all pigs were initially provided with parenteral nutrition (PN) via the umbilical catheter, supplemented with minimal enteral nutrition (MEN) with formula via the orogastric tube. The PN solution was based on a commercially-available product (Kabiven, Fresenius Kabi, Bad Homburg, Germany), and its composition was adjusted to meet nutritional requirements of preterm pigs, as previously described.7 The formula diet consisted of three products commonly used for infants (per litre of water: 75 g Liquigen MCT, 80 g Pepdite and 70 g Arla DI-9224; from Nutricia, Allerød, Denmark, and Arla Food Ingredients, Viby J, Denmark, respectively). The provision of MEN was initiated within 5 h of delivery as boluses of 3 ml/kg every 3 h on days 1–2. On day 3, PN supply was stopped and total enteral nutrition with formula was provided as boluses of 15 ml/kg every 3 h until euthanasia and tissue collection on day 5.
2.2. Tissue collection, intestinal morphology and inflammatory cytokine analysis
The pigs were euthanized with sodium pentobarbital (200 mg/kg, i.a.). The gastrointestinal tract was immediately removed, and the small intestine was carefully emptied of its contents and weighed. Pieces of the distal small intestine (83% along the length of the small intestine) were snap frozen in liquid nitrogen and kept at −80°C for subsequent analysis of the DNA methylome and inflammatory cytokines. IL-8 levels in the distal small intestine were analysed with ELISA (R&D Systems, Abingdon, Oxfordshire, UK) following the manufacturer’s instructions and measured as picograms per milligram of wet tissue. In addition, two 1-cm pieces were collected and fixed in 4% paraformaldehyde for histological analysis. The paraformaldehyde-fixed samples were embedded in paraffin, sectioned (5 μm), mounted on slides and stained with haematoxylin and eosin. Representative cross-sections were selected from each pig, and at least 10 well-oriented crypts and villi were measured using an Axiophot microscope (Carl Zeiss, Oberkochen, Germany) and NIH image software version 1.60 (softWoRx Explorer version 1.1; Applied Precision, Issaquah, WA). To estimate the epithelial cell proportion in the small intestine tissue from each group, we examined cross sections of the distal small intestine by immunohistochemistry using the epithelial cell marker, cytokeratin (DAKO M3515). Staining was developed with UltraVision LP Detection System (ThermoFisher Scientific). The sections were counterstained with Mayer’s haematoxylin. Images were acquired using the OlyVIA software (OLYMPUS, version 2.9) and the proportion (%) of the positive staining for cytokeratin in cross-sectional area was analysed by the IHC toolbox in ImageJ.
2.3. Organic acids and microbiota analysis
The luminal contents of the colon were subjected to organic acid (short-chain fatty acid, SCFA) concentration measurements, using gas chromatography and total bacterial load was quantified using RT-qPCR, as previously described.13 The luminal contents of the distal small intestine were collected for the analysis of gut microbiota. In brief, total DNA was extracted from the distal small intestinal contents, and the v3-v4 hypervariable regions of the 16S rRNA sequence were amplified with PCR. The resultant amplicons were sequenced using the Illumina MiSeq system (Illumina, San Diego, CA), producing paired-end reads. The microbiota community structure was analysed with Mothur, based on the official protocol developed by the Mothur developers.14 The optimized reads were clustered into operational taxonomic units (OTUs), and representative sequences were aligned to the SILVA reference (version: SILVA123). The OTU annotation results were employed to determine the microorganism composition in each sample.
2.4. Reduced representation bisulphite sequencing (RRBS)
Genomic DNA was extracted from the 14 rinsed intact pieces of distal intestine, using the DNeasy Blood & Tissue Kit (Qiagen) and subjected to RRBS library preparation, as previously described.15 We used whole intestinal tissue samples because whole tissue responses likely better represented the in vivo state of the intestine, with all the interacting cell types, than just isolated cell types (e.g. enterocytes, goblet cells, enteroendocrine cells). In brief, 1.5 µg of genomic DNA was digested with the MspI enzyme (NEB), followed by end repair, A-base tailing and 5-methylcytosine-modified adapter ligation. Size selection was performed to obtain DNA fractions of MspI-digested products in the range of 40–250 bp. Subsequently, bisulphite treatment was conducted using the ZYMO EZ DNA Methylation-Gold Kit following the manufacturer’s instructions. Twelve cycles of PCR were performed to enrich the DNA fragments, in which each library was integrated with the DNA index. The libraries were analysed using an Agilent 2100 Bioanalyzer and qPCR for quality control. The libraries were then subjected to paired-end 125 bp multiplex sequencing on the HiSeq 2500 platform. Raw sequencing data were processed via the Illumina base-calling pipeline. Low-quality reads that contained more than 30% ‘N’s or showed a low-quality value (quality value <20) in over 10% of the sequence were omitted from the data analysis. The bisulphite sequence MAPping program (BSMAP)16 was used for sequence alignment to the Ensembl pig reference genome (Sscrofa10.2). The methylation levels of individual cytosines were calculated as the ratio of the sequenced depth of the ascertained methylated CpG cytosines to the total sequenced depth of individual CpG cytosines.
2.5. Illumina hiseq sequencing-based bisulphite sequencing PCR (BSP)
Gene-specific DNA methylation was assessed by BSP, according to a previously published method.17 IN brief, BSP primers were designed using the online MethPrimer software and listed in Supplementary Table S1. Genomic DNA (500 ng) was converted using the ZYMO EZ DNA Methylation-Gold Kit (ZYMO) and one-tenth of the elution products were used as templates for PCR amplification. For each sample, BSP products of multiple genes were generated, pooled equally and subjected to adaptor ligation. Barcoded libraries from all 14 samples were sequenced on the Hiseq platform using paired-end 250 bp strategy. Data were processed and analysed using BSMAP, as described above.
