

Abstract
The dysregulation of the inflammatory response after trauma leads to significant morbidity and mortality. Monocytes and macrophages play a central role in the orchestration of the inflammatory response after injury. Serum interleukin-6 (IL-6) concentration correlates with poor outcomes after injury. Tumor necrosis factor-α (TNF-α) is a proinflammatory cytokine that plays a crucial role in the pathogenesis of multiple organ dysfunction syndrome. Furthermore, in the presence of C5a, monocytes and macrophages have potentiated responses, but the mechanisms underlying this response remain largely unknown. Peripheral blood mononuclear cells (PBMCs) were isolated from healthy volunteers and pretreated with C5a (100 ng/mL) for 1 h before adding lipopolysaccharide (LPS) (10 ng/mL) for up to 20 h. Inhibitors for the mitogen-activated protein kinases (MAPKs) were added 1 h before adding C5a. C5a primes monocytes for LPS-induced IL-6 and TNF-α production. Treatment of PBMCs with C5a leads to a rapid activation of the 3 MAPK pathways. SP600125 (inhibitor of c-Jun NH2-terminal kinase MAPK) and PD98059 (inhibitor of extracellular signal–regulated kinase MAPK) did not affect the C5a priming of the LPS-induced IL-6 and TNF-α production, whereas SB203580, a specific inhibitor of p38 MAPK, did suppress the C5a priming effect. These results demonstrate that C5a primes adherent PBMCs and modulates LPS-induced IL-6 and TNF-α production. Results from extracellular signal–regulated kinase and c-Jun NH2-terminal kinase MAPK blockade suggest that these signaling pathways have minimal or no role in reprogramming LPS-mediated IL-6 and TNF-α production. On the contrary, in PBMCs, C5a activates the p38 cascade, and this pathway plays a major role in the C5a enhancement of LPS-induced IL-6 and TNF-α production.
Keywords: Inflammation, C5a, LPS, monocytes, p38 MAPK, IL-6, TNF-α, signaling pathway
INTRODUCTION
Dysregulation of the inflammatory response leads to acute respiratory distress syndrome (ARDS) and multiple organ dysfunction syndrome (MODS), with a concomitant significant morbidity and mortality for the injured patient. Monocytes and macrophages are major participants in the innate immune response to injury and play a central role in the development of the inflammatory response, but the specific mechanisms leading to the excessive systemic inflammatory response syndrome remain unclear. One of the major challenges is early identification of patients at risk for MODS and ARDS to allow early intervention with potential new therapeutic targets.
After trauma, a number of cellular and humoral systems are activated in response to injury. Several lines of evidence have suggested that injury can activate the innate immune system (1) through pattern recognition receptors, such as the toll-like receptors (TLRs), that traditionally have been implicated in the host response to infection. The stimulation of TLR4 by lipopolysaccharide (LPS), a major component of the gramnegative bacteria cell wall, triggers a cascade of intracellular events, including activation of the nuclear factor kappa B family of transcription factors and the mitogen-activated protein kinase (MAPK) family, leading to the production of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) by macrophages (2).
Interleukin 6 is a cytokine with a wide range of biological effects and can serve as a prognostic factor for outcome in septic patients (3). After trauma, IL-6 release correlates with injury severity, complications, and mortality (4, 5). TNF-α is one of the major proinflammatory cytokines involved in the early response to injury, and administration of TNF-α to humans or animals can reproduce the major features of septic and endotoxin shock (6, 7).
In addition to the increase in levels of proinflammatory cytokines, the complement system is also activated after injury, releasing the anaphylatoxins C3a and C5a, essential for the development of optimal inflammatory response (8). C5a is a potent proinflammatory peptide with a multitude of functions and signals through its receptor, C5aR, which activates phosphokinase A, phospholipase C, and the MAPK family in neutrophils (8). Early after trauma, high levels of C5a are associated with injury severity, ARDS, increased multiorgan failure, and reduced survival (9).
The activation of the complement system and of the TLR signaling pathway at the same time raises the possibility for cross talk or synergistic interaction between the 2 pathways (10). The “2-hit” theory or the priming of the immune system by an initial injury leading to exaggerated inflammatory responses after a second hit seems to be linked to enhanced TLR activation (11, 12). In the presence of high levels of C5a, monocytes and macrophages have a potentiated response to LPS: alveolar macrophages, Kupffer cells, and peripheral blood mononuclear cells (PBMCs) stimulated by LPS in the presence of C5a showed an enhanced production of TNF-α or IL-6 (13–17), and IL-6 has been shown to be responsible for the upregulation of the C5a receptor in various organs during sepsis (18). Although the MAPK pathways have been implicated in the C5a-induced enhancement of LPS-induced IL-6 production (19), most of the mechanisms underlying the synergistic effect of C5a and LPS in monocytes and macrophages are unknown.
Here we demonstrate that C5a stimulation of adherent PBMCs triggers a rapid and transient activation of the extracellular signal–regulated kinase (ERK), p38, and c-Jun NH2-terminal kinase (JNK) MAPK pathways and enhances LPS-induced IL-6 and TNF-α production. Preincubation with an inhibitor of p38 MAPK (SB203580) abrogated the priming effect of C5a on LPS-induced IL-6 and TNF-α production. Surprisingly, the C5a priming effect was not affected by the blockade of the ERK MAPK pathway (PD98059) or by a JNK inhibitor (SP600125).
MATERIALS AND METHODS
PBMC preparation
Venous blood (60 mL) was collected from healthy human volunteers in syringes containing 5 mL of 3.6% sodium citrate. Each aliquot of 15 mL of whole blood was diluted with 20 mL of the Dulbecco phosphate-buffered saline (DBPS, without calcium or magnesium; BioWhittaker, Walkersville, Md) and underlay with 15 mL of Ficoll-Paque Plus (Amersham Bioscience, Piscataway, NJ). Tubes were then centrifuged at room temperature for 30 min at 1200 rpm, and the buffy coat was retrieved and washed with RPMI (Gibco/Invitrogen, Carlsbad, Calif) supplemented with 0.1 mg/mL of gentamycin (BioWhittaker). The PBMCs were then allowed to adhere on tissue culture plates by incubation at 37°C for 2 h. The cells were then rinsed 2 times with RPMI-gentamycin and incubated for 30 min at 37°C with RPMI-gentamycin + 10% adult bovine serum (Donor Adult Bovine Serum; HyClone, Logan, Utah).
