

SUMMARY
Single-molecule fluorescence is widely used to study conformational complexity in proteins, and has proven especially valuable with intrinsically disordered proteins (IDPs). Protein studies using dual-color single-molecule Förster resonance energy transfer (smFRET) are now quite common, but many could benefit from simultaneous measurement of multiple distances through multi-color labeling. Such studies, however, have suffered from limitations in site-specific incorporation of more than two dyes per polypeptide. Here we present a fully site-specific three-color labeling scheme for α-synuclein, an IDP with important putative functions and links to Parkinson’s disease. The convergent synthesis combines native chemical ligation with regiospecific cysteine protection of expressed protein fragments to permit highly controlled labeling via standard cysteine-maleimide chemistry, enabling more global smFRET studies. Furthermore, this modular approach is generally compatible with recombinant proteins and expandable to accommodate even more complex experiments, such as by labeling with additional colors.
Keywords: smFRET, Native chemical ligation, Intrinsically disordered proteins, Protein labeling, Alpha-synuclein
In Brief
Lee and Moran et al. present a strategy for fully site-specific labeling of the intrinsically disordered protein α-synuclein with three fluorophores for multi-color single-molecule FRET. This expandable method uses standard maleimide-cysteine chemistry, and enables more global investigation of complex proteins and their dynamic interactions.

INTRODUCTION
Much light has been shed on protein structure and dynamics by single-molecule Förster resonance energy transfer (smFRET). The strong distance dependence of smFRET has proven especially useful in flexible biomolecules that are hard to study by conventional structural biology methods such as crystallography. (Deniz, et al., 2000; Joo, et al., 2008; Kim and Ha, 2013; Schuler and Hofmann, 2013; Weiss, 1999) Averaging over an ensemble can lead to loss of information by these traditional ensemble approaches, while the single-molecule approach can directly detect conformational subpopulations and dynamics that are often of functional significance. smFRET has therefore been widely used in a two-color format for measurements of a number of protein, RNA, and DNA systems. IDPs, which tend to have complex interaction and folding landscapes, have particularly benefited from the implementation of such studies. (Banerjee and Deniz, 2014; Brucale, et al., 2014)
While two-color smFRET has contributed significantly to numerous protein studies, the method is capable of providing only one distance in a molecule for a given pair of labeling positions. To expand the scope of information available from smFRET, a few studies have begun to explore the incorporation of additional dyes (three or more) for multi-color smFRET. (Gambin and Deniz, 2010; Hohng, et al., 2014; Juette, et al., 2014) In a three-color smFRET scheme, for example, a single protein can contain a donor (D) and two acceptors (A1 and A2) to report simultaneously on 3 distances (D-A1, D-A2, A1-A2). Such an experiment could provide information about coupling between conformational properties in two parts or domains of the protein, imparting a more global, comprehensive view of the protein conformation with potentially important links to function. However, a major constraint on exploring the utility of such multi-color smFRET studies has been limitations in the ability to label site-specifically with three or more dyes to yield a single protein species.
Statistical cysteine labeling, where dyes are restricted to cysteine side-chain locations, but are not directed to a particular cysteine on the polypeptide chain, has worked well for two-color smFRET in a number of protein systems. However, adding a third labeling position would result in a practically unmanageable array of up to 27 labeled isomers, hugely complicating the interpretation of already challenging three-color smFRET experiments. Therefore, previous three-color smFRET studies have been restricted to either oligonucleotides (Clamme and Deniz, 2005; Hohng, et al., 2004; Lee, et al., 2010), multiple dyes distributed on multiple molecules for protein-ligand binding studies (Ferrie, et al., 2017; Kastantin, et al., 2017; Kim, et al., 2013; Munro, et al., 2010; Roy, et al., 2009), multimeric proteins with separate labeling on each monomer (Ernst, et al., 2012; Kim, et al., 2014), combination of fluorescent protein conjugation and chemical labeling (Voss, et al., 2014), or on a single protein with only partial site-specificity (Milles, et al., 2012). Since none of these methods yields fully specific three-color labeling on a single protein, they all inherently limit the questions that can be addressed about protein conformation using single molecule techniques. Numerous orthogonal chemistries have been developed for protein labeling, that can be incorporated, for example, using noncanonical amino acids (Tyagi and Lemke, 2015), or by chemical synthesis (Hejjaoui, et al., 2011). However, incorporation of three unique chemical moieties still remains a significant challenge. Furthermore, thiolmaleimide chemistry still prevails as the most widely used labeling chemistry, due to high yields and an abundance of commercially available maleimide-modified dyes.