2.6. Statistical analysis
For phenotypic values, bacterial load, SCFA and cytokine data, comparisons between the two groups were conducted using Student’s t-test, and a two-tailed P-value <0.05 was considered statistically significant. The correlation between the log-transformed relative abundance of the microbiota and concentration of SCFAs was assessed using Spearman’s rank correlation test. A two-sided P-value <0.05 was regarded as statistically significant. Differentially methylated positions (DMPs) were identified according to the methylation levels of cytosines between samples from the two groups using the Mann–Whitney U-test. DMPs showing a mean methylation difference >0.2 and a P-value <0.01 were considered statistically significant. To identify differentially methylated regions (DMRs), pairs of significant DMPs were used to delimit regions exhibiting homogeneous methylation changes, and the interval methylation levels between the two groups were tested using the Mann–Whitney U-test, with a false discovery rate of <0.05. For gene expression analysis, RT-qPCR analysis was performed using QuantiTect SYBR Green PCR Kit (Qiagen) on LightCycler 480 (Roche). Relative quantification of target genes was normalized to housekeeping gene HPRT1 (Supplementary Table S2). Comparisons were made using the Student’s t-test and a two-tailed P-value <0.05 was considered as statistically significant.
3. Results
3.1. Phenotypic effects of AB treatment in preterm pigs
Pigs from the two groups in this study were subgroups of pigs from a larger study where detailed clinical characteristics and phenotypic variables have been published previously.8 At autopsy, none of the AB pigs had any NEC lesions in their intestines, while two of seven CON pigs were diagnosed as NEC according to our macroscopic NEC evaluation system.18 One of these NEC pigs showed colon lesions (haemorrhage, local necrosis, pneumatosis intestinalis, NEC score 5) while the other NEC pig showed both colon and distal intestine lesions (severe, extensive pneumatosis intestinalis, haemorrhage, necrosis, NEC score 6). All the AB pigs were given a NEC score of 1, reflecting no visible lesions or abnormalities (Fig. 1a). Histological analysis of the distal small intestine revealed clear villus atrophy in the pig diagnosed as NEC in the distal intestine (Fig. 1b). Compared with CON pigs, the AB pigs showed a higher growth velocity and lower crypt depth (both P < 0.05), but no change in villus height (Fig. 1c). To assess the approximate proportion of epithelial cells, immunohistochemical analyses of the epithelial marker cytokeratin was performed on cross sections of distal small intestine. No significant difference in the proportion of epithelial cells was observed between AB and CON pigs (44.1 vs. 42.2%, P = 0.38, Supplementary Fig. S1).
Figure 1.
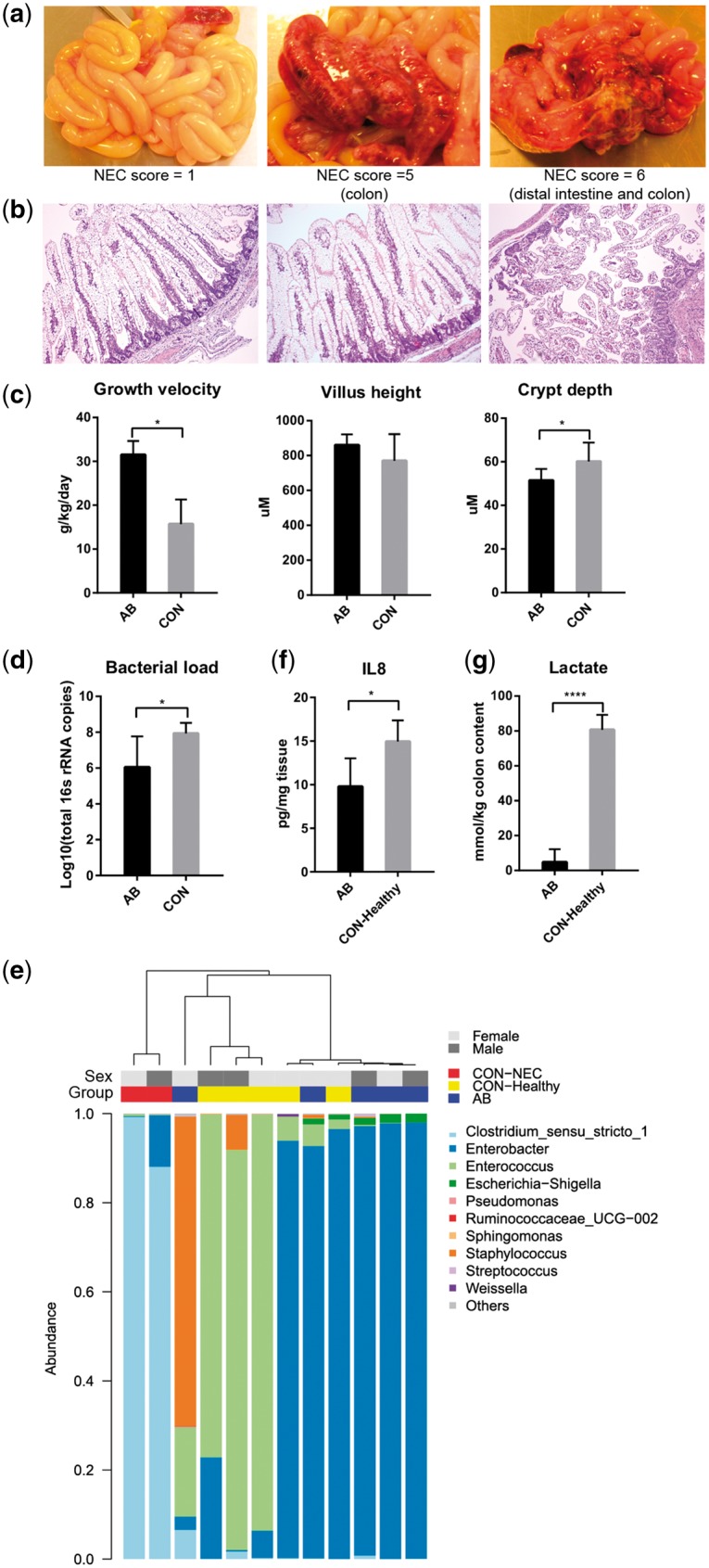
Phenotypic and microbial characteristics of preterm pigs. (a) Representative photographs and (b) histopathology of the small intestine in preterm pigs receiving AB (AB pigs, NEC score 1) or CON-raised pigs and with NEC lesions (CON-NEC pigs, NEC scores 5–6). (c) Growth velocity of preterm pigs, and villus height and crypt depth in the distal intestine. (d) Bacterial load and (e) dendrogram illustrating the microbial composition in the distal small intestine of preterm pigs. (f) IL-8 expression in the distal intestine. (g) Lactate concentration in the colon. Bar charts were presented as the mean values ± SEM (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Relative to CON pigs, the AB pigs had a lower mean total bacterial load (106 versus 108 in bacterial counts, Fig. 1d), with two of the AB pigs having values similar to that in CON pigs. We then analysed the microbial composition from their intestinal contents using 16S rRNA