Cytokine measurements
The PBMCs were treated either with inhibitors SB230580, SP600125, PD98059 (Biosource, Camarillo, Calif), and/or C5a (Calbiochem, La Jolla, Calif), and/or LPS (LPS from Escherichia coli 0111:B4; Sigma, St Louis, Mo) for the appropriate amount of time. Cell supernatants (from 106 PBMCs per condition) were collected after 20 h of LPS treatment and frozen at −80°C. The TNF-α and IL-6 concentrations were measured by using the BD OptEIA enzyme-linked immunosorbent assay kits (BD Bioscience, San Jose, Calif) specific to each cytokine using the manufacturer’s protocol.
Western blot
Cells (3 × 106) were lysed in 200 μL of lysis buffer (20 mmol/L Tris pH 8.0, 137 mmol/L NaCl, 2 mmol/L ethylenediaminetetraacetic acid, 10% glycerol, 1% Triton X-100, 10 μL/mL Protease Inhibitor Cocktail II (Sigma), 10 μL/mL Phosphatase Inhibitor cocktail II (Sigma), 0.2 mmol/L phenylmethylsulfonyl fluoride, 0.5 mmol/L dithiothreitol). Lysates were then vortexed and incubated for 15 min on ice before being cleared by centrifugation (5 min; 4°C; 12,000 rpm). Protein concentrations of the supernatants were measured using the bicinchoninic acid protein assay (Pierce, Rockford, Ill); 2× loading buffer was added to the supernatants. Samples were boiled for 5 min and stored at −70°C; 20 to 40 μg of total cellular proteins was separated onto a sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto Hybond-Nitrocellulose membrane (Amer-sham Pharmacia, Piscataway, NJ) for 2 h. Immunoblotting was performed as directed by the manufacturer of the primary antibodies: phospho-p44/42 MAP kinase (Thr202/Tyr204) (p-ERK; Cell Signaling, Danvers, Mass), phospho-p38 MAP Kinase (Thr180/Tyr182) (p-p38; Cell Signaling), phospho-JNK MAP kinase (Thr183/Tyr185) (pJNK; Promega, Madison, Wis), p-cJun (Ser63) (Cell Signaling), and p-CREB (Ser133) (Cell Signaling). Equal loading was confirmed using an antibody to ERK2 (Santa Cruz Biotechnology, Santa Cruz, Calif). Antirabbit immunoglobulin G labeled with horseradish peroxidase (Santa Cruz Biotechnology) was used as the secondary antibody, and the signal was visualized using the Super Signal West Pico detection system (Pierce). Densitometry was performed using ImageJ 1.33u (W.S. Rasband, ImageJ, US National Institutes of Health, Bethesda, Md; http://rsb.info.nih.gov/ij/, 1997–2005).
Fluorescence-activated cell sorter
Peripheral blood mononuclear cells were prepared as described earlier. Adherent cells (after being treated with C5a for 20 h) were rinsed 2 times with 10 mL of DPBS without Ca++ or Mg++ and then with 10 mL of Versene (0.2 mg/mL ethylenediaminetetraacetic acid; BioWhittaker) and incubated with 3 mL of TrypLE Express (Gibco, Grand Island, NY). As soon as the cells detached, the PBMCs were resuspended in 10 mL of 0.1% bovine serum albumin (BSA)–DPBS and rinsed 2 times with 0.1% BSA-DPBS. The cells were then incubated at 4°C for 30 min with anti-CD88 fluorescein isothiocyanate (FITC), anti-CD14 phycoerythrin (PE), or their corresponding isotypic control antibodies (FITC- or PE-labeled). All antibodies were purchased from Serotec (Raleigh, NC). The cells were rinsed 2 times with 0.1% BSA-DPBS and fixed in 1% formaldehyde (Fisher Scientific, Pittsburgh, Pa). Data were collected and analyzed by using CELLQuest software and a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
Real-time reverse-transcriptase polymerase chain reaction
Peripheral blood mononuclear cells (5 × 106) were lysed and total RNA was extracted with an RNeasy kit (Qiagen, Valencia, Calif) according to the manufacturer’s protocol. Reverse transcription was performed using an Omniscript RT kit (Qiagen). Gene expression quantification was done using the TaqMan Gene Expression Assays listed below and a TaqMan Universal Polymerase Chain Reaction (PCR) Master Mix (No AmpErase UNG) from Applied Biosystems (Foster City, CA). The PCR experiments were carried out in an ABI 7900HT sequence detection system (Applied Biosystems) with the following conditions: a single step at 95°C for 10 min, 40 cycles at 95°C for 15 s, and a single step at 60°C for 1 min. The PCR results were analyzed with the SDS 2.1 software (Applied Biosystems). The following TaqMan gene expression assays were used: Hs99999905_m1 (glyceraldehyde-3-phosphate dehydrogenase) and Hs00356609_g1 (C5aR).
Statistics
All values of cytokine data represent mean and SEM. Data were analyzed with a paired Student t test using Prism 4 for Macintosh (GraphPad Software, San Diego, Calif).