Here, we describe a general scheme to site-specifically attach three unique dyes to a protein using exclusively cysteine-maleimide chemistry and recombinant protein expression. We demonstrate this method using α-synuclein, which is a well-studied and biologically important IDP. This Parkinson’s disease linked protein has a complex folding landscape with multiple folding and interaction states, making it an excellent model protein for proof-of-principle triple dye-labeling modification as described here. (Ferreon, et al., 2009; McClendon, et al., 2009; Trexler and Rhoades, 2010; Ulmer, et al., 2005; Wu, et al., 2009) Moreover, site-specific labeling may open the door to future smFRET studies of correlated rearrangement and dynamics in α-synuclein.
RESULTS
To achieve site-specificity, we combined the expression of protein fragments, regioselective N-terminal cysteine protection and fragment assembly by native chemical ligation (NCL) (Hackeng, et al., 1999; Villain, et al., 2001). This strategy allows for the straightforward labeling of polypeptide intermediates at a single reactive Cys residue. In this way, attachment of the fluorophores is directed with a high level of accuracy to achieve a truly site-specifically, triple-labeled protein. To demonstrate the approach, we designed a triple Cys mutant of α-synuclein (S9C, A85C,*141C) where *141C is a C-terminal addition to the 140 amino acids in wildtype α-synuclein. (Figure 1) Labeling positions for this proof-of-principle construct were chosen to provide information about both N- and C-terminal portions of the protein of interest (also see Methods). To facilitate assembly by NCL, α-synuclein was expressed as two protein fragments, αS1–84 with a C-terminal thioester and αS85–141 with an N-terminal Cys. The lengths of these fragments were chosen to facilitate ligation at the second Cys residue in the protein sequence (in this case, A85C).
Figure 1.

Site-specific three-color labeling scheme for α-synuclein using native chemical ligation. “Dextran” indicates dextran-aldehyde resin.
The C-terminal thioester of αS1–84 was obtained by conjugating commercially available intein protein on the C-terminus of the fragment, whereas the conjugated intein connected to the protein fragment via thioester bond created by N-S acyl transfer can be substituted with other thiols. (Muir, 2003) By using 2-mercaptoethanesulfonic acid (MESNA), which forms a stable thioester to substitute the intein, the C-terminal thioester for the native chemical ligation could be prepared. (Evans, et al., 1998) Following expression as an intein fusion protein and removal of the intein by the thiol substitution, the αS1–84 thioester fragment could be labeled by a maleimide Alexa Fluor (AF) 680 dye (A2) at position S9C without competing hydrolysis or thiolactone formation due to the neutral pH of the reaction.
The second protein fragment, αS85–141 containing two cysteine residues was expressed as an N-terminal maltose binding protein (MBP) fusion protein with factor Xa protease cleavage site linking the C-terminal α-synuclein fragment. Following cleavage by factor Xa, whose digestion severs the amide bond at the C-terminus of the recognition site, so that the resulting fragment can have the desired N-terminal residue, the resulting N-terminal Cys residue of αS85–141 was covalently captured by an aldehyde resin, which selectively and reversibly forms a thiazolidine linkage. (Villain, et al., 2001) In addition to immobilizing the desired αS85–141 cleavage product, the thiazolidine chemically protects the N-terminal cysteine thiol from other reactions. As a result, subsequent, on resin maleimide treatment selectively modified only *141C. After labeling, excess dye was washed away and the single-labeled αS85–141*141C (AF488, D) protein fragment was chemically released from the resin at pH 4.2 in the presence of methoxylamine.
To form the final triple-labeled α-synuclein, mono labeled intermediates αS1–84A9C(AF680)-thioester and αS85–141*141C(AF488) were combined at neutral pH to undergo NCL yielding a full-length protein. (Hackeng, et al., 1999; Muir, 2003) After purification by reverse-phase HPLC, full-length α-synuclein was obtained, which was site-specifically labeled at positions 9C and 141C while retaining a single free cysteine residue at position 85 that could be modified further. (Kochendoerfer, et al., 2003; Muir, et al., 1997) Treatment of this intermediate with AF594 (A1) maleimide yielded the desired triple-labeled αS. (Figure 1; Mass confirmed by mass spectrometry (Figure S1)).
