

Abstract
Metformin is the first-line treatment for type 2 diabetes (T2D). Although widely prescribed, the glucose-lowering mechanism for metformin is incompletely understood. Here, we used a genome-wide association approach in a diverse group of individuals with T2D from the Action to Control Cardiovascular Risk in Diabetes (ACCORD) clinical trial to identify common and rare variants associated with HbA1c response to metformin treatment and followed up these findings in four replication cohorts. Common variants in PRPF31 and CPA6 were associated with worse and better metformin response, respectively (P < 5 × 10−6), and meta-analysis in independent cohorts displayed similar associations with metformin response (P = 1.2 × 10−8 and P = 0.005, respectively). Previous studies have shown that PRPF31(+/−) knockout mice have increased total body fat (P = 1.78 × 10−6) and increased fasted circulating glucose (P = 5.73 × 10−6). Furthermore, rare variants in STAT3 associated with worse metformin response (q <0.1). STAT3 is a ubiquitously expressed pleiotropic transcriptional activator that participates in the regulation of metabolism and feeding behavior. Here, we provide novel evidence for associations of common and rare variants in PRPF31, CPA6, and STAT3 with metformin response that may provide insight into mechanisms important for metformin efficacy in T2D.
Introduction
The incidence of type 2 diabetes (T2D) is increasing and the Centers for Disease Control and Prevention estimate that 11% of U.S. adults, aged 20 years or older, have diagnosed or undiagnosed T2D, and 35% of people in the same age-group have prediabetes based on fasting glucose or hemoglobin A1c (HbA1c) levels (1). T2D is now considered a global epidemic with prevalence increasing from 108 million in 1980 to 422 million in 2014 (2). Individuals with T2D are at an increased risk of developing blindness and kidney failure and are at risk for lower-limb amputations. Furthermore, individuals with T2D are 2–4 times more likely to develop cardiovascular disease (CVD), including heart attack and stroke (3).
Metformin, a member of the biguanide class of drugs, is now considered first-line therapy for individuals with T2D (4). Despite intensive investigation, the molecular mechanisms mediating metformin’s beneficial effects on glycemic control remain controversial and poorly understood. Studies performed predominantly in animal models have implicated various mechanisms and molecules as participating in metformin’s beneficial effects, including direct inhibitory effects on mitochondrial function, activation of hepatic AMPK, and alterations in glucagon signaling pathways (5–10). The relative importance of these mechanisms for metformin’s beneficial effects on glycemic control in humans is unclear.
There is significant interindividual variability in response to metformin (11–13). This suggests that a better understanding of the mechanisms by which metformin functions might allow for more tailored and precise treatment. Studies suggest that heritable factors contribute to this variability, which provides an opportunity to identify causal genetic contributors through genetic association studies (11). Recent genome-wide association studies (GWAS) have identified common variants that affected metformin response in the ATM locus, which has been shown to activate AMPK, and the SLC2A2 locus, which encodes the facilitated glucose transporter, GLUT2 (12,13). Here, we use a GWAS approach in a large cohort of individuals with T2D in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) clinical trial to test for associations of both common and rare variant single nucleotide polymorphisms (SNPs) with change in HbA1c in response to metformin treatment.
The ACCORD trial followed 10,251 participants for up to 8 years at 77 clinical centers in the U.S. and Canada to compare the benefits and risks of treatment strategies for intensively targeting glycemia, blood pressure, and dyslipidemia versus standard targets in individuals with T2D at high risk for CVD (14–17). No overall benefit and possible harms were observed in the combined primary CVD end points with intensive glucose-lowering therapy. However, significant variability in treatment response was also observed, highlighting the potential for identifying genetic markers of drug response that may lead to the development of improved and personalized treatment strategies. The results presented here point to novel mechanisms of metformin drug response and potential therapeutic targets for the treatment of T2D.
Research Design and Methods
Study Participants
The ACCORD trial (ClinicalTrials.gov identifier: NCT00000620) was a double 2 × 2 factorial design comparing intensive versus standard treatment approaches for controlling glycemia, blood pressure, and dyslipidemia that enrolled 10,251 patients with T2D with a history of CVD or at least two known risk factors for CVD, such as documented atherosclerosis, albuminuria, dyslipidemia, hypertension, smoking, or obesity (14). Additional details about randomization and selection criteria for the various arms of the ACCORD trial can be found in the Supplementary Data. Participants in ACCORD were given an option to provide a blood sample for future genetic studies, and over 80% of participants agreed to do so. A workflow describing the selection of subjects can be found in Fig. 1. Additional demographic information can be found in Table 1.
Figure 1.

Study design and quality control (QC) workflow. Boxes represent the steps taken in the workflow. The number of remaining subjects after a quality control step can be found in the relevant attached circle.
Table 1.
Cohort demographics and concomitant medications
| All races combined (N = 1,312) | White subjects only (N = 845) | Black subjects only (N = 222) | |
|---|---|---|---|
| Intensive glycemia arm |
56.33 |
59.41 |
53.15 |
| Standard glycemia arm |
43.67 |
40.59 |
46.85 |
| Intensive blood pressure arm |
23.86 |
23.67 |
34.23 |
| Standard blood pressure arm |
23.78 |
21.89 |
33.33 |
| Fibrate lipid treatment arm |
25.99 |
25.68 |
14.41 |
| Placebo lipid treatment arm |
26.37 |
28.76 |
18.01 |
| Female |
39.48 |
36.33 |
52.70 |
| Age (mean [95% CI]) |
62.72 [62.36, 63.10] |
63.14 [62.70, 63.58] |
62.25 [61.37, 63.12] |
| BMI (mean [95% CI]) |
32.43 [32.13, 32.73] |
33.18 [32.82, 33.54] |
32.67 [31.94, 33.39] |
| Years with T2D (mean [95% CI]) |
10.10 [9.67, 10.51] |
9.73 [9.23, 10.24] |
10.82 [9.76, 11.87] |
| History of cardiovascular disease |
33.54 |
35.27 |
29.28 |
| Concomitant medications* | |||
| Angiotensin type 2 antagonists |
16.39 |
15.98 |
16.67 |
| ACE inhibitors |
46.19 |
46.98 |
52.25 |
| α-Glucosidase inhibitors |
0.53 |
0.47 |
0.45 |
| Cholesterol absorption inhibitors |
2.13 |
2.49 |
1.80 |
| Statin |
49.70 |
51.83 |
30.18 |
| Lisinopril |
16.46 |
16.45 |
25.23 |
| Loop diuretics |
8.23 |
8.99 |
10.36 |
| Meglitinides |
2.97 |
2.37 |
3.15 |
| Nitrates |
5.72 |
6.51 |
3.60 |
| Other diabetes treatments |
2.67 |
2.84 |
4.05 |
| Sulfonylureas |
52.44 |
51.83 |
45.05 |
| Thiazolidinediones |
31.63 |
33.02 |
29.28 |
| Insulin | 38.11 | 38.11 | 45.95 |
Data are %, unless stated otherwise.