microbiome sequencing. Data from two pigs in the AB group were omitted due to insufficient reads for OTU annotation. Based on the remaining 12 samples, there was lower inter-individual diversity of the intestinal microbiota in AB versus CON pigs. The two NEC pigs from the CON group was dominated by Clostridium and showed marked compositional difference from the other pigs, as indicated by hierarchical clustering (Fig. 1e). The five healthy pigs from CON group were dominated by Enterococcus and Enterobacter, while most AB pigs were dominated by Enterobacter, except for one pig that was dominated by Staphylococcus (Fig. 1e). Hence, in addition to a decreased total bacterial count, the AB pigs had less abundance of Gram-positive bacteria (Clostridium and Enterococcus) than the NEC and healthy pigs from the CON group, i.e. CON–NEC and CON–Healthy pigs, respectively. Previous studies show that stool samples from preterm infants that develop NEC are dominated by Firmicutes, especially Enterococcus in the early postnatal period.19,20 Therefore, the dominant Enterococcus in CON–Healthy pigs might imply a risk of developing NEC, although normal intestinal morphology was observed in these pigs at autopsy. The level of interleukin 8 (IL-8), a biomarker of NEC in both human infants and rats,21–23 was elevated in the distal intestine of these CON–Healthy pigs (P < 0.05, Fig. 1f). Finally, colonic lactate concentrations, known to be elevated in association with NEC,18 was markedly increased in the CON–Healthy pigs (P < 0.0001, Fig. 1g). The lactate concentration was positively correlated with the relative abundance of Enterococcus (ρ = 0.61, P < 0.05).
3.2. DNA methylation profile in preterm pig intestine
Next, we evaluated the DNA methylome of the small intestine in the AB and CON groups via RRBS, which was developed to measure the DNA methylation of high-CG regions at a single base-pair resolution. As we applied a 125-bp paired-end sequencing strategy, MspI-digested fragments of the RRBS library were expanded to 40–250 bp. As a result, we generated a total of 57.65 gigabases (Gb) of clean bases from 14 libraries after quality control analyses. Using the BSMAP, we found that 68.0% of the clean reads could be mapped to the pig reference genome, reaching an average read depth of 8.71–13.59 per strand for each sample. The bisulphite conversion rate of C–to-T reached 99.6%, as calculated based on the methylated level of lambda DNA (Supplementary Table S3). As RRBS enriches high-CpG regions in the genome and mammalian DNA methylation occurs almost exclusively at CGs, we focused on analyses of CpG methylation. Only the CpG sites showing 4× or more coverage per strand were analysed to maintain a high accuracy level for methylation levels, resulting in an average of 4.66 million CpG sites being covered in the analyses for each of the samples. The pig genome contains 56.06 million CpGs, and we therefore managed to examine ∼8.31% of all CpGs in the pig genome using this representation strategy.
DNA methylation might be biased towards specific alleles in specific genomic regions owing to gene imprinting.24 To address this issue, we applied two methods to analyse allele-specific methylation (ASM) across 14 samples, including an AMR-based method25 and an SNP calling-based method referred to as “SMAP”.26 By integrating the results from these two methods, we sought to reveal genomic regions that most likely showed differential methylation between paternal and maternal genomes, and we checked whether these regions of genomic imprinting could be affected by AB usage. However, no significant differences in ASM levels were observed between the two groups, indicating that the administered oral AB and the different colonization levels had no effect on ASM (Supplementary Fig. S2, data not shown).