RESULTS
C5aR is present on the surface of human monocytes
The anaphylatoxin C5a binds to its receptor (C5aR/CD88), which belongs to the G-protein coupled superfamily of rhodopsin-like receptors (20). Complement activation contributes to the inflammatory response, and targeting the C5aR can improve the outcome of sepsis in mice (21). The C5aR has been shown to be present on a multitude of cells, including neutrophils and monocytes, and C5a has been shown to influence the level of cytokines, like TNF-α, IL-1β, and IL-6 (13–17, 22). We confirmed the presence of C5aR on human PBMCs by FACS (Fig. 1, A–D) and Western blot (Fig. 1E). Adherent PBMCs treated with 100 nmol/L C5a for 20 h were stained with anti-C5aR FITC and with anti-CD14 PE, a monocyte surface receptor (Fig. 1, A–D). The cells were gated on the basis of their CD14+ staining (Fig. 1A, region R3). On average, 94.62% of the gated cells were CD14+ (Fig. 1B), 85.67% were C5aR+ (Fig. 1C), and 78.91% were CD14+C5aR+ (Fig. 1D). This demonstrates that the C5aR is present at the surface of the cells in adherent PBMCs. Western blot analysis of the C5aR demonstrated that the expression of the C5aR remains constant after C5a treatment because the relative amount of C5aR did not change (C5aRunstimulated/C5aRC5a-20h ratio is 1.05; Fig. 1E). The relative amount of CD14 present does not seem to change either (CD14unstimulated/CD14C5a-20h ratio is 0.9, Fig. 1E). Absolute quantification of the C5aR mRNA also showed that the C5aR mRNA is present in adherent PBMCs and after 20 h of C5a stimulation in similar levels (data not shown).
Fig. 1. The C5a receptor (CD88) is present at the surface of human PBMCs.
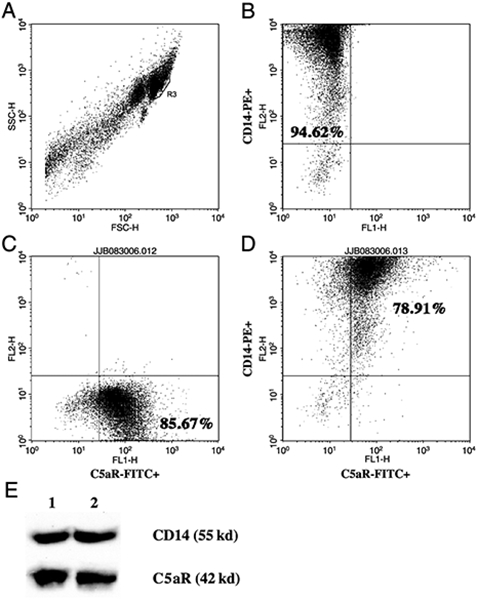
A–D, Flow cytometric analysis of adherent PBMCs treated with C5a for 20 h from a representative healthy volunteer. A, Forward scatter/side scatter of peripheral blood leucocytes. Cells in the R3 region were regarded as CD14+ monocytes. B, Representative dot blot depicting log fluorescence of FL-2 (CD14-PE) on the y axis and FL-1 (FITC) on the x axis. Cells in the upper left quadrant are CD14-PE+. The percentage of positive cells represents the mean of 3 independent experiments. C, Representative dot blot depicting log fluorescence of FL-2 (PE) on the y axis and FL-1 [C5aR (CD88)-FITC] on the x axis. Cells in the lower right quadrant are C5aR (CD88)-FITC+. The percentage of positive cells represents the mean of 3 independent experiments. D, Representative dot blot depicting log fluorescence of FL-2 (CD14-PE) on the y axis and FL-1 [C5aR (CD88)-FITC] on the x axis. Cells in the upper right quadrant are CD14-PE+ and C5aR (CD88)-FITC+. The percentage of positive cells represents the mean of 3 independent experiments. E, Western blot of adherent PBMCs untreated (lane 1) or treated with C5a (100 ng/mL) for 20 h (lane 2). Membranes were probed with anti-CD14 and anti-C5aR (Cd88) antibodies.
C5a enhances LPS-induced IL-6 and TNF-α production by human monocytes
Peripheral blood mononuclear cells were isolated from healthy volunteers and subjected to pretreatment with an optimal dose of C5a (100 nmol/L) for 1 h. The cells were then incubated with 10 or 100 ng/mL of LPS for 20 h. In the absence of C5a, LPS stimulates IL-6 and TNF-α production (Fig. 2; IL-6, white bars; TNF-α, black bars). The IL-6 and TNF-α production was significantly increased, by 36% and 25% (Fig. 2), respectively, in PBMCs incubated in the presence of C5a and LPS (10 ng/mL) compared with cells stimulated only by the same concentration of LPS. A higher LPS concentration (100 ng/mL) does not permit observation of the priming effect of C5a on TNF-α (Fig. 2) and shows a lesser priming effect on IL-6 (Fig. 2, 19%) probably because of near-maximal cytokine production achieved by the high dose of LPS.
Fig. 2. Effect of C5a on LPS-induced IL-6 and TNF-α production in human PBMCs.
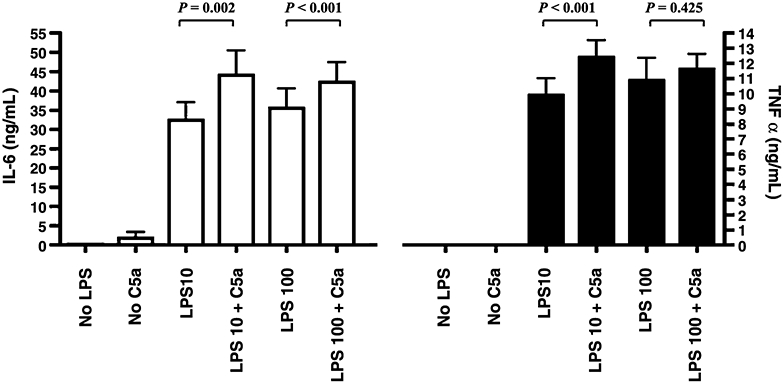
The PBMCs were treated with or without C5a (100 ng/mL) for an hour and then stimulated with LPS (10 or 100 ng/mL) as indicated. White bars represent IL-6 levels; and black bars, TNF-α. Data are represented as mean ± SEM. Results were analyzed using a paired t test.