With full-length α-synuclein labeled site-specifically with all three fluorophores, preliminary proof-of-principle three-color smFRET experiments were carried out on freely diffusing molecules in solution. We used 488 nm laser excitation of D and simultaneous detection of D, A1 and A2 (see Experimental Procedures and Figure S2 for more details on the smFRET setup). Simultaneous bursts of fluorescence on all three channels revealed triply labeled molecules traversing the focal volume (Figure 2a). A simple data analysis method described previously was used to analyze the bursts. (Clamme and Deniz, 2005) Proximity coordinate (PC, Figure 2b) histograms were plotted, with the x-axis denoting normalized emission from A1, y-axis normalized emission from A2, and the color-coding denoting the number of events (Figure 2b). First, a PC histogram for the construct at neutral pH shows low emission from A1 and almost no emission from A2 (Figure 2c, d). This is consistent with a relatively extended C-terminal tail (residues ~95 to 140), which is consistent with the high proportion of negatively charged tail-residues at neutral pH. Upon addition of SDS micelles to the solution, new population density appears at a somewhat higher y-axis position and low on the x-axis, signaling increased emission by A2 and decreased emission by A1 (Figure 2d). This observation is consistent with A1 and A2 being in closer proximity (and therefore a higher A1-A2 FRET efficiency) due to formation of a hairpin-like helical structure in this condition, as previously reported. (Ferreon, et al., 2009; Ulmer, et al., 2005) The increase of A1-A2 FRET was further confirmed by two-color smFRET control experiments (Figure S3).
Figure 2.
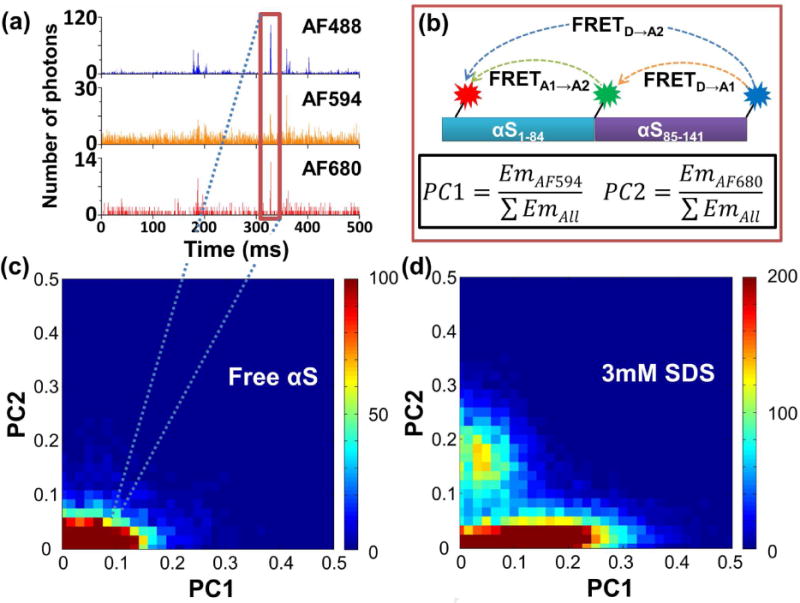
Three-color smFRET data. (a) Example of three-color single-molecule fluorescence time-trace. Note that the y-axis scales are smaller for the A1/A2 plots to provide a clearer visualization of the smaller bursts on these channels. (b) Scheme showing the three possible FRET pairs on the protein and the formulae for PC1/PC2. Em in the formulae stands for Emission. (c) Three-color smFRET data for the unbound protein plotted as a PC1/PC2 2D-histogram. The color indicates the number of molecules within the designated bin as shown in the scale bar on the right of the graph. The maximum of the scale was set for better visualization of bins with smaller number of events. (d) 2D-histogram for measurement in 3mM SDS.
DISCUSSION
The labeling method developed here represents a powerful approach for obtaining a fully site-specific three-color labeled protein, which opens the door for future studies using the strengths of three-color smFRET. In the case of α-synuclein, detailed experiments can now be carried out to probe correlations between conformational distributions in multiple regions of the protein during coupled binding and folding. Use of more complex data acquisition and analysis such as alternating-laser excitation (ALEX) will be important for these more detailed studies, which would reveal a more complete view of the complex binding-folding nature of this important protein. (Lee, et al., 2007).