*Tabulated percentages represent the percentage of subjects with at least one record of taking the medication or a medication in the drug class during the metformin treatment window. This is not an exhaustive list of medications recorded in ACCORD, but rather a representative list of the commonly used medications in ACCORD.
Phenotype Definitions
Glycemic response to metformin was evaluated in subjects who began taking metformin while enrolled in ACCORD; subjects who reported taking metformin or another biguanide prior to enrollment in the trial were excluded. Some subjects were taking additional medications and were accounted for as described in the Supplementary Data and Graham et al. (18). Subjects were scheduled for study visits every 1 or 4 months based on randomization to the intensive or standard glycemia arms, respectively. HbA1c was recorded every 4 months. Metformin response was calculated as on-treatment HbA1c minus pretreatment HbA1c. Pretreatment HbA1c levels were recorded no more than 30 days prior to the start of metformin. On-treatment HbA1c levels were defined as the first recorded measurement acquired after at least 90 days and no more than 270 days from the start of metformin treatment. This interval was chosen to allow for HbA1c levels to stabilize after starting metformin, while limiting changes in glycemia levels due to additional modifications to treatment regimens. Medication compliance and other details regarding the phenotype definition can be found in the Supplementary Data.
Genotyping
Briefly, 6,085 unique samples from ACCORD participants who consented to genetic studies conducted by any investigator were genotyped at the University of Virginia on Illumina HumanOmniExpressExome-8 v1.0 chips (Set 1); 8,174 unique samples, including the above 6,085 samples plus 2,089 samples from ACCORD participants who consented to genetic studies only if conducted by ACCORD investigators, were genotyped at the University of North Carolina on Affymetrix Axiom Biobank1 chips (Set 2). The data were then merged, resulting in one data set consisting of 5,971 samples genotyped at a total of 1,240,656 individual SNPs, and another with an additional 2,083 samples genotyped at 583,613 SNPs. Additional details can be found in the Supplementary Data and Marvel et al. (19).
Data Processing
Covariate Selection
We implemented a variable selection procedure to address potential confounding. Some variables were included in the model based on previous studies or expert knowledge, whereas other variables were selected based on a backward selection approach and Bayesian information criteria to identify covariates specific to the ACCORD data set. All covariate names and descriptions before variable selection can be found in Supplementary Table 1. A substantial proportion of the cohort was taking other medications during the metformin treatment response time frame and these concomitant medications were incorporated into the model as described in the Supplementary Data. To prevent confounding due to population substructure, principal component analysis was performed based on the genotype data using EIGENSTRAT (v4.2) and is described in the Supplementary Data. The final models after covariate selection can be found in Supplementary Tables 2–4. A workflow detailing each step of the analysis can be found in Supplementary Fig. 4.
Common Variant Analysis
Associations between a phenotype, covariates, and single common variant (minor allele frequency [MAF] >3%) were tested using the linear regression model. Genotyped variants were tested as an additive variable using PLINK, where 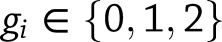 is the number of minor alleles for the
is the number of minor alleles for the 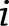 th individual. Imputed variants were tested using a linear regression model in the statistical programming language, R, where
th individual. Imputed variants were tested using a linear regression model in the statistical programming language, R, where 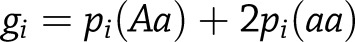 is the dosage score computed from the posterior probabilities for genotypes
is the dosage score computed from the posterior probabilities for genotypes 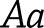 and
and 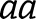 (20). For SNPs that were only genotyped in Set 1 subjects and were imputed in Set 2 subjects, association tests were calculated for each set separately and the results were combined by meta-analysis using PLINK (21,22). Tables and figures specify whether each result derives from a SNP that was genotyped in all subjects, imputed in all subjects, or represents a combination of genotyped and imputed data using the meta-analysis approach described.
(20). For SNPs that were only genotyped in Set 1 subjects and were imputed in Set 2 subjects, association tests were calculated for each set separately and the results were combined by meta-analysis using PLINK (21,22). Tables and figures specify whether each result derives from a SNP that was genotyped in all subjects, imputed in all subjects, or represents a combination of genotyped and imputed data using the meta-analysis approach described.
Rare Variant Analysis
The rare variant analysis approach has been previously described (19). Briefly, all variants (MAF ≤3%) were mapped to gene annotation from Ensemble (GRCh37.p13). A suite of five rare variant tests comprised of burden and nonburden were then used to assess associations with metformin response. We combined the set of five P values from each test into a single P value for each gene using the procedure described by Dai et al. (23). Subsequently, the combined P value was corrected for multiple comparisons with a false discovery rate approach (24,25), and a threshold of q <0.1 was used for statistical significance.
