

Abstract
Phenotypic screens, which focus on measuring and quantifying discrete cellular changes rather than affinity for individual recombinant proteins, have recently attracted renewed interest as an efficient strategy for drug discovery. In this article, we describe the discovery of a new chemical probe, bisamide (CCT251236), identified using an unbiased phenotypic screen to detect inhibitors of the HSF1 stress pathway. The chemical probe is orally bioavailable and displays efficacy in a human ovarian carcinoma xenograft model. By developing cell-based SAR and using chemical proteomics, we identified pirin as a high affinity molecular target, which was confirmed by SPR and crystallography.
Introduction
Despite the recent extraordinary progress seen in cancer therapy using molecularly targeted drugs, the disease commonly remains resistant to effective long-term treatment. Even when excellent responses to drugs are initially observed, resistance is almost inevitable and patients are then left with few treatment options1 as the discovery of targeted therapies in oncology has focused on relatively few protein families.2 To break this cycle and expand the treatment options for cancer patients, new approaches are needed to discover novel druggable protein targets.3
In phenotypic screens, small molecules undergo high-throughput screening against intact cells, rather than recombinant proteins, and discrete phenotypic changes in the cell are measured and quantified.4 Interest in phenotypic screens has increased significantly in recent years due to their potential to effectively discover new drugs.5 Phenotypic screens have several advantages over screens using recombinant proteins. Hits from a phenotypic screen will, by definition, be cell permeable and have cellular activity, potentially reducing optimization cycles and timelines. Also, because the screening approach is unbiased, established knowledge of the biology of molecular targets is not required. Finally, polypharmacology is often observed with small molecules and structurally related protein families; this can be crucial for efficacy and is perfectly compatible with a phenotypic screening approach.6
In contrast, progressing hits from a phenotypic screen can generate a number of unique challenges. Discovering pharmacodynamic (PD) biomarkers in vivo for use in animals can be difficult when developing hits from phenotypic screens, as the pathways commonly need to be activated with an external stimulus.7 Cell-based screens are typically more expensive and time-consuming and so may require a greater commitment prior to the screening campaign.8 Furthermore, molecular target identification, deconvolution, and validation are crucial steps if new chemical probes9 and drugs are to be discovered. These are often a bottleneck in phenotypic screening.10 Even with successful target deconvolution, the target discovered may not be of interest; for example, the target may already be drugged or be a known antitarget. Finally, polypharmacology may be a serious impediment to compound progression because the interaction with multiple structurally related protein targets may prove impossible to deconvolute.11 These challenges alter the balance between prioritizing druglike properties of compounds and an efficient target identification strategy.12
To execute a successful phenotypic screening campaign, it is critical to select an appropriate phenotype for small molecule intervention. HSF1 is a transcription factor and the master regulator of the ancient, canonical heat shock response.13 A large body of work has verified the importance of HSF1 to tumorigenesis and cancer progression.14 HSF1 has been proposed to be activated by various elements of the cancer state, potentially reprogramming the transcriptome in a way that is overlapping with, but distinct from, the heat shock response.15 Also, a strong correlation has been reported between the expression of activated HSF1 in tumors and adverse clinical outcomes.16 This evidence indicates that the inhibition of HSF1-mediated transcription could be a viable strategy in cancer treatment.17 Moreover, inhibiting the HSF1 stress pathway would represent an attempt at targeting non-oncogene addiction and proteotoxic stress, which has been proposed to be advantageous.18 However, HSF1 is a ligandless transcription factor and so is unlikely to be amenable to standard drug discovery strategies and direct inhibition with small molecules. Therefore, we proposed that an inhibitor of HSF1-mediated transcription, which antagonized the HSF1 pathway but without necessarily binding directly to HSF1, could be discovered and developed via a cell-based phenotypic screen.
Results
HSF1 Phenotypic Assay
To observe HSF1-mediated transcription in an in vitro setting, the HSF1 pathway is activated by a validated heat shock protein 90 (HSP90) inhibitor,19 or another form of external stress,20 which initiates the heat shock response. Commonly, the output of the heat-shock response is quantified by measuring the induction of heat shock 70 kDa protein 1 (HSP72) expression, the stress-inducible HSP70 isoform.19 HSF1 pathway inhibitors are then defined by their ability to block the induction of HSP72. Several HSF1-mediated HSP72 induction inhibitors have been discovered via this method with different proposed molecular mechanisms of action (Figure 1).21,28
Figure 1.

Inhibitors of HSF1-mediated HSP72 induction.
With the aim of discovering novel hit-matter that inhibits HSF1-mediated transcription, we previously carried out a cell-based high-throughput phenotypic screen in U2OS human osteosarcoma cells of ∼200000 compounds, including ∼35000 compounds from a kinase-focused library.22 The screen quantified the inhibition of HSP72 induction using the Arrayscan assay28 following treatment with the HSP90 inhibitor tanespimycin (17-AAG).23
The Bisamide Series
Using this screen, we identified a potent hit from the kinase-focused deck, bisamide 1 (CCT245232) (Figure 2).24 Following resynthesis, the screening hit was confirmed and displayed a pIC50 = 8.55 ± 0.09 (IC50 = 2.8 nM, n = 49)25 in our HSP72 cell-based enzyme-linked immunosorbent assay (ELISA) in U2OS cells. The HSP72 cell-based ELISA assay is an alternative assay format to the Arrayscan assay for quantifying the induced expression of HSP72 and was used as our primary phenotypic pathway assay throughout the study.26 The IC50 was defined as the concentration that inhibited the signal to 50% of the 17-AAG (250 nM) induced HSP72 expression, relative to the control 17-AAG alone (see Supporting Information for details). The clear structural feature defining this chemotype was the N,N′-4-methyl-1,3-phenylenediamide core (Figure 2).
Figure 2.

HSF1 pathway inhibitor, bisamide 1.
We had previously demonstrated that pan cyclin-dependent kinase (CDK) inhibitors, and potent CDK9 inhibitors in particular that act as transcription antagonists,27 can inhibit the HSF1-mediated HSP72 induction phenotype; we initially suspected a similar mechanism for this chemotype.28 However, upon biochemical screening of bisamide 1 against CDK2 and CDK9, no inhibition was observed (<10% inhibition at 1 μM, see Supporting Information). Therefore, we hypothesized that bisamide 1 was acting through a different mechanism of action and consequently 1 was submitted for further characterization.
Hit Characterization: Kinase Activity
HSF1 is regulated by multiple post-translational phosphorylations,29 so we hypothesized that kinase inhibition, other than CDK2 and CDK9, was causing the observed HSF1-mediated HSP72 induction inhibition. Bisamide 1 was screened against a broad kinase set using the KINOMEscan biochemical screening platform, a binding assay which gave single-point percentage inhibition values at 1 μM (DiscoverX, http://www.discoverx.com).30 Analysis of this data set for 442 kinases revealed that bisamide 1 inhibited only nine kinases >50% and four kinases >90% (see Supporting Information for details). To validate the kinase hits, orthogonal functional assays were carried out on the four kinases that displayed >90% inhibition. Of these, KIT, PDGFRA, and PDGFRB failed to confirm in their respective functional assays, all returning pIC50 < 5 (IC50 > 10000 nM). Bisamide 1 did display modest activity against BRAF, returning a pIC50 = 6.38 (IC50 = 420 nM, n = 1).31 Although it seemed unlikely that the modest biochemical BRAF activity would translate into cellular activity, seven commercially available32 potent BRAF inhibitors of differing chemotypes were tested in our cellular HSF1-mediated HSP72 induction inhibition assay for comparison and none displayed pIC50 > 5 (IC50 < 10000 nM).33 This indicated that BRAF inhibition was not an important contributor to the observed HSF1 transcription inhibition phenotype (see Supporting Information).
Hit Characterization: Cellular
To establish whether bisamide 1 displays cellular activity without activation of the heat shock response using the HSP90 inhibitor 17-AAG, the growth inhibitory effects were assayed in the U2OS cell line using the CellTiter Blue (CTB) assay (Promega). Following 4 days of treatment, bisamide 1 displayed a highly potent pGI50 = 7.74 ± 0.08 (GI50 = 18 nM, n = 13), when compared to vehicle. To assess the broader single agent cellular activity of 1, the compound was assayed for growth inhibition against a large, genetically diverse panel of human cancer cell lines (Genomics of Drug Sensitivity in Cancer, www.cancerrxgene.org).34 Of the 635 cell lines assayed, 628 (99%) displayed a pGI50 > 6 (GI50 < 1000 nM), 455 (72%) a pGI50 > 7 (GI50 < 100 nM), and 18 (2.8%) a pGI50 > 8 (GI50 < 10 nM), with no clear selectivity against any specific tissue type (see Supporting Information for details). These results suggested that bisamide 1 had wide-ranging anticancer activity and that activation of the HSF1 pathway with an HSP90 inhibitor was not required for 1 to inhibit cancer cell growth.
Cell-Based SAR
Given the complexity of factors underlying cell-based structure-activity relationships (SAR), we focused on matched pair changes to establish which features were crucial for the cellular activity of the bisamide chemotype. There were two primary aims to our medicinal chemistry strategy. First, because our biochemical kinase screening had failed to reveal any potential kinase targets, a broader target identification strategy would need to be developed. Second, although bisamide 1 had excellent cellular potency, the compound had poor aqueous solubility (kinetic solubility <2 μM), which would need to be addressed to validate HSF1 pathway inhibition as an anticancer strategy in an in vivo human tumor xenograft model. Both goals required us to improve our understanding of the cellular SAR, particularly to identify a vector-to-solvent through which either a chemical probe linker or a solubilizing group could be attached, without disrupting the ability of the compound to bind to the primary pharmacological target or targets.
To develop the cell-based SAR, we switched to the human ovarian carcinoma cell line SK-OV-3, which was relatively sensitive to growth inhibition and is commonly used in translational drug discovery both in vitro35 and in vivo.36 When the activity of bisamide 1 in the SK-OV-3 cell line was assayed, it gave a pIC50 = 7.17 ± 0.07 (IC50 = 68 nM, n = 4) using the HSP72 cell-based ELISA assay induced with 250 nM 17-AAG. Bisamide 1 also inhibited the proliferation of SK-OV-3 cells in the CTB assay with a pGI50 = 8.08 ± 0.12 (GI50 = 8.4 nM, n = 12).
We first focused the exploration on the benzodioxane motif (Scheme 1). The left-hand ring system analogues (Table 1, entries 1–6) were synthesized via a three-step procedure from the commercially available 2-methyl-5-nitroaniline 2. Following amide bond formation, through the reaction of 2-methyl-5-nitroaniline 2 with 2-methylquinoline-6-carboxylic acid 3 and subsequent reduction of the nitro-group using iron, the second amide bond was generated using the corresponding carboxylic acid in a HATU-mediated coupling to afford the various benzodioxane analogues in good to moderate yields.
Scheme 1. Synthesis of the Benzodioxane Bisamide Replacements.

Reagents and conditions: (i) oxalyl chloride, DMF, DCM, then, pyridine, DCM; (ii) Fe, NH4Cl, EtOH/H2O; (iii) RPhCO2H, HATU, DIPEA, DMF.
Table 1. Bisamide Analogues.

All results are quoted as the geometric mean ± SEM, pIC50/pGI50/pKD = −log IC50/GI50/KD (M). The number of repeats, n, are described in parentheses. ND = not determined.
Cell-based ELISA assay for inhibition of HSP72 induction; SK-OV-3 cells were pretreated with compound at the relevant concentration for 1 h before the addition of 250 nM 17-AAG. HSP72 levels were then quantified after 18 h.
Growth inhibition was measured after 96 h of treatment and compared to vehicle control.
pKD values were measured through analysis of the sensorgram at equilibrium where possible. Values were then fitted to a one-site specific binding model using Graphpad Prism Version 6.
Changing the 5-substituted dioxane isomer 1 (Table 1, entry 1) to the 6-substituted dioxane isomer 6 (Table 1, entry 2) resulted in a complete loss of cellular activity (pIC50 < 5), indicating that the isomer of the benzodioxane was crucial. The acyclic dimethoxy analogue 7 (Table 1, entry 3) also displayed a complete loss of cellular activity. Next, we investigated the role of the two oxygen atoms of the dioxane ring. Removing the para-oxygen of the dioxane to give the meta-chroman 8 (Table 1, entry 4) resulted in a modest 5-fold decrease in cellular HSF1-mediated HSP72 induction inhibition and a 7-fold decrease in growth inhibition compared to 1; while the para-chroman isomer 9 (Table 1, entry 5) displayed a further 6-fold decrease in HSF1-mediated HSP72 induction inhibition and an 8-fold decrease in growth inhibition compared to 8, which represents a 37- and 51-fold decrease in cellular activity respectively compared to the original hit, bisamide 1. Surprisingly, isochroman 10 (Table 1, entry 6) displayed no measurable cellular activity. From these results, it was clear that the benzodioxane moiety was crucial for the cellular activity of the bisamide series. Because the SAR surrounding the benzodioxane was steep and complex, we decided that this region was unlikely to be solvent exposed and so would be incompatible with linker and solubilizing group attachment.
Second, we investigated the role of the central ring and amide moieties in the cellular activity of the bisamide chemotype. We hypothesized that the amide groups could play an important role in hydrogen bonding with a potential target and in controlling the overall shape of the ligand, while substitution at the central ring could lead to the discovery of solvent-exposed vectors. The synthesis of these analogues (Table 1, entries 7–10) was similar to that described in Scheme 1. Substitution at both the 6-position (Table 1, entry 7) and the 4-position (Table 1, entry 8) of the central benzene ring resulted in a complete loss of cellular activity, suggesting that neither of these vectors represented a viable route to solvent. Owing to the potential for the toluene methyl group to affect the conformation of the right-hand amide, we decided to leave this group unchanged. N-Methyl substitution of the left-hand amide (Table 1, entry 9) and an attempted sulfonamide bioisosteric replacement (Table 1, entry 10) also resulted in a complete loss of cellular activity. Again, owing to the steep SAR in this region of the chemotype, it was clearly incompatible with linker and solubilizing group attachment.
Finally, we examined the role of the 2-methylquinoline ring in the cellular activity of the bisamide series. All analogues (Table 1, entries 11–13) were synthesized via a method similar to that described in Scheme 1. First, we removed the 2-methyl group, which is both lipophilic and has the potential to be a weak hydrogen bond donor, to give quinoline 15 (Table 1, entry 11). Removal of this group resulted in no significant change in the cellular activity when compared to 1 (Table 1, entry 1). Next we moved the quinoline nitrogen from the 1- to the 3-position to give isoquinoline 16 (Table 1, entry 12). Again, no significant change in cellular activity was observed when compared to bisamide 1. Finally, we partially reduced the quinoline ring to give tetrahydroquinoline 17 (Table 1, entry 13). By changing to tetrahydroquinoline 17, the nitrogen atom now acts as a hydrogen bond donor rather than an acceptor. Despite this reversal in anticipated binding properties, only a 3.5-fold decrease in HSF1-mediated HSP72 induction inhibition and no significant change in antiproliferative activity was observed when compared to bisamide 1. The broad SAR of the quinoline region of the molecule suggested that this moiety is solvent exposed and so could be exploited for solubilizing group and linker attachment.
Exploiting Vectors-to-Solvent: Solubilizing Group Optimization
Owing to the complexities of the cellular SAR, attaching a solubilizing group to the solvent-exposed region of the ligand was considered a more expeditious strategy to generate an in vivo chemical probe suitable for use in animal models rather than attempting to optimize the intrinsic solubility of the bisamide series without insight from structural information. To address this objective, we adopted an iterative strategy to exploit the rapid synthesis of analogues that would exhibit improved physicochemical properties and demonstrate the appropriate mouse pharmacokinetic (PK) parameters for in vivo study (Scheme 2).
Scheme 2. Synthesis of Solubilizing Group Analogues.

Reagents and conditions: (i) oxalyl chloride, DMF, 1,4-benzodioxane-6-carboxylic acid, DCM, then, 4-methyl-3-nitroaniline, pyridine, DCM, RT, then Pd/C (10%), H2 (1 atm), EtOH; (ii) NaH, RCH2OH, THF, 0 °C to reflux; (iii) n-BuLi, CO2(s), THF, −78 °C; (iv) HATU, DIPEA, DMF, RT.
The optimal route to the synthesis of these analogues would be to introduce the solubilizing group via nucleophilic aromatic substitution on the quinoline as the final step; however, this gave consistently low yields. Therefore, the solubilizing group was introduced at the start of the synthesis. Formation of the precursor carboxylic acid 19 was achieved via lithium halogen exchange of the corresponding aryl bromide 20. The resulting bisamides were then assayed for cellular activity and in vitro PK parameters (Table 2).
Table 2. Optimization of the Bisamide Solubilizing Group.


pGI50 = −log GI50 (M); geometric mean ± SEM, n = number of biological repeats in parentheses. For the corresponding SK-OV-3 HSP72 cell-based ELISA data See Supporting Information.
Mouse liver microsome (MLM) assay was carried out at Cyprotex, arithmetic mean of n = 2. In vitro Clint is calculated from the half-life using standard procedures and assumes the fraction unbound in the assay is 1.37
Kinetic solubility (KS) measured via an in-house HPLC method from phosphate buffer at pH 7.4, all values quoted to 1 SF, the dynamic range of the assay is 1–100 μM, arithmetic mean of n = 2.
Measured via an in-house HPLC method, arithmetic mean of n = 2, all values quoted to 2 SF.38
MoKa version 2.5.2, all values quoted to 2 SF. See Supporting Information for details.
We first attached the oxygen-linked ether chain to give glycol 21 (Table 2, entry 2). No significant decrease in either HSF1-mediated HSP72 induction inhibition (see Supporting Information) or the antiproliferative activity was observed when compared to lead bisamide 1 (Table 2, entry 1). This suggested first that the oxygen linker had no detrimental effect on activity and, second, that this region of the molecule was indeed solvent exposed. To carry out multiparameter optimization of the solubilizing group, we used four in vitro properties to assess each compound’s potential for in vivo mouse PK: microsomal stability, kinetic solubility at pH 7.4, lipophilicity as measured by LogD7.4, and the predicted basicity of the solubilizing group. The glycol 21 displayed an increase in lipophilicity compared to lead bisamide 1; this increase was reflected in the 2.4-fold decrease in microsomal stability with a modest increase in kinetic solubility. However, glycol 21 remained a low solubility compound, which is inconsistent with good oral bioavailability. From these data, we concluded that the compound would need to be charged to deliver the correct balance of properties. To introduce a charged moiety, we first focused on the dimethylamine group with a 3-carbon linker. Dimethylamine 22 (Table 2, entry 3) displayed no significant change in cellular activity despite this analogue being predominately charged at physiological pH, according to the calculated pKa. Consistent with its cationic character, dimethylamine 22 displayed a 1.4 log unit decrease in lipophilicity when compared to 21, which was accompanied by an 18-fold improvement in kinetic solubility. Unfortunately, the decrease in lipophilicity was not reflected in an improvement in microsomal stability. We hypothesized that this was due to oxidation of the N-methyl groups, so we moved to cyclic amines to reduce the CYP450 mediated degradation. Morpholine 23 (Table 2, entry 4) showed little improvement in microsomal stability or kinetic solubility when compared to 21. Analysis of the LogD7.4 and calculated pKa for morpholine 23 suggested that the solubilizing group was not sufficiently basic to balance the physicochemical properties of the compound. Moving to piperidine 24 (Table 2, entry 5), we observed a better balance in physicochemical properties, with the reduction in lipophilicity reflected in improvements in both microsomal stability and kinetic solubility. However, piperidine 24 was predicted to be highly basic and we were concerned that this would have a detrimental effect on permeability and therefore on oral bioavailability. To reduce the basicity of the solubilizing group without increasing lipophilicity, we reduced the linker length to give 25 (Table 2, entry 6), thereby exploiting the inductive effect of the oxygen. Despite the 0.9 log unit decrease in predicted basicity, the kinetic solubility and lipophilicity were unchanged. Unfortunately, 25 displayed a decrease in microsomal stability, so we reduced the ring size to give pyrrolidine 26 (CCT251236, Table 2, entry 7),24 which displayed the desired balance of in vitro properties, while maintaining excellent cellular activity with a pIC50 = 7.73 ± 0.07 (IC50 = 19 nM, n = 15, see Supporting Information, Table S3 for details) for inhibition of HSF1-mediated HSP72 induction. The free GI50 in SK-OV-3 cells was then calculated from the free fraction in the cell assay, which gave a free GI50 = 1.1 nM.39 Western blotting confirmed that pyrrolidine 26 blocked the HSF1-mediated induction of both HSP72 and HSP27 as representative heat shock proteins, following treatment with the HSP90 inhibitor 17-AAG. Also, qPCR analysis demonstrated that 26 inhibited the induction of HSP72 at the mRNA level, clearly blocking the induction of HSPA1A gene expression with a pIC50 = 7.40 (IC50 = 40 nM, n = 1, see Supporting Information). Taking these results together, pyrrolidine 26 was selected for in vivo study.