3.3. Genome-wide DNA methylation changes
Based on the above analyses, we next sought to infer the changes in intestinal genomic methylation in response to delayed microbial colonization by AB treatment just after preterm birth. To avoid the potentially confounding influence of X chromosome inactivation on DNA methylation patterns between male and female neonates, only autosomal data were used in this study hereafter. We first examined the global pattern of genome-wide methylation. Hierarchical clustering and principal component analysis (PCA) of the methylation levels of all CpG sites were performed to examine the whole-genome methylation status of these samples. Both clustering and PCA results indicated that the two groups were not clearly separated into two clusters owing to individual epigenomic variation (Fig. 2a and Supplementary Fig. S3). However, based on the average methylation levels observed across all samples, genic regions showed clear divergence between the two groups, suggesting differences in specific genomic regions or CpG sites (Supplementary Fig. S4). Thereby we next carried out pair-wise comparisons to screen for DMPs between the two groups across the whole genome. This revealed 3,850 CpG sites to be DMPs exhibiting methylation level differences of greater than 20% (Fig. 2b). Among these DMPs, 46.6% were distributed within genic regions, including gene body regions or putative regulatory elements of promoters (2 kb upstream and 500 bp downstream from the transcription start sites—TSS).
Figure 2.

Changes in DNA methylation in response to microbial colonization. (a) PCA using genome-wide DNA methylation data. (b) Scatter plot of the methylation levels of DMPs in the CON and AB groups, showing the density at each point. (c) Genomic distribution of DMRs. (d) Correlation between RRBS and BSP data. (e) Heat map depicting the hierarchical clustering of DMR-associated genes for each preterm pig.
Based on the identification of DMPs, we further screened for key genome-wide DMRs that can potentially affect gene functions (see Materials and methods). A total of 87 DMRs were identified in the AB versus CON groups (Supplementary Table S4). Compared with the CON group, 47 DMRs were hypomethylated in the AB group. These DMRs were distributed across all the autosomes, with an average length of 47 base pairs. Based on the current gene annotation for the pig genome, 48 DMRs were associated with putative promoters or intragenic regions (Supplementary Table S4 and Fig. 2c). There were 39 genes with gene symbols that were ascertainable from the Ensembl database. Two genes (NNAT and MEST) were recognized as imprinted genes according to an online imprinting gene database (http://www.geneimprint.com (11 January 2018, date last accessed)) and were excluded from the subsequent analyses. To confirm the observed DNA methylation variations, BSP was performed on five genes with DMRs in either putative promoter or gene body. Examination on all individual cytosines within the five genes revealed significant correlation between the RRBS and BSP data (P < 0.0001, R2 = 0.78, Fig. 2d and Supplementary Fig. S5). According to hierarchical clustering by individual methylation level of the DMRs in these 37 genes, the AB and CON pigs were grouped into two distinct clusters (Fig. 2e). Within the CON group, the two NEC pigs and five healthy pigs were not clearly separated. This suggests that NEC lesions observed on day 5 did not markedly affect the intestinal methylation status.
3.4. Integrated methylome–proteome analyses
We next sought to evaluate the functional significance of the identified DMRs. Considering DNA methylation plays an important role for transcription regulation, it is expected that divergent DNA methylation will ultimately lead to changes of protein expression and further trigger changes of cellular functions via protein–protein interactions. To address this possibility, we applied a previously published proteome data set with a series of differentially expressed proteins following antibiotic treatment of preterm pigs for five days after birth27 (Supplementary Table S5). This study used an identical treatment protocol except that the AB were provided both orally and systemically. As DNA methylation at TSS is associated with transcription silencing while the mechanism of methylation in other genomic regions is less known, we selected genes (ARAP1, CPN1, DHCR7, HOXA2, TRMU and ssc-mir-126) containing DMRs adjacent to TSSs and searched BioGRID database for their potential interactions with the divergently expressed proteins obtained from the proteome data. We found that four genes (ARAP1, CPN1, DHCR7 and HOXA2) were involved in extensive protein–protein interaction networks, in which 78 proteins showed either direct or indirect interaction with each other (Fig. 3a). Among these four genes, CPN1 showed direct interaction with complement component 3 (C3), which is an important constituent of the innate immune system that enhances phagocytosis to clear microbes. As CPN1 functions as a suppressor of C3,28 and was hypomethylated in its promoter region (Fig. 3b), it could be therefore up-regulated, leading to decreased C3 expression in the AB group. To test this, we examined the mRNA expression levels of both CPN1 and C3 from a larger set of samples from the original study,8 including 14 pigs from the AB group and 14 pigs from the CON group (of which 8 had NEC and 6 were healthy). Relative to the CON pigs (both CON–healthy and CON–NEC), the AB pigs showed significantly decreased C3 level (Fig. 3c). However, in agreement with the gene expression database,29 the qPCR results showed very low CPN1 expression levels in the small intestine, preventing detailed comparisons between groups (Supplementary Fig. S6). Nevertheless, consistent with that C3 was reduced in the AB pigs, lipopolysaccharide binding protein (LBP) and neutrophil chemotactic factor IL-8 were also reduced in the AB pigs (Fig. 3d). We examined the proportion of neutrophils by immunohistochemical analyses on cross sections of distal small intestine, using the neutrophil marker, myeloperoxidase (MPO). The number of MPO-positive cells among the overall cell population was small and 3.6 versus 4.7 per villus in the AB and CON groups, respectively (P = 0.08, Supplementary Fig. S7). Together with the reduced IL-8 levels, this suggests that that innate immune response was less activated in the AB pigs.
Figure 3.