C5a activates the ERK, p38, and JNK MAPK pathways in human monocytes
Activation of the complement cascade plays a key role in the response to injury. However, little is known about the downstream signaling events activated by C5a in monocytes/macrophages. The MAPK superfamily, composed of the ERK, p38 MAPK, and JNK, is activated by many extracellular stimuli and plays a crucial role in the inflammatory response (23).
Preincubation of adherent PBMCs with C5a caused the activation of the ERK, p38, and JNK MAPK pathways, as shown by Western blotting of their phosphorylated form (active forms). p-p38, p-JNK, and p-ERK are rapidly (within 5 min) stimulated after addition of C5a (Fig. 3, A–D).
Fig. 3. Activation of the MAPK pathway by C5a.
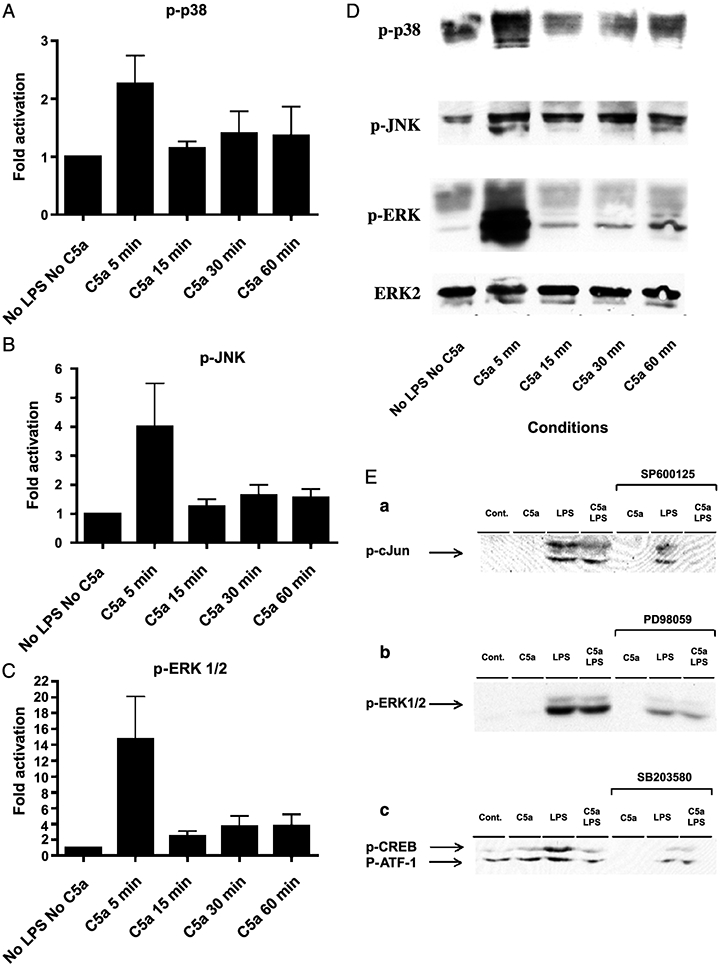
Adherent PBMCs were treated with C5a (100 ng/mL) for different amounts of time, as indicated. Cell extracts were prepared and analyzed by Western blotting as described in “Materials and Methods.” Graphs represent densitometric data, normalized to ERK-2, expressed as fold increase from the controls (No LPS, No C5a) for (A) p-p38, (B) p-JNK, and (C) p-ERK1/2, respectively. Representative immunoblots probed with the respective phosphoantibody (D). The PBMCs were treated for 1 h with C5a and/or with LPS for 30 min (E). Downregulation of each pathway by their respective inhibitor was assessed by immunoblotting against (a) p-cJun for the JNK pathway, (b) pERK1/2 for the ERK pathway, and (c) p-CREB for the p38 cascade. The p-CREB antibody also recognizes the phosphorylated form of the CREB-related protein ATF-1.
The C5a priming effect on LPS-induced IL-6 and TNF-α is only dependent upon one of the MAPK: the p38 MAPK
To address the role of each cascade in the priming effect of C5a on LPS-induced IL-6 and TNF-α, we used selective inhibitors for each pathway. The p38 MAPK inhibitor, SB203580 (1 μmol/L), strongly inhibits the C5a effect on monocytes. SB203580 is a selective inhibitor of p38 MAPK, which acts by competitively inhibiting ATP binding and therefore inhibiting the p38 MAPK–dependent phosphorylation of its substrates, such as CREB (24, 25) (as shown in Fig. 3E, C). No significant differences in LPS-dependent IL-6 and TNF-α production between PBMCs treated with or without C5a were seen after addition of SB203580 to the cells (Fig. 4). Although the LPS-induced IL-6 levels are independent of the inhibitor treatment (Fig. 4), there is still a difference in the amount of IL-6 produced by LPS and C5a with or without inhibitor (Fig. 4), confirming that the SB203580 inhibitor affects the C5a priming effect on IL-6. Because blockade of the p38 pathway strongly attenuated the LPS-induced TNF-α production (Fig. 4), it is hard to interpret the difference between the LPS + C5a with or without inhibitor.
Fig. 4. Inhibition of the C5a reprogramming effect on LPS-induced IL-6 and TNF-α production by SB203580, a p38 MAPK inhibitor.
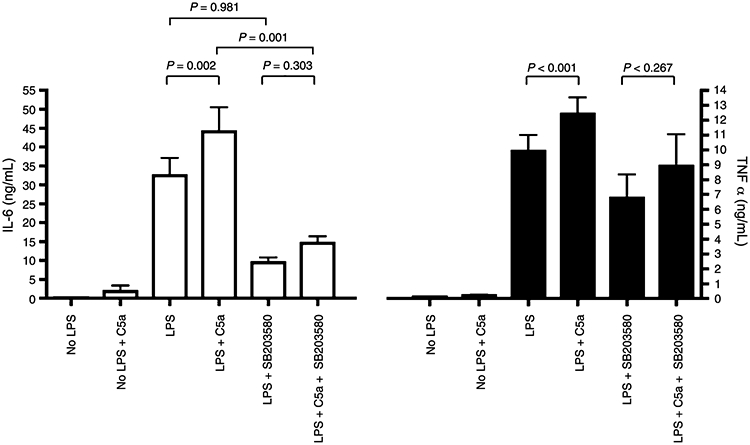
The PBMCs were treated with or without SB203580 for an hour and with or without C5a (100 ng/mL) for an hour, then stimulated with LPS (10 ng/mL) as indicated. White bars represent IL-6 levels; and black bars, TNF-α. Data are represented as mean ± SEM. Results were analyzed using a paired t test.