Mouse Pharmacokinetics (PK)
To assess the potential of bisamide 26 as an in vivo chemical tool to study HSF1-mediated transcriptional activity inhibition, the compound was dosed in BALB/c mice at 5 mg/kg as an oral solution and iv bolus. Blood concentrations were then measured over a 24 h period (Table 3).
Table 3. Mouse Blood PK Parameters for Bisamide 26a.
| mouse | dose po/iv (mg/kg) | AUC0–24hPO (h·nM)b | blood Cl (mL/min/kg)b | Vss (L/kg)b | half-life (h) | %F | fubc | AUCu,0–24hPO (h·nM)f | Clu (mL/min/kg)g | Vdu (L/kg)h |
|---|---|---|---|---|---|---|---|---|---|---|
| BALB/c | 5/5 | 5800 (8100–4200) | 9.2 (12–7.3) | 4.2 (6.3–2.8) | 5.3 | 39 | 0.0083d | 48 | 1100 | 510 |
| athymic | 20/0 | 1900 (2800–1400) | NA | NA | NA | NA | 0.015e | 29 | NA | NA |
All values are quoted to 2 SF. The 90% confidence intervals (CI) are in parentheses. NA = not applicable.
The geometric mean of n = 3 individual mice.
fub = fup/B:P.
fup = 0.010 (0.011–0.0097), B:P@1 μM = 1.2:1 (1.4–1.0).
fup = 0.025 (0.030–0.020), B:P@1 μM = 1.7:1 (1.7–1.7).
AUCu = AUC·fub.
Clu = Cl/fub.
Vdu = Vss/fub.
Analysis of the mouse PK data revealed that bisamide 26 possessed low total blood clearance (10% hepatic blood flow)40 and moderate oral bioavailability, with a half-life sufficient to allow once-daily dosing. In vitro assessment of the plasma protein binding and the blood to plasma ratio revealed that 26 was highly bound to plasma proteins (∼99%); therefore, the unbound clearance was high, with a low free exposure from the 5 mg/kg oral dose, equivalent to a free Cav0–24h = 2.0 nM.41,42 The high unbound volume of distribution indicates that 26 readily binds to tissues, consistent with the basicity of the compound.43
Bisamide 26 Displays Efficacy in a Human Ovarian Carcinoma Xenograft Model
Despite the high unbound clearance of bisamide 26 in mouse, the good oral bioavailability, half-life, and excellent in vitro cellular activity (free GI50 = 1.1 nM) encouraged us to test the potential efficacy of 26 in an in vivo human tumor xenograft model. On the basis of the free Cav0–24h = 2.0 nM observed in nontumor bearing immunocompetent BALB/c mice following the 5 mg/kg po qd dose, a 20 mg/kg po qd dose in immunodeprived athymic mice was selected to cover ∼10 times the in vitro free GI50 in SK-OV-3 cells.44 To our surprise, following this 20 mg/kg po dose, the free exposure was actually a disappointing AUCu0–24h = 29 h·nM, equivalent to a free Cav0–24h = 1.2 nM (Table 3, see Supporting Information for details). Despite the lower than expected free exposure of bisamide 26, this dose still represented coverage of the in vitro free GI50 in SK-OV-3 cells and was well tolerated in a mouse multidose tolerability study;45 therefore, the 20 mg/kg po qd dose was selected for further investigation.46
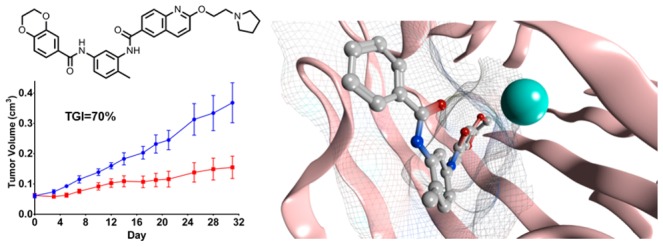
SK-OV-3 cells were injected subcutaneously into athymic mice for tumor formation. Once tumors were established, the mice were randomized into treatment and control groups and were dosed orally once-a-day with either vehicle or 20 mg/kg of bisamide 26. Tumor volumes and mouse body weights were measured throughout and tumor weights were measured at the end of the study, while total tumor concentrations were measured 2 and 6 h post final dose (Figure 3 and Supporting Information).
Figure 3.
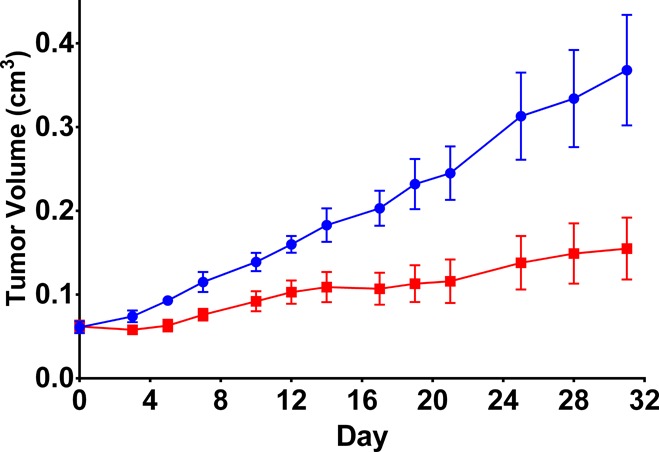
Efficacy of bisamide 26 against SK-OV-3 human ovarian carcinoma xenograft model. Blue, vehicle control, n = 8; red, 26, 20 mg/kg po qd, n = 8 (vehicle = 10% DMSO, 90% of a 25% (2-hydroxypropyl)-β-cyclodextrin in 50 mM citrate buffer pH 5). Error bars: arithmetic mean ± SEM. Dosing breaks were carried out on days 5–12, 14, 16, 18, 20, 22, 24, 26, 29, 31.
The mice were dosed intermittently throughout the study with 26 to maintain their condition, as assessed through monitoring body weights (see Supporting Information). Clear therapeutic efficacy was observed with bisamide 26, with a tumor growth inhibition (%TGI)47 of 70% based on final tumor volumes. The study was terminated after 33 days, and comparison of the control and treated arms indicated a 64% reduction in mean tumor weights (p = 0.015)48 with total tumor concentrations of 26 as high as 940 nM, consistent with the compound’s basicity and high volume of distribution (see Supporting Information).
Target Identification
Following the successful efficacy study, the potential for bisamide 26 as a chemical probe to study the effects of HSF1 transcription inhibition, both in vitro and in vivo, were clear. However, the variety of HSF1 transcription inhibitors in the literature49 suggests there are multiple mechanisms through which this phenotype may be observed, each with differing potential for drug discovery.
To decipher the molecular mechanism of the bisamide series, we needed to discover their protein targets. The potency of the bisamide 26 in cell-based assays suggests that only high affinity efficacy and epistatic targets would be of interest.11 Our biochemical kinase screening had indicated that there were no high affinity kinase targets. We therefore expanded our biochemical screening to include other protein families. Bisamide 26 was submitted to the Cerep Diversity Screen (Cerep, http://www.cerep.fr) comprising 98 molecular targets, including receptors and enzymes, measured at 10 μM.50 Analysis of these screening data revealed that bisamide 26 displays a good selectivity profile. Only six targets (the receptors: adenosine A2A and A3, histamine H2 and H3, muscarinic, and the enzyme acetylcholine esterase) displayed >80% inhibition, and these generally represented the highly promiscuous receptor protein targets (see Supporting Information for details). No hits displayed sufficient activity to clearly relate the molecular target to the efficacious free concentrations achieved in vitro or in vivo.
Because no clear protein families had been revealed from our biochemical screening, a different approach was necessary. We decided to exploit a chemical proteomics strategy and using our knowledge of the cellular SAR, we designed a protein pull-down chemical probe to identify high affinity molecular targets from a human cancer cell lysate (Figure 4).
Figure 4.

Tool compounds for target identification. All quoted cellular activities are in the SK-OV-3 cell line, the numbers of repeats are in parentheses.
To carry out the chemical proteomics pull-down strategy, the bisamide warhead would need to be attached via a linker to a solid-phase bead, without disrupting binding to the molecular target or targets. The solubilizing group vector was the obvious choice for the linker attachment, but to test whether cellular activity was maintained, amide 27 was designed as a cell-permeable mimic of the solid-phase probe. Pleasingly, amide 27 maintained excellent cellular potency (SK-OV-3 pGI50 = 8.04 ± 0.06, GI50 = 9.1 nM, n = 4), so amine 29 was used for attachment to the bead via amide bond formation. In addition to bisamide 26, tetrahydroquinoline 17 was selected as a second positive control due to its distinct structural difference. These two compounds would be used to displace specific molecular targets from the solid-phase probe. Also, to help distinguish false positives from the pull-down experiment, a physicochemically matched negative control was designed. The dioxane isomer 6 (Table 1, entry 2) had previously been shown to lose all cellular activity. To physicochemically match the dioxane isomer 6 to bisamide 26, the same ethoxy pyrrolidine solubilizing group was attached to give isomer 28, which, as expected, displayed no measurable cellular activity.
With all four probe molecules in hand, the compounds were submitted to the stable isotope labeling by amino acids in cell culture (SILAC) mass-spectrometry based pull-down assay (Evotec, https:www.evotec.com) to identify and quantify molecular targets of the bisamide series from the lysate of SK-OV-3 cells. Each protein captured by the bead, whether specific or nonspecific, was analyzed in this methodology by quantitative mass spectrometry. By using SILAC to quantify the relative amounts of protein captured from the lysate, this approach can, in principle, determine the apparent affinity of every stable protein in the lysate for the surface-bound bisamide probe, although particularly low abundance proteins may not be discovered via this method and proteins that are unstable to the lysis conditions would not be detected. Displacement of the captured proteins with the free active bisamides 26 and 17 then allowed for estimation of their apparent affinities for specific protein targets. Proteins that were apparently displaced from the bead-bound probe by the inactive isomer 28 were considered nonspecific or irrelevant for the cellular activity and discarded. The putative protein targets could then be ranked based on their affinity for further investigation (Table 4, see Supporting Information for details).51
Table 4. Molecular Targets from the Pull-down Assay Using the Bisamide Probes in SK-OV-3 Cell Lysatea.

| protein | protein EC50 (μM)b | 26 AC50 (μM)c | 26Kiapp (μM)d | 17 AC50 (μM)c | 17 Kiapp (μM)d | 28 AC50 (μM)c |
|---|---|---|---|---|---|---|
| GSK3β | 1.3 | 1.2 | NA | 10 | NAe | NAb |
| Pirin | 1.3 | 1.5 | 0.028 | 10 | 0.19 | NAb |
(A) SK-OV-3 cell lysate protein binding curves for immobilized 29. (B) Protein displacement curves for pyrrolidine 26. Curves are an average of two different mixing conditions. EC50 and AC50 are determined from analysis of the curves without limits.
EC50 is the apparent immobilized bisamide probe concentration at which 50% of the available protein is bound. The probe bisamide is assumed to be >10-fold molar excess over proteins in the lysate.
AC50 is the apparent bisamide concentration at which 50% of the protein is displaced from the immobilized bisamide probe. At least 2-fold enrichment of bound proteins when compared to the matrix.
Kiapp values are estimated using the Cheng–Prusoff equation, apparent [29-immobilized] = 67 μM,52 assuming no mass transport limitation, interactions are purely competitive and all interactions are direct.
Not applicable (NA). Kiapp cannot be calculated if the interaction with the probe is indirect.
Analysis of these data from the quantified chemical proteomics pull-down experiment revealed very few molecular targets of the bisamides, consistent with the selectivity observed with our biochemical screening. After excluding highly promiscuous proteins commonly observed in pull-down experiments,53 three putative targets were identified. PDE6D displayed only weak apparent affinity for the bead-bound probe and little selectivity between active (26 and 17) and inactive (28) probes and so was not considered further. GSK3β displayed apparent high affinity for both active analogues, with no apparent affinity for the inactive analogue. However, GSK3β had previously been assayed in our biochemical kinase screening and both binding and functional assays (see Supporting Information) had demonstrated that the bisamide series have no measurable affinity for this kinase; we therefore concluded that GSK3β was potentially an indirect target of the bisamide series. The final putative protein target was pirin. The binding curve for pirin to the bead-bound bisamide probe was quite shallow; however, the displacement curves for pirin, which were very similar to the displacement curves for GSK3β with both cell active probe ligands, gave robust data with an apparent Ki = 28 nM for 26 and no affinity was observed with the inactive control. Using this method on a SK-OV-3 cell lysate, pirin emerged as the only remaining putative target of the bisamide series.54
Pirin Validation
Very little is known about pirin, and few papers have been published discussing its possible function. Pirin was first identified in 1997 by Wendler et al. from a yeast two-hybrid screen to discover interactors of the transcription factor NFI/CTF1. Pirin was described as an iron-binding, metal-dependent protein, predominately localized within the nucleus, highly conserved across species, and ubiquitously expressed in all tissue types.55 Following this initial discovery, Scheidereit et al. reported that human pirin interacts with the proto-oncoprotein, BCL3, linking pirin with the NFκB pathway.56 The role of pirin in the NFκB pathway was further demonstrated by Lui et al.; using SPR, they revealed the metal-dependent formation of a pirin/p65/DNA complex and hypothesized the role of human pirin to be a redox-sensing transcription factor regulator.57 The redox activity of pirin has previously been discussed, as the expression level of pirin was proposed to be under the control of the transcription factor NRF2 through changes in cellular oxidative stress.58 Wang et al. previously reported that human pirin interacts with the tumor suppressor protein EAF2/U19 and that exogenous overexpression of pirin increased colony formation in LNCaP human prostate cancer cells.59 In the human melanoma cell line WM266.4, Alcalay et al. demonstrated that shRNA knockdown of pirin could induce a senescence phenotype and suppress colony formation.60 Also in the WM266.4 cell-line, Osada et al. demonstrated that siRNA knockdown of pirin could suppress cell migration.61,62 These results are consistent with the observation by Simizu et al. that siRNA knockdown of pirin suppressed migration of human adenocarcinoma HeLa cells and that pirin was important for epithelial–mesenchymal transition (EMT).63 However, little evidence has emerged of an antiproliferative phenotype from genetic inhibition of pirin expression and our in-house data using siRNA in SK-OV-3 cells was consistent with this finding.64 Nonetheless, because our original screening paradigm was designed to discover inhibitors of transcription, the proposed role of pirin as a redox-sensitive transcription factor regulator was certainly intriguing.
To confirm pirin as a high affinity molecular target of the bisamide series, we needed to assess the affinity of the compounds and establish SAR in an orthogonal assay format. Pirin has no known catalytic function and no known endogenous ligand in mammalian cells, consequently we decided to focus on surface plasmon resonance (SPR) to measure the affinity of the bisamide series for pirin. Purified recombinant human pirin was attached to the SPR chip through a standard amide coupling. The affinity of the bisamide ligand was assessed by equilibrium analysis of the resulting sensorgram (Figure 5).
Figure 5.

Representative SPR sensorgram and binding isotherm of bisamide 26 bound to recombinant pirin. The dotted-line represents the time-point the equilibrium response was measured. The binding isotherm was fitted to a one-site specific binding model using Graphpad Prism 6.07.
The known pirin ligand TPh A61 was used as a positive control and to protect the binding site during amine coupling to the SPR chip but displayed only modest affinity in our hands (pKD = 5.77 ± 0.04, KD = 1700 nM, n = 4).65 The SPR sensorgram showed that bisamide 26 is indeed a tight-binding ligand of pirin. Equilibrium analysis revealed that bisamide 26 had a pKD = 7.36 ± 0.01 (KD = 44 nM, n = 3). However, the ratio of theoretical Rmax to measured Rmax from the binding isotherm was 0.40, indicating apparent substoichiometric binding. This could be due to the coupling of pirin to the SPR chip impairing protein folding or because pirin is a metal-dependent protein, and the metal was leaching out into the running buffer; both effects could negatively impact the apparent binding affinity and stoichiometry.
To establish the SAR surrounding pirin binding, several bisamide analogues were selected for further study. We first focused on the two other analogues used in target identification. The cell active analogue tetrahydroquinoline 17 (Table 1, entry 13) was also found to display tight-binding affinity for pirin; in contrast, the negative control dioxane isomer 28 (pIC50 < 5.00, IC50 > 10000 nM, n = 2) displayed no measurable affinity. These data suggested that pirin SAR and the cellular SAR were linked. Increasing the steric bulk of the solubilizing group, as exemplified by piperidine 24 (Table 2, entry 4), again returned a high affinity pirin ligand (pKD = 7.52 ± 0.02, KD = 30 nM, n = 3), consistent with the potent cellular activity. However, structural changes to the benzodioxane amide motif proved more complex. The original hit, bisamide 1 (Table 1, entry 1), was a tight-binding pirin ligand; whereas the cell inactive analogues, methylated amide 13 (Table 1, entry 9) and dimethoxy 7 (Table 1, entry 3), displayed no affinity for pirin. However, the two chroman isomers 8 and 9 (Table 1, Entries 4 and 5) were tight-binding ligands for pirin but their target affinity did not reflect their decrease in cellular activity when compared to bisamide 1. Finally, the isochroman analogue 10 (Table 1, entry 6) was still a high affinity ligand for pirin, despite displaying no cellular activity (see Supporting Information, Table S11 for details).
Pirin is a member of the cupin super family of proteins, which are so-called because they all possess a conserved β-barrel.66 However, due to minimal sequence homology between members of the cupin family, crystallography is often needed to identify them. The cupins display a huge array of functionality, including oxidases and isomerases.67 To better understand pirin SAR, we determined the crystal structure of efficacious bisamide 26 bound to pirin (Figure 6).
Figure 6.

X-ray crystal structure of bisamide 26. PDB 5JCT, Pymol image of pirin (light-blue cartoon representation) in complex with bisamide 26 (cyan stick representation), 2Fo – Fc map contoured at 1.0 σ (blue mesh) and Pymol image of pirin (magenta, blue stick representation) in complex with bisamide 26 (cyan stick representation), distances shown are in Å, 2Fo – Fc contoured at 1.0 σ (blue mesh). Crystals belonged to the space group P212121 and diffracted to a resolution of 1.73 Å.
Pirin is a metal-binding bicupin characterized by the two β-barrels that are clearly visible in the crystal structure.68 Pirin binds a single metal-ion in one of the β-barrels, which is where a deep, small molecule binding pocket is located. The metal-ion is clearly visible in the electron density, although it is unclear which metal-ion is actually bound. The metal-ion binding site is formed of three histidine residues (His56, His58, and His101), a glutamic acid (Glu103) and two water molecules, showing the metal-ion possesses octahedral coordination. Bisamide 26 forms no direct interactions with the metal-ion, instead a water-mediated interaction is created by the two amide functional groups, which form a hydrogen bonding pincer around this metal-coordinated water molecule, revealing why the two amides are crucial for binding. The methyl-distal amide also acts as a hydrogen-bond donor with an aspartic acid (Asp43) at the base of the binding site, explaining why methylation of that group is not tolerated. The methylpyridine ring of the quinoline substituent is clearly solvent exposed and the solubilizing group cannot be resolved due to flexibility, consistent with the broad SAR at this position and the eventual success of the pull-down probe. The SAR around the benzodioxane ring is less clear. From the cellular SAR, the oxygen atoms of the dioxane are crucial for activity. The narrow binding site is consistent with the cellularly inactive dioxane isomer 28 and the cellularly inactive dimethoxy analogue 7 having low affinity for pirin (Table 1 and Supporting Information, Table S11), as both these groups would cause a steric clash. However, there are no clear interactions between the protein and the dioxane ring oxygens, so it is unsurprising that changing the positions of the oxygen atoms has little effect on pirin affinity, such that chromans 8, 9, and isochroman 10 (Table 1, entries 4–6) still display high affinity for pirin, in contrast to their complex cellular SAR.