Methylome-proteome network analyses. (a) Visualization of potential interactions between genes associated with DMRs and genes encoding differentially expressed proteins according to the BioGRID interaction data set. (b) Individual methylation level of each CpG cytosine within the DMR at CPN1 promoter. (c, d) Relative expression of genes in the distal intestine, presented as mean values ± SEM (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Furthermore, we employed the genes that were differentially expressed in protein level together with all the DMR-associated genes to perform functional enrichment analysis. In agreement with the above results, we found that 2 of the 11 significantly enriched KEGG pathways (Supplementary Table S6) were related to ‘bacterial infection’, including ‘legionellosis’ and ‘pathogenic Escherichia coli infection’. These two pathways were both related to Gram-negative bacteria, supporting the change in LBP expression (Fig. 3d). Interestingly, we also revealed five metabolism-related pathways and four pathways related to vascular functions. Previous studies indicated that the recruitment of immune cells and phagocytosis might result in local depletion of oxygen, which could further trigger a stress response that increases angiogenesis and induces metabolic changes to compensate for oxygen deficits.30–32 We found that, within the glycolysis and gluconeogenesis pathways, the glycolytic enzyme aldolase A (encoded by ALDOA) was down-regulated in the AB group. In contrast, the gluconeogenesis regulatory enzymes fructose-1, 6-bisphosphatase 1 (encoded by FBP1) and mitochondrial PEP-carboxykinase (encoded by PCK2) were up-regulated in the AB group (Supplementary Fig. S8). Together, the enriched KEGG pathways indicate that delayed bacterial colonization following AB treatment may affect intestinal oxygenation, vascular function and tissue metabolism.
3.5. Hypoxia-associated vascular endothelial functions
Based on above results, we further studied in greater detail some genes related to possible hypoxia and altered vascular functions of the immature intestine of AB and CON pigs. This is relevant as innate immunity is linked with hypoxia, and hypoxia induces angiogenesis.33,34 Under hypoxic conditions, the expression of hypoxia-inducible factor 1-alpha (HIF1A) will be triggered to mediate the hypoxia response of cells.33,34 We therefore quantified the mRNA expression of HIF1A in the small intestines. Consistent with this, HIF1A expression was reduced in AB versus CON pigs (Fig. 4a). Increased HIF1A expression may stimulates vascular endothelial growth factor receptor 2 (VEGFR2) signalling in order to trigger angiogenesis and compensate for an oxygen deficit.31 In this study, we did not observe any differences in VEGFA and VEGFR2 expression between CON and AB pigs (Fig. 4a). Previous studies suggested that lack of VEGFR2 signalling facilitated NEC.35 This result might indicate these CON pigs also failed to activate the VEGFR2 signalling pathway under hypoxic condition. Another effect of hypoxia is on the vascular tone pathway. Our KEGG analyses had indicated several DMR-associated genes involved in the vasodilation-related pathways, including GNAS and MRVI1 (Supplementary Table S6), which belong to the G protein-coupled receptor (GPCR) signalling pathway. GNAS encodes for stimulatory G-protein alpha subunit (Gs-α) while MRVI1 (also called IRAG), had been shown to prevent calcium release within the GPCR signalling and thus contributes to vasodilation.36,37 RT-qPCR results showed the mRNA expression of GNAS and MRVI1 also tended to be decreased in the AB pigs versus NEC pigs, though not significantly (Supplementary Fig. S6).
Figure 4.

Expression of genes related to hypoxia and vascular function. (a) The relative expression of HIF1A (related to hypoxia) and VEGFA, VEGFR2, ssc-mir-126 and PTPRE (related to vascular function). Values are presented as mean ± SEM (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). (b) Three genes (ssc-mir-126, PTPRE, LRP8), related to endothelial homeostasis showed hypomethylation in the promoters or gene bodies in the AB pigs.
Finally, we investigated in detail three DMR-associated genes potentially related to endothelial homeostasis. MicroRNA-126 is essential for vascular integrity and inhibits haemorrhage,38 PTPRE negatively regulates endothelial cell proliferation39 and LRP8 initiates endothelial antiapoptosis.40 The DMRs in these genes were all hypomethylated in the AB versus CON pigs (Supplementary Table S4). The DMR within the putative promoter region of MicroRNA-126 was located in the intron of EGFL7 and contained four CpGs (Fig. 4b). Hypomethylation of this region may increase the transcription of MicroRNA-126 and there was a tendency to up-regulation of MicroRNA-126 in the AB pigs (P = 0.29, Fig. 4a). In contrast, PTPRE and LRP8 contained the DMRs within their intron and exon, respectively (Fig. 4b). The DMR in LRP8 also co-localized with a CpG island. The expression of PTPRE was decreased in AB versus CON pigs especially when compared with NEC pigs (P < 0.05, Fig. 4a). The expression of LRP8 was too low to be accurately measured in the small intestine in both groups (Supplementary Fig. S6), as confirmed by the gene database.29
4. Discussion
The initial bacterial colonization of gut in early life of infants is critical because the microbiota shapes development of immunity and has effects on metabolism.1 The molecular mechanisms whereby this occurs remain obscure but bacterial products, such as fermentation metabolites, may affect host cells via epigenetic modifications.41 Using preterm pigs as a model for preterm infants, we have demonstrated that differences in bacterial colonization of the immature small intestine induce marked changes in gene expression that are regulated by epigenetic mechanisms. A delay in bacterial colonization, resulting from a relatively modest AB-induced reduction in bacterial density in the small intestine over the first 5 days, caused changes to DNA methylation for intestinal genes related to improved innate immune response, hypoxia-related vascular function and tissue metabolism. These apparent beneficial effects are targets to help prevent the immature intestine from detrimental responses to invading bacteria after preterm birth. Among the CON-reared pigs, the observed intestinal methylation and gene expression levels were similar for pigs with and without NEC lesions, indicating that even moderate differences in intestinal bacterial colonization and density (e.g. 106 versus 108 bacteria), not NEC lesions, was the main factor affecting methylation of genes in the immature intestine just after birth.