Replication of ACCORD Results
SNPs that associated with metformin response in the common variant analyses (P < 5 × 10−6) were tested for associations in the following cohorts. There was one cohort from European ancestry, which included combined samples from Genetics of Diabetes Audit and Research in Tayside Scotland (GoDARTS) and PMET1-EU (Pharmacogenomics of Metformin, cohort 1, of European ancestry; PMET1 was previously named PMT2) (REP1) (n = 6,963). There were two cohorts from African American ancestry, referred to as REP2 (n = 646) and REP3 (n = 369). Briefly, the GoDARTS cohort in REP1 has been described before by the Wellcome Trust Case Control Consortium 2 (13). The REP2 cohort was collected from two main clinical sites, Kaiser Permanente Northern California and Kaiser Permanente South East, as previously described (26). The PMET1-EU in REP1 and the REP3 cohorts were collected from the Research Program on Genes, Environment and Health (RPGEH), based at Kaiser Permanente Northern California (27). Metformin response for REP1 was defined as pretreatment HbA1c minus posttreatment HbA1c, resulting in effects that are in the opposite direction of those in ACCORD. Because of this, the additive inverse of effect sizes in REP1 was used in the analysis with ACCORD results. For REP2 and REP3, metformin response was calculated as posttreatment HbA1c minus pretreatment HbA1c.
To provide additional support for the role of the lead SNP, rs57081354, in modulating metformin action, we performed an association test between rs57081354 and baseline HbA1c in ACCORD individuals that were already on metformin at the start of the trial (REP4) and compared those results to the association of baseline HbA1c in individuals not on metformin at the start of the trial. All individuals in REP4 were excluded from the discovery cohort due to the lack of a recorded pretreatment HbA1c, making it impossible to calculate a change in HbA1c with treatment. For this analysis, an association test was performed as described in the common variant analysis with the exception of the phenotype being baseline level of HbA1c rather than change in HbA1c.
Each SNP with P < 5 × 10−8 in the discovery cohort or with P < 5 × 10−6 in the discovery cohort and q <0.01 in at least one replication cohort were meta-analyzed across available cohorts using PLINK (21,22). Meta-analysis with P < 0.05 was considered to be statistically significant. Additional information regarding each cohort can be found in the Supplementary Data, and a workflow describing how the replication was evaluated can be found in Supplementary Fig. 4.
Dietary Analysis
Dietary questionnaires were assessed for 1,600 participants that both consented to genotyping and had dietary records as part of the Health-Related Quality of Life substudy within ACCORD. Scores in response to the dietary questionnaires were tested for association with the lead SNP, rs57081354, using a linear regression model and the same covariate selection process as described above. Details regarding this analysis can be found in the Supplementary Data.
Results
A total of 1,312, 845, and 222 subjects were included in the common and rare variant analyses for all races combined, white, and black cohorts, respectively (Fig. 1). Other racial groups were included in the all races combined group, but sample sizes were too small to perform stratified analyses. Variation was observed in HbA1c response (Fig. 2). The mean change in HbA1c was −1.42% (95% CI −1.49, −1.36) for all races combined, −1.47% (−1.55, −1.39) for white subjects only, and −1.35% (−1.50, −1.19) for black subjects only. The difference in the change in HbA1c between white and black subjects was not statistically significant (P = 0.16). Variables selected as being significantly associated with metformin response explained approximately 55% of the variation in metformin response and were subsequently included in the common and rare variant analyses (Supplementary Tables 2–4). Briefly, assignment to the intensive glycemia arm of the ACCORD trial, years with T2D, and pretreatment HbA1c were significantly associated with metformin response in all tested groups.
Figure 2.

Distributions of HbA1c response to metformin treatment. A: HbA1c response when all races are combined. B: HbA1c response in only white participants. C: HbA1c response in only black participants.
Common Variant Analysis
A total of 316,203 genotyped, 547,639 meta-analyzed, and 7,567,403 imputed variants had MAF >3% and were included in the common variant analysis when all races were combined. A total of 1, 3, and 7 SNPs were significantly associated with HbA1c response in all races combined, white subjects only, and black subjects only, respectively (P < 5 × 10−8). In all subjects combined, the lead SNP rs57081354 located within an intron in the NBEA gene associated with HbA1c response (Fig. 3). Sixty-five SNPs in 16 loci on 14 different chromosomes (in genes NBEA, LOC105374140, CXCL13, MCC, FAM189A1, LOC107984525, LOC105379109, KIAA1024, RBFOX1, LOC643339, UBOX5, NR2C1, BPHL, PRPF31, LOC105377165, and ADAMTS9-AS2) reached the threshold for suggestive significance (P < 5 × 10−6). For white subjects only, 3 SNPs in KIAA1024 were associated with HbA1c response at genome-wide levels of significance (P < 5 × 10−8), and 23 SNPs in 7 loci (in genes KIAA1024, LOC283177, LOC105377165, ADAMTS9-AS2, CPA6, NR2C1, and DPYD) reached the threshold for suggestive significance (P < 5 × 10−6) (Supplementary Fig. 1). When black subjects were analyzed separately, 2 SNPs in the LOC102724874 locus were associated with HbA1c response at genome-wide levels (P < 5 × 10−8), and 53 SNPs in 8 loci (in genes LOC102724874, LOC105374308, LOC105369406, SLC35D3, RHPN2, ETS1, SDK2, and GPC6) reached the threshold for suggestive significance (P < 5 × 10−6) (Supplementary Fig. 2). Lead SNPs associated with HbA1c response (P < 5 × 10−6) are presented in Tables 2–4.
Figure 3.

HbA1c response to metformin treatment is diminished by SNPs in NBEA. A: Manhattan plot of HbA1c response to metformin treatment when all races were combined (N = 1,312). The horizontal dashed lines represent suggestive statistical significance (P = 5 × 10−6) (bottom line) and genome-wide significance (P = 5 × 10−8) (top line). Each point represents a single SNP. Genotyped SNPs are represented with black dots, imputed SNPs are represented by blue dots, and SNPs meta-analyzed from subjects genotyped in Set 1 subjects and imputed in Set 2 subjects are represented by pink dots. B: A LocusZoom (48) plot demonstrating the lead SNP, rs57081354, association with change in HbA1c when all races are combined. Linkage disequilibrium was calculated using the 1000 Genomes reference panel using the hg19/1000 Genomes Nov 2014 EUR selection, matching the panel used to perform imputation in ACCORD. C: Forest plot demonstrating associations of rs57081354 in available replication cohorts. The β values for REP1 were changed to the additive inverse to maintain a consistent direction of effect. A larger β value means a diminished metformin response. Data for rs57081354 were not available for REP2 and REP3. Bars represent the 95% CIs. D: Change in HbA1c by rs57081354 genotype as represented by violin plots.