The apparent disconnect between the pirin affinity SAR and the antiproliferative cellular SAR led us to hypothesize that either simply binding to pirin was not enough to recapitulate the cellular phenotype or that binding to a second molecular target was also crucial. For example, in the study by Lui et al.57 the formation of protein-protein interactions with pirin was under the allosteric control of the redox-state of the iron bound in the protein. Ligands bound in the metal-binding site are therefore likely to act in an allosteric manner to regulate the function of pirin. It is commonly known that allosteric modulators display complex SAR, where small structural changes can have little effect on protein affinity but can drastically alter the observed phenotype, switching ligands between positive, negative, and neutral allosteric modulation.69,70 The alternative hypothesis consistent with the observed complex cellular SAR was that a second high affinity protein target was required to explain the antiproliferative activity of the bisamide 26 and was not detected in our target-ID campaign due to either low abundance or poor stability in the lysate. The dual targeting of proteins is often observed; for example, CDK4 and CDK6, where simultaneous inhibition of both kinases is necessary to observe an antiproliferative phenotype71 and owing to the similarity of their respective binding sites, small-molecule inhibitors inevitably inhibit both proteins.72 RNAi knockdown of pirin by several groups has demonstrated very limited effects on cell proliferation, and as pirin has no known enzymatic function, discovering and validating a cellular biomarker to investigate its role in the antiproliferative phenotype and in vivo efficacy is an ongoing challenge.
One phenotype that has previously been associated with a small-molecule binding to pirin is cancer cell migration.61 Miyazaki et al. demonstrated that their pirin ligand, TPh A, inhibited the migration of the melanoma cell line, WM266.4.61 To investigate whether our tool compound, bisamide 26, phenocopied this distinct pirin-ligand chemotype, we decided to investigate its antimigratory activity using a scratch-wound assay. The scratch-wound assay is commonly used to assess the effects of small-molecules on cell migration.73 Bisamide 26 demonstrated excellent antiproliferative activity against the WM266.4 cell line (pGI50 = 7.92 ± 0.10, GI50 = 12 nM, n = 11). To exclude the effect of inhibiting proliferation on migration in the scratch-wound assay, the cells were plated at high confluency and the timeframe of the assay was reduced to 30 h. The relative wound-density was then measured at various concentrations and time-points. The negative control isomer 28 displayed no measurable antimigratory activity in stark contrast to the potent pirin ligand bisamide 26, which was able to strongly inhibit the migration of WM266.4 cells at 100 nM (Figure 7).
Figure 7.

Antimigratory activity of chemical probe bisamide 26. (A) Wound healing images of WM266.4 cells; after 30 h, the wound has almost completely healed in the control in contrast to 100 nM 26. (B) Quantification of the relative wound density reveals the maximum antimigratory activity is achieved at 100 nM 26. Each point represents the arithmetic mean ±SEM of the study carried out in triplicate.
Discussion
Bisamide 26 displays excellent and wide-ranging in vitro cellular potency, inhibiting both HSF1-mediated transcriptional activity (defined by the inhibition of the induced expression of the heat-shock proteins HSP72 and HSP27 and HSPA1A mRNA) and cancer cell proliferation. Also, 26 has good mouse PK and demonstrated its anticancer effects in vivo at low free exposures with clear tumor growth inhibition at tolerable doses. Kinase screening and broader proteome screening, in addition to the chemical proteomics using SILAC quantified pull-down with rigorous controls and follow-up testing, indicated that pirin was the sole specific protein target that could be identified to date. SPR analysis of the binding of the bisamide series to pirin confirms that these compounds are high affinity ligands, with most aspects of the pirin SAR being consistent with the cellular SAR. However, there was an apparent disconnect surrounding the dioxane group of the chemotype. This requires further investigation to determine whether pirin is crucial for the in vitro antiproliferative activity and in vivo efficacy via an allosteric modulation mechanism or whether binding to a second high affinity protein target is needed to fully account for the observed phenotype. Because the SILAC quantified pull-down assay can only identify protein targets that are stable to cell lysis, additional in-cell target-ID methods are currently under investigation to probe for a potential second target. Nevertheless, the tool compound, bisamide 26, demonstrated the previously proposed anticancer phenotype of pirin ligands by inhibiting the migration of WM266.4 melanoma cells in vitro. The role of pirin in the bisamide phenotype and the cellular effects of modulating the HSF1 pathway with bisamide 26 are currently also under investigation and will be reported subsequently.
Conclusions
Using a high-throughput screen to identify inhibitors of the HSF1-mediated stress pathway, we have discovered an extremely potent inhibitor of human cancer cell proliferation in vitro from the bisamide chemotype. By exploring the SAR from the cellular assays, we designed a chemical probe, bisamide 26, which is highly potent and displays an appropriate mouse pharmacokinetic profile to significantly inhibit growth in a human ovarian carcinoma xenograft model. The chemical probe 26 was also used to design a chemical proteomic pull-down experiment, which identified the putative transcription factor regulator, pirin, as a protein target. The high affinity of chemical probe 26 for pirin was confirmed by SPR. Comparison of the biophysical with the cellular data indicated that active molecules bind pirin but that the cellular SAR is more complex, although 26 did display a potent inhibitory effect on the migration of human melanoma cells, consistent with the putative pirin cancer phenotype. Despite this, we propose that bisamide 26, in combination with the physicochemically matched negative control dioxane isomer 28, are promising chemical probes to investigate the role of HSF1 pathway inhibition and pirin binding in vitro and in vivo.
Experimental Section
Experimental Procedures (Chemistry)
All final compounds were screened through our in-house computational PAINS filter and gave no structural alerts as potential assay interference compounds.74 Unless otherwise stated, reactions were conducted in oven-dried glassware under an atmosphere of nitrogen or argon using anhydrous solvents. All commercially obtained reagents and solvents were used as received. Thin layer chromatography (TLC) was performed on precoated aluminum sheets of silica (60 F254 nm, Merck) and visualized using short-wave UV light. Flash column chromatography was carried out on Merck silica gel 60 (partial size 40–65 μm). Column chromatography was also performed on a Biotage SP1 purification system using Biotage Flash silica cartridges (SNAP KP-Sil). Ion exchange chromatography was performed using acidic Isolute Flash SCX-II columns. Semipreparative HPLC was performed on an Agilent 6120 system, flow 20 mL/min, eluents 0.1% acetic acid in water and 0.1% acetic acid in methanol, gradient of 10–100% organic phase. 1H NMR spectra were recorded on Bruker AMX500 (500 MHz) spectrometers using an internal deuterium lock. Chemical shifts are quoted in parts per million (ppm) using the following internal references: CDCl3 (δH 7.26), MeOD (δH 3.31), and DMSO-d6 (δH 2.50). Signal multiplicities are recorded as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), doublet of doublets (dd), doublet of doublet of doublets (ddd), broad (br), or obscured (obs). Coupling constants, J, were measured to the nearest 0.1 Hz. 13C NMR spectra were recorded on Bruker AMX500 spectrometers at 126 MHz using an internal deuterium lock. Chemical shifts are quoted to 0.01 ppm, unless greater accuracy was required, using the following internal references: CDCl3 (δC 77.0), MeOD (δC 49.0), and DMSO-d6 (δC 39.5). High resolution mass spectra were recorded on an Agilent 1200 series HPLC and diode array detector coupled to a 6210 time-of-flight mass spectrometer with dual multimode APCI/ESI source or on a Waters Acquity UPLC and diode array detector coupled to a Waters G2 QToF mass spectrometer fitted with a multimode ESI/APCI source. Analytical separation was carried out according to the methods listed below. The mobile phase was a mixture of methanol (solvent A) and water (solvent B), both containing formic acid at 0.1%, UV detection was at 254 nm. Method I: Agilent 1200 series HPLC, Merck Purospher STAR (RP-18e, 30 mm × 4 mm) column using a flow rate of 1.5 mL/min in a 4 min gradient elution. Gradient elution was as follows: 10:90 (A/B) to 90:10 (A/B) over 2.5 min, 90:10 (A/B) for 1 min, and then reversion back to 10:90 (A/B) over 0.3 min, finally 10:90 (A/B) for 0.2 min. Method II: Agilent 1200 series HPLC, Merck Chromolith flash column (RP-18e, 25 mm × 2 mm) at 30 °C using a flow rate of 0.75 mL/min in a 4 min gradient elution. Gradient elution was as follows: 5:95 (A/B) to 100:0 (A/B) over 2.5 min, 100:0 (A/B) for 1 min, and then reversion back to 5:95 (A/B) over 0.1 min, finally 5:95 (A/B) for 0.4 min. Method III: Waters Acquity UPLC, Phenomenex Kinetex XB-C18 column (30 mm × 2.1 mm, 1.7 μ, 100 A) at 30 °C using flow rate of 0.3 mL/min in a 4 min gradient elution. Gradient elution was as follows: 10:90 (A/B) to 90:10 (A/B) over 3 min, 90:10 (A/B) for 0.5 min, and then reversion back to 10:90 (A/B) over 0.3 min, finally 10:90 (A/B) for 0.2 min; Method IV: Waters Acquity UPLC, Phenomenex Kinetex C18 column (30 mm × 2.1 mm, 2.6 μ, 100A), flow rate and gradient elution according to Method III. The following reference masses were used for HRMS analysis: Agilent 1200 series, caffeine [M + H]+ 195.087652, hexakis(1H,1H,3H-tetrafluoropentoxy)phosphazene [M + H]+ 922.009798, and hexakis(2,2-difluoroethoxy)phosphazene [M + H]+ 622.02896 or reserpine [M + H]+ 609.280657. Waters Acquity UPLC: leucine enkephalin fragment ion [M + H]+ 397.1876. All compounds were >95% purity by LCMS analysis unless otherwise stated.
General Synthetic Procedures
Method A
Oxalyl chloride (1.2–1.7 equiv) was added dropwise to a solution of the carboxylic acid (1.0–1.5 equiv) and DMF in anhydrous dichloromethane. The reaction mixture was stirred at room temperature for 3–4 h and then concentrated. The remaining residue was redissolved in dichloromethane and concentrated again. Then, the aniline (1.0 equiv) was added to the remaining residue and the combined solids were dissolved in anhydrous pyridine or a mixture of anhydrous pyridine and dichloromethane. The resulting reaction mixture was stirred at room temperature overnight.
Method B
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU, 1.2–1.3 equiv) was added to a solution of the carboxylic acid (1.0–1.2 equiv) and N,N-diisopropylethylamine (2.0–5.0 equiv) in anhydrous DMF. The reaction mixture was stirred for 10 min before the aniline (1.0 equiv) was added. The reaction mixture was stirred at room temperature overnight. The reaction mixture was then diluted with water. The resulting precipitate was isolated by filtration and washed with water.
Method C
The (3-nitrophenyl)amide (1.0 equiv), iron powder (9–11 equiv), and ammonium chloride (9–11 equiv) in ethanol/water (4/1) were added to a round-bottom flask and heated to reflux overnight. Then the reaction mixture was filtered over a short pad of Celite or silica gel. The filtrate was reduced in vacuo until dryness. The remaining residue was resuspended in dichloromethane and washed with satd NaHCO3 (aq), water, and brine. The organic layer was dried over sodium sulfate or magnesium sulfate and reduced in vacuo until dryness to afford the corresponding (3-aminophenyl)amide.
Method D
Palladium (10% on activated carbon, 42–68 mg/1 mmol) was added to a suspension of the nitro compound in a 1:1 mixture of ethanol/ethyl acetate and stirred under H2 (1 atm.) at 28 °C overnight. The reaction mixture was then filtered through Celite and the Celite pad washed with further ethyl acetate. The filtrate was concentrated in vacuo to afford the corresponding aniline.
Method E
NaH (60% in mineral oil, 1.1–2.2 equiv) was added to a solution of the alcohol (1.2–2.8 equiv) in anhydrous THF at 0 °C. The reaction was allowed to stir at 0 °C for 10 min, then at room temperature for 30 min. 6-Bromo-2-chloroquinoline was added and the resulting suspension heated to reflux for 1–16 h. The reaction was allowed to cool to room temperature, then diluted with water/satd NaHCO3 (aq and extracted with dichloromethane (3×)). The organic layers were combined, washed with water, dried over magnesium sulfate, and concentrated in vacuo to afford the crude product.
Method F
n-BuLi (1.2–2.2 equiv, 2.5 M in hexanes, freshly titrated before use using standard procedures) was added dropwise to a solution of the aryl bromide in anhydrous THF at −78 °C under nitrogen. The reaction was stirred at −78 °C for 40 min before solid CO2 was added. After stirring for 5 min, the reaction was allowed to warm to room temperature. The reaction was quenched with water and concentrated under reduced pressure to remove the THF. The aqueous layer was washed with ethyl acetate, acidified to pH 3 by the addition of 2 M HCl (aq), and concentrated in vacuo to afford the crude product.
Preparation of Compounds in Table 1
2-Methyl-N-(2-methyl-5-nitrophenyl)quinoline-6-carboxamide 4
2-Methylquinoline-6-carboxylic acid 3 (3.69 g, 19.72 mmol), oxalyl chloride (1.8 mL, 20.15 mmol), and DMF (310 μL, 4.00 mmol) in anhydrous dichloromethane (35 mL) were reacted according to method A. 2-Methyl-5-nitroaniline 2 (3.0 g, 19.72 mmol), and the acid chloride were then reacted in anhydrous pyridine (35 mL). The reaction mixture was reduced in vacuo until dryness. The remaining residue was triturated with diethyl ether. The crude product was purified by column chromatography (Biotage, gradient of 0–50% ethanol in dichloromethane) to afford the product as a yellow amorphous solid (5.57 g, 88%). IR (thin film): νmax 1661, 1527, 1342, 1260 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.63 (d, J = 1.8 Hz, 1H), 8.43 (d, J = 8.4 Hz, 1H), 8.40 (d, J = 2.4 Hz, 1H), 8.25 (dd, J = 8.7, 2.0 Hz, 1H), 8.07 (d, J = 2.9 Hz, 1H), 8.05 (d, J = 3.0 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 2.71 (s, 3H), 2.44 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.54, 161.05, 146.20, 144.90, 143.38, 142.15, 137.76, 132.04, 130.08, 129.93, 129.31, 126.62, 126.09, 124.16, 121.00, 120.94, 23.98, 18.82. HRMS (ESI+): calcd for C18H15NaN3O3 (M + Na)+, 344.1006; found, 344.0999.
N-(5-Amino-2-methylphenyl)-2-methylquinoline-6-carboxamide 5
2-Methyl-N-(2-methyl-5-nitrophenyl)quinoline-6-carboxamide 4 (4.0 g, 12.45 mmol), iron powder (6.95 g, 124.0 mmol), and ammonium chloride (2.12 g, 124.0 mmol) in ethanol (50 mL) and water (12.5 mL) were reacted according to method C to afford the product as a light-yellow amorphous solid (1.95 g, 54%). IR (thin film): νmax 3340, 3252, 1645, 1582, 1487, 1278 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.61–8.51 (m, 1H), 8.38 (d, J = 8.4 Hz, 1H), 8.21 (dd, J = 8.8, 1.7 Hz, 1H), 8.00 (d, J = 8.8 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.65 (d, J = 2.0 Hz, 1H), 6.42 (dd, J = 8.1, 2.2 Hz, 1H), 4.94 (s, 2H), 2.70 (s, 3H), 2.09 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.18, 161.09, 148.86, 147.31, 137.58, 137.01, 132.25, 130.87, 128.72, 128.49, 128.38, 125.84, 123.38, 120.72, 112.75, 112.61, 25.49, 17.50. HRMS (ESI+): calcd for C18H18N3O (M + H)+, 292.1444; found, 292.1446.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-methylquinoline-6-carboxamide 1
2-Methyl-6-quinolinecarboxylic acid (600 mg, 3.21 mmol), HATU (1.46 g, 3.85 mmol), and N-(5-amino-2-methylphenyl)-2-methylquinoline-6-carboxamide 5 (910 mg, 3.21 mmol) were reacted in N,N-diisopropylethylamine (1.12 mL, 6.41 mmol) and anhydrous DMF (25 mL) according to method B to afford the product as an off-white amorphous solid (1.38 g, 95%). IR (solid): νmax 2922, 1645, 1582, 1496, 1284, 1258, 1063 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.14 (s, 1H), 10.08 (s, 1H), 8.61 (s, 1H), 8.41 (br d, J = 7.3 Hz, 1H), 8.24 (d, J = 8.7 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.87 (d, J = 2.1 Hz, 1H), 7.64–7.56 (m, 2H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.24 (d, J = 8.5 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.35–4.26 (m, 4H), 2.71 (s, 3H), 2.24 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.44, 164.82, 161.21, 148.93, 146.80, 143.39, 137.77, 137.62, 136.74, 131.97, 130.59, 129.23, 128.81, 128.64, 128.38, 128.14, 125.86, 123.44, 121.66, 119.07, 118.63, 117.30, 117.12, 64.86, 64.48, 25.51, 17.94. HRMS (ESI+): calcd for C27H24N3O4 (M + H)+, 454.1761; found, 454.1733.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-5-carboxamido)-2-methylphenyl)-2-methylquinoline-6-carboxamide 6
2,3-Dihydrobenzo[b][1,4]dioxine-5-carboxylic acid (46 mg, 0.26 mmol), HATU (127 mg, 0.34 mmol), and N-(5-amino-2-methylphenyl)-2-methylquinoline-6-carboxamide 5 (75 mg, 0.26 mmol) in N,N-diisopropylethylamine (0.14 mL, 0.82 mmol) and anhydrous DMF (2.5 mL) were reacted according to method B to afford the product as a white amorphous solid (95 mg, 81%). IR (solid): νmax 3370, 3256, 1644, 1598, 1532, 1450 1091, 743 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.16 (s, 1H), 10.09 (s, 1H), 8.61 (s, 1H), 8.41 (d, J = 8.4 Hz, 1H), 8.27–8.21 (m, 2H), 8.03 (d, J = 8.7 Hz, 1H), 7.82 (br s, 1H), 7.58–7.52 (m, 2H), 7.25 (d, J = 8.3 Hz, 1H), 7.18–7.11 (m, 2H), 7.02–6.98 (m, 1H), 6.92 (t, J = 7.8 Hz, 1H), 4.40–4.27 (m, 4H), 2.71 (s, 3H), 2.24 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.43, 164.26, 161.23, 148.94, 144.11, 141.65, 137.62, 137.52, 136.89, 131.90, 130.74, 129.44, 128.81, 128.67, 128.38, 126.14, 125.85, 123.45, 121.73, 121.17, 119.51, 118.44, 118.00, 64.93, 64.21, 25.51, 17.97. HRMS (ESI+): calcd for C27H24N3O4 (M + H)+, 454.1761; found, 454.1760.
N-(5-(3,4-Dimethoxybenzamido)-2-methylphenyl)-2-methylquinoline-6-carboxamide 7
3,4-Dimethoxybenzoic acid (34 mg, 0.189 mmol), HATU (82 mg, 0.215 mmol), and N-(5-amino-2-methylphenyl)-2-methylquinoline-6-carboxamide 5 (50 mg, 0.172 mmol) in N,N-diisopropylethylamine (0.075 mL, 0.429 mmol) and anhydrous DMF (1.2 mL) were reacted according to method B. The crude product was purified by flash column chromatography (1–5% methanol in dichloromethane) to afford compound 7 as a white amorphous solid (52 mg, 67%). IR (thin film): νmax 1648, 1601, 1508, 1418, 1268, 1225 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.13 (s, 1H), 10.09 (s, 1H), 8.61 (d, J = 2.0 Hz, 1H), 8.41 (d, J = 8.4 Hz, 1H), 8.24 (dd, J = 8.8, 2.0 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.85 (d, J = 2.2 Hz, 1H), 7.62 (td, J = 8.8, 2.2 Hz, 2H), 7.55 (d, J = 2.1 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.26 (d, J = 8.5 Hz, 1H), 7.08 (d, J = 8.5 Hz, 1H), 3.85 (s, 3H), 3.84 (s, 3H), 2.71 (s, 3H), 2.26 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.01, 164.74, 160.76, 151.58, 148.48, 148.26, 137.32, 137.16, 136.27, 131.51, 130.15, 128.75, 128.37, 128.16, 127.91, 126.92, 125.40, 122.98, 121.03, 118.76, 118.32, 110.98, 110.91, 55.67, 55.62, 25.04, 17.46. HRMS (ESI+): calcd for C27H26N3O (M + H)+, 456.19178; found, 456.19189.