The generality of the scheme also allows it to be extended to other IDPs and proteins, and to address functionally important problems such as allostery. (Csizmok, et al., 2016; Ferreon, et al., 2013; Motlagh, et al., 2014; Tompa, 2012; Wright and Dyson, 2015) In this context, we note that many biologically important IDPs are large, and undergo functionally important interactions and conformational changes involving different subdomains, making the labeling strategy described and multi-color smFRET measurements of particular importance. Furthermore, such enhanced protein labeling methods would also extend the strengths of multicolor smFRET to protein structure/function studies in more complex environments. An interesting example is that of protein liquid droplets (Banani, et al., 2017; Brangwynne, et al., 2015; Mitrea, et al., 2016). These droplets are the subject of an increasing number of studies aimed at understanding the physical basis of formation, dynamics and substructure of membraneless cellular organelles, which are critical in many cellular functions and disease. Since IDPs and their multidomain interactions have been shown to be important components of these organelles and their function, we anticipate that the discussed advantages of the multicolor-smFRET method will be particularly useful for such studies in this biologically important but less understood context. The modularity of the labeling scheme reported here also allows for extension to four or more colors with dual-cysteine labeling in N-terminal fragments similar to that of the C-terminal fragment, additional ligation steps, or utilization of an orthogonal chemical moiety by methods such as unnatural amino acid incorporation. Similar approaches could be used to engineer constructs for combined single-molecule FRET and pulling experiments. (Hohng, et al., 2007) Overall, our presented labeling method is expected to expand the scope of multicolor smFRET experiments to better understand the complex and biologically significant folding landscapes of proteins. (Sevcsik, et al., 2011)
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ashok A. Deniz (deniz@scripps.edu).
Experimental Model and Subject Details
Peptide expression in E. coli
BL21(DE3) Chemically Competent E. coli (Invitrogen) cells were purchased and used to express protein fragments cloned in plasmids. The cells were transformed by standard heat shock methods and inoculated on LB-Agar plate containing 100mg/L ampicillin for colony formation. Single colony was picked and grown over night in LB at 37°C with 250 RPM shaking and then stored at −80°C in 25% glycerol. For the expression of peptides, the glycerol stock was picked with inoculation loop and inoculated in 1mL of LB with ampicillin, and grown over night in the same condition as above to be used as the starter culture. Then the starter culture was used further to grow cells in 1L of LB with 100mg/L ampicillin at 37°C with 250 RPM shaking. The cells were grown to ODλ=600nm~0.6, induced with 400μM IPTG, and then grown for an additional 2 hours. Cells were collected by centrifugation at 9,000 g for 10 minutes, and stored at −80°C before further purification.
Method Details
Cloning for α-synuclein peptides
Fragments of α-synuclein were expressed independently as intein-fusion constructs from commercially available plasmid vectors according to published protocols with minor modification (IMPACT™ Kit, pMAL™ Protein Fusion & Purification system, New England Biolabs (NEB)). Briefly, DNA cassettes encoding α-synuclein amino acids 1–84 and 85–141 (with codon 141 being an appendage to yield a C-terminal cysteine) were assembled (annealed and ligated) from suitable purchased oligomers (Integrated DNA Technologies) (See Table S1).
Gene fragment cassettes and plasmid vectors (pTXB1 for αS1–84 and pMAL-c5X for αS85–141) were digested with SapI and NdeI (pTXB1) restriction endonucleases or XmnI and PstI (pMAL-c5X; αS85–141 was digested only with PstI) to yield complementarity 5ʹ overhang ends or blunt end (XmnI) for subsequent DNA ligation with T4 DNA ligase (NEB). Then the recombinant plasmids were used to transform chemically competent DH5α cells (Invitrogen) and screened using LB-Agar plates with ampicillin. Single colonies from the LB-Agar plate were picked and grown over night at 37°C in LB, then the amplified plasmids were extracted using plasmid miniprep kits (Qiagen). Construct sequences were confirmed by Sanger-based sequencing (Eton Bioscience Inc., San Diego), using T7 promoters (αS1–84) and pMAL vector sequencing primers as suggested in pMAL™ Protein Fusion & Purification system protocol (αS85–141).