Table 2.
Lead SNPs associated with HbA1c response in common variant analysis in all races combined (P < 5 × 10−6)
| SNP | Chromosome | Position | Gene | Type | MAF | β | P value |
|---|---|---|---|---|---|---|---|
| rs7371352 |
2 |
205916609 |
NA |
IMPU |
0.461 |
0.166 |
1.29E-06 |
| rs12694529 |
2 |
220685415 |
NA |
META |
0.131 |
0.232 |
1.06E-06 |
| rs2371651 |
3 |
64899735 |
ADAMTS9-AS2 |
IMPU |
0.346 |
0.171 |
4.89E-06 |
| rs36050186 |
3 |
73884807 |
LOC105377165 |
IMPU |
0.061 |
0.315 |
4.46E-06 |
| rs28535480 |
3 |
144238665 |
LOC105374140 |
IMPU |
0.447 |
0.184 |
4.12E-07 |
| rs7669299 |
4 |
77525989 |
CXCL13 |
GENO |
0.106 |
−0.348 |
5.88E-07 |
| rs79827403 |
4 |
140248372 |
NA |
IMPU |
0.046 |
−0.386 |
2.19E-06 |
| rs6884548 |
5 |
104706194 |
LOC105379109 |
IMPU |
0.038 |
0.439 |
2.64E-06 |
| rs56061109 |
5 |
113100738 |
MCC |
IMPU |
0.048 |
0.394 |
6.00E-07 |
| rs6903843 |
6 |
3125135 |
BPHL |
IMPU |
0.031 |
0.587 |
3.36E-06 |
| rs79573377 |
6 |
91226278 |
NA |
IMPU |
0.033 |
0.437 |
3.19E-06 |
| rs59506474 |
6 |
136927217 |
NA |
IMPU |
0.066 |
−0.355 |
1.57E-06 |
| rs2578120 |
10 |
78453110 |
NA |
IMPU |
0.358 |
−0.160 |
2.53E-06 |
| rs7307035 |
12 |
53778761 |
LOC107984525 |
META |
0.033 |
−0.440 |
1.77E-06 |
| rs12300320 |
12 |
93077216 |
LOC643339 |
IMPU |
0.053 |
−0.386 |
3.01E-06 |
| rs12827634 |
12 |
95026750 |
NR2C1 |
IMPU |
0.362 |
0.154 |
3.18E-06 |
| rs57081354 |
13 |
35202456 |
NBEA |
GENO |
0.077 |
0.326 |
4.02E-08 |
| rs4144603 |
14 |
96729576 |
NA |
META |
0.405 |
0.159 |
1.36E-06 |
| rs7173199 |
15 |
29424654 |
FAM189A1 |
IMPU |
0.046 |
0.419 |
1.77E-06 |
| rs182384419 |
15 |
79438983 |
KIAA1024 |
IMPU |
0.043 |
0.394 |
2.66E-06 |
| rs74007109 |
16 |
6852072 |
RBFOX1 |
IMPU |
0.040 |
0.407 |
2.92E-06 |
| rs2529698 |
17 |
12350355 |
NA |
META |
0.061 |
0.331 |
9.24E-07 |
| rs254271 |
19 |
54630757 |
PRPF31 |
IMPU |
0.330 |
0.163 |
3.79E-06 |
| rs6139020 | 20 | 3156571 | UBOX5 | IMPU | 0.102 | 0.257 | 3.08E-06 |
Table 4.
Lead SNPs associated with HbA1c response in common variant analysis in black subjects only (P < 5 × 10−6)
| SNP | Chromosome | Position | Gene | Type | MAF | β | P value |
|---|---|---|---|---|---|---|---|
| rs7573365 |
2 |
183278246 |
NA |
META |
0.325 |
−0.379 |
3.67E-06 |
| rs12496540 |
3 |
32062976 |
NA |
IMPU |
0.352 |
0.388 |
4.98E-06 |
| rs62284968 |
3 |
197429996 |
LOC105374308 |
IMPU |
0.040 |
1.066 |
1.20E-07 |
| rs9295608 |
6 |
23977533 |
NA |
GENO |
0.208 |
−0.462 |
1.35E-06 |
| rs111792250 |
6 |
136927930 |
SLC35D3a |
IMPU |
0.192 |
−0.528 |
7.97E-07 |
| rs4413694 |
7 |
142430649 |
NA |
IMPU |
0.458 |
−0.346 |
4.43E-06 |
| rs201739756 |
8 |
73149128 |
NA |
IMPU |
0.054 |
0.875 |
2.04E-06 |
| rs145554029 |
8 |
77485920 |
LOC102724874b |
IMPU |
0.035 |
−1.221 |
8.76E-09 |
| rs10897557 |
11 |
80135250 |
LOC105369406 |
IMPU |
0.319 |
−0.432 |
6.73E-07 |
| rs7130041 |
11 |
128552747 |
ETS1 |
IMPU |
0.348 |
−0.406 |
3.02E-06 |
| rs76853142 |
13 |
93580057 |
GPC6 |
IMPU |
0.041 |
−0.940 |
4.54E-06 |
| rs1449836 |
14 |
42994192 |
NA |
META |
0.471 |
−0.353 |
1.43E-06 |
| rs62072150 |
17 |
73412120 |
SDK2 |
IMPU |
0.451 |
−0.423 |
4.12E-06 |
| rs185341538 |
19 |
32981426 |
RHPN2 |
IMPU |
0.291 |
−0.428 |
2.99E-06 |
| rs34600526 | 19 | 33504770 | NA | IMPU | 0.295 | −0.395 | 4.49E-06 |
ars111792250 does not fall in SLC35D3, but rs7763578 (P = 1.47 × 10−6) is in the same peak and is located in SLC35D3.
brs145554029 does not fall in LOC102724874, but rs75121529 (P = 1.22 × 10−8) is in the same peak and is located in LOC102724874.