N-(5-(Chroman-7-carboxamido)-2-methylphenyl)-2-methylquinoline-6-carboxamide 8
2-Methyl-6-quinolinecarboxylic acid (75 mg, 0.42 mmol), HATU (182 mg, 0.48 mmol), and N-(5-amino-2-methylphenyl)-2-methylquinoline-6-carboxamide 5 (111 mg, 0.38 mmol) in N,N-diisopropylethylamine (0.20 mL, 1.15 mmol) and anhydrous DMF (3.5 mL) were reacted according to method B. The crude product was purified by column chromatography (Biotage, gradient of 0–10% methanol in dichloromethane) to afford an off-white amorphous solid. This material was triturated with diethyl ether to afford the product as a white amorphous solid (51 mg, 30%). IR (thin film): νmax 2957, 1651, 1599, 1527, 1499, 1310, 1283 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.14 (s, 1H), 10.13 (s, 1H), 8.61 (d, J = 1.8 Hz, 1H), 8.41 (d, J = 8.4 Hz, 1H), 8.24 (dd, J = 8.8, 1.9 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.89 (d, J = 2.0 Hz, 1H), 7.60 (dd, J = 8.3, 2.1 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.43 (dd, J = 7.9, 1.7 Hz, 1H), 7.36 (d, J = 1.6 Hz, 1H), 7.25 (d, J = 8.4 Hz, 1H), 7.19 (d, J = 7.9 Hz, 1H), 4.23–4.14 (m, 2H), 2.80 (t, J = 6.4 Hz, 2H), 2.71 (s, 3H), 2.25 (s, 3H), 1.94 (p, J = 6.3 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.44, 165.31, 161.21, 154.79, 148.93, 137.70, 137.63, 136.73, 134.25, 131.96, 130.61, 130.29, 129.32, 128.81, 128.64, 128.38, 126.70, 125.86, 123.44, 119.61, 119.08, 118.64, 115.94, 66.57, 25.51, 24.73, 22.00, 17.95. HRMS (ESI+): calcd for C28H26N3O3 (M + H)+, 452.1969; found, 452.1956.
N-(5-(Chroman-6-carboxamido)-2-methylphenyl)-2-methylquinoline-6-carboxamide 9
Chroman-6-carboxylic acid (40 mg, 0.227 mmol), HATU (98 mg, 0.257 mmol), and N-(5-amino-2-methylphenyl)-2-methylquinoline-6-carboxamide 5 (60 mg, 0.206 mmol) in N,N-diisopropylethylamine (0.088 mL, 0.505 mmol) and anhydrous DMF (1.5 mL) were reacted according to method B. The crude product was purified by flash column chromatography (1–3.5% methanol in dichloromethane) to afford the product as a white amorphous solid (62 mg, 62%). IR (thin film): νmax 1647, 1600, 1528, 1495, 1318, 1256 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.13 (s, 1H), 10.04 (s, 1H), 8.61 (d, J = 2.0 Hz, 1H), 8.41 (d, J = 8.4 Hz, 1H), 8.24 (dd, J = 8.7, 2.0 Hz, 1H), 8.03 (d, J = 8.7 Hz, 1H), 7.87 (d, J = 2.2 Hz, 1H), 7.75 (d, J = 2.3 Hz, 1H), 7.72 (dd, J = 8.5, 2.3 Hz, 1H), 7.59 (dd, J = 8.3, 2.2 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.24 (d, J = 8.6 Hz, 1H), 6.84 (d, J = 8.5 Hz, 1H), 4.23–4.17 (m, 2H), 2.82 (t, J = 6.4 Hz, 2H), 2.71 (s, 3H), 2.24 (s, 3H), 2.00–1.91 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 164.98, 164.86, 160.75, 157.36, 148.47, 137.46, 137.16, 136.25, 131.51, 130.12, 129.78, 128.63, 128.36, 128.17, 127.91, 127.00, 126.26, 125.40, 122.98, 122.03, 118.54, 118.11, 116.07, 66.36, 25.05, 24.26, 21.54, 17.47. HRMS (ESI+): calcd for C28H26N3O (M + H)+, 452.19687; found, 452.19758.
N-(5-(Isochroman-7-carboxamido)-2-methylphenyl)-2-methylquinoline-6-carboxamide 10
7-Bromoisochroman (152 mg, 0.71 mmol), Herrmann’s palladacycle (33.4 mg, 0.04 mmol), and tri-tert-butylphosphonium tetrafluoroborate (41 mg, 0.14 mmol) were combined with 2-(trimethylsilyl)ethanol (7.0 mL) in a microwave vial. Molybdenum hexacarbonyl (377 mg, 1.427 mmol) was then added, followed by DBU (1.0 M in THF, 2.57 mL, 2.57 mmol), and the vial promptly sealed. The reaction mixture was heated to 130 °C for 1 h in a microwave. After this time, further 2-(trimethylsilyl)ethanol (3.0 mL) was added and the reaction heated to 130 °C for 1 h under microwave irradiation. The reaction mixture was concentrated under high vacuum. The resulting residue was purified by column chromatography (Biotage, gradient of 0–100% ethyl acetate in cyclohexane) to afford 2-(trimethylsilyl)ethyl isochroman-7-carboxylate compound as a yellow oil (108 mg, 54%). This oil was only 80% pure but was used directly in the next step.75
To a stirring solution of 2-(trimethylsilyl)ethyl isochroman-7-carboxylate (105 mg, 0.302 mmol) in THF (3 mL) at room temperature under argon was dropwise added TBAF 1.0 M in THF (0.45 mL, 0.45 mmol). The reaction was allowed to stir at room temperature for 2 h. The reaction mixture was then diluted with water (25 mL) and the THF removed in vacuo. The aqueous layer was washed with dichloromethane (15 mL). TLC/LCMS showed that there was product in both the aqueous and organic layers. Therefore, the organic layer was extracted with a portion of 1 M NaOH (15 mL), followed by water (15 mL). The combined aqueous layers were acidified (to ∼ pH 2) using 1 M HCl (aq) and extracted with dichloromethane (3 × 15 mL). The combined organic layer was concentrated in vacuo to afford isochroman-7-carboxylic acid as a colorless amorphous solid. This material was used in the next step without further purification.
Isochroman-7-carboxylic acid (50 mg, 0.28 mmol), HATU (130 mg, 0.34 mmol), and N-(5-amino-2-methylphenyl)-2-methylquinoline-6-carboxamide 5 (87 mg, 0.30 mmol) in N,N-diisopropylethylamine (0.14 mL, 0.82 mmol) and anhydrous DMF (2.5 mL) were reacted according to method B. The crude product was purified by column chromatography (Biotage, gradient of 0–5% methanol in dichloromethane) to afford a pale-yellow amorphous solid (78 mg, 63%). IR (thin film): νmax 2965, 1650, 1600, 1528, 1499, 1285 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.19 (s, 1H), 10.14 (s, 1H), 8.61 (d, J = 2.0 Hz, 1H), 8.41 (d, J = 8.4 Hz, 1H), 8.24 (dd, J = 8.8, 2.0 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.88 (d, J = 2.1 Hz, 1H), 7.78 (dd, J = 8.0, 1.8 Hz, 1H), 7.67–7.65 (m, 1H), 7.60 (dd, J = 8.3, 2.2 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.26 (d, J = 8.6 Hz, 1H), 4.76 (s, 2H), 3.91 (t, J = 5.7 Hz, 2H), 2.86 (t, J = 5.7 Hz, 2H), 2.71 (s, 3H), 2.25 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.58, 165.46, 161.22, 148.91, 137.70, 137.66, 137.49, 136.76, 135.49, 132.87, 131.96, 130.65, 129.34, 129.26, 128.80, 128.65, 128.40, 125.96, 125.86, 124.37, 123.46, 119.05, 118.63, 67.49, 64.82, 28.24, 25.50, 17.95. HRMS (ESI+): calcd for C28H26N3O3 (M + H)+, 452.1969; found, 452.1964.
N-(3-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2,6-dimethylphenyl)-2-methylquinoline-6-carboxamide 11
2,6-Dimethylaniline (4.07 mL, 32.7 mmol) and sulfuric acid (30 mL) were added to a round-bottom flask. The solution was cooled on an ice bath and nitric acid (2.6 mL, 43.1 mmol, 69%) was added dropwise to the stirred solution. The resulting reaction mixture was warmed to room temperature and stirred for further 30 min at room temperature. The reaction mixture was then poured onto cold water and neutralized by addition of solid sodium hydroxide. 2,6-Dimethyl-3-nitroaniline was isolated by filtration and washed with copious amounts of water. The crude product was purified by column chromatography (Biotage, gradient of 0–40% ethyl acetate in cyclohexane) to afford the product as a brown amorphous solid (1.82 g, 33%). IR (solid): νmax 3421, 3349, 2919, 1634, 1513, 1460, 1348 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 6.99 (d, J = 8.1 Hz, 1H), 6.93 (d, J = 8.1 Hz, 1H), 5.21 (s, 2H), 2.15 (s, 3H), 2.14 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 150.03, 146.44, 127.90, 126.09, 113.71, 111.03, 18.71, 13.41. HRMS (ESI+): calcd for C8H11N2O2 (M + H)+, 167.0815; found, 167.0831.
2-Methylquinoline-6-carboxylic acid 3 (1.27 g, 6.77 mmol), oxalyl chloride (750 μL, 8.40 mmol), and DMF (110 μL, 1.42 mmol) in anhydrous dichloromethane (15 mL) were reacted according to method A. The resulting acid chloride and 2,6-dimethyl-3-nitroaniline (1.5 g, 3.01 mmol) were then reacted in anhydrous pyridine (20 mL). The reaction mixture was concentrated in vacuo. The remaining residue was redissolved in dichloromethane and washed with satd NaHCO3 (aq), water, and brine. The organic layer was dried over sodium sulfate and reduced in vacuo until dryness to afford N-(2,6-dimethyl-3-nitrophenyl)-2-methylquinoline-6-carboxamide as a dark-red to brown amorphous solid (1.69 g). The crude product was used in the next synthetic step without further purification.
N-(2,6-Dimethyl-3-nitrophenyl)-2-methylquinoline-6-carboxamide (1.50 g, 4.47 mmol), iron powder (2.50 g, 44.7 mmol), and ammonium chloride (762 mg, 44.7 mmol) in ethanol (40 mL) and water (10 mL) were reacted according to method C. The crude product was purified by column chromatography (Biotage, gradient of 0–30% ethanol in dichloromethane) to afford the product as a brown amorphous solid (1.14 g, 84%). IR (solid): νmax 3232, 2916, 1649, 1621, 1487, 1281 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 9.84 (s, 1H), 8.60 (d, J = 1.9 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 8.25 (dd, J = 8.8, 2.0 Hz, 1H), 8.01 (d, J = 8.7 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 6.56 (d, J = 8.1 Hz, 1H), 4.72 (s, 2H), 2.70 (s, 3H), 2.07 (s, 3H), 1.95 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.08, 161.05, 148.86, 145.57, 137.57, 135.64, 132.08, 128.77, 128.40, 128.27, 127.51, 125.89, 123.38, 123.09, 120.01, 113.46, 25.49, 18.15, 12.84. HRMS (ESI+): calcd for C19H20N3O (M + H)+, 306.1606; found, 306.1597.
2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxylic acid (415 mg, 2.30 mmol), oxalyl chloride (350 μL, 3.92 mmol), and DMF (50 μL, 646 mmol) in anhydrous dichloromethane (10 mL) were reacted according to method A. N-(3-Amino-2,6-dimethylphenyl)-2-methylquinoline-6-carboxamide (703 mg, 2.30 mmol) and the acid chloride were then reacted in anhydrous pyridine. The reaction mixture was diluted with diethyl ether. The product precipitated from the solution, isolated by filtration, and washed with water and diethyl ether. The crude product was purified by column chromatography (Biotage, gradient of 0–10% ethanol in dichloromethane) to afford compound 11 as a beige amorphous solid (237.4 mg, 22%). IR (thin film): νmax 1649, 1584, 1487, 1290, 1067 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.80 (s, 1H), 8.63 (s, 1H), 8.41 (d, J = 8.4 Hz, 1H), 8.27 (d, J = 10.8 Hz, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.55 (d, J = 2.0 Hz, 1H), 7.54–7.53 (m, 1H), 7.53–7.50 (m, 1H), 7.18 (m, 2H), 6.98 (d, J = 8.4 Hz, 1H), 4.32–4.29 (m, 4H), 2.71 (s, 3H), 2.24 (s, 3H), 2.10 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.22, 164.92, 161.17, 148.93, 146.73, 143.41, 137.60, 136.13, 135.30, 133.96, 132.95, 131.78, 128.86, 128.55, 128.27, 127.84, 127.38, 126.24, 125.90, 123.44, 121.61, 117.31, 117.12, 64.85, 64.47, 25.51, 18.59, 13.94. HRMS (ESI+): calcd for C28H26N3O4 (M + H)+, 468.1923; found, 428.1932.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2,4-dimethylphenyl)-2-methylquinoline-6-carboxamide 12
2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxylic acid (542 mg, 3.01 mmol), oxalyl chloride (325 μL, 4.71 mmol), and DMF (50 μL, 0.646 mmol) in anhydrous dichloromethane (10 mL) were reacted according to method A. 2,4-Dimethyl-5-nitroaniline (500 mg, 3.01 mmol) and the acid chloride were then reacted in anhydrous pyridine (15 mL). The reaction mixture was concentrated in vacuo. The remaining residue was triturated with diethyl ether and dried in vacuo to give N-(2,4-dimethyl-5-nitrophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide as a brown amorphous solid (1.11 g), which was used in the next synthetic step without further purification.
N-(2,4-Dimethyl-5-nitrophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (988 mg, 2.08 mmol), ammonium chloride (394 mg, 23.1 mmol), and iron powder (1.29 g, 23.1 mmol) in ethanol (10 mL) and water (2.5 mL) were reacted according to method C to afford N-(5-amino-2,4-dimethylphenyl)-2,3-dihydrobenz[b][1,4]dioxine-6-carboxamide as a brown amorphous solid (353 mg, 57% over two steps). IR (solid): νmax 3352, 3265, 2918, 1634, 1613, 1489, 1288 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 9.43 (s, 1H), 7.49 (d, J = 2.0 Hz, 1H), 7.47 (dd, J = 8.3, 2.1 Hz, 1H), 6.95 (d, J = 8.3 Hz, 1H), 6.77 (s, 1H), 6.58 (s, 1H), 4.65 (s, 2H), 4.40–4.10 (m, 4H), 2.02 (s, 3H), 2.00 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 164.53, 146.53, 144.85, 143.36, 134.81, 131.82, 128.22, 121.45, 121.11, 119.64, 117.23, 117.00, 113.13, 64.81, 64.46, 17.41, 17.29. HRMS (ESI+): calcd for C17H19N2O3 (M + H)+, 299.1396; found, 299.1555.
2-Methylquinoline-6-carboxylic acid 3 (188 mg, 1.00 mmol), oxalyl chloride (80 μL, 0.896 mmol), and DMF (20 μL, 0.258 mmol) in anhydrous dichloromethane (5 mL) were reacted according to method A. The resulting acid chloride and N-(5-amino-2,4-dimethylphenyl)-2,3-dihydrobenz[b][1,4]dioxine-6-carboxamide (201 mg, 0.674 mmol) were then reacted in anhydrous pyridine (10 mL). The reaction mixture was then diluted with dichloromethane and washed with satd NaHCO3 (aq), water, and brine. The organic layer was dried over sodium sulfate and reduced in vacuo until dryness. The crude product was purified by column chromatography (Biotage, gradient of 0–5% ethanol in dichloromethane). The product was further purified by semipreparative HPLC, according to the method specified in the general procedures, to afford a beige amorphous solid (30.4 mg, 9.7%). IR (solid): νmax 1664, 1620, 1531, 1497, 1291 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.09 (s, 1H), 9.71 (s, 1H), 8.59 (s, 1H), 8.40 (d, J = 8.4 Hz, 1H), 8.29–8.16 (m, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.64–7.43 (m, 3H), 7.35 (s, 1H), 7.17 (s, 1H), 6.98 (d, J = 8.3 Hz, 1H), 4.39–4.15 (m, 4H), 2.70 (s, 3H), 2.25 (s, 3H), 2.20 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.48, 164.83, 161.18, 148.91, 146.74, 143.41, 137.60, 134.65, 134.46, 132.25, 132.01, 131.85, 131.63, 128.77, 128.61, 128.40, 127.87, 125.84, 125.00, 123.43, 121.60, 117.31, 117.11, 64.84, 64.48, 25.50, 17.97, 17.89. HRMS (ESI+): calcd for C28H26N3O4 (M + H)+, 468.1923; found, 468.1914.
2-Methyl-N-(2-methyl-5-(N-methyl-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)quinoline-6-carboxamide 13
4-Methyl-3-nitroaniline 2 (5.15 g, 33.8 mmol) was dissolved in DMF (20 mL), and iodomethane (1.8 mL, 28.9 mmol) was added. Then, satd NaHCO3 (aq) (20 mL, 28.2 mmol) was added portionwise to the stirred reaction mixture. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was partitioned between dichloromethane and water. The aqueous layer was extracted with dichloromethane. The combined organic layers were then washed with water and brine. The crude product was purified by column chromatography (Biotage, gradient of 0–20% ethyl acetate in cyclohexane) to afford N,4-dimethyl-3-nitroaniline as a red amorphous solid (1.55 g, 33% yield). IR (thin film): νmax 3401, 2926, 1690, 1526, 1493, 1321, 1286 cm–1. 1H NMR (500 MHz, CDCl3) δ 7.18 (d, J = 2.6 Hz, 1H), 7.10 (d, J = 8.3 Hz, 1H), 6.75 (dd, J = 8.3, 2.6 Hz, 1H), 3.91 (s, 1H), 2.87 (s, 3H), 2.47 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 149.77, 148.06, 133.16, 121.34, 117.63, 106.97, 30.65, 19.53. HRMS (ESI+): calcd for C8H11N2O2 (M + H)+, 167.0820; found, 167.0825.
2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxylic acid (1.63 g, 9.03 mmol), oxalyl chloride (970 μL, 10.86 mmol), and DMF (140 μL, 1.81 mmol) in anhydrous dichloromethane (15 mL) were reacted according to method A. The resulting acid chloride and N-4-dimethyl-3-nitroaniline (1.5 g, 9.03 mmol) were then reacted in anhydrous dichloromethane (15 mL) and pyridine (3.0 mL, 34.0 mmol). The reaction mixture was diluted with dichloromethane and washed with satd NH4Cl (aq). The aqueous layer was extracted with dichloromethane. The combined organic layers were washed with satd NH4Cl (aq), water, and brine. The organic layer was dried over sodium sulfate and reduced in vacuo until dryness to give N-methyl-N-(4-methyl-3-nitrophenyl)-2,3-dihydrobenz[b][1,4]dioxine-6-carboxamide (2.51 g), which was used in the next synthetic step without further purification.
N-Methyl-N-(4-methyl-3-nitrophenyl)-2,3-dihydrobenz[b][1,4]dioxine-6-carboxamide (2.50 g, 7.61 mmol), iron powder (4.56 g, 82.0 mmol), and ammonium chloride (1.36 g, 80 mmol) in ethanol (20 mL) and water (5 mL) were reacted according to method C. The crude product was resuspended in water. The product was isolated by filtration and washed with water and diethyl ether. The product was isolated as a beige amorphous solid (1.18 g, 52%). IR (solid): νmax 3447, 3354, 2934, 1600, 1579, 1509, 1426, 1282 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 6.85–6.82 (m, 1H), 6.80 (d, J = 7.8 Hz, 1H), 6.78–6.72 (m, 1H), 6.66 (d, J = 8.4 Hz, 1H), 6.40–6.32 (m, 1H), 6.23–6.13 (m, 1H), 4.92 (s, 2H), 4.18 (d, J = 8.0 Hz, 4H), 3.23 (s, 3H), 1.97 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 168.76, 147.70, 144.89, 144.04, 142.72, 130.83, 129.67, 122.40, 119.85, 118.09, 116.54, 114.85, 112.22, 64.58, 64.32, 38.98, 17.45. HRMS (ESI+): calcd for C17H19N2O3 (M + H)+, 300.1474; found, 299.1401.