Although, the ERK MAPK pathway is activated in monocytes by C5a, preincubation of the cells with a specific ERK MAPK inhibitor, PD98059 (5 μmol/L), significantly decreased the LPS-induced IL-6 production by 69% (Fig. 5) but did not abrogate the C5a reprogramming effect, as pretreatment of the PBMCs with C5a in addition to LPS upregulated IL-6 production (Fig. 5). Similar results were obtained with TNF-α (Fig. 5), although the blockade of the ERK MAPK pathway decreased TNF-α release to a lesser extent (Fig. 5, −30%). Because the inhibitor attenuated both the LPS-dependent IL-6 and TNF-α (Fig. 5), the action of PD98059 on the C5a reprogramming effect could not be confirmed by comparing the LPS + C5a with or without inhibitor treatment (Fig. 5). We confirmed by Western blot that PD98059 selectively blocked the activation of MEK, hence inhibiting the phosphorylation and activation of ERK1/2 (Fig. 3E, B).
Fig. 5. PD98059, an ERK MAPK inhibitor, does not block the C5a effect on LPS-dependent IL-6 and TNF-α production in human PBMCs.
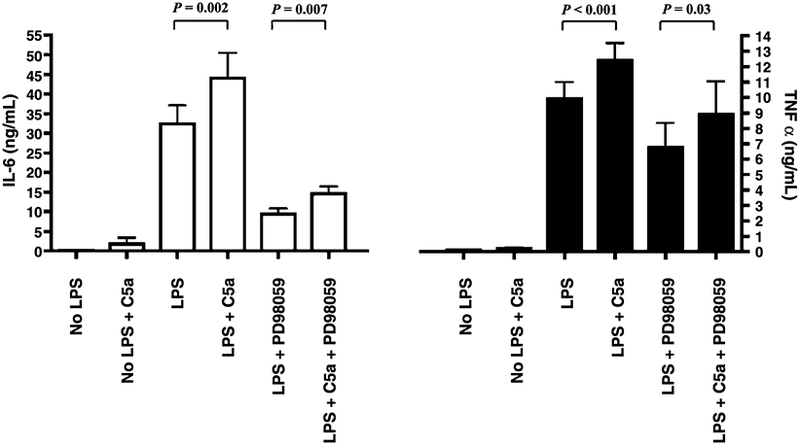
The PBMCs were treated with or without PD98059 for an hour and with or without C5a (100 ng/mL) for an hour, then stimulated with LPS (10 ng/mL) as indicated. White bars represent IL-6 levels; and black bars, TNF-α. Data are represented as mean ± SEM. Results were analyzed using a paired t test.
Inhibition of the JNK pathway led to similar results. SP600125 selectively inhibits JNK1, 2, and 3; the phosphorylation of c-Jun (Fig. 3E, A); and the expression of some inflammatory genes. Inhibiting the JNK pathway with SP600125, (5 μmol/L) did not affect the C5a reprogramming (Fig. 6) for either IL-6 or TNF-α production.
Fig. 6. A JNK inhibitor, SP600125, does not abrogate the C5a effect on LPS-dependent IL-6 and TNF-α production in human PBMCs.
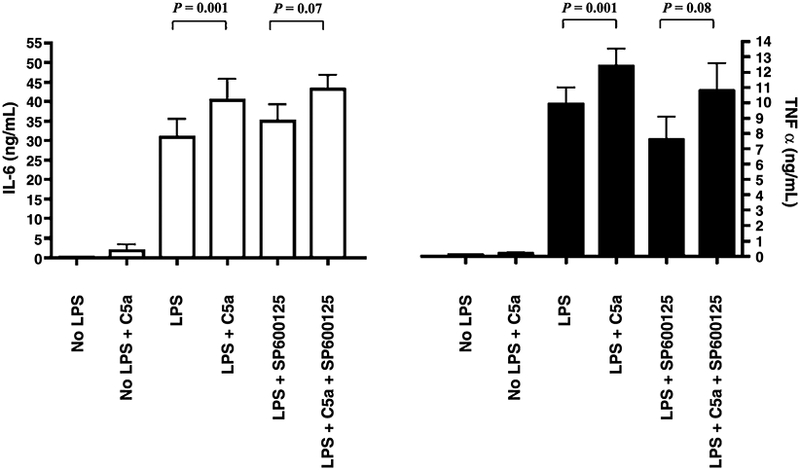
The PBMCs were treated with or without SP600125 for an hour and with or without C5a (100 ng/mL) for an hour, then stimulated with LPS (10 ng/mL) as indicated. White bars represent IL-6 levels; and black bars, TNF-α. Data are represented as mean ± SEM. Results were analyzed using a paired t test.
DISCUSSION
The proinflammatory peptide C5a signals through its G protein–coupled rhodopsin-like receptor, C5aR. Our FACS and Western blot analysis of C5aR on human PBMCs showed that C5aR is expressed at the surface of human PBMCs (Fig. 1) and that, in our system, C5aR is still expressed at the surface of the cells after treatment with C5a for 20 h. This is in contradiction with previous data demonstrating that C5a decreased the monocyte C5aR expression by 20% in whole blood (26). However, our results do correlate with clinical data showing that, in septic patients, C5aR expression does not change throughout the course of the disease (27).