Table 3.
Lead SNPs associated with HbA1c response in common variant analysis in white subjects only (P < 5 × 10−6)
| SNP | Chromosome | Position | Gene | Type | MAF | β | P value |
|---|---|---|---|---|---|---|---|
| rs12047072 |
1 |
97401162 |
DPYD |
META |
0.184 |
−0.215 |
4.66E-06 |
| rs13034779 |
2 |
234091216 |
NA |
IMPU |
0.305 |
0.239 |
6.24E-07 |
| rs2371651 |
3 |
64899735 |
ADAMTS9-AS2 |
IMPU |
0.431 |
0.192 |
3.71E-06 |
| rs36050186 |
3 |
73884807 |
LOC105377165 |
IMPU |
0.082 |
0.332 |
2.47E-06 |
| rs71140472 |
3 |
144248959 |
NA |
IMPU |
0.461 |
0.177 |
3.30E-06 |
| rs2929535 |
8 |
67754375 |
CPA6a |
META |
0.307 |
−0.188 |
3.47E-06 |
| rs111976192 |
8 |
83716899 |
NA |
IMPU |
0.267 |
0.195 |
4.99E-06 |
| rs10904150 |
10 |
3970713 |
NA |
IMPU |
0.072 |
−0.336 |
4.66E-06 |
| rs4567482 |
11 |
134474052 |
LOC283177 |
META |
0.123 |
−0.286 |
1.66E-07 |
| rs7298631 |
12 |
95063515 |
NR2C1 |
IMPU |
0.397 |
0.176 |
4.51E-06 |
| rs4775474 |
15 |
62150069 |
NA |
META |
0.355 |
0.189 |
2.07E-06 |
| rs182384419 | 15 | 79438983 | KIAA1024 | IMPU | 0.054 | 0.503 | 1.53E-08 |
ars2929535 does not fall in CPA6, but rs2162145 (P = 4.04 × 10−6) is in the same peak and is located in CPA6.
Rare Variant Analysis
Rare variants (MAF ≤0.03) in a total of 17,078 genes were tested for association with HbA1c in all subjects combined and separately in white and black subjects. In the combined subject analysis, rare variants in STAT3 were significantly associated with HbA1c response (q <0.1) (Supplementary Fig. 3). Five rare variants were available for testing in STAT3: rs146620441 (monomorphic), rs140604473 (missense), rs149214040 (missense), rs114401618 (intronic), and rs17882069 (synonymous) (28). Importantly, rs146620441 was monomorphic and the other four SNPs only showed variation in black subjects, although there were no statistically significant rare variant associations observed in race-based analyses (q >0.2). We tested whether STAT3 failed to yield significant associations in the stratified analysis due to the small number of black subjects (n = 222) by oversampling black subjects until we had the same number of subjects as when all races were combined (n = 1,312). Oversampling to build a larger cohort resulted in a q = 0.0016 versus a q = 1 in the original cohort of black subjects, supporting the premise that this rare variant finding is relevant for black subjects even though it was detected when all subjects were combined due to an increase in statistical power.
Association With Dietary Phenotypes
NBEA has been previously reported to influence sugary food preference in animal models (29), and we further assessed whether the lead SNP in this locus, rs57081354, was associated with dietary scores. Data were available in 1,553 ACCORD subjects (Supplementary Table 6). The C-allele in rs57081354 that associated with worse metformin effect also associated with increased consumption of dessert (q = 0.12). Subjects with the C-allele in rs57081354 were more likely to answer “Yes” to the question, “Did you eat dessert?” within past 3 months (β = 0.044 [95% CI 0.012, 0.075]).
Replication
A total of 134 unique SNPs were associated with metformin response (P < 5 × 10−6) when all races were combined, in white subjects only, and in black subjects only. Of these SNPs, 81, 64, and 61 were available in REP1, REP2, and REP3 cohorts, respectively. The most significant finding in the discovery cohort was rs57081354 located in the NBEA gene (P = 4.02 × 10−8, β = 0.323). This finding did not replicate in REP1 (P > 0.05) and was not available for testing in REP2 or REP3. Out of the 26 SNPs in NBEA with P < 1 × 10−6, only rs1337379 was tested in REP2 and REP3 but was not associated with metformin response (P > 0.05). SNPs in NBEA were not significantly associated with metformin response in ACCORD when black or white subjects were tested separately, only when all races were combined. Interestingly, in the set of subjects who were already on metformin when they enrolled in ACCORD (REP4), rs57081354, was marginally associated with higher baseline HbA1c (P = 0.09), whereas rs57081354 was not associated with baseline variation in HbA1c in those not on metformin (P = 0.63). Although the phenotypes for REP1 and REP4 are not identical, meta-analysis of the ACCORD discovery cohort with REP1 and REP4 produced an association representing higher HbA1c on metformin (P = 0.009) (Fig. 3).
In ACCORD, rs254271, located in an intron in pre-mRNA processing factor 31 (PRPF31), was associated with worse metformin response when all races were combined (P = 3.79 × 10−6). In the REP1 cohort, rs254271 was associated with worse metformin response when corrected for baseline HbA1c (P = 6.21 × 10−5) and without adjusting for baseline HbA1c (P = 1.57 × 10−2). Genotypes for rs254271 were not available for analysis in the African American replication cohorts, REP2 and REP3. Meta-analysis of ACCORD with REP1 produced a significant association for rs254271 with worse metformin response (P = 1.2 × 10−8) (Fig. 4).
Figure 4.

Forest plot of associations with replicated SNPs in REP1, REP2, and REP3. A: Discovery and replication results for rs2162145, located in the CPA6 gene. B: Discovery and replication results for rs254271, an intronic variant in PRPF31. A larger β value means a diminished metformin response. The β values for REP1 were changed to the additive inverse to maintain a consistent direction of effect. Bars represent the 95% CIs.