2-Methylquinoline-6-carboxylic acid 3 (345 mg, 1.84 mmol), oxalyl chloride (180 μL, 2.06 mmol), and DMF (300 μL, 3.87 mmol) in anhydrous dichloromethane (10 mL) were reacted according to method A. The resulting acid chloride and N-(3-amino-4-methylphenyl)-N-methyl-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (500 mg, 1.68 mmol) were then reacted in anhydrous dichloromethane (13 mL) and pyridine (1.2 mL, 14.89 mmol). The reaction mixture was diluted with water. The product was isolated by filtration and washed with water and diethyl ether. The crude product was then purified with a cation exchange sorbent (Isolute SCX-II, washing first with methanol, and then with 10% 2 M ammonia in methanol). The product was further purified by semipreparative HPLC according to the method specified in the general procedures to afford a yellow amorphous solid (22.4 mg, 2.9%). IR (solid): νmax 2927, 1624, 1493, 1282 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.59 (d, J = 1.8 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 8.21 (dd, J = 8.8, 1.9 Hz, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.36 (d, J = 2.2 Hz, 1H), 7.18 (d, J = 8.2 Hz, 1H), 6.90 (dd, J = 8.1, 2.2 Hz, 1H), 6.87 (d, J = 2.0 Hz, 1H), 6.76 (d, J = 2.0 Hz, 1H), 6.68 (d, J = 8.4 Hz, 1H), 4.27–4.14 (m, 4H), 3.17 (s, 3H), 2.70 (s, 3H), 2.23 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 169.06, 161.29, 148.95, 144.99, 143.28, 142.94, 137.62, 137.39, 131.93, 131.75, 131.25, 129.44, 128.80, 128.75, 128.39, 125.81, 125.05, 124.52, 123.47, 122.42, 118.16, 116.71, 64.60, 64.33, 49.06, 38.94, 25.51, 18.04. HRMS (ESI+): calcd for C28H26N3O4 (M + H)+, 468.1923; found, 468.1919.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-sulfonamido)-2-methylphenyl)-2-methylquinoline-6-carboxamide 14
2-Methylquinoline-6-carboxylic acid (3.69 g, 19.72 mmol), oxalyl chloride (1.8 mL, 20.15 mmol), and DMF (310 μL, 4.00 mmol) were reacted in anhydrous dichloromethane (35 mL) according to method A. The resulting acid chloride was reacted with 2-methyl-5-nitroaniline (3.0 g, 19.72 mmol) in anhydrous pyridine (35 mL). The reaction mixture was reduced in vacuo until dryness. The remaining residue was triturated with diethyl ether and dried in vacuo. The crude product was purified by column chromatography (Biotage, gradient of 0–50% ethanol in dichloromethane) to afford the product as a yellow amorphous solid (5.57 g, 88%). IR (thin film): νmax 1661, 1527, 1342, 1260 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.63 (d, J = 1.8 Hz, 1H), 8.43 (d, J = 8.4 Hz, 1H), 8.40 (d, J = 2.4 Hz, 1H), 8.25 (dd, J = 8.7, 2.0 Hz, 1H), 8.07 (d, J = 2.9 Hz, 1H), 8.05 (d, J = 3.0 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 2.71 (s, 3H), 2.44 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.54, 161.05, 146.20, 144.90, 143.38, 142.15, 137.76, 132.04, 130.08, 129.93, 129.31, 126.62, 126.09, 124.16, 121.00, 120.94, 23.98, 18.82. HRMS (ESI+): calcd for C18H15NaN3O3 (M + Na)+, 344.1006; found, 344.0999.
2-Methyl-N-(2-methyl-5-nitrophenyl)quinoline-6-carboxamide (4.0 g, 12.45 mmol), iron powder (6.95 g, 124.0 mmol), and ammonium chloride (2.12 g, 124.0 mmol) in ethanol (50 mL) and water (12.5 mL) were reacted according to method C to afford N-(5-amino-2-methylphenyl)-2-methylquinoline-6-carboxamide as a light-yellow amorphous solid (1.95 g, 48% over two steps). IR (thin film): νmax 3340, 3252, 1645, 1582, 1487, 1278 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.61–8.51 (m, 1H), 8.38 (d, J = 8.4 Hz, 1H), 8.21 (dd, J = 8.8, 1.7 Hz, 1H), 8.00 (d, J = 8.8 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.65 (d, J = 2.0 Hz, 1H), 6.42 (dd, J = 8.1, 2.2 Hz, 1H), 4.94 (s, 2H), 2.70 (s, 3H), 2.09 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.18, 161.09, 148.86, 147.31, 137.58, 137.01, 132.25, 130.87, 128.72, 128.49, 128.38, 125.84, 123.38, 120.72, 112.75, 112.61, 25.49, 17.50. HRMS (ESI+): calcd for C18H18N3O (M + H)+, 292.1444; found, 292.1446.
N-(5-Amino-2-methylphenyl)-2-methylquinoline-6-carboxamide (500 mg, 1.60 mmol) and 2,3-dihydrobenzo[b][1,4]dioxine-6-sulfonyl chloride (591 mg, 2.39 mmol) and anhydrous pyridine (7 mL) were added to a round-bottom flask. The resulting reaction mixture was stirred at room temperature under argon overnight. The reaction mixture was diluted with diethyl ether. The product was isolated by filtration and washed with water and diethyl ether. The crude product was purified by column chromatography (Biotage, gradient of 0–10% ethanol in dichloromethane) to afford the product as a beige amorphous solid (90.7 mg, 12%). IR (thin film): νmax 1651, 1529, 1494, 1328, 1285, 1254, 1153 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.14 (s, 1H), 10.06 (s, 1H), 8.57 (d, J = 1.9 Hz, 1H), 8.40 (d, J = 8.4 Hz, 1H), 8.20 (dd, J = 8.8, 2.0 Hz, 1H), 8.01 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.30–7.23 (m, 2H), 7.22 (d, J = 2.2 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 6.94 (dd, J = 8.2, 2.2 Hz, 1H), 4.53–3.98 (m, 4H), 2.70 (s, 3H), 2.16 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.30, 161.25, 147.61, 143.69, 137.73, 137.72, 137.30, 136.26, 132.43, 131.78, 131.30, 129.59, 128.75, 128.72, 128.41, 125.83, 123.47, 120.81, 118.35, 118.00, 117.84, 116.11, 64.80, 64.48, 25.46, 17.83. HRMS (ESI+): calcd for C26H24N3O5S (M + H)+, 490.1436; found, 490.1436.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)quinoline-6-carboxamide 15
6-Quinolinecarboxylic acid (0.064 g, 0.369 mmol), HATU (0.160 g, 0.422 mmol), and N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide 18 (0.100 g, 0.352 mmol) in N,N-diisopropylethylamine (0.123 mL, 0.703 mmol) and anhydrous DMF (2.5 mL) were reacted according to method B to afford a white amorphous solid (141 mg, 91%). IR (thin film): νmax 1649, 1599, 1528, 1504, 1318, 1290, 1260, 1066 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.18 (s, 1H), 10.08 (s, 1H), 9.02 (dd, J = 4.2, 1.9 Hz, 1H), 8.67 (d, J = 1.9 Hz, 1H), 8.55 (dd, J = 8.5, 1.9 Hz, 1H), 8.29 (dd, J = 8.8, 2.2 Hz, 1H), 8.15 (d, J = 8.8 Hz, 1H), 7.88 (d, J = 2.1 Hz, 1H), 7.65 (dd, J = 9.6, 2.2 Hz, 1H), 7.59 (dd, J = 8.5, 2.2 Hz, 1H), 7.54 (d, J = 2.2 Hz, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.25 (d, J = 8.5 Hz, 1H), 6.99 (d, J = 8.5 Hz, 1H), 4.33–4.29 (m, 4H), 2.25 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.38, 164.83, 152.68, 149.25, 146.82, 143.40, 137.80, 137.67, 136.67, 132.83, 130.63, 129.61, 129.24, 128.95, 128.47, 128.14, 127.61, 122.73, 121.67, 119.06, 118.68, 117.31, 117.13, 64.86, 64.49, 17.95. HRMS (ESI+): calcd for C26H22N3O4 (M + H)+, 440.1605; found, 440.1768.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)isoquinoline-7-carboxamide 16
Isoquinoline-7-carboxylic acid (50 mg, 0.290 mmol), HATU (130 mg, 0.343 mmol), and N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide 18 (75 mg, 0.264 mmol) in N,N-diisopropylethylamine (0.101 mL, 0.580 mmol) and anhydrous DMF (1.8 mL) were reacted according to method B. The crude product was purified by trituration and brief sonication in ethyl acetate, then isolated by filtration and washed with ethyl acetate to afford the product as a white amorphous solid (52 mg, 45%). IR (thin film): νmax 1641, 1599, 1531, 1504, 1319, 1295, 1243, 1065 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.23 (s, 1H), 10.08 (s, 1H), 9.48 (s, 1H), 8.81 (s, 1H), 8.62 (d, J = 5.7 Hz, 1H), 8.30 (dd, J = 8.6, 1.7 Hz, 1H), 8.12 (d, J = 8.6 Hz, 1H), 7.94 (d, J = 5.7 Hz, 1H), 7.89 (d, J = 2.1 Hz, 1H), 7.59 (dd, J = 8.3, 2.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.25 (d, J = 8.6 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.32–4.29 (m, 4H), 2.25 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.08, 164.82, 153.21, 146.81, 143.39, 143.20, 137.80, 137.37, 136.53, 134.19, 130.65, 130.19, 129.23, 128.72, 128.11, 127.81, 127.48, 121.65, 121.41, 119.02, 118.74, 117.30, 117.12, 64.85, 64.48, 17.93. HRMS (ESI+): calcd for C26H22N3O4 (M + H)+, 440.1605; found, 440.1604.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-1,2,3,4-tetrahydroquinoline-6-carboxamide 17
A suspension of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2 methylphenyl)quinoline-6-carboxamide 15 (0.097 g, 0.22 mmol) and Pd/C (10%, 0.025 g) in methanol (8 mL), ethyl acetate (6 mL), and 10 drops of glacial acetic acid was stirred under 1 atm hydrogen at 40 °C overnight, filtered through Celite with ethyl acetate, and concentrated. The residue was purified by flash column chromatography using a gradient of 10–22% ethyl acetate in dichloromethane to afford the title compound (72 mg, 74%) as a white amorphous solid. IR (solid): νmax 2924, 1634, 1604, 1579, 1504, 1291, 1248, 1065, 888 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.02 (s, 1H), 9.35 (s, 1H), 7.77 (d, J = 2.2 Hz, 1H), 7.57–7.52 (m, 4H), 7.50 (dd, J = 8.5, 2.2 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.47 (d, J = 9.0 Hz, 1H), 6.35 (br s, 1H), 4.34–4.27 (m, 4H), 3.27–3.20 (m, 2H), 2.72 (t, J = 6.2 Hz, 2H), 2.17 (s, 3H), 1.85–1.76 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.65, 164.74, 148.79, 146.76, 143.38, 137.56, 137.50, 130.31, 129.43, 129.00, 128.21, 127.29, 121.64, 120.44, 119.09, 119.06, 117.93, 117.28, 117.11, 112.41, 64.85, 64.48, 41.05, 27.27, 21.50, 17.95. HRMS (ESI+): calcd for C26H26N3O4 (M + H)+, 444.1718; found, 444.1812.
N-(3-Amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide 18
1,4-Benzodioxane-6-carboxylic acid (2.486 g, 13.80 mmol), oxalyl chloride (1.40 mL, 16.6 mmol), and DMF (0.027 mL, 0.34 mmol) in anhydrous dichloromethane (34 mL) were reacted according to method A. The resulting acid chloride was dissolved in anhydrous dichloromethane (12 mL) and added dropwise to a solution of 4-methyl-3-nitroaniline 2 (2.100 g, 13.80 mmol) and pyridine (2.23 mL, 27.6 mmol) in anhydrous dichloromethane (35 mL). The reaction mixture was stirred at room temperature for 2 h and then concentrated. The resulting amorphous solid was suspended in methanol, diluted with water, and then isolated by filtration and washed with water to afford N-(4-methyl-3-nitrophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (4.24 g, 98%) as a pale-tan colored amorphous solid, which was used in the next step without further purification.
Palladium (10% on activated carbon, 0.567 g) and N-(4-methyl-3-nitrophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (4.237 g, 13.48 mmol) in ethanol (90 mL) and ethyl acetate (90 mL) were reacted according to method D to afford the desired product (3.803 g, 99%) as a pale-yellow amorphous solid. IR (thin film): νmax 1641, 1611, 1582, 1289, 1064 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 9.70 (s, 1H), 7.49 (d, J = 2.2 Hz, 1H), 7.46 (dd, J = 8.3, 2.2 Hz, 1H), 7.10 (d, J = 2.0 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 8.1 Hz, 1H), 6.79 (dd, J = 8.1, 2.0 Hz, 1H), 4.81 (s, 2H), 4.32–4.26 (m, 4H), 2.01 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 164.48, 146.89, 146.54, 143.30, 138.09, 130.04, 128.60, 121.54, 117.18, 117.05, 109.26, 106.88, 64.82, 64.47, 17.46. (1 carbon missing) HRMS (ESI+): calcd for C16H17N2O3 (M + H)+, 285.1234; found, 285.1233.
Preparation of Compounds in Table 2
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(2-methoxyethoxy)quinoline-6-carboxamide 21
NaH (60% in mineral oil, 0.198 g, 4.950 mmol) was added to 2-methoxyethanol (4.0 mL, 50.7 mmol) at 0 °C under inert atmosphere. The reaction mixture was stirred at 0 °C for 10 min then at room temperature for 20 min before 6-bromo-2-chloroquinoline (0.400 g, 1.649 mmol) was added. This suspension was then gradually heated to 70 °C, the amorphous solid dissolved as heated, and the resulting solution was allowed to stir overnight. The reaction mixture was cooled to room temperature and diluted with water and the resulting precipitate isolated by filtration. The precipitate was washed with water and dried to afford 6-bromo-2-(2-methoxyethoxy)quinoline as a dull-pink amorphous solid (0.497 g), which was used in the next step without further purification.
n-BuLi (1.84 M in hexanes) (0.578 mL, 1.063 mmol) was added dropwise to a solution of 6-bromo-2-(2-methoxyethoxy)quinoline (0.250 g, 0.886 mmol) in anhydrous THF (3.0 mL) at −78 °C and the resulting brown-yellow mixture was stirred at −78 °C for 35 min. Solid CO2 was added, and the resulting orange mixture was stirred at −78 °C for 5 min, then it was allowed to warm to room temperature and concentrated under reduced pressure. The residue was dissolved in brine (5.0 mL) and washed with dichloromethane (1 × 5.0 mL). The aqueous phase was acidified with 2M HCl (aq) to pH 3, and the resulting precipitate was isolated by filtration and washed with water to afford 2-(2-methoxyethoxy)quinoline-6-carboxylic acid as a pale pink amorphous solid (0.127 g), which was used in next step without further purification.
2-(2-Methoxyethoxy)quinoline-6-carboxylic acid (0.048 g, 0.193 mmol), HATU (0.084 g, 0.220 mmol), and N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide 18 (0.050 g, 0.176 mmol) in N,N-diisopropylethylamine (0.068 mL, 0.387 mmol) and anhydrous DMF (1.25 mL) were reacted according to method B to afford the product 21 as a white amorphous solid (87 mg, 56% over 3 steps). IR (thin film): νmax 2925, 1642, 1602, 1582, 1526, 1502, 1345, 1286, 1261, 816, 784 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.07 (app. d, J = 3.0 Hz, 2H), 8.57 (d, J = 2.0 Hz, 1H), 8.39 (d, J = 8.8 Hz, 1H), 8.22 (dd, J = 8.7, 2.0 Hz, 1H), 7.89–7.84 (m, 2H), 7.58 (dd, J = 8.3, 2.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.24 (d, J = 8.6 Hz, 1H), 7.14 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.61–4.55 (m, 2H), 4.32–4.29 (m, 4H), 3.77–3.72 (m, 2H), 3.33 (s, 3H), 2.24 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.39, 164.81, 163.13, 147.93, 146.80, 143.38, 140.80, 137.75, 136.78, 130.57, 129.22, 128.90, 128.58, 128.15, 127.21, 126.76, 124.52, 121.65, 119.07, 118.57, 117.29, 117.12, 114.39, 70.56, 65.40, 64.85, 64.48, 58.58, 17.94. HRMS (ESI+): calcd for C29H28N3O6 (M + H)+, 514.1973; found, 514.1969.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(3-(dimethylamino)propoxy)quinoline-6-carboxamide 22
NaH (60% in mineral oil, 0.109 g, 2.72 mmol), 4-(2-hydroxyethyl)morpholine (0.337 mL, 2.78 mmol), and 6-bromo-2-chloroquinoline (0.3 g, 1.237 mmol) in anhydrous THF (4 mL) were reacted according to method E. The reaction mixture was heated for 1 h. The crude product was purified by flash column chromatography (4–6% methanol in dichloromethane) to afford 3-((6-bromoquinolin-2-yl)oxy)-N,N-dimethylpropan-1-amine as a pale-salmon colored oil (0.346 g), which was used directly in the next step.
n-BuLi (1.62 M in hexanes, 0.788 mL, 1.277 mmol) and 3-((6-bromoquinolin-2-yl)oxy)-N,N-dimethylpropan-1-amine (0.329 g, 1.064 mmol) in anhydrous THF (3.5 mL) were reacted according to method F to afford 2-(3-(dimethylamino)propoxy)quinoline-6-carboxylic acid as a white amorphous solid (0.170 g), which was used in the next step without further purification.
2-(3-(Dimethylamino)propoxy)quinoline-6-carboxylic acid (91 mg, 0.220 mmol), HATU (104 mg, 0.275 mmol), and N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide 18 (50 mg, 0.176 mmol) in N,N-diisopropylethylamine (0.154 mL, 0.879 mmol) and anhydrous DMF (1.5 mL) were reacted according to method B. The crude product was purified by flash column chromatography (3.5–6.6% 2M NH3-methanol in dichloromethane) to afford the product 22 as a light-tan amorphous solid (0.078 g, 38% over 3 steps). IR (thin film): νmax 2945, 1647, 1603, 1583, 1528, 1502, 1286, 1262 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 8.56 (d, J = 2.0 Hz, 1H), 8.37 (d, J = 8.8 Hz, 1H), 8.22 (dd, J = 8.7, 2.0 Hz, 1H), 7.88–7.84 (m, 2H), 7.58 (dd, J = 8.3, 2.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.24 (d, J = 8.5 Hz, 1H), 7.10 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.47 (t, J = 6.7 Hz, 2H), 4.34–4.27 (m, 4H), 2.39 (t, J = 7.1 Hz, 2H), 2.24 (s, 3H), 2.17 (s, 6H), 1.93 (p, J = 6.8 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 164.94, 164.35, 162.88, 147.63, 146.34, 142.93, 140.17, 137.29, 136.33, 130.10, 130.01, 128.76, 128.39, 128.12, 127.69, 126.73, 123.98, 121.19, 118.62, 118.11, 116.84, 116.66, 113.98, 64.38, 64.02, 55.76, 45.21, 26.55, 17.48. HRMS (ESI+): calcd for C31H33N4O5 (M + H)+, 541.2446; found, 541.24423.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(3-morpholinopropoxy)quinoline-6-carboxamide 23
4-(3-Hydroxypropyl)morpholine (0.171 mL, 1.24 mmol), NaH (60% mineral oil, 0.047 g, 1.19 mmol), and 6-bromo-2-chloroquinoline (0.250 g, 1.03 mmol) in anhydrous THF (3.50 mL) were reacted according to method E. The reaction mixture was heated for 16 h. Purification using flash column chromatography (1.5–3.5% methanol in dichloromethane) gave 4-(3-((6-bromoquinolin-2-yl)oxy)propyl)morpholine (221 mg) as a white amorphous solid, which was used directly in the next step.
n-BuLi (1.56 M in hexanes, 0.460 mL, 0.717 mmol) and 4-(3-((6-bromoquinolin-2-yl)oxy)propyl)morpholine (0.210 g, 0.598 mmol) in anhydrous THF (2.00 mL) were reacted according to method F to afford 2-(3-morpholinopropoxy)quinoline-6-carboxylic acid (0.273 g) as a yellow amorphous solid, which was used in the next step without further purification.