Aldehyde-dextran resin synthesis
The aldehyde-functionalized dextran resin was prepared starting from CM Sephadex C-50 resin (GE Healthcare Life Sciences) with a carboxylic acid substitution of approximately 0.45 mmol/g of dry resin, using the general protocol (Villain, et al., 2001). 1 g of resin (1 equiv., 4.5 mmol –COOH functionality) was swelled according to the manufacturer’s instructions: 24 hours in 200mM monosodium phosphate (Fisher), pH 6.5. 1 g of resin swelled to approximately 30 mL of wet volume. The carboxylic acid was activated by the addition of a water solution containing 1.42 g of N-hydroxysuccinimide (NHS, Oakwood) (2 equiv., 9 mmol) and 3.47 g of 1′-ethyl-(3′-dimethylaminopropyl)-carbodiimide-HCl (EDC, Bachem) (4 equiv., 18 mmol) for 10 minutes. The resin was then washed thoroughly with water and then incubated for 1 hour with a water solution of 1 M 2-amino-acetaldehyde diethyl acetal (Sigma Aldrich) and 200 mM 2-morpholino-ethanesulfonic acid (MES, Fisher), adjusted to pH 6.4 with HCl (Fisher). The resin was then washed thoroughly with water, after which any unreacted sites were blocked by adding a solution of 1 M ammonium acetate (Fisher), pH 7.5 for 10 minutes. Following another wash with water, the resin was rinsed thoroughly with 10 mM HCl to liberate the aldehyde functionality. The finished resin was then equilibrated with 0.1 M acetic acid (Fisher) prior to storage at 4°C.
αS1–84 peptide purification
The general protocol for the IMPACT™ Kit was followed. Briefly, cells were lysed with a microfluidizer (M110Y, Microfluidics) in lysis buffer (20mM HEPES, 500mM NaCl, 1mM EDTA, pH 8.5). The lysed cells were centrifuged at 27,143g for 45 minutes to separate the pellet. The supernatant (~100 ml) from the centrifugation was bound to the chitin resin (NEB, 5mL bed volume) on a self-packed gravity column with flow rate ~0.5ml/min. Once the binding was complete, the column was washed with 20 column volumes of the lysis buffer (flow rate ~2ml/min). On-column cleavage was initiated by washing the column with 3 column volumes of cleavage buffer (lysis buffer with 50mM MESNA (2-sulfanylethanesulfonate)) and stopping the flow. The column was incubated at room temperature ~17 hours. After the incubation, the sample was eluted with 5 column volumes of cleavage buffer. The samples were run on an SDS-PAGE gel (16%) to determine which fraction contained the peptide. These fractions were pooled together. This sample was stored at −20°C with added Guanidinium chloride (GdmCl) (to 2M) and MESNA (to 50mM).
αS85–141 peptide purification
The general protocol for the pMAL Protein Fusion & Purification System (NEB) was followed. Briefly, cells were lysed with sonication (Fisher Scientific Sonic Dismembrator Model 500) in lysis buffer (20mM Tris-Cl, 200mM NaCl, 1mM EDTA, 1mM DTT, pH 7.4). The lysed cells were centrifuged at 27,143g for 45 minutes to separate the pellet. The supernatant (~50mL) was loaded onto an amylose resin (NEB, 15mL bed volume) on a self-packed gravity column with flow rate ~5ml/min. The column was washed with 12 column volumes of lysis buffer, and eluted with 5 column volumes of the elution buffer (lysis buffer with 10mM maltose). The samples were run on an SDS-PAGE gel (16%) to determine which fractions contained the peptide. These fractions were pooled together and stored at −80 °C after freezing in a dry ice-ethanol bath.