Preliminary evidence for a dynamic crosstalk between the intestinal methylome and bacterial colonization after birth has been derived from rodents.12,42 The preterm pig is the only model of preterm infants that combines a high sensitivity to intestinal disorders (NEC, feeding intolerance) with many other physiological signs of preterm birth (e.g. respiratory dysfunction, cardiovascular impairments, metabolic dysfunction).18 Recent studies have demonstrated a high similarity of both DNA methylation patterns and gut microbiomes between pigs and humans, supporting that pigs are highly relevant biomedical models for study of human diseases.43–45 In this study, we investigated sections of the distal small intestine because this intestinal region is most commonly affected by NEC lesions, highly populated with bacteria and critical for immune development and bacterial tolerance. Maladaptation to feeding and bacterial colonization in preterm neonates (e.g. NEC) involves all cell types and layers of the small intestine, hence we investigated whole tissue rather than isolated cell populations. Five days after preterm birth, when intestinal lesions were relatively mild in control pigs, the proportion of epithelial cells was similar in the two groups, as indicated by our staining of intestinal cross sections. The short-term AB treatment did not induce any marked change in the already low proportion of neutrophils in the epithelium, and in a previous study a similar AB treatment did not affect the proportion of intestinal goblet cells 7. The observed DNA methylation changes are therefore unlikely to result from microbiota-dependent changes in the relative cell proportions in the immature intestinal mucosa during the first 5 days after birth.
Microbial community dysbiosis is an important factor for neonatal gut health and preterm infants with a gut dominated by Firmicutes in the first days after birth may later develop NEC.19,20 In this study, the CON-reared pigs were also dominated by Firmicutes (Enterococcus and Clostridium) on day 5, while all AB-treated pigs were protected from NEC lesions and dominated by Enterobacter. Compared with CON pigs, LBP, IL-8 and C3 were reduced in the distal intestine of AB-treated pigs. LBP is a pattern recognition receptor transferring a variety of ligands from both Gram-positive and Gram-negative bacteria to the host through toll-like receptors (e.g. TLR2 and TLR4).46 The chemokine IL-8 can be induced both by TLR247 and TLR448 and recruits immune cells towards the site of infection. C3 labels the pathogen to facilitate phagocytosis to clear bacteria, including Enterococcus.49 Reduced expression of these genes indicates less need to mount an innate immune response in the AB-treated pigs.
Our results demonstrated clear effects of an AB-induced delay in bacterial colonization on intestinal DNA methylation and expression of selected genes. The integrative methylome–proteome analysis revealed that genes involved in bacterial infection, vasodilation-related pathways and metabolic pathways were most affected. These affected functions may be closely associated with differential degree of intestinal hypoxia, induced by variable innate immune response to invading bacteria. Normally, immune cell recruitment to sites of infection and induction of phagocytosis consume excessive oxygen, and induce angiogenesis and glycolysis to compensate for the oxygen deficit.30–32 Accordingly, we observed decreases in C3 and HIF1A transcription in the AB versus CON group. The suppressor of C3, CPN1, might be more expressed due to promoter hypomethylation in response to AB treatment. Similarly, MicroRNA-126 expression tended to be increased in the AB group, probably due to promoter hypomethylation. In mice and zebra fish, loss of MicroRNA-126 impacts endothelial cell proliferation and vascular integrity, resulting in fragile and leaky vessels.50,51 On the other hand, the intragenic DMR of PTPRE was hypomethylated in the AB group together with reduced expression levels. This may be explained by the general positive correlation between gene-body methylation and gene expression observed previously.52 PTPRE is highly abundant in endothelial cells and its down-regulation may indicate increased endothelial proliferation in AB versus CON pigs.39
Finally, our results indicated that AB treatment reduced glycolysis and increased gluconeogenesis-related gene expression in the AB group (Supplementary Fig. S8). These effects may be a direct consequence of the decreased tissue hypoxia and less need for activation of innate immune response to combat invading bacteria in the AB group. Effects on tissue metabolism were also supported by DNA methylation differences in genes related to metabolism. For example, the putative promoters of DHCR7 and TRMU showed hypomethylation in the AB group, indicating up-regulation of these two genes. DHCR7 encodes for 7-dehydrocholesterol reductase that produces cholesterol using NADPH, a cofactor used in anabolic reactions. Similarly, TRMU encodes for the mitochondrial tRNA-specific 2-thiouridylase 1 that is closely related with mitochondrial function.53 Thus, the potential up-regulation of DHCR7 and TRMU could be associated with active energy consumption in the intestine of AB pigs without hypoxic stress. In conclusion, a delay in bacterial colonization by oral AB treatment just after preterm birth may provide lower innate immune response, less hypoxic stress, better vascular integrity and increased metabolism in the immature intestine via epigenetic mechanisms.
Supplementary Material
Acknowledgements
We thank Thomas Thymann, Elin Skytte, Kristina Møller, Jane Povlsen and Karina Ryom for their technical support with animal procedures and laboratory analyses.
Funding
This study was supported by the Danish Strategic Research Council [NEOMUNE program, 12-132401], the Agricultural Science and Technology Innovation Program (ASTIP), and the China Scholarship Council [Scholarship No. 201406150073 to X.P.].
Data availability
All RRBS sequencing and processed data were deposited in the Gene Expression Omnibus (GEO) with accession GSE88697. Microbiome data have been submitted to DNA Data Bank of Japan (Accession number LC333600 to LC333727).
Conflict of interest
None declared.
Supplementary data
Supplementary data are available at DNARES online.