2-(3-Morpholinopropoxy)quinoline-6-carboxylic acid hydrochloride (0.076 g, 0.188 mmol), HATU (0.089 g, 0.234 mmol), and N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide 18 (0.040 g, 0.141 mmol) in N,N-diisopropylethylamine (0.139 mL, 0.797 mmol) and anhydrous DMF (1.35 mL) were reacted according to method B. The crude product was purified by flash column chromatography to afford compound 23 as a white amorphous solid (51.0 mg, 47% over 3 steps). IR (film): νmax 3189, 2934, 1636, 1504, 1293, 1066, 819 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 8.56 (d, J = 2.1 Hz, 1H), 8.37 (d, J = 8.9 Hz, 1H), 8.22 (dd, J = 8.7, 2.0 Hz, 1H), 7.88–7.83 (m, 2H), 7.58 (dd, J = 8.4, 2.2 Hz, 1H), 7.54 (d, J = 2.2 Hz, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.24 (d, J = 8.3 Hz, 1H), 7.10 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.49 (t, J = 6.6 Hz, 2H), 4.34–4.27 (m, 4H), 3.59 (t, J = 4.7 Hz, 4H), 2.50–2.44 (m, 2H), 2.39 (br s, 4H), 2.24 (s, 3H), 1.97 (p, J = 6.7 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 164.95, 164.36, 162.88, 147.62, 146.35, 142.94, 140.21, 137.30, 136.34, 130.12, 130.03, 128.77, 128.42, 128.14, 127.70, 126.72, 124.00, 121.21, 118.63, 118.12, 116.85, 116.67, 114.00, 66.20, 64.40, 64.33, 64.03, 54.93, 53.34, 25.47, 17.49 (2 carbons missing). HRMS (ESI+): calcd for C33H35N4O6 (M + H)+, 583.2551; found, 583.2586.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(3-(piperidin-1-yl)propoxy)quinoline-6-carboxamide 24
1-Piperidinepropanol (0.235 mL, 1.55 mmol), NaH (60% in mineral oil, 0.059 g), and 6-bromo-2-chloroquinoline (0.300 g, 1.24 mmol) in anhydrous THF (4.00 mL) were reacted according to method E. The reaction was heated for 5 h. Purification using flash column chromatography (2.5–6% methanol in dichloromethane) isolated 6-bromo-2-(3-(piperidin-1-yl)propoxy)quinoline (329 mg) as a yellow oil, which was used directly in the next step.
n-BuLi (1.56 M in hexanes, 0.732 mL, 1.14 mmol) and 6-bromo-2-(3-(piperidin-1-yl)propoxy)quinoline (0.319 g, 0.913 mmol) in anhydrous THF (3.10 mL) were reacted according to method F to afford the crude product, which was used in the next step without further purification.
2-(3-(Piperidin-1-yl)propoxy)quinoline-6-carboxylic acid hydrochloride (76 mg, 0.188 mmol), HATU (89 mg, 0.234 mmol), and N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide 18 (40 mg, 0.141 mmol) in N,N-diisopropylethylamine (0.139 mL, 0.797 mmol) and anhydrous DMF (1.35 mL) were reacted according to method B. The crude product was purified by flash column chromatography (3.5–15% methanol in dichloromethane) to afford compound 24 as a white amorphous solid (53 mg, 49% over 3 steps). IR (film): νmax 3257, 2928, 1601, 1526, 1284, 1064, 818 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 8.57 (d, J = 2.1 Hz, 1H), 8.38 (d, J = 8.9 Hz, 1H), 8.23 (dd, J = 8.8, 2.1 Hz, 1H), 7.89–7.83 (m, 2H), 7.58 (dd, J = 8.3, 2.1 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.49 (t, J = 6.5 Hz, 2H), 4.34–4.26 (m, 4H), 2.70–2.30 (m, 6H), 2.24 (s, 3H), 2.02 (br s, 2H), 1.56 (br s, 4H), 1.42 (br s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 164.94, 164.37, 162.80, 147.59, 146.35, 142.94, 140.25, 137.31, 136.33, 130.12, 130.06, 128.78, 128.44, 128.15, 127.69, 126.72, 124.03, 121.20, 118.64, 118.14, 116.85, 116.67, 113.96, 64.40, 64.21, 64.03, 54.82, 53.67, 24.99, 23.34, 17.48, (3 carbons missing). HRMS (ESI+): calcd for C34H37N4O5 (M + H)+, 581.2758; found, 581.2759.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(2-(piperidin-1-yl)ethoxy)quinoline-6-carboxamide 25
4-(2-Hydroxyethyl)piperidine (0.197 mL, 1.49 mmol), NaH (60% in mineral oil, 0.057 g, 1.42 mmol), and 6-bromo-2-chloroquinoline (0.300 g, 1.24 mmol) in anhydrous THF (4 mL) were reacted according to method E. The reaction mixture was heated for 5 h. Purification by flash column chromatography (4–5% methanol in dichloromethane) isolated 334 mg of a pale-yellow oil, which was used directly in the next step.
n-BuLi (2.28 M in hexanes, 0.437 mL, 0.996 mmol) and 6-bromo-2-(2-(piperidin-1-yl)ethoxy)quinoline (0.204 g, 0.664 mmol) in anhydrous THF (2.20 mL) were reacted according to method F (a precipitate formed after addition of n-BuLi therefore further THF (1.0 mL) was added to facilitate stirring) to afford the crude product, which was used in the next step without further purification.
2-(2-(Piperidin-1-yl)ethoxy)quinoline-6-carboxylic acid hydrochloride (85%, 93 mg, 0.234 mmol), HATU (111 mg, 0.293 mmol), and N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide 18 (50 mg, 0.176 mmol) in N,N-diisopropylethylamine (0.174 mL, 0.997 mmol) and anhydrous DMF (1.5 mL) were reacted according to method B. The crude product was purified by flash column chromatography (3.5–10% methanol in dichloromethane) to afford compound 25 as a white amorphous solid (75 mg, 75%). IR (film): νmax 3230, 2932, 1636, 1504, 1291, 1040, 822 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 8.57 (d, J = 1.9 Hz, 1H), 8.38 (d, J = 8.9 Hz, 1H), 8.22 (dd, J = 8.7, 2.0 Hz, 1H), 7.88–7.84 (m, 2H), 7.58 (dd, J = 8.3, 2.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.24 (d, J = 8.5 Hz, 1H), 7.11 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.60–4.54 (m, 2H), 4.34–4.27 (m, 4H), 2.75 (br s, 2H), 2.61–2.36 (br s, 4H), 2.24 (s, 3H), 1.51 (br s, 4H), 1.39 (br s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.39, 164.81, 163.15, 148.00, 146.80, 143.39, 140.71, 137.75, 136.79, 130.57, 130.52, 129.23, 128.88, 128.57, 128.15, 127.21, 124.48, 121.65, 119.08, 118.58, 117.30, 117.12, 114.47, 64.86, 64.48, 63.73, 57.50, 54.76, 25.95, 24.30, 17.94 (2 carbons missing). HRMS (ESI+): calcd for C33H35N4O5 (M + H)+, 567.2602; found, 567.2598.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(2-(pyrrolidin-1-yl)ethoxy)quinoline-6-carboxamide 26
NaH (60% in mineral oil, 57 mg, 1.42 mmol), 4-(2-hydroxyethyl)pyrrolidine (171 mg, 1.48 mmol), and bromo-2-chloroquinoline (300 mg, 1.24 mmol) were reacted according to method E. The reaction was heated for 4 h. Purification by flash column chromatography using a gradient of 2–5% methanol in dichloromethane afforded a pale-yellow oil (329 mg), which was used directly in the next step.
n-BuLi (1.62 M in hexanes, 0.76 mL, 1.23 mmol) and 6-bromo-2-(2-(pyrrolidin-1-yl)ethoxy)quinoline (38 mg, 0.990 mmol) in anhydrous THF (3.3 mL) were reacted according to method F to afford the crude product, which was used in the next step without further purification.
2-(2-(Pyrrolidin-1-yl)ethoxy)quinoline-6-carboxylic acid hydrochloride (89 mg, 0.23 mmol), HATU (111 mg, 0.29 mmol), and N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (50 mg, 0.18 mmol) in N,N-diisopropylethylamine (0.17 mL, 1.0 mmol) and anhydrous DMF (1.5 mL) was reacted according to method B. The crude product was purified by flash column chromatography using a gradient of 5–12% methanol in dichloromethane to afford the product 26 as a white amorphous solid (66 mg, 70%). IR (solid): νmax 1637, 1601, 1581, 1289, 1065, 819 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 2H), 8.57 (d, J = 2.0 Hz, 1H), 8.38 (d, J = 8.8 Hz, 1H), 8.22 (dd, J = 8.7, 2.0 Hz, 1H), 7.88–7.84 (m, 2H), 7.58 (dd, J = 8.3, 2.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.24 (d, J = 8.5 Hz, 1H), 7.12 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.57 (t, J = 5.8 Hz, 2H), 4.34–4.27 (m, 4H), 2.89 (br s, 2H), 2.58 (br s, 4H), 2.23 (s, 3H), 1.70 (br s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 165.39, 164.81, 163.15, 148.00, 146.80, 143.39, 140.72, 137.76, 136.79, 130.57, 130.52, 129.23, 128.88, 128.58, 128.15, 127.21, 124.49, 121.65, 119.08, 118.58, 117.30, 117.12, 114.48, 65.03, 64.86, 64.48, 54.58, 54.42, 40.32, 40.15, 23.61, 17.95. HRMS (ESI+): calcd for C32H33N4O5 (M + H)+, 553.2446; found, 553.2467.
Preparation of Compounds in Figure 4
N-(4-Methyl-3-(6-(2-propionamidoethoxy)-2-naphthamido)phenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide 27
Propionyl chloride (2.5 μL, 0.029 mmol) was added to a solution of 2-(2-aminoethoxy)-N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)quinoline-6-carboxamide 29 (0.013 g, 0.026 mmol) in anhydrous dichloromethane (0.75 mL) and DMF (0.15 mL). The reaction mixture was stirred at room temperature for 2.5 h, concentrated to remove dichloromethane, and then diluted with water, extracted with dichloromethane (3×), dried over magnesium sulfate, filtered, and concentrated. Heptane was added and concentrated (2×) to remove residual DMF. The crude material was purified by flash column chromatography using a gradient of 2–3.5% methanol in dichloromethane to afford the title compound 27 (10 mg, 71%) as a white amorphous solid. IR (thin film): νmax 2934, 1645, 1529, 1502, 1286, 1262, 1065 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.08 (s, 1H), 10.07 (s, 1H), 8.57 (d, J = 2.0 Hz, 1H), 8.39 (d, J = 8.8 Hz, 1H), 8.23 (dd, J = 8.7, 2.1 Hz, 1H), 8.04 (br t, J = 5.5 Hz, 1H), 7.88–7.84 (m, 2H), 7.58 (dd, J = 8.4, 2.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.11 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.47 (t, J = 5.7 Hz, 2H), 4.34–4.27 (m, 4H), 3.51 (q, J = 5.7 Hz, 2H), 2.24 (s, 3H), 2.10 (q, J = 7.6 Hz, 2H), 0.99 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 173.66, 165.39, 164.81, 163.13, 147.95, 146.80, 143.39, 140.76, 137.75, 136.79, 130.58, 129.23, 128.91, 128.59, 128.15, 127.21, 124.54, 121.66, 119.08, 118.59, 117.30, 117.12, 114.42, 65.03, 64.86, 64.49, 38.44, 28.86, 17.95, 10.36, (1 carbon missing). HRMS (ESI+): calcd for C31H31N4O6 (M + H)+, 555.2238; found, 555.2228.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-5-carboxamido)-2-methylphenyl)-2-(2-(pyrrolidin-1-yl)ethoxy)quinoline-6-carboxamide 28
2,3-Dihydrobenzo[b][1,4]dioxine-5-carboxylic acid (980 mg, 5.44 mmol), oxalyl chloride (0.552 mL, 6.53 mmol), and DMF (0.011 mL, 0.14 mmol) in anhydrous dichloromethane (14 mL) were reacted according to method A. The resulting acid chloride was dissolved in anhydrous dichloromethane (9 mL) and added dropwise to a solution of 4-methyl-3-nitroaniline 2 (828 mg, 5.44 mmol) and pyridine (0.88 mL, 10.88 mmol) in anhydrous dichloromethane (15 mL). A precipitate formed during the addition. The reaction mixture was stirred at room temperature overnight and then concentrated. The resulting amorphous solid was suspended in methanol, diluted with water, and then isolated by filtration and washed with water to afford N-(4-methyl-3-nitrophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide as a pale-brown amorphous solid (1.57 g, 92%). 1H NMR (500 MHz, DMSO-d6) δ 10.45 (s, 1H), 8.53 (d, J = 2.2 Hz, 1H), 7.88 (dd, J = 8.3, 2.2 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.16 (dd, J = 7.6, 1.6 Hz, 1H), 7.04 (dd, J = 8.1, 1.6 Hz, 1H), 6.97–6.92 (m, 1H), 4.39–4.36 (m, 2H), 4.34–4.30 (m, 2H), 2.50 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 164.91, 149.06, 144.14, 141.73, 138.33, 133.53, 127.95, 125.57, 124.69, 121.71, 121.25, 119.85, 115.23, 64.96, 64.23, 19.68. HRMS (ESI+): calcd for C16H15N2O5 (M + H)+, 315.0975; found, 315.0976.
Palladium (10% on activated carbon, 0.325 g) and N-(4-methyl-3-nitrophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (1.50 g, 4.77 mmol) in ethanol (32 mL) and ethyl acetate (32 mL) were reacted according to method D to afford N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (1.30 g, 96%) as a white amorphous solid. 1H NMR (500 MHz, CDCl3) δ 9.35 (s, 1H), 7.80 (dd, J = 7.8, 1.6 Hz, 1H), 7.34 (br s, 1H), 7.03 (dd, J = 8.0, 1.7 Hz, 1H), 7.01–6.92 (m, 2H), 6.76 (dd, J = 8.0, 2.0 Hz, 1H), 4.49–4.44 (m, 3H), 4.36–4.30 (m, 3H), 3.54 (br s, 2H). 13C NMR (126 MHz, CDCl3) δ 162.58, 145.09, 143.61, 141.73, 137.17, 130.51, 124.23, 122.59, 121.51, 121.00, 118.39, 110.47, 107.22, 65.13, 63.59, 16.91. HRMS (ESI+): calcd for C16H17N2O3 (M + H)+, 286.1265; found, 286.1263: calcd for C16H17N2NaO3 (M + Na)+, 307.1053; found, 307.1048.
2-(2-(Pyrrolidin-1-yl)ethoxy)quinoline-6-carboxylic acid hydrochloride (95 mg, 0.253 mmol), HATU, (104 mg, 0.274 mmol), and N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide (0.060 g, 0.211 mmol) in N,N-diisopropylethylamine (0.147 mL, 0.844 mmol) and anhydrous DMF (2 mL) were reacted according to method B. The crude product was purified by flash column chromatography using a gradient of 5–10% methanol in dichloromethane to afford the title compound 28 as a cream amorphous solid (61 mg, 52%). IR (thin film): νmax 2926, 1651, 1619, 1601, 1527, 1470, 1282 cm–1. 1H NMR (500 MHz, DMSO-d6) δ 10.10 (s, 1H), 10.09 (s, 1H), 8.57 (d, J = 2.0 Hz, 1H), 8.38 (d, J = 8.8 Hz, 1H), 8.23 (dd, J = 8.7, 2.0 Hz, 1H), 7.86 (d, J = 8.7 Hz, 1H), 7.81 (d, J = 2.1 Hz, 1H), 7.53 (dd, J = 8.3, 2.1 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.15 (dd, J = 7.6, 1.6 Hz, 1H), 7.12 (d, J = 8.8 Hz, 1H), 7.01 (dd, J = 8.0, 1.7 Hz, 1H), 6.92 (t, J = 7.8 Hz, 1H), 4.57 (t, J = 5.8 Hz, 2H), 4.40–4.35 (m, 2H), 4.30 (dt, J = 3.8, 2.3 Hz, 2H), 2.91 (br s, 2H), 2.60 (br s, 4H), 2.23 (s, 3H), 1.71 (s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 165.38, 164.26, 163.15, 148.01, 144.11, 141.65, 140.73, 137.51, 136.95, 130.72, 130.45, 129.44, 128.90, 128.62, 127.20, 126.15, 124.49, 121.72, 121.17, 119.50, 118.45, 117.95, 114.48, 64.93, 64.21, 54.55, 54.42, 23.60, 17.98 (2 carbons missing). HRMS (ESI+): calcd for C32H33N4O5 (M + H)+, 553.2445; found, 553.2424.
2-(2-Aminoethoxy)-N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)quinoline-6-carboxamide 29
NaH (60% in mineral oil, 0.112 g, 2.79 mmol), tert-butyl N-(2-hydroxyethyl)carbamate (0.43 mL, 2.79 mmol), and 6-bromo-2-chloroquinoline (0.564 g, 2.33 mmol) in anhydrous THF (13 mL) were reacted according to method E. The reaction was heated for 5 h. Purification by flash column chromatography using a gradient of 20–25% diethyl ether in petroleum ether afforded tert-butyl (2-((6-bromoquinolin-2-yl)oxy)ethyl)carbamate as a white amorphous solid (248 mg, 29%). 1H NMR (500 MHz, CDCl3) δ 7.92 (d, J = 8.9 Hz, 1H), 7.88 (d, J = 1.6 Hz, 1H), 7.74–7.66 (m, 2H), 6.94 (d, J = 8.8 Hz, 1H), 5.14 (br s, 1H), 4.56 (t, J = 5.1 Hz, 2H), 3.61 (br s, 2H), 1.46 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 145.03, 137.93, 132.86, 129.48, 128.93, 117.42, 113.99, 65.52, 40.14, 28.40, (2 carbons missing). HRMS (ESI+): calcd for C16H2079BrN2O3 (M + H)+, 367.0652; found, 367.0649.
n-BuLi (2.12 M in hexanes, 0.350 mL, 0.743 mmol) and tert-butyl (2-((6-bromoquinolin-2-yl)oxy)ethyl)carbamate (0.124 g, 0.338 mmol) in anhydrous THF (3.0 mL) were reacted according to method F. The reaction was quenched with water and the reaction mixture concentrated to remove THF. The resulting aqueous solution was washed with ethyl acetate (1×) (brine was added to help clear the emulsion formed) and then acidified with 2M HCl to pH 2–3. The aqueous layer was extracted with dichloromethane (3×) and the combined organic layer dried over magnesium sulfate to afford 2-(2-((tert-butoxycarbonyl)amino)ethoxy)quinoline-6-carboxylic acid as a white amorphous solid (52 mg, 46%). 1H NMR (500 MHz, DMSO-d6) δ 13.06 (br s, 1H), 8.56 (d, J = 1.9 Hz, 1H), 8.41 (d, J = 8.9 Hz, 1H), 8.13 (dd, J = 8.7, 2.0 Hz, 1H), 7.80 (d, J = 8.8 Hz, 1H), 7.07 (d, J = 8.9 Hz, 1H), 7.04 (t, J = 5.7 Hz, 1H), 4.43 (t, J = 5.8 Hz, 2H), 3.37 (q, J = 5.6 Hz, 2H), 1.37 (s, 9H). HRMS (ESI+): calcd for C17H21N2O5 (M + H)+, 333.1445; found, 333.1447.
2-(2-((tert-Butoxycarbonyl)amino)ethoxy)quinoline-6-carboxylic acid (46 mg, 0.138 mmol), HATU (66 mg, 0.173 mmol), and N-(3-aminophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (39 mg, 0.138 mmol) in N,N-diisopropylethylamine (0.073 mL, 0.415 mmol) and anhydrous DMF (2 mL) were reacted according to method B. The crude material was purified by flash column chromatography using a gradient of 1–3% methanol in dichloromethane to afford the product as an off-white amorphous solid (77 mg, 56%). 1H NMR (500 MHz, DMSO-d6) δ 10.07 (s, 1H), 10.07 (s, 1H), 8.57 (d, J = 2.1 Hz, 1H), 8.39 (d, J = 8.8 Hz, 1H), 8.22 (dd, J = 8.8, 2.1 Hz, 1H), 7.86 (d, J = 8.8 Hz, 1H), 7.86 (d, J = 2.2 Hz, 1H), 7.58 (dd, J = 8.5, 2.2 Hz, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.24 (d, J = 8.5 Hz, 1H), 7.09 (d, J = 8.9 Hz, 1H), 7.06 (t, J = 5.6 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 4.45 (t, J = 5.7 Hz, 2H), 4.33–4.28 (m, 2H), 3.39 (ap q, J = 5.7 Hz, 2H), 2.24 (s, 3H), 1.38 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 165.39, 164.81, 163.12, 147.95, 146.80, 143.39, 140.71, 137.76, 136.79, 130.57, 129.23, 128.88, 128.58, 128.15, 127.21, 124.53, 121.66, 119.08, 118.58, 117.30, 117.12, 114.47, 78.21, 65.13, 64.86, 64.48, 39.72, 28.70, 17.95 (2 carbons missing). HRMS (ESI+): calcd for C33H35N4O7 (M + H)+, 599.2500; found, 599.2495.