Complement activation during sepsis and other systemic stress leads to production of C5a, which is a potent anaphylatoxin, and induces numerous cellular responses, including chemotactic migration, smooth muscle contraction, release of granular enzymes from phagocytic cells, and reactive oxygen species production. Toll-like receptors have traditionally been implicated in the host response to infection, by recognizing pathogen-associated molecular patterns displayed by a variety of microorganisms and their cell wall components, such as LPS. Recent reports have suggested that, in addition to pathogen-associated molecular patterns, TLRs are also involved in mediating responses to endogenous danger signals, such as necrotic cells, fibrinogen, and heat-shock proteins (1), suggesting that injury also activates the TLR system. Thus, the possibility for synergism and cross talk exists because human PBMCs have both TLR4 and C5a receptors in abundance on their surface.
A primary response to exposure of human PBMCs to LPS is the induction of a proinflammatory response characterized by cell activation and production of inflammatory cytokines such as TNF-α and IL-6. In addition, LPS-dependent IL-6 and TNF-α production has been shown to be synergistically stimulated by pretreatment of human PBMCs with C5a (Fig. 2). Our data confirm earlier studies showing an enhancement of both IL-6 and TNF-α production using both stimuli, C5a and LPS, together in circulating human monocytes (13, 15, 16). However, this pattern is not ubiquitous, and although there is a similar synergistic increase for IL-6, TNF-α has not been shown to increase in rat Kuppfer cells (17). C5a also stimulates LPS-dependent IL-6 release in rat neutrophils but suppresses, rather than stimulates, TNF-α production. In contrast, in rat alveolar macrophages, costimulation by C5a and LPS clearly enhanced TNF-α production (14, 19). Our results showed a 36% increase in LPS-induced IL-6 production and a 25% upregulation in LPS-triggered TNF-α release after C5a-induced reprogramming of human PBMCs. Although it seems that the rat Kuppfer cells and neutrophils behave in the same way as human monocytes in regard to IL-6 regulation by LPS and C5a, these cells have a different regulation of the production of TNF-α, which is a major component of the host systemic inflammatory response. These results suggest major differences between rat neutrophils and Kuppfer cells and human PBMCs and may explain, in part, the increased tolerance in the rat to septic challenges.
Exposure of macrophages to LPS activates the ERK, p38, and JNK MAPK pathways (reviewed in (23)). C5a stimulation of PBMCs also triggers phosphorylation of all 3 MAPK cascades (Fig. 3). ERK MAPK induction by C5a has been shown in neutrophils (14, 19, 28) and murine RAW 264.7 macrophages (29), as was the C5a-triggered p38 MAPK cascade (19, 29). C5a stimulates upregulation of pERK and pJNK in elutriated human monocytes (30). Thus, the 3 major MAPK pathways seem to be activated by C5a in most inflammatory cell types, including human PBMCs, as our data demonstrate, although differences might exist in the spatial and temporal regulation of the MAPK cascades and of their targets. Only more selective testing will shed light on this issue. Interestingly though, activation of the 3 MAPK pathways by C5a does not prove that they have a causal role in the synergy between C5a and LPS in monocytes, and only p38 MAPK seems to participate predominantly in the overproduction of LPS-induced IL-6 and TNF-α by C5a. Activation of ERK by C5a might be linked to its chemotactic effect, as inhibition of ERK phosphorylation by the nonsteroidal antiinflammatory drug benzydamine inhibits human monocyte migration (31). In 2004, C5a-activated ERK was linked to the chemotactic migration of RAW macrophages (29, 32). In alveolar macrophages, JNK and ERK activation by C5a contributed to the generation of reactive oxygen species by the respiratory burst (33). In neutrophils, C5a is able to inhibit apoptosis via the ERK cascade and the phosphorylation of Bad (34), and ERK activation by C5a might also promote survival in human PBMCs. In contrast, in human monocytes, C5a has been shown to downregulate LPS-induced IL-12 production, and inhibition of JNK abolished this effect (30). The activation by C5a of the ERK and JNK pathway in human monocytes is therefore likely to have a function not yet uncovered.
We (35, 36) and others (for review, see (37)) have demonstrated that the p38 MAPK pathway plays a major role in inflammatory responses. In addition, our laboratory showed that sustained p38 basal activity in alveolar macrophages in critically ill patients was highly prognostic of outcome. Patients with bronchoalveolar lavage cells having a maximal baseline p38 activity and being unresponsive to LPS stimulation had a greater risk for longer ICU and hospital stays, more ventilator days, and higher MOD scores (38). Recently, it was demonstrated that the increased TLR4 reactivity after injury, leading to an enhanced inflammatory response by macrophages, was mediated by the increased activation of the p38 MAPK cascade (39). Furthermore, in a human model of endotoxemia, blockade of p38 MAPK leads to a strong inhibition of cytokine production (including TNF-α and IL-6) and diminished leukocyte responses (40). In a murine model, blockade of p38 MAPK offered a protective effect from C5a-induced acute lung injury (41). More recently, inhibition of the p38 pathway has been shown to reduce dermal inflammation after burn (42). Our results are consistent with these data because only inhibition of the p38 MAPK pathway abolishes the C5a-enhanced IL-6 and TNF-α release in human monocytes stimulated with LPS (Fig. 4). The results should be a reduced systemic inflammatory response and improved survival to sepsis and severe C5a-induced injury.