In white subjects in ACCORD, rs2162145, located in carboxypeptidase A6 (CPA6), was associated with better metformin response (P = 4.04 × 10−6). This finding was consistent with findings in African American subjects in REP2 (P = 0.006). Although, rs2162145 was not significantly associated with metformin response in black subjects in ACCORD (P = 0.14), it is possible that we were underpowered to detect an association due to the relatively small number of black subjects available (n = 222) and that meta-analysis could increase statistical power through the inclusion of additional subjects. Meta-analysis of black subjects in ACCORD and those in REP2 and REP3 demonstrated the most significant association of rs2162145 with better metformin response (P = 0.005) compared with meta-analysis of all white subjects (ACCORD white subjects and REP1) and all cohorts (ACCORD, REP1–3) with P = 0.22 and P = 0.09, respectively (Fig. 4).
Discussion
T2D adversely affects the quality of life for millions of individuals and places a significant burden on the health care system in the U.S. and globally (30,31). Although many treatment options for T2D are available, metformin has remained the first-line treatment for T2D for decades. Nevertheless, the mechanisms by which metformin lowers blood glucose are not well understood, and factors that may predict which patients will have optimal responses to metformin are also not well understood. Here, we use a GWAS approach in a large cohort of individuals with T2D in the ACCORD clinical trial to test for both common and rare variant SNPs that associate with change in HbA1c level in response to metformin treatment. To support our initial findings, we explored these associations in multiple independent cohorts.
A previous study conducted in a large cohort using electronic medical records found that African American patients on metformin had a larger reduction in HbA1c than European Americans (32). We failed to find a significant difference between metformin response in white subjects and black subjects in ACCORD, with changes in HbA1c of −1.47% and −1.35%, respectively (P = 0.16). It is possible that enrollment in a more structured clinical trial impacts these outcomes differently than the far less structured care reflected in electronic medical record data. Additional research will be needed to definitely characterize the response of metformin across ethnicities. Previous GWAS have identified rs11212617 in the ATM locus and rs8192675 in SLC2A2 as being significantly associated with metformin response (12,13). However, neither SNP was significantly associated with metformin response here (P > 0.1). These SNPs may have failed to replicate in the current study due to cohort differences and, in particular, the sample size of the ACCORD cohort and the use of concomitant medications. Information regarding the prescribed dose for metformin was not recorded in ACCORD and may have impacted our findings. However, as described in research design and methods, we sought a minimal time window to assess the response of metformin while limiting the likelihood of other medication changes. Additional details about how these studies compare with ACCORD can be found in the Supplementary Data.
In the analysis of subjects from all racial groups combined (n = 1,312) (Fig. 1), those with the C-allele of rs254271, an intronic variant in PRPF31, had a worse metformin response at suggestive significance levels (P = 3.79 × 10−6, β = 0.16). The effect of this variant was also observed in REP1, consisting of subjects of European ancestry (P = 6.21 × 10−5, β = −0.073). Interestingly, rs254271 is an expression quantitative trait locus in several tissues for PRPF31 and nearby genes, including NADH:ubiquinone oxidoreductase (NDUFA3), which codes for a subunit of the complex I of the respiratory chain (33–35). Metformin is known to inhibit complex I of the respiratory chain to cause AMPK activation (5). In addition, previous studies have shown that PRPF31(+/−) knockout mice have increased total body fat (P = 1.78 × 10−6) and increased fasted circulating glucose (P = 5.73 × 10−6) (36).
Genotypes for rs254271 were not available for analysis in the African American replication cohorts, REP2 and REP3. The phenotype in REP1 represents HbA1c reduction, so the direction of the effect for rs254271 is consistent between ACCORD and REP1 and represents worse HbA1c response in individuals with the C-allele (Fig. 4). Mutations in PRPF31 have been associated with an autosomal dominant rare Mendelian disease, retinitis pigmentosa (37). PRPF31 was previously identified as a candidate gene for diabetic retinopathy in a genome-wide linkage study in Mexican Americans with T2D (38). We tested rs254271 for association with eye disorders in ACCORD; however, no significant associations were found (P > 0.05) (data not shown). Further research will be needed to determine what role SNPs in PRPF31 may have in metformin drug response or in diabetic retinopathy.
In the analysis of only white subjects in ACCORD, rs2162145, in the CPA6 gene, was associated with metformin response at suggestive significance levels (P = 4.04 × 10−6, β = −0.197). This same SNP was associated with metformin response in the REP2 cohort (P = 0.006, β = −0.127), but not in REP1 or REP3 (P > 0.05) (Fig. 4). To our knowledge, SNPs in CPA6 have not previously been associated with metformin response; however, rs2162145 has a weak significant association with fasting glucose (P = 0.02) (39). In addition, a meta-analysis of T2D in 17 GWAS comprising 8,284 case and 15,543 control subjects in African Americans identified rs7003257, an intron variant in CPA6, as being associated with T2D (P = 1.17 × 10−6) (40), and this SNP has also been previously reported to affect 2-h glucose levels (41). We were underpowered to detect an association in black subjects. However, meta-analyzing all available black subjects in ACCORD, REP2, and REP3 improved the statistical power so an association was detectable (P = 0.005). It will be necessary for additional studies to investigate the extent to which CPA6 is involved with metformin drug response.