Trifluoroacetic acid (0.75 mL) was added to a solution of tert-butyl (2-((6-((5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)carbamoyl)quinolin-2-yl)oxy)ethyl)carbamate (0.039 g, 0.065 mmol) in anhydrous dichloromethane (0.75 mL), and the reaction mixture was stirred at room temperature for 75 min, concentrated, dichloromethane added, and concentrated again, diluted with a small amount of methanol, and then a 1:1 mixture of satd NaHCO3 (aq) was slowly added. The resulting precipitate was isolated by filtration and washed with water. The crude material was purified by flash column chromatography using 10% methanol in dichloromethane then a gradient of 5–9% 2M NH3 methanol/DCM to afford the title compound (22 mg, 68%) as an off-white amorphous solid. 1H NMR (500 MHz, DMSO-d6) δ 10.08 (s, 2H), 8.57 (d, J = 2.0 Hz, 1H), 8.39 (d, J = 8.9 Hz, 1H), 8.22 (dd, J = 8.7, 2.1 Hz, 1H), 7.89–7.84 (m, 2H), 7.58 (dd, J = 8.3, 2.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.12 (d, J = 8.7 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 4.42 (t, J = 5.8 Hz, 2H), 4.33–4.28 (m, 4H), 2.99 (t, J = 5.9 Hz, 2H), 2.25 (s, 3H), 2.06 (br s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 165.41, 164.81, 163.40, 148.07, 146.81, 143.39, 140.64, 137.76, 136.80, 130.58, 130.48, 129.23, 128.86, 128.58, 128.15, 127.20, 124.48, 121.66, 119.08, 118.58, 117.31, 117.12, 114.51, 68.85, 64.86, 64.49, 41.13, 17.96. HRMS (ESI+): calcd for C28H27N4O5 (M + H)+, 499.1976; found, 499.1974.
Acknowledgments
This work was supported by Cancer Research UK grant numbers C309/A8274 and C309/A11566. We acknowledge CRUK Centre funding and NHS funding to the NIHR Biomedical Research Centre at The Institute of Cancer Research, the Royal Marsden Hospital, and funding from Cancer Research Technology Pioneer Fund and Battle Against Cancer Investment Trust. Professor Paul Workman is a CRUK Life Fellow. We thank Dr Nora Cronin and the staff of the European Synchrotron Radiation facility for their support during data collection.
Glossary
Abbreviations Used
- CTB
Cell Titer Blue
- ELISA
enzyme-linked immunosorbent assay
- HSF1
heat shock transcription factor 1
- HSP70
70 kDa heat shock proteins
- SAR
structure-activity relationship
- HSP72
heat shock 70 kDa protein 1
- HSP90
heat shock protein 90
- 17-AAG
tanespimycin (17-N-allylamino-17-demethoxygeldanamycin)
- BRAF
B-Raf proto-oncogene, serine/threonine kinase
- KIT
tyrosine protein kinase KIT
- PDGFRA
platelet-derived growth factor receptor alpha polypeptide
- PDGFRB
β-type platelet-derived growth factor receptor
- HTS
high-throughput screen
- nM
nanomolar
- μM
micromolar
- IC50
half-minimal (50%) inhibitory concentration
- GI50
concentration for 50% of maximal inhibition of cell proliferation
- CDK2
cyclin-dependent kinase 2
- CDK9
cyclin-dependent kinase 9
- SEM
standard error of the mean
- K
dissociation constant
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- MLM
mouse liver microsomes
- KS
kinetic solubility
- HBF
hepatic blood flow
- SPR
surface plasmon resonance
- GPCR
G protein-coupled receptors
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- EAF2/U19
ELL-associated factor 2
- NRF2
nuclear factor erythroid 2 related factor 2
- BCL3
B-cell lymphoma 3-encoded protein
- NFI/CTF1
nuclear factor I
- p65
nuclear factor NF-kappa-B p65 subunit
- EMT
epithelial–mesenchymal transition
- GSK3β
glycogen synthase kinase 3 beta
- PDE6D
retinal rod rhodopsin-sensitive cGMP 3′,5′-cyclic phosphodiesterase subunit delta
- SILAC
stable isotope labeling by amino acids in cell culture
- PK
pharmacokinetics
- AUC
area under the curve
- AUCu
unbound area under the curve
- Cav0–24 h
average concentration over a 24 h period
- po
per os, oral dose
- qd
quaqe die, once a day dose
- Cl
clearance
- Clu
unbound clearance
- Vss
volume of distribution at steady-state
- Vdu
unbound volume of distribution
- %F
percentage oral bioavailability
- fup
fraction unbound in plasma
- fub
fraction unbound in blood
- fua
fraction unbound in the assay
- B:P
blood to plasma ratio
- CYP450
cytochrome P450
- DMF
dimethylformamide
- DCM
dichloromethane
- DIPEA
diisopropylethylamine
- THF
tetrahydrofuran
- satd
saturated
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01055.
NMR spectra of final compounds, CDK inhibition assay, Cancergene cell line profiling, DiscoveRx KinomeScan, kinase inhibition assays, BRAF inhibitors, solubilizing group optimization, mouse pharmacokinetics, in vivo mouse efficacy studies, Cerep Diversity Screen, SILAC target identification, surface plasmon resonance (SPR), pirin crystallography, full experimental procedures (PDF)
SMILES molecular formula strings (CSV)
1%DMSO WM266.4 migration assay VC (MOV)
Bisamide 26 WM266.4 migration assay 100 nM (MOV)
Bisamide 28 WM266.4 migration assay 100 nM (MOV)
Accession Codes
Atomic coordinates and structure factors for the crystal structures of pirin with compound 26 can be accessed using PDB code 5JCT. Authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The Institute of Cancer Research has a potential financial interest in inhibitors of the HSF1 pathway and operates a Rewards to Discoverers.
Supplementary Material
References
- a Holohan C.; Van Schaeybroeck S.; Longley D. B.; Johnston P. G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]; b Zahreddine H.; Borden K. L. B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. 10.3389/fphar.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Garraway L. A.; Janne P. A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discovery 2012, 2, 214–226. 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]; d Al-Lazikani B.; Banerji U.; Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- a Kinch M. S. An analysis of FDA-approved drugs for oncology. Drug Discovery Today 2014, 19, 1831–1835. 10.1016/j.drudis.2014.08.007. [DOI] [PubMed] [Google Scholar]; b Patel M. N.; Halling-Brown M. D.; Tym J. E.; Workman P.; Al-Lazikani B. Objective assessment of cancer genes for drug discovery. Nat. Rev. Drug Discovery 2013, 12, 35–50. 10.1038/nrd3913. [DOI] [PubMed] [Google Scholar]
- a Kotz J. Phenotypic screening, take two. SciBX 2012, 5, 15. 10.1038/scibx.2012.380. [DOI] [Google Scholar]; b Tang H.; Duggan S.; Richardson P. L.; Marin V.; Warder S. E.; McLoughlin S. M. Target identification of compounds from a cell viability phenotypic screen using a bead/lysate-based affinity capture platform. J. Biomol. Screening 2016, 21, 201–211. 10.1177/1087057115622431. [DOI] [PubMed] [Google Scholar]
- a Moffat J. G.; Rudolph J.; Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat. Rev. Drug Discovery 2014, 13, 588–602. 10.1038/nrd4366. [DOI] [PubMed] [Google Scholar]; b Vincent F.; Loria P.; Pregel M.; Stanton R.; Kitching L.; Nocka K.; Doyonnas R.; Steppan C.; Gilbert A.; Schroeter T.; Peakman M. Developing predictive assays: the phenotypic screening “rule of 3”. Sci. Transl. Med. 2015, 7, 293ps15. 10.1126/scitranslmed.aab1201. [DOI] [PubMed] [Google Scholar]; c Swinney D. C.; Anthony J. How were new medicines discovered?. Nat. Rev. Drug Discovery 2011, 10, 507–519. 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]; d Cheeseman M. D.; Faisal A.; Rayter S.; Barbeau O.; Kalusa A.; Westlake M.; Burke R.; Swan M.; van Montfort R.; Linardopoulos S.; Jones K. Targeting the PPM1D phenotype; 2,4-bisarylthiazoles cause highly selective apoptosis in PPM1D amplified cell-lines. Bioorg. Med. Chem. Lett. 2014, 24, 3469–3474. 10.1016/j.bmcl.2014.05.067. [DOI] [PubMed] [Google Scholar]
- a Zheng W.; Thorne N.; McKew J. C. Phenotypic screens as a renewed approach for drug discovery. Drug Discovery Today 2013, 18, 1067–1073. 10.1016/j.drudis.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lee J. A.; Uhlik M. T.; Moxham C. M.; Tomandl D.; Sall D. J. Modern phenotypic drug discovery is a viable, neoclassic pharma strategy. J. Med. Chem. 2012, 55, 4527–4538. 10.1021/jm201649s. [DOI] [PubMed] [Google Scholar]; c Eder J.; Sedrani R.; Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat. Rev. Drug Discovery 2014, 13, 577–587. 10.1038/nrd4336. [DOI] [PubMed] [Google Scholar]
- a Anighoro A.; Bajorath J.; Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]; b Peters J. Polypharmacology – foe or friend?. J. Med. Chem. 2013, 56, 8955–8971. 10.1021/jm400856t. [DOI] [PubMed] [Google Scholar]; c Medina-Franco J. L.; Giulianotti M. A.; Welmaker G. S.; Houghten R. A. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discovery Today 2013, 18, 495–501. 10.1016/j.drudis.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jalencas X.; Mestres J. On the origins of drug polypharmacology. MedChemComm 2013, 4, 80–87. 10.1039/C2MD20242E. [DOI] [Google Scholar]
- Swinney D. C. The value of translational biomarkers to phenotypic assays. Front. Pharmacol. 2014, 5, 171. 10.3389/fphar.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Guan N.; Li T.; Mais D. E.; Wang M. Quality control of cell-based high-throughput drug screening. Acta Pharm. Sin. B 2012, 2, 429–438. 10.1016/j.apsb.2012.03.006. [DOI] [Google Scholar]
- a Arrowsmith C. H.; Audia J. E.; Austin C.; Baell J.; Bennett J.; Blagg J.; Bountra C.; Brennan P. E.; Brown P. J.; Bunnage M. E.; Buser-Doepner C.; Campbell R. M.; Carter A. J.; Cohen P.; Copeland R. A.; Cravatt B.; Dahlin J. L.; Dhanak D.; Edwards A. M.; Frederiksen M.; Frye S. V.; Gray N.; Grimshaw C. E.; Hepworth D.; Howe T.; Huber K. V.; Jin J.; Knapp S.; Kotz J. D.; Kruger R. G.; Lowe D.; Mader M. M.; Marsden B.; Mueller-Fahrnow A.; Müller S.; O’Hagan R. C.; Overington J. P.; Owen D. R.; Rosenberg S. H.; Roth B.; Ross R.; Schapira M.; Schreiber S. L.; Shoichet B.; Sundström M.; Superti-Furga G.; Taunton J.; Toledo-Sherman L.; Walpole C.; Walters M. A.; Willson T. M.; Workman P.; Young R. N.; Zuercher W. J. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Workman P.; Collins I. Probing the probes: fitness factors for small molecule tools. Chem. Biol. 2010, 17, 561–577. 10.1016/j.chembiol.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bunnage M. E.; Piantnitski Chekler E. L.; Jones L. H. Target validation using chemical probes. Nat. Chem. Biol. 2013, 9, 195–199. 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- a Ziegler S.; Pries V.; Hedberg C.; Waldmann H. Target identification for small bioactive molecules: finding the needle in the haystack. Angew. Chem., Int. Ed. 2013, 52, 2744–2792. 10.1002/anie.201208749. [DOI] [PubMed] [Google Scholar]; b Rix U.; Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 2009, 5, 616–624. 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- Schirle M.; Jenkins J. L. Identifying compound efficacy targets in phenotypic drug discovery. Drug Discovery Today 2016, 21, 82–89. 10.1016/j.drudis.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Hann M. M.; Keseru G. M. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discovery 2012, 11, 355–365. 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- a Vihervaara A.; Sistonen L. HSF1 at a glance. J. Cell Sci. 2014, 127, 261–266. 10.1242/jcs.132605. [DOI] [PubMed] [Google Scholar]; b Anckar J.; Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu. Rev. Biochem. 2011, 80, 1089–1115. 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]; c Jiang S.; Tu K.; Fu Q.; Schmitt D. C.; Zhou L.; Lu N.; Zhao Y. Multifaceted roles of HSF1 in cancer. Tumor Biol. 2015, 36, 4923–4931. 10.1007/s13277-015-3674-x. [DOI] [PubMed] [Google Scholar]
- a Whitesell L.; Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin. Ther. Targets 2009, 13, 469–478. 10.1517/14728220902832697. [DOI] [PubMed] [Google Scholar]; b Moore C. L.; Dewal M. B.; Nekongo E. E.; Santiago S.; Lu N. B.; Levine S. S.; Shoulders M. D. Transportable, chemical genetic methodology for the small molecule-mediated inhibition of heat shock factor 1. ACS Chem. Biol. 2016, 11, 200–210. 10.1021/acschembio.5b00740. [DOI] [PMC free article] [PubMed] [Google Scholar]; c de Billy E.; Travers J.; Workman P. Shock about heat shock in cancer. Oncotarget 2012, 3, 741–743. 10.18632/oncotarget.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendillo M. L.; Santagata S.; Koeva M.; Bell G. W.; Hu R.; Tamimi R. M.; Fraenkel E.; Ince T. A.; Whitesell L.; Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Santagata S.; Hu R.; Lin N. U.; Mendillo M. L.; Collins L. C.; Hankinson S. E.; Schnitt S. J.; Whitesell L.; Tamimi R. M.; Lindquist S.; Ince T. A. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 18378–18383. 10.1073/pnas.1115031108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fang F.; Chang R.; Yang L. Heat shock factor 1 promotes invasion and metastasis of hepatocellular carcinoma in vitro and in vivo. Cancer 2012, 118, 1782–1794. 10.1002/cncr.26482. [DOI] [PubMed] [Google Scholar]; c Scott K. L.; Nogueira C.; Heffernan T. P.; van Doorn R.; Dhakal S.; Hanna J. A.; Min C.; Jaskelioff M.; Xiao Y.; Wu C.-J.; Cameron L. A.; Perry S. R.; Zeid R.; Feinberg T.; Kim M.; Vanede Woude G.; Granter S. R.; Bosenberg M.; Chu G. C.; DePinho R. A.; Rimm D. L.; Chin L. Pro-invasion metastasis drivers in early stage melanoma are oncogenes. Cancer Cell 2011, 20, 92–103. 10.1016/j.ccr.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a discussion of transcription inhibitors in oncology, see:; a Bensaude O. Inhibiting eukaryotic transcription, which compound to choose? how to evaluate its activity?. Transcr. 2011, 2, 103–108. 10.4161/trns.2.3.16172. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stellrecht C. M.; Chen L. S. Transcription inhibition as a therapeutic target for cancer. Cancers 2011, 3, 4170–4190. 10.3390/cancers3044170. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kwiatkowski N.; Zhang T.; Rahl P. B.; Abraham B. J.; Reddy J.; Ficarro S. B.; Dastur A.; Amzallag A.; Ramaswamy S.; Tesar B.; Jenkins C. E.; Hannett N. M.; McMillin D.; Sanda T.; Sim T.; Kim N. D.; Look T.; Mitsiades C. S.; Weng A. P.; Brown J. R.; Benes C. H.; Marto J. A.; Young R. A.; Gray N. S. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Nguyen T. K.; Grant S. Dinaciclib (SCH727965) inhibits the unfolded protein response through a CDK1- and 5-dependent mechanism. Mol. Cancer Ther. 2014, 13, 662–674. 10.1158/1535-7163.MCT-13-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lam F.; Abbas A. Y.; Shao H.; Teo T.; Adams J.; Li P.; Bradshaw T. D.; Fischer P. M.; Walsby E.; Pepper C.; Chen Y.; Ding J.; Wang S. Targeting RNA transcription and translation in ovarian cancer cells with pharmacological inhibitor CDKI-73. Oncotarget 2014, 5, 7691–7704. 10.18632/oncotarget.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Wang S.; Griffiths G.; Midgley C. A.; Barnett A. L.; Cooper M.; Grabarek J.; Ingram L.; Jackson W.; Kontopidis G.; McClue S. J.; McInnes C.; McLachlan J.; Meades C.; Mezna M.; Stuart I.; Thomas M. P.; Zheleva D. I.; Lane D. P.; Jackson R. C.; Glover D. M.; Blake D. G.; Fischer P. M. Discovery and characterization of 2-anilino-4-(thiazol-5-yl)pyrimidine transcriptional CDK inhibitors as anticancer agents. Chem. Biol. 2010, 17, 1111–1121. 10.1016/j.chembiol.2010.07.016. [DOI] [PubMed] [Google Scholar]; g Villicana C.; Cruz G.; Zurita M. The basal transcription machinery as a target for cancer therapy. Cancer Cell Int. 2014, 14, 18. 10.1186/1475-2867-14-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solimini N. L.; Luo J.; Elledge S. J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- a Zou J.; Guo Y.; Guettouche T.; Smith D. F.; Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 1998, 94, 471–480. 10.1016/S0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]; b Doubrovin M.; Che J. T.; Serganova I.; Moroz E.; Solit D. B.; Ageyeva L.; Kochetkova T.; Pillarsetti N.; Finn R.; Rosen N.; Blasberg R. G. Monitoring the induction of heat shock factor 1/heat shock protein 70 expression following 17-allylamino-17-demethoxygeldanamycin treatment by positron emission tomography and optical reporter gene imaging. Mol. Imaging 2012, 11, 67–76. 10.2310/7290.2011.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Au Q.; Zhang Y.; Barber J. R.; Ng S. C.; Zhang B. Identification of inhibitors of HSF1 functional activity by high-content target-based screening. J. Biomol. Screening 2009, 14, 1165–1175. 10.1177/1087057109347472. [DOI] [PubMed] [Google Scholar]
- West J. D.; Wang Y.; Morano K. A. Small molecule activators of the heat shock response: chemical properties, molecular targets, and therapeutic promise. Chem. Res. Toxicol. 2012, 25, 2036–2053. 10.1021/tx300264x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Santagata S.; Mendillo M. L.; Tang Y.; Subramanian A.; Perley C. C.; Roche S. P.; Wong B.; Narayan R.; Kwon H.; Koeva M.; Amon A.; Golub T. R.; Porco J. A. Jr.; Whitesell L.; Lindquist S. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 2013, 341, 1238303. 10.1126/science.1238303. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Triptolide:Westerheide S. D.; Kawahara T. L. A.; Orton K.; Morimoto R. I. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J. Biol. Chem. 2006, 281, 9616–9622. 10.1074/jbc.M512044200. [DOI] [PubMed] [Google Scholar]; c BEZ235 and SNS-032:Acquaviva J.; He S.; Sang J.; Smith D. L.; Sequeira M.; Zhang C.; Bates R. C.; Proia D. A. mTOR inhibition potentiates HSP90 inhibitor activity via cessation of HSP synthesis. Mol. Cancer Res. 2014, 12, 703–713. 10.1158/1541-7786.MCR-13-0605. [DOI] [PubMed] [Google Scholar]; d For Dinaciclib and SNS-032 see ref (28)
- The compound library was provided by AstraZeneca, see ref (28).
- Powers M. V.; Valenti M.; Miranda S.; Maloney A.; Eccles S. A.; Thomas G.; Clarke P. A.; Workman P. Mode of cell death induced by the HSP90 inhibitor 17-AAG (tanespimycin) is dependent on the expression of pro-apoptotic BAX. Oncotarget 2013, 4, 1963–1965. 10.18632/oncotarget.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K.; Rye C.; Chessum N.; Cheeseman M.; Pasqua A. E.; Pike K. G.; Faulder P. F.. Fused 1,4-dihydrodioxin derivatives as inhibitors of heat shock transcription factor 1. WO 2015/049535 A1, April 9, 2015.
- pIC50 = −log10 IC50 (M). All results are quoted to three significant figures as geometric mean ± standard error of the mean (SEM). The geometric mean and the number of repeats are quoted in the parenthesis.