References
- 1. Castanys-Munoz E., Martin M.J., Vazquez E.. 2016, Building a beneficial microbiome from birth, Adv. Nutr., 7, 323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rook G.A. 2012, Hygiene hypothesis and autoimmune diseases, Clin. Rev. Allerg. Immunol., 42, 5–15. [DOI] [PubMed] [Google Scholar]
- 3. Beck S., Wojdyla D., Say L., et al. 2010, The worldwide incidence of preterm birth: a systematic review of maternal mortality and morbidity, Bull. World Health Org., 88, 31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neu J., Pammi M.. 2017, Pathogenesis of NEC: impact of an altered intestinal microbiome, Semin. Perinatol., 41, 29–35. [DOI] [PubMed] [Google Scholar]
- 5. Grylack L.J., Scanlon J.W.. 1978, Oral gentamicin therapy in the prevention of neonatal necrotizing enterocolitis. A controlled double-blind trial, Am. J. Dis. Child., 132, 1192–4. [DOI] [PubMed] [Google Scholar]
- 6. Egan E.A., Mantilla G., Nelson R.M., Eitzman D.V.. 1976, A prospective controlled trial of oral kanamycin in the prevention of neonatal necrotizing enterocolitis, J. Pediatr., 89, 467–70. [DOI] [PubMed] [Google Scholar]
- 7. Jensen M.L., Thymann T., Cilieborg M.S., et al. 2014, Antibiotics modulate intestinal immunity and prevent necrotizing enterocolitis in preterm neonatal piglets, Am. J. Physiol. Gastrointest. Liver Physiol., 306, G59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Birck M.M., Nguyen D.N., Cilieborg M.S., et al. 2016, Enteral but not parenteral antibiotics enhance gut function and prevent necrotizing enterocolitis in formula-fed newborn preterm pigs, Am. J. Physiol. Gastrointest. Liver Physiol., 310, G323–33. [DOI] [PubMed] [Google Scholar]
- 9. Paul B., Barnes S., Demark-Wahnefried W., et al. 2015, Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases, Clin. Epigenet., 7, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ye J., Wu W., Li Y., Li L.. 2017, Influences of the gut microbiota on DNA methylation and histone modification, Dig. Dis. Sci., 62, 1155–64. [DOI] [PubMed] [Google Scholar]
- 11. Reik W. 2007, Stability and flexibility of epigenetic gene regulation in mammalian development, Nature, 447, 425–32. [DOI] [PubMed] [Google Scholar]
- 12. Yu D.H., Gadkari M., Zhou Q., et al. 2015, Postnatal epigenetic regulation of intestinal stem cells requires DNA methylation and is guided by the microbiome, Genome Biol., 16, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hansen C.H., Holm T.L., Krych L., et al. 2013, Gut microbiota regulates NKG2D ligand expression on intestinal epithelial cells, Eur. J. Immunol., 43, 447–57. [DOI] [PubMed] [Google Scholar]
- 14. Schloss P.D., Westcott S.L., Ryabin T., et al. 2009, Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities, Appl. Environ. Microbiol., 75, 7537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gao F., Zhang J., Jiang P., et al. 2014, Marked methylation changes in intestinal genes during the perinatal period of preterm neonates, BMC Genomics, 15, 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xi Y., Li W.. 2009, BSMAP: whole genome bisulfite sequence MAPping program, BMC Bioinformatics, 10, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao F., Liang H., Lu H., et al. 2015, Global analysis of DNA methylation in hepatocellular carcinoma by a liquid hybridization capture-based bisulfite sequencing approach, Clin. Epigenet., 7, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sangild P.T., Thymann T., Schmidt M., Stoll B., Burrin D.G., Buddington R.K.. 2013, Invited review: the preterm pig as a model in pediatric gastroenterology, J. Anim. Sci., 91, 4713–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mai V., Young C.M., Ukhanova M., et al. 2011, Fecal microbiota in premature infants prior to necrotizing enterocolitis, PLoS ONE, 6, e20647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morrow A.L., Lagomarcino A.J., Schibler K.R., et al. 2013, Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants, Microbiome, 1, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benkoe T.M., Mechtler T.P., Weninger M., Pones M., Rebhandl W., Kasper D.C.. 2014, Serum levels of interleukin-8 and gut-associated biomarkers in diagnosing necrotizing enterocolitis in preterm infants, J. Pediatr. Surg., 49, 1446–51. [DOI] [PubMed] [Google Scholar]
- 22. Bergholz R., Zschiegner M., Eschenburg G., et al. 2013, Mucosal loss with increased expression of IL-6, IL-8, and COX-2 in a formula-feeding only neonatal rat model of necrotizing enterocolitis, J. Pediatr. Surg., 48, 2301–7. [DOI] [PubMed] [Google Scholar]
- 23. Neunhoeffer F., Jansen H., Goelz R., et al. 2015, Combination of excessive weight gain and interleukin-8: a possible predictor of necrotising enterocolitis in neonates?, Z Geburtshilfe Neonatol., 219, 221–5. [DOI] [PubMed] [Google Scholar]
- 24. Ferguson-Smith A.C. 2011, Genomic imprinting: the emergence of an epigenetic paradigm, Nat. Rev. Genet., 12, 565–75. [DOI] [PubMed] [Google Scholar]
- 25. Fang F., Hodges E., Molaro A., Dean M., Hannon G.J., Smith A.D.. 2012, Genomic landscape of human allele-specific DNA methylation, Proc. Natl Acad. Sci. USA, 109, 7332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao S., Zou D., Mao L., et al. 2015, SMAP: a streamlined methylation analysis pipeline for bisulfite sequencing, Gigascience, 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang P., Jensen M.L., Cilieborg M.S., et al. 2012, Antibiotics increase gut metabolism and antioxidant proteins and decrease acute phase response and necrotizing enterocolitis in preterm neonates, PLoS One, 7, e44929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Campbell W.D., Lazoura E., Okada N., Okada H.. 2002, Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N, Microbiol. Immunol., 46, 131–4. [DOI] [PubMed] [Google Scholar]