A genotyped SNP in the NBEA gene on chr13 rs57081354 was associated with change in HbA1c levels at genome-wide significance (P = 4.02 × 10−8, β = 0.323) in our discovery cohort (Fig. 3). SNPs in NBEA did not replicate in REP1–3 cohorts. rs57081354 was marginally associated with baseline HbA1c levels in individuals who were taking metformin at the time they enrolled in ACCORD, with the C-allele displaying higher HbA1c values (P = 0.09) (Fig. 3C). NBEA is a protein kinase A anchor protein that is expressed at low levels ubiquitously and at higher levels in the brain, pituitary, and β-cells in the pancreas (42). Interestingly, haplotype-insufficient Nbea(+/−) mice prefer sugary foods (glucose and fructose) more than their wild-type controls, gain more weight per unit of food than wild-type mice, and have higher insulin and leptin levels (29). Subsequently, we tested for associations between rs57081354 and 19 dietary scores recorded for a subset of 1,553 ACCORD subjects and found that rs57081354 was associated with an increase in dessert consumption (q <0.2) in those with C-allele (Supplementary Table 6). An expanded summary of the literature regarding NBEA can be found in the Supplementary Data.
Additionally, we tested rare variants for associations with change in HbA1c, and STAT3 when all races were combined, was the only gene that achieved statistical significance for metformin treatment response (q <0.1). However, as described in results, black subjects were the only population with rare variation in STAT3, and STAT3 was not detected in the stratified analysis likely due to the small number of subjects available for analysis (n = 222). In this same cohort, rare variants in STAT3 were not associated with baseline HbA1c levels (q >0.2). STAT3 encodes a ubiquitously expressed transcription factor with broad activities in metabolism, immunity, and cell growth (43). STAT3 is an essential signal transducer for the key metabolic hormone leptin and the pleiotropic cytokine interleukin 6 with broad effects on feeding behavior and fuel homeostasis. Interestingly, metformin has been shown to interact with STAT3 signaling in chemotherapeutic applications and in an inflammatory bowel disease model (44–47). To our knowledge, interactions between metformin and STAT3 signaling have not been investigated with regard to glucose homeostasis.
Here, we present evidence for PRPF31, CPA6, and STAT3 being involved in novel glucose-lowering mechanisms for metformin. Additional investigation will also be required to confirm the clinical impact of the identified variants with regard to metformin therapy in subjects with diabetes. Although prior studies suggest roles for NBEA and STAT3 genes in glucose metabolism, additional research will be required to investigate mechanisms by which they might interact with metformin. Such studies are likely to provide new insight into mechanisms regulating glucose metabolism and may point the way toward novel therapeutic targets for more precise interventions in T2D.
Supplementary Material
Article Information
Funding. This research was supported by National Heart, Lung, and Blood Institute grants 5R01HL110380-04 to the University of North Carolina at Chapel Hill and R01 HL110400 to the Joslin Diabetes Center and the University of Virginia. S.W.Y. and K.M.G. were supported by grants from the National Institutes of Health (GM61390 and GM117163). E.R.P. holds a Wellcome Trust Investigator Award (102820/Z/13/Z).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. D.M.R., S.W.Y., M.A.H., H.L.M., M.J.W., J.B.B., and A.A.M.-R. wrote the article. D.M.R., M.M.H., M.K., J.C.M., H.L.M., A.D., K.M.G., E.R.P., M.J.W., J.B.B., and A.A.M.-R. designed the research. D.M.R., S.W.Y., K.Z., S.W.M., H.S.H., T.M.H., H.G., M.J.W., and A.A.M.-R. performed the research. D.M.R., S.W.Y., K.Z., S.W.M., and J.R.J. analyzed the data. A.A.M.-R. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the American Society for Clinical Pharmacology & Therapeutics Annual Meeting, Washington, DC, 15–18 March 2017.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db17-1164/-/DC1.
References
- 1.Centers for Disease Control and Prevention National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. Atlanta, GA, U.S. Department of Health and Human Services, 2014 [Google Scholar]
- 2.World Health Organization. Global report on diabetes [Internet], 2017. Available from http://www.who.int/diabetes/global-report/en/. Accessed 28 April 2018
- 3.Mozaffarian D, Benjamin EJ, Go AS, et al.; American Heart Association Statistics Committee; Stroke Statistics Subcommittee. Heart disease and stroke statistics—2016 update. A report from the American Heart Association. Circulation 2016;133:e38–e360 [DOI] [PubMed] [Google Scholar]
- 4.Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia 2013;56:1898–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou G, Myers R, Li Y, et al. . Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001;108:1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foretz M, Hébrard S, Leclerc J, et al. . Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest 2010;120:2355–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buse JB, DeFronzo RA, Rosenstock J, et al. . The primary glucose-lowering effect of metformin resides in the gut, not the circulation: results from short-term pharmacokinetic and 12-week dose-ranging studies. Diabetes Care 2016;39:198–205 [DOI] [PubMed] [Google Scholar]
- 8.McCreight LJ, Bailey CJ, Pearson ER. Metformin and the gastrointestinal tract. Diabetologia 2016;59:426–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baur JA, Birnbaum MJ. Control of gluconeogenesis by metformin: does redox trump energy charge? Cell Metab 2014;20:197–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrannini E. The target of metformin in type 2 diabetes. N Engl J Med 2014;371:1547–1548 [DOI] [PubMed] [Google Scholar]
- 11.Zhou K, Donnelly L, Yang J, et al.; Wellcome Trust Case Control Consortium 2 . Heritability of variation in glycaemic response to metformin: a genome-wide complex trait analysis. Lancet Diabetes Endocrinol 2014;2:481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou K, Yee SW, Seiser EL, et al.; MetGen Investigators; DPP Investigators; ACCORD Investigators . Variation in the glucose transporter gene SLC2A2 is associated with glycemic response to metformin. Nat Genet 2016;48:1055–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou K, Bellenguez C, Spencer CC, et al.; GoDARTS and UKPDS Diabetes Pharmacogenetics Study Group; Wellcome Trust Case Control Consortium 2; MAGIC Investigators . Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet 2011;43:117–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buse JB, Bigger JT, Byington RP, et al.; ACCORD Study Group . Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol 2007;99(Suppl.):21i–33i [DOI] [PubMed] [Google Scholar]