- Hardcastle A.; Boxall K.; Richards J.; Tomlin P.; Sharp S.; Clarke P.; Workman P.; Aherne W. Solid-phase immunoassays in mechanism-based drug discovery: their application in the development of inhibitors of the molecular chaperone heat-shock protein 90. Assay Drug Dev. Technol. 2005, 3, 273–285. 10.1089/adt.2005.3.273. [DOI] [PubMed] [Google Scholar]
- Laitem C.; Zaborowska J.; Isa N. F.; Kufs J.; Dienstbier M.; Murphy S. CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II-transcribed genes. Nat. Struct. Mol. Biol. 2015, 22, 396–405. 10.1038/nsmb.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye C. S.; Chessum N. E. A.; Lamont S.; Pike K. G.; Faulder P.; Demeritt J.; Kemmitt P.; Tucker J.; Zani L.; Cheeseman M. D.; Isaac R.; Goodwin L.; Boros J.; Raynaud F.; Hayes A.; Henley A.; de Billy E.; Lynch C. J.; Sharp S. Y.; te Poele R.; O’ Fee L.; Foote K. M.; Green S.; Workman P.; Jones K. Discovery of 4,6-disubstituted pyrimidines as potent inhibitors of the heat shock factor (HSF1) stress pathway and CDK9. MedChemComm 2016, 7, 1580–1586. 10.1039/C6MD00159A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Guettouche T.; Boellmann F.; Lane W. S.; Voellmy R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005, 6, 4. 10.1186/1471-2091-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Budzynski M. A.; Puustinen M. C.; Joutsen J.; Sistonen L. Uncoupling stress-inducible phosphorylation of heat shock factor 1 from its activation. Mol. Cell. Biol. 2015, 35, 2530–2540. 10.1128/MCB.00816-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KINOMEscan—World’s Largest Kinase Assay Panel; DiscoverX Corporation: San Diego, CA, 2016; https://www.discoverx.com/services/drug-discovery-development-services/kinase-profiling/kinomescan (accessed August 31, 2016).
- SelectScreen Screening and Profiling Services; Thermo Fisher Scientific Inc., 2016; http://www.thermofisher.com/uk/en/home/products-and-services/services/custom-services/screening-and-profiling-services/selectscreen-profiling-service.html?icid=fr-dd-1 (accessed August 31, 2016).
- All BRAF inhibitors were purchased from Selleckchem and were used without further purification, http://www.selleckchem.com (accessedAugust 31, 2016). See Supporting Information for details.
- All BRAF inhibitors were screened in the human ovarian carcinoma cell-line SK-OV-3.
- Garnett M. J.; Edelman E. J.; Heidorn S. J.; Greenman C. D.; Dastur A.; Lau K. W.; Greninger P.; Thompson I. R.; Luo X.; Soares J.; Liu Q.; Iorio F.; Surdez D.; Chen L.; Milano R. J.; Bignell G. R.; Tam A. T.; Davies H.; Stevenson J. A.; Barthorpe S.; Lutz S. R.; Kogera F.; Lawrence K.; Mclaren-Douglas A.; Mitropoulos X.; Mironenko T.; Thi H.; Richardson L.; Zhou W.; Jewitt F.; Zhang T.; O’Brien P.; Boisvert J. L.; Price S.; Hur W.; Yang W.; Deng X.; Butler A.; Choi H. G.; Chang J. W.; Baselga J.; Stamenkovic I.; Engelman J. A.; Sharma S. V.; Delattre O.; Saez-Rodriguez J.; Gray N. S.; Settleman J.; Futreal P. A.; Haber D. A.; Stratton M. R.; Ramaswamy S.; Mcdermott U.; Benes C. H. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATCC:; a Huang R. Y-J; Kuay K. T.; Tan T. Z.; Asad M.; Tang H. M.; Ng A. H. C.; Ye J.; Chung V. Y.; Thiery J. P. Functional relevance of a six mesenchymal gene signature in epithelial-mesenchymal transition (EMT) reversal by the triple angiokinase inhibitor, nintedanib (BIBF1120). Oncotarget 2015, 6, 22098–22113. 10.18632/oncotarget.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang Z.; Xie Z.; Sun G.; Yang P.; Li J.; Yang H.; Xiao S.; Liu Y.; Qiu H.; Qin L.; Zhang C.; Zhang F.; Shan B. Reversing drug resistance of cisplatin by HSP90 inhibitors in human ovarian cancer cells. Int. J. Clin. Exp. Med. 2015, 8, 6687–6701. [PMC free article] [PubMed] [Google Scholar]; c Wu W.; Yu L.-H.; Ma B.; Xu M.-J. The inhibitory effect of doxycycline on cisplatin-sensitive and -resistant epithelial ovarian cancer. PLoS One 2014, 9, e89841. 10.1371/journal.pone.0089841. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; d Fekete M.; Santiskulvong C.; Eng C.; Dorigo O. Effect of PI3K/AKT pathway inhibition-mediated G1 arrest on chemosensitization in ovarian cancer cells. Anticancer Res. 2012, 32, 445–452. [PubMed] [Google Scholar]; e Tan L.; Wang J.; Tanizaki J.; Huang Z. F.; Aref A. R.; Rusan M.; Zhu S. J.; Zhang Y. Y.; Ercan D.; Liao R. G.; Capelletti M.; Zhou W. J.; Hur W.; Kim N.; Sim T.; Gaudet S.; Barbie D. A.; Yeh J. R. J.; Yun C. H.; Hammerman P. S.; Mohammadi M.; Janne P. A.; Gray N. S. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E4869–E4877. 10.1073/pnas.1403438111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of SK-OV-3 murine xenograft studies, see:; a Shaw T. J.; Senterman M. K.; Dawson K.; Crane C. A.; Vanderhyden B. C. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol. Ther. 2004, 10, 1032–1042. 10.1016/j.ymthe.2004.08.013. [DOI] [PubMed] [Google Scholar]; b Faratian D.; Zweemer A. J. M.; Nagumo Y.; Sims A. H.; Muir M.; Dodds M.; Mullen P.; Um I.; Kay C.; Hasmann M.; Harrison D. J.; Langdon S. P. Trastuzumab and pertuzumab produce changes in morphology and estrogen receptor signaling in ovarian cancer xenografts revealing new treatment strategies. Clin. Cancer Res. 2011, 17, 4451–4461. 10.1158/1078-0432.CCR-10-2461. [DOI] [PubMed] [Google Scholar]; c Zhang A. L.; Shen G. D.; Zhao T.; Zhang G. H.; Liu J.; Song L. H.; Wei W.; Bing L.; Wu Z. S.; Wu Q. Augmented inhibition of angiogenesis by combination of HER2 antibody ChA21 and trastuzumab in human ovarian carcinoma xenograft. J. Ovarian Res. 2010, 3, 20. 10.1186/1757-2215-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Li W.; Ji Z.-L.; Zhuo G.-C.; Xu R.-J.; Wang J.; Jiang H.-R. Effects of a selective cyclooxygenase-1 inhibitor in SKOV-3 ovarian carcinoma xenograft-bearing mice. Med. Oncol. 2010, 27, 98–104. 10.1007/s12032-009-9179-y. [DOI] [PubMed] [Google Scholar]; e Bauerschlag D. O.; Schem C.; Tiwari S.; Egberts J. H.; Weigel M. T.; Kalthoff H.; Jonat W.; Maass N.; Meinhold-Heerlein I. Sunitinib (SU11248) inhibits growth of human ovarian cancer in xenografted mice. Anticancer Res. 2010, 30, 3355–3360. [PubMed] [Google Scholar]
- Obach R. S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [PubMed] [Google Scholar]
- The in-house HPLC method to measure LogD7.4 is based on:Kerns E. H.; Di L.; Petusky S.; Kleintop T.; Huryn D.; McConnell O.; Carter G. Pharmaceutical profiling method for lipophilicity and integrity using liquid chromatography-mass spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2003, 791, 381–388. 10.1016/S1570-0232(03)00250-2. [DOI] [PubMed] [Google Scholar]
- The free-fraction in the cell assay was measured by dialysis using standard techniques, fua = 0.47 (90%CI = 0.54–0.42). Free GI50 = GI50·fua
- Hepatic blood flow (HBF) in mouse was assumed to be 90 mL/min/kg.Sanoh S.; Horiguchi A.; Sugihara K.; Kotake Y.; Tayama Y.; Ohshita H.; Tateno C.; Horie T.; Kitamura S.; Ohta S. Prediction of in vivo hepatic clearance and half-life of drug candidates in human using chimeric mice with humanized liver. Drug Metab. Dispos. 2012, 40, 322–328. 10.1124/dmd.111.040923. [DOI] [PubMed] [Google Scholar]
- Smith D. A.; Di L.; Kerns E. H. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discovery 2010, 9, 929–939. 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- Cav0–24h = AUC0–24h/24.
- Smith D. A.; Beaumont K.; Maurer T. S.; Di Li. Volume of distribution in drug design. J. Med. Chem. 2015, 58, 5691–5698. 10.1021/acs.jmedchem.5b00201. [DOI] [PubMed] [Google Scholar]
- Assumes linear scaling of free exposure (AUCu) between different doses.
- Two mice were dosed at 30 mg/kg po qd for 4 days and were then monitored for the following 3 days. Both mice retained good condition over that period as assessed by %body weight. See Supporting Information for details.
- Smith M. A.; Houghton P. A proposal regarding reporting of in vitro testing results. Clin. Cancer Res. 2013, 19, 2828–2833. 10.1158/1078-0432.CCR-13-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The tumor growth inhibition is defined as %TGI = (((Ct – C0) – (Tt – T0))/(Ct – C0)) × 100; Tt = arithmetic mean volume of treated tumors at time = t; T0 = arithmetic mean volume of treated tumors at time = 0; Ct = arithmetic mean volume of control tumors at time = t; C0 = arithmetic mean volume control tumors at time = 0.
- The p-value refers to the comparison of untransformed arithmetic means of the treated and control arms using an unpaired Student’s t test with Welch’s correction. The value is reported to two significant figures.
- de Billy E.; Powers M. V.; Smith J. R.; Workman P. Drugging the heat shock factor 1 pathway: exploitation of the critical cancer cell dependence on the guardian of the proteome. Cell Cycle 2009, 8, 3806–3808. 10.4161/cc.8.23.10423. [DOI] [PubMed] [Google Scholar]
- Diversity Profile—P9; Eurofins, 2016; http://www.cerep.fr/cerep/users/pages/catalog/profiles/DetailProfile.asp?profile=2121 (accessed August 31, 2016).
- a Target Identification and Validation; Evotec, 2016; https://www.evotec.com/uploads/media_library/36/2015-09_DDUp-Services_Target__identification.pdf (accessed August 31, 2016).; b Sharma K.; Weber C.; Bairlein M.; Greff Z.; Kéri G.; Cox J.; Olsen J. V.; Daub H. Proteomics strategy for quantitative protein interaction profiling in cell extracts. Nat. Methods 2009, 6, 741–744. 10.1038/nmeth.1373. [DOI] [PubMed] [Google Scholar]
- Cheng Y.-C.; Prusoff W. H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L.; Boulon S.; Lam Y. W.; Urcia R.; Boisvert F.-M.; Vandermoere F.; Morrice N. A.; Swift S.; Rothbauer U.; Leonhardt H.; Lamond A. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 2008, 183, 223–239. 10.1083/jcb.200805092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In-house data using a biotinylated bisamide derivative in a protein pull-down experiment also identified Pirin as a molecular target of the cellularly active bisamide series (data not shown).
- Wendler W. M. F.; Kremmer E.; Forster R.; Winnacker E. L. Identification of pirin, a novel highly conserved nuclear protein. J. Biol. Chem. 1997, 272, 8482–8489. 10.1074/jbc.272.13.8482. [DOI] [PubMed] [Google Scholar]
- Dechend R.; Hirano F.; Lehmann K.; Heissmeyer V.; Ansieau S.; Wulczyn F. G.; Scheidereit C.; Leutz A. The BCL-3 oncoprotein acts as a bridging factor between NF-κB/REL and nuclear co-regulators. Oncogene 1999, 18, 3316–3323. 10.1038/sj.onc.1202717. [DOI] [PubMed] [Google Scholar]
- Liu F.; Rehmani I.; Esaki S.; Fu R.; Chen L.; de Serrano V.; Liu A. Pirin is an iron-dependent redox regulator of NF-κB. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9722–9727. 10.1073/pnas.1221743110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Brzoska K.; Stepkowski T. M.; Kruszewski M. Basal PIR expression in HeLa Cells is driven by NRF2 via evolutionary conserved antioxidant response element. Mol. Cell. Biochem. 2014, 389, 99–111. 10.1007/s11010-013-1931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gelbman B. D.; Heguy A.; O’Connor T. P.; Zabner J.; Crystal R. G. Upregulation of pirin expression by chronic cigarette smoking is associated with bronchial epithelial cell apoptosis. Respir. Res. 2007, 8, 10. 10.1186/1465-9921-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Z.; Wang D.; Hahn J.; Ai J.; Wang Z. Pirin down-regulates the EAF2/U19 protein and alleviates its growth inhibition in prostate cancer cells. Prostate 2014, 74, 113–120. 10.1002/pros.22729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licciulli S.; Luise C.; Scafetta G.; Capra M.; Giardina G.; Nuciforo P.; Bosari S.; Viale G.; Mazzarol G.; Tonelli C.; Lanfrancone L.; Alcalay M. Pirin inhibits cellular senescence in melanocytic cells. Am. J. Pathol. 2011, 178, 2397–2406. 10.1016/j.ajpath.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki I.; Simizu S.; Okumura H.; Takagi S.; Osada H. A small-molecule inhibitor shows that pirin regulates migration of melanoma cells. Nat. Chem. Biol. 2010, 6, 667–673. 10.1038/nchembio.423. [DOI] [PubMed] [Google Scholar]
- Yamaoka H.; Sumiyoshi H.; Higashi K.; Nakao S.; Minakawa K.; Sumida K.; Saito K.; Ikoma N.; Mabuchi T.; Ozawa A.; Inagaki Y. A novel small compound accelerates dermal wound healing by modifying infiltration, proliferation and migration of distinct cellular components in mice. J. Dermatol. Sci. 2014, 74, 204–213. 10.1016/j.jdermsci.2014.03.002. [DOI] [PubMed] [Google Scholar]
- a Komai K.; Niwa Y.; Sasazawa Y.; Simizu S. Pirin regulates epithelial to mesenchymal transition independently of BCL3-SLUG signaling. FEBS Lett. 2015, 589, 738–743. 10.1016/j.febslet.2015.01.040. [DOI] [PubMed] [Google Scholar]; b Marcucci F.; Stassi G.; De Maria R. Epithelial–mesenchymal transition: a new target in anticancer drug discovery. Nat. Rev. Drug Discovery 2016, 15, 311–325. 10.1038/nrd.2015.13. [DOI] [PubMed] [Google Scholar]
- In-house studies using siRNA to inhibit pirin synthesis in SK-OV-3 cells demonstrated only limited effects on proliferation. Further studies are ongoing.
- TPh A was purchased from Sigma-Aldrich: http://www.sigmaaldrich.com/catalog/product/sigma/t6205?lang=en®ion=GB (August 31, 2016).
- a Dunwell J. M.; Culham A.; Carter C. E.; Sosa-Aguirre C. R.; Goodenough P. W. Evolution of functional diversity in the cupin superfamily. Trends Biochem. Sci. 2001, 26, 740–746. 10.1016/S0968-0004(01)01981-8. [DOI] [PubMed] [Google Scholar]; b Dunwell J. M.; Purvis A.; Khuri S. Cupins: the most functionally diverse protein superfamily?. Phytochemistry 2004, 65, 7–17. 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- a Khuri S.; Bakker F. T.; Dunwell J. M. Phylogeny, function, and evolution of the cupins, a structurally conserved, functionally diverse superfamily of proteins. Mol. Biol. Evol. 2001, 18, 593–605. 10.1093/oxfordjournals.molbev.a003840. [DOI] [PubMed] [Google Scholar]; b Uberto R.; Moomaw E. W. Protein similarity networks reveal relationships among sequence, structure, and function within the cupin superfamily. PLoS One 2013, 8, e74477. 10.1371/journal.pone.0074477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PDB 1J1L:Pang H.; Bartlam M.; Zeng Q.; Miyatake H.; Hisano T.; Miki K.; Wong L. L.; Gao G. F.; Rao Z. Crystal structure of human pirin an iron-binding nuclear protein and transcription cofactor. J. Biol. Chem. 2004, 279, 1491–1498. For other examples of Pirin crystal structures, see: PDB 3ACL, ref (61); PDBs 3ACL, 4ERO, 4EWA, 4EWD, 4EWE, 4GUL, and 4HLT, ref (57) 10.1074/jbc.M310022200. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W. 2013 Philip S. Portoghese Medicinal Chemistry Lectureship: Drug Discovery Targeting Allosteric Sites. J. Med. Chem. 2014, 57, 7485–7498. 10.1021/jm5011786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosa P. I.; Amin E. A. Tactical approaches to interconverting GPCR agonists and antagonists. J. Med. Chem. 2016, 59, 810–840. 10.1021/acs.jmedchem.5b00982. [DOI] [PubMed] [Google Scholar]
- Blomen V. A.; Májek P.; Jae L. T.; Bigenzahn J. W.; Nieuwenhuis J.; Staring J.; Sacco R.; van Diemen F. R.; Olk N.; Stukalov A.; Marceau C.; Janssen H.; Carette J. E.; Bennett K. L.; Colinge J.; Superti-Furga G.; Brummelkamp T. R. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- Wang T.; Birsoy K.; Hughes N. W.; Krupczak K. M.; Post Y.; Wei J. J.; Lander E. S.; Sabatini D. M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Staben S. T.; Feng J. A.; Lyle K.; Belvin M.; Boggs J.; Burch J. D.; Chua C.; Cui H.; DiPasquale A. G.; Friedman L. S.; Heise C.; Koeppen H.; Kotey A.; Mintzer R.; Oh A.; Roberts D. A.; Rouge L.; Rudolph J.; Tam C.; Wang W.; Xiao Y.; Young A.; Zhang Y.; Hoeflich K. P. Back pocket flexibility provides group II p21-activated kinase (PAK) selectivity for type I 1/2 kinase inhibitors. J. Med. Chem. 2014, 57, 1033–1045. 10.1021/jm401768t. [DOI] [PubMed] [Google Scholar]; b Ingrassia L.; Lefranc F.; Dewelle J.; Pottier L.; Mathieu V.; Spiegl-Kreinecker S.; Sauvage S.; El Yazidi M.; Dehoux M.; Berger W.; Van Quaquebeke E.; Kiss R. Structure–activity relationship analysis of novel derivatives of narciclasine (an amaryllidaceae isocarbostyril derivative) as potential anticancer agents. J. Med. Chem. 2009, 52, 1100–1114. 10.1021/jm8013585. [DOI] [PubMed] [Google Scholar]; c Singh J. P.; Tamang S.; Rajamohanan P. R.; Jima N. C.; Chakraborty G.; Kundu G. C.; Gaikwad S. M.; Khan M. I. Isolation, structure, and functional elucidation of a modified pentapeptide, Cysteine protease inhibitor (CPI-2081) from streptomyces species 2081 that exhibit inhibitory effect on cancer cell migration. J. Med. Chem. 2010, 53, 5121–5128. 10.1021/jm9014179. [DOI] [PubMed] [Google Scholar]; d Nagababu P.; Barui A. K.; Thulasiram B.; Devi C. S.; Satyanarayana S.; Patra C. R.; Sreedhar B. Antiangiogenic activity of mononuclear copper(II) polypyridyl complexes for the treatment of cancers. J. Med. Chem. 2015, 58, 5226–5241. 10.1021/acs.jmedchem.5b00651. [DOI] [PubMed] [Google Scholar]; e Amin H.; Wani N. A.; Farooq S.; Nayak D.; Chakraborty S.; Shankar S.; ur Rasool R.; Koul S.; Goswami A.; Rai R. Inhibition of invasion in pancreatic cancer cells by conjugate of EPA with β3,3-Pip-OH via PI3K/Akt/NF-kB pathway. ACS Med. Chem. Lett. 2015, 6, 1071–1074. 10.1021/acsmedchemlett.5b00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In-house PAINS filter protocol: Biovia Pipeline Pilot version 9.5.
- Lagerlund O.; Larhed M. Microwave-promoted aminocarbonylations of aryl chlorides using Mo(CO)6 as a solid carbon monoxide source. J. Comb. Chem. 2006, 8, 4–6. 10.1021/cc050102r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.