- 29. Kapushesky M., Emam I., Holloway E., et al. 2010, Gene expression atlas at the European bioinformatics institute, Nucleic Acids Res., 38, D690–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Colgan S.P., Taylor C.T.. 2010, Hypoxia: an alarm signal during intestinal inflammation, Nat. Rev. Gastroenterol. Hepatol., 7, 281–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krock B.L., Skuli N., Simon M.C.. 2011, Hypoxia-induced angiogenesis: good and evil, Genes Cancer, 2, 1117–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zeitouni N.E., Chotikatum S., von Kockritz-Blickwede M., Naim H.Y.. 2016, The impact of hypoxia on intestinal epithelial cell functions: consequences for invasion by bacterial pathogens, Mol. Cell. Pediatr., 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rius J., Guma M., Schachtrup C., et al. 2008, NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha, Nature, 453, 807–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pugh C.W., Ratcliffe P.J.. 2003, Regulation of angiogenesis by hypoxia: role of the HIF system, Nat. Med., 9, 677–84. [DOI] [PubMed] [Google Scholar]
- 35. Yan X., Managlia E., Liu S.X., et al. 2016, Lack of VEGFR2 signaling causes maldevelopment of the intestinal microvasculature and facilitates necrotizing enterocolitis in neonatal mice, Am. J. Physiol. Gastrointest. Liver Physiol., 310, G716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dudzinski D.M., Michel T.. 2007, Life history of eNOS: partners and pathways, Cardiovasc. Res., 75, 247–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schlossmann J., Desch M.. 2011, IRAG and novel PKG targeting in the cardiovascular system, Am. J. Physiol. Heart Circ. Physiol., 301, H672–82. [DOI] [PubMed] [Google Scholar]
- 38. Herbert S.P., Stainier D.Y.. 2011, Molecular control of endothelial cell behaviour during blood vessel morphogenesis, Nat. Rev. Mol. Cell Biol., 12, 551–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thompson L.J., Jiang J., Madamanchi N., Runge M.S., Patterson C.. 2001, PTP-epsilon, a tyrosine phosphatase expressed in endothelium, negatively regulates endothelial cell proliferation, Am. J. Physiol. Heart Circ. Physiol., 281, H396–403. [DOI] [PubMed] [Google Scholar]
- 40. Sinha R.K., Yang X.V., Fernandez J.A., Xu X., Mosnier L.O., Griffin J.H.. 2016, Apolipoprotein E receptor 2 mediates activated protein C-induced endothelial Akt activation and endothelial barrier stabilization, Arterioscler. Thromb. Vasc. Biol., 36, 518–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagy-Szakal D., Kellermayer R.. 2011, The remarkable capacity for gut microbial and host interactions, Gut Microbes, 2, 178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cortese R., Lu L., Yu Y., Ruden D., Claud E.C.. 2016, Epigenome-microbiome crosstalk: a potential new paradigm influencing neonatal susceptibility to disease, Epigenetics, 11, 205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xiao L., Estelle J., Kiilerich P., et al. 2016, A reference gene catalogue of the pig gut microbiome, Nat. Microbiol., 1, 16161. [DOI] [PubMed] [Google Scholar]
- 44. Choi M., Lee J., Le M.T., et al. 2015, Genome-wide analysis of DNA methylation in pigs using reduced representation bisulfite sequencing, DNA Res., 22, 343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schachtschneider K.M., Madsen O., Park C., Rund L.A., Groenen M.A., Schook L.B.. 2015, Adult porcine genome-wide DNA methylation patterns support pigs as a biomedical model, BMC Genomics, 16, 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schroder N.W., Heine H., Alexander C., et al. 2004, Lipopolysaccharide binding protein binds to triacylated and diacylated lipopeptides and mediates innate immune responses, J. Immunol., 173, 2683–91. [DOI] [PubMed] [Google Scholar]
- 47. Thornton N.L., Cody M.J., Yost C.C.. 2012, Toll-like receptor 1/2 stimulation induces elevated interleukin-8 secretion in polymorphonuclear leukocytes isolated from preterm and term newborn infants, Neonatology, 101, 140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wheeler D.S., Chase M.A., Senft A.P., Poynter S.E., Wong H.R., Page K.. 2009, Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4, Respir. Res., 10, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leendertse M., Willems R.J., Flierman R., de Vos A.F., Bonten M.J., van der Poll T.. 2010, The complement system facilitates clearance of Enterococcus faecium during murine peritonitis, J. Infect. Dis., 201, 544–52. [DOI] [PubMed] [Google Scholar]
- 50. Fish J.E., Santoro M.M., Morton S.U., et al. 2008, miR-126 regulates angiogenic signaling and vascular integrity, Dev. Cell., 15, 272–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang S., Aurora A.B., Johnson B.A., et al. 2008, The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis, Dev. Cell, 15, 261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ball M.P., Li J.B., Gao Y., et al. 2009, Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells, Nat. Biotechnol., 27, 361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Armengod M.E., Meseguer S., Villarroya M., et al. 2014, Modification of the wobble uridine in bacterial and mitochondrial tRNAs reading NNA/NNG triplets of 2-codon boxes, RNA Biology, 11, 1495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RRBS sequencing and processed data were deposited in the Gene Expression Omnibus (GEO) with accession GSE88697. Microbiome data have been submitted to DNA Data Bank of Japan (Accession number LC333600 to LC333727).