- 15.Goff DC Jr, Gerstein HC, Ginsberg HN, et al.; ACCORD Study Group . Prevention of cardiovascular disease in persons with type 2 diabetes mellitus: current knowledge and rationale for the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007;99:4i–20i [DOI] [PubMed] [Google Scholar]
- 16.Gerstein HC, Riddle MC, Kendall DM, et al.; ACCORD Study Group . Glycemia treatment strategies in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007;99:34i–43i [DOI] [PubMed] [Google Scholar]
- 17.Ginsberg HN, Bonds DE, Lovato LC, et al.; ACCORD Study Group . Evolution of the lipid trial protocol of the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007;99:56i–67i [DOI] [PubMed] [Google Scholar]
- 18.Graham HT, Rotroff DM, Marvel SW, et al.; ACCORD/ACCORDion Investigators. Incorporating concomitant medications into genome-wide analyses for the study of complex disease and drug response. Front Genet 2016;7:138 [DOI] [PMC free article] [PubMed]
- 19.Marvel SW, Rotroff DM, Wagner MJ, et al.; ACCORD/ACCORDion Investigators. Common and rare genetic markers of lipid variation in subjects with type 2 diabetes from the ACCORD clinical trial. PeerJ 2017;5:e3187 [DOI] [PMC free article] [PubMed]
- 20.R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria, 2014
- 21.Purcell S, Neale B, Todd-Brown K, et al. . PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaun Purcell. PLINK v1.07 [Internet], 2009. Available from http://zzz.bwh.harvard.edu/plink/. Accessed 28 April 2018
- 23.Dai H, Leeder JS, Cui Y. A modified generalized Fisher method for combining probabilities from dependent tests. Front Genet 2014;5:32 [DOI] [PMC free article] [PubMed]
- 24.Dabney A, Storey JD, Warnes G. Q-value estimation for false discovery rate control. Medicine (Baltimore) 2004;344:539–548 [Google Scholar]
- 25.Storey JD. A direct approach to false discovery rates. J R Stat Soc Series B Stat Methodol 2002;64:479–498 [Google Scholar]
- 26.Goswami S, Yee SW, Xu F, et al. . A longitudinal HbA1c model elucidates genes linked to disease progression on metformin. Clin Pharmacol Ther 2016;100:537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banda Y, Kvale MN, Hoffmann TJ, et al. . Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics 2015;200:1285–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.National Center for Biotechnology Information, National Library of Medicine. Database of single nucleotide polymorphisms (dbSNP) [Internet], 2017. Available from http://www.ncbi.nlm.nih.gov/SNP/. Accessed 12 November 2017
- 29.Olszewski PK, Rozman J, Jacobsson JA, et al. . Neurobeachin, a regulator of synaptic protein targeting, is associated with body fat mass and feeding behavior in mice and body-mass index in humans. PLoS Genet 2012;8:e1002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.da Rocha Fernandes J, Ogurtsova K, Linnenkamp U, et al. . IDF Diabetes Atlas estimates of 2014 global health expenditures on diabetes. Diabetes Res Clin Pract 2016;117:48–54 [DOI] [PubMed] [Google Scholar]
- 31.Danaei G, Finucane MM, Lu Y, et al.; Global Burden of Metabolic Risk Factors of Chronic Diseases Collaborating Group (Blood Glucose) . National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 2011;378:31–40 [DOI] [PubMed] [Google Scholar]
- 32.Williams LK, Padhukasahasram B, Ahmedani BK, et al. . Differing effects of metformin on glycemic control by race-ethnicity. J Clin Endocrinol Metab 2014;99:3160–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeller T, Wild P, Szymczak S, et al. . Genetics and beyond--the transcriptome of human monocytes and disease susceptibility. PLoS One 2010;5:e10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.GTEx Consortium The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000;348:607–614 [PMC free article] [PubMed] [Google Scholar]
- 36.Brown SDM, Moore MW; The International Mouse Phenotyping Consortium . The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mamm Genome 2012;23:632–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waseem NH, Vaclavik V, Webster A, Jenkins SA, Bird AC, Bhattacharya SS. Mutations in the gene coding for the pre-mRNA splicing factor, PRPF31, in patients with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2007;48:1330–1334 [DOI] [PubMed] [Google Scholar]
- 38.Hallman DM, Boerwinkle E, Gonzalez VH, Klein BEK, Klein R, Hanis CL. A genome-wide linkage scan for diabetic retinopathy susceptibility genes in Mexican Americans with type 2 diabetes from Starr County, Texas. Diabetes 2007;56:1167–1173 [DOI] [PubMed] [Google Scholar]
- 39.Dupuis J, Langenberg C, Prokopenko I, et al.; DIAGRAM Consortium; GIANT Consortium; Global BPgen Consortium; Procardis Consortium; MAGIC Investigators . New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010;42:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ng MCY, Shriner D, Chen BH, et al.; FIND Consortium; eMERGE Consortium; DIAGRAM Consortium; MuTHER Consortium; MEta-analysis of type 2 DIabetes in African Americans Consortium . Meta-analysis of genome-wide association studies in African Americans provides insights into the genetic architecture of type 2 diabetes. PLoS Genet 2014;10:e1004517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saxena R, Hivert M-F, Langenberg C, et al.; GIANT consortium; MAGIC Investigators . Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 2010;42:142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uhlén M, Fagerberg L, Hallström BM, et al. . Proteomics. Tissue-based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 43.Johnston PA, Grandis JR. STAT3 signaling: anticancer strategies and challenges. Mol Interv 2011;11:18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng X-S, Wang S, Deng A, et al. . Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle 2012;11:367–376 [DOI] [PubMed] [Google Scholar]
- 45.Feng Y, Ke C, Tang Q, et al. . Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis 2014;5:e1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan X-L, Bhattacharyya KK, Dutta SK, et al. . Metformin suppresses pancreatic tumor growth with inhibition of NFκB/STAT3 inflammatory signaling. Pancreas 2015;44:636–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee S-Y, Lee SH, Yang E-J, et al. . Metformin ameliorates inflammatory bowel disease by suppression of the STAT3 signaling pathway and regulation of the between Th17/Treg balance. PLoS One 2015;10:e0135858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pruim RJ, Welch RP, Sanna S, et al. . LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010;26:2336–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.