IL-6 is a pleiotropic cytokine with a multiple functions: it is a major proinflammatory mediator that coordinates the acute-phase response to trauma and infection. Blockade of IL-6 signaling has improved survival in 2 mice sepsis models (18, 43) and has proven to be highly effective in humans for treating chronic inflammatory diseases such as Crohn disease or rheumatoid arthritis (44–46). At the same time, IL-6 exhibits anti-inflammatory proprieties and is able to stimulate the production of other anti-inflammatory cytokines like IL-10 and IL-1ra (47). Because IL-6 plays a protective role in the immune response against gram-negative bacteria (48), it seems that it is the overexpression of IL-6 that is detrimental more than its mere presence, and the solution seems to be to reduce the levels of IL-6 without completely inhibiting its production (for review, see (47)). Similarly, preclinical studies with p38 MAPK inhibitors have shown a crucial role for this kinase in inflammation. Several p38 MAPK inhibitory compounds were assessed in human clinical trials, and results showed that the inhibitors might be efficacious in the treatment of sepsis, Crohn disease, or rheumatoid arthritis (for review, see (49)). Our data suggest that blocking p38 MAPK will only slightly inhibit the IL-6 release (hence inhibiting only the overproduction of IL-6), which may be of survival benefit. Elucidation of the mechanisms involved in the p38 cross talk between the C5a and LPS pathways in monocytes should contribute to the early identification of patients at risk for developing MODS and may serve as a potential therapeutic target.
ACKNOWLEDGMENTS
We thank Keir Warner, Laura Hennessy, and Pat Klotz for excellent technical support. This work was supported by grants from the National Institutes of Health (R01 GM45813 and T32 GM07037).
REFERENCES
- 1.Murphy TJ, Paterson HM, Mannick JA, Lederer JA: Injury, sepsis, and the regulation of Toll-like receptor responses. J Leukoc Biol 75:400–407, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K: Toll-like receptor signalling. Nat Rev Immunol 4:499–511, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Hack CE, De Groot ER, Felt-Bersma RJ, Nuijens JH, Strack Van Schijndel RJ, Eerenberg-Belmer AJ, Thijs LG, Aarden LA: Increased plasma levels of interleukin-6 in sepsis. Blood 74:1704–1710, 1989. [PubMed] [Google Scholar]
- 4.Gebhard F, Pfetsch H, Steinbach G, Strecker W, Kinzl L, Bruckner UB: Is interleukin 6 an early marker of injury severity following major trauma in humans? Arch Surg 135:291–295, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Giannoudis PV, Hildebrand F, Pape HC: Inflammatory serum markers in patients with multiple trauma. Can they predict outcome? J Bone Joint Surg Br 86:313–323, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Beutler B, Cerami A: Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature 320:584–588, 1986. [DOI] [PubMed] [Google Scholar]
- 7.van der Poll T, Lowry SF: Tumor necrosis factor in sepsis: mediator of multiple organ failure or essential part of host defense? Shock 3:1–12, 1995. [DOI] [PubMed] [Google Scholar]
- 8.Ward PA: The dark side of C5a in sepsis. Nat Rev Immunol 4:133–142, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Yao YM, Redl H, Bahrami S, Schlag G: The inflammatory basis of trauma/shock-associated multiple organ failure. Inflamm Res 47:201–210, 1998. [DOI] [PubMed] [Google Scholar]
- 10.Hawlisch H, Kohl J: Complement and Toll-like receptors: key regulators of adaptive immune responses. Mol Immunol 43:13–21, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Paterson HM, Murphy TJ, Purcell EJ, Shelley O, Kriynovich SJ, Lien E, Mannick JA, Lederer JA: Injury primes the innate immune system for enhanced Toll-like receptor reactivity. J Immunol 171:1473–1483, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Murphy TJ, Paterson HM, Kriynovich S, Zang Y, Kurt-Jones EA, Mannick JA, Lederer JA: Linking the “two-hit” response following injury to enhanced TLR4 reactivity. J Leukoc Biol 77:16–23, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Cavaillon JM, Fitting C, Haeffner-Cavaillon N: Recombinant C5a enhances interleukin 1 and tumor necrosis factor release by lipopolysaccharide-stimulated monocytes and macrophages. Eur J Immunol 20:253–257, 1990. [DOI] [PubMed] [Google Scholar]
- 14.Riedemann NC, Guo RF, Bernacki KD, Reuben JS, Laudes IJ, Neff TA, Gao H, Speyer C, Sarma VJ, Zetoune FS, et al. : Regulation by C5a of neutrophil activation during sepsis. Immunity 19:193–202, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Montz H, Koch KC, Zierz R, Gotze O: The role of C5a in interleukin-6 production induced by lipopolysaccharide or interleukin-1. Immunology 74:373–379, 1991. [PMC free article] [PubMed] [Google Scholar]
- 16.Gross V, Andus T: Human recombinant C5a enhances lipopolysaccharide-induced synthesis of interleukin-6 by human monocytes. Eur J Clin Invest 22:271–276, 1992. [DOI] [PubMed] [Google Scholar]
- 17.Mack C, Jungermann K, Gotze O, Schieferdecker HL: Anaphylatoxin C5a actions in rat liver: synergistic enhancement by C5a of lipopolysaccharide-dependent alpha(2)-macroglobulin gene expression in hepatocytes via IL-6 release from Kupffer cells. J Immunol 167:3972–3979, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Riedemann NC, Neff TA, Guo RF, Bernacki KD, Laudes IJ, Sarma JV, Lambris JD, Ward PA: Protective effects of IL-6 blockade in sepsis are linked to reduced C5a receptor expression. J Immunol 170:503–507, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Riedemann NC, Guo RF, Hollmann TJ, Gao H, Neff TA, Reuben JS, Speyer CL, Sarma JV, Wetsel RA, Zetoune FS, et al. : Regulatory role of C5a in LPS-induced IL-6 production by neutrophils during sepsis. FASEB J 18:370–372, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Gerard NP, Gerard C: The chemotactic receptor for human C5a anaphylatoxin. Nature 349:614–617, 1991. [DOI] [PubMed] [Google Scholar]
- 21.Riedemann NC, Guo RF, Neff TA, Laudes IJ, Keller KA, Sarma VJ, Markiewski MM, Mastellos D, Strey CW, Pierson CL, et al. : Increased C5a receptor expression in sepsis. J Clin Invest 110:101–108, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okusawa S, Dinarello CA, Yancey KB, Endres S, Lawley TJ, Frank MM, Burke JF, Gelfand JA: C5a induction of human interleukin 1. Synergistic effect with endotoxin or interferon-gamma. J Immunol 139:2635–2640, 1987. [PubMed] [Google Scholar]
- 23.Cuschieri J, Maier RV: Mitogen-activated protein kinase (MAPK). Crit Care Med 33:S417–S419, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ: FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J 15:4629–4642, 1996. [PMC free article] [PubMed] [Google Scholar]
- 25.Yu CT, Shih HM, Lai MZ: Multiple signals required for cyclic AMP-responsive element binding protein (CREB) binding protein interaction induced by CD3/CD28 costimulation. J Immunol 166:284–292, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Furebring M, Hakansson L, Venge P, Sjolin J: C5a, interleukin-8 and tumour necrosis factor-alpha–induced changes in granulocyte and monocyte expression of complement receptors in whole blood and on isolated leukocytes. Scand J Immunol 63:208–216, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Furebring M, Hakansson LD, Venge P, Nilsson B, Sjolin J: Expression of the C5a receptor (CD88) on granulocytes and monocytes in patients with severe sepsis. Crit Care 6:363–370, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buhl AM, Avdi N, Worthen GS, Johnson GL: Mapping of the C5a receptor signal transduction network in human neutrophils. Proc Natl Acad Sci USA 91:9190–9194, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiou WF, Tsai HR, Yang LM, Tsai WJ: C5a differentially stimulates the ERK1/2 and p38 MAPK phosphorylation through independent signaling pathways to induced chemotactic migration in RAW264.7 macrophages. Int Immunopharmacol 4:1329–1341, 2004. [DOI] [PubMed] [Google Scholar]
- 30.la Sala A, Gadina M, Kelsall BL: G(i)-protein-dependent inhibition of IL-12 production is mediated by activation of the phosphatidylinositol 3-kinase-protein 3 kinase B/Akt pathway and JNK. J Immunol 175:2994–2999, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Riboldi E, Frascaroli G, Transidico P, Luini W, Bernasconi S, Mancini F, Guglielmotti A, Milanese C, Pinza M, Sozzani S, et al. : Benzydamine inhibits monocyte migration and MAPK activation induced by chemotactic agonists. BrJ Pharmacol 140:377–383, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai HR, Yang LM, Tsai WJ, Chiou WF: Andrographolide acts through inhibition of ERK1/2 and Akt phosphorylation to suppress chemotactic migration. Eur J Pharmacol 498:45–52, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Iles KE, Dickinson DA, Watanabe N, Iwamoto T, Forman HJ: AP-1 activation through endogenous H(2)O(2) generation by alveolar macrophages. Free Radic Biol Med 32:1304–1313, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Perianayagam MC, Balakrishnan VS, Pereira BJ, Jaber BL: C5a delays apoptosis of human neutrophils via an extracellular signal-regulated kinase and Bad-mediated signalling pathway. Eur J Clin Invest 34:50–56, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Arbabi S, Rosengart MR, Garcia I, Jelacic S, Maier RV: Priming interleukin 8 production: role of platelet-activating factor and p38. Arch Surg 134: 1348–1353, 1999. [DOI] [PubMed] [Google Scholar]
- 36.Arbabi S, Garcia I, Bauer GJ, Maier RV: Alcohol (ethanol) inhibits IL-8 and TNF: role of the p38 pathway. J Immunol 162:7441–7445, 1999. [PubMed] [Google Scholar]
- 37.Herlaar E, Brown Z: p38 MAPK signalling cascades in inflammatory disease. Mol Med Today 5:439–447, 1999. [DOI] [PubMed] [Google Scholar]
- 38.Rosengart MR, Nathens AB, Arbabi S, Neff MJ, Garcia I, Martin TR, Maier RV: Mitogen-activated protein kinases in the intensive care unit: prognostic potential. Ann Surg 237:94–100, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maung AA, Fujimi S, Miller ML, MacConmara MP, Mannick JA, Lederer JA: Enhanced TLR4 reactivity following injury is mediated by increased p38 activation. J Leukoc Biol 78:565–573, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Branger J, van den Blink B, Weijer S, Madwed J, Bos CL, Gupta A, Yong CL, Polmar SH, Olszyna DP, Hack CE, et al. : Anti-inflammatory effects of a p38 mitogen-activated protein kinase inhibitor during human endotoxemia. J Immunol 168:4070–4077, 2002. [DOI] [PubMed] [Google Scholar]
- 41.Nash SP, Heuertz RM: Blockade of p38 map kinase inhibits complement-induced acute lung injury in a murine model. Int Immunopharmacol 5:1870–1880, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Ipaktchi K, Mattar A, Niederbichler AD, Hoesel LM, Hemmila MR, Su GL, Remick DG, Wang SC, Arbabi S: Topical p38MAPK inhibition reduces dermal inflammation and epithelial apoptosis in burn wounds. Shock 26:201–209, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Gennari R, Alexander JW: Anti-interleukin-6 antibody treatment improves survival during gut-derived sepsis in a time-dependent manner by enhancing host defense. Crit Care Med 23:1945–1953, 1995. [DOI] [PubMed] [Google Scholar]
- 44.Mihara M, Kasutani K, Okazaki M, Nakamura A, Kawai S, Sugimoto M, Matsumoto Y, Ohsugi Y: Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int Immunopharmacol 5:1731–1740, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Mihara M, Nishimoto N, Ohsugi Y: The therapy of autoimmune diseases by anti–interleukin-6 receptor antibody. Expert Opin Biol Ther 5:683–690, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Nishimoto N: Interleukin-6 in rheumatoid arthritis. Curr Opin Rheumatol 18:277–281, 2006. [DOI] [PubMed] [Google Scholar]
- 47.Jones SA: Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol 175:3463–3468, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Dalrymple SA, Slattery R, Aud DM, Krishna M, Lucian LA, Murray R: Interleukin-6 is required for a protective immune response to systemic Escherichia coli infection. Infect Immun 64:3231–3235, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar S, Boehm J, Lee JC: p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2: 717–726, 2003. [DOI] [PubMed] [Google Scholar]