αS1–84 AF680 labeling
Peptide released from the chitin binding resin was further purified by using an anion exchange column (HiTrap Q HP 5mL, GE Healthcare Life Sciences) to remove impurities prior to labeling. First, the sample was buffer exchanged into 20mM Tris-Cl, 10mM MESNA, pH 8.0 buffer using an Amicon Ultra Centrifugal Filter (3k MWCO; referred as spin filter here on) to remove excess GdmCl and MESNA used for storage. Then the sample was quickly loaded onto a Q column at room temperature using a peristaltic pump (Minipuls 2, Gilson), and the flow-through was collected (15mL). The flow-through, which contains pure αS1–84 peptide, was then buffer-exchanged into pro-NCL (native chemical ligation) buffer (100mM Na2HPO4/NaH2PO4, 6M GdmCl, pH 7.2) using a spin filter at 4°C while concentrating the sample to ~1.5 ml. Then the peptide concentration was measured using Absλ=280nm (NanoDrop 2000c, Thermo Scientific) (~60 nmoles). 6 fold molar excess of Alexa Fluor 680 C2 maleimide dye (Thermo Fisher Scientific) was added to the sample, and incubated at the room temperature for 1.5 hours. After the incubation, MESNA was added to 50 mM to quench the labeling reaction as well as to protect the thioester C-terminus of the peptide. Then the sample was buffer-exchanged again using a spin filter at 4°C into the pro-NCL buffer to remove excess free dye and to concentrate the sample as much as possible (~100 μl). The sample was immediately used for native chemical ligation.
αS85–141 AF488 labeling on aldehyde resin
The concentration of the MBP- αS85–141 conjugate from the amylose resin purification was determined by using Absλ=280nm of MBP. The protein was subjected to Factor Xa (NEB) digestion, where 10μl of Factor Xa was added to the 100 nmoles of the protein. The sample was incubated exactly for 72 minutes on ice, and Factor Xa was deactivated to prevent non-specific digestion by adding 5 fold volume excess of aldehyde binding buffer (100 mM MES, 6M GdmCl, 10mM TCEP, pH 5.2) to the sample. The sample was added to the aldehyde-dextran resin (200μl bed volume) in a gravity column, and the sample was incubated at room temperature with shaking overnight ~19 hours. After the binding reaction, the sample was flushed with 25 column volumes of labeling buffer (100 mM MES, 6M GdmCl, pH 6.7). A 3-fold molar excess of Alexa Fluor 488 C5 maleimide dye (Thermo Fisher Scientific) was added to the column with 1.3 mL labeling buffer. The labeling reaction was carried out at room temperature for 3 hours. After the labeling reaction, the resin was washed with 25 column-volumes of labeling buffer, followed by a 10-column volume wash with 100 mM sodium acetate pH 4.2 buffer to remove residual free dye as well as to get the sample ready for release from the aldehyde resin. Then, 1.5 mL of release buffer (100 mM sodium acetate, 400 mM methoxyamine, pH 4.2) was added to the resin and incubated at room temperature with shaking for ~ 24 hours. The flow-through was collected after the release reaction and the resin was further flushed with 10 column volumes of release buffer, and the flush was collected as well. Then the sample was buffer-exchanged into pro-NCL buffer by using the spin filter to remove excess methoxyamine and to concentrate the sample as much as possible for native chemical ligation. Note: We found that AF680 lost its fluorescence (half-life ~12 hours) when it was exposed to the above peptide release condition. Reversal to other buffer conditions did not result in recovery of fluorescence. Therefore, use of AF680 in the initial labeling step was avoided, to be noted in design of such constructs.
Native chemical ligation
Labeled peptides were pooled together in a molar ratio of 3:1 αS1–84 to αS85–141. The solution was further concentrated using a spin filter to 100 μl. Thiophenol and α-toluenethiol were added to the sample to 1.5% (v/v) each. The sample was incubated at room temperature with shaking by vortexer for about 30 hours. Then, the sample was diluted in Buffer A (20 mM Tris-Cl, pH 7.5) to reduce the thiol concentration below 0.1%, and exchanged into Buffer A with 10 mM DTT using spin filter to remove excess thiols and GdmCl. The sample was loaded onto an anion exchange column (HiTrap Q HP 1mL) using a peristaltic pump to remove excess αS1–84 peptide, and eluted with Buffer B (20 mM Tris-Cl, 1M NaCl, pH 7.5). 1 mL fractions were collected for elution. The fractions containing sample were pooled together and concentrated with 1 mM DTT in solution for disulfide bond reduction. The concentrated sample was purified using HPLC with C4 reverse-phase column (Jupiter 5 μm C4 300 Å 250 × 10 mm, Phenomenex) with a buffer gradient from 100% HPLC Buffer A (H2O with 0.1% TFA) to 55% HPLC Buffer A and 45% HPLC Buffer B (Acetonitrile with 0.1% TFA) for 60 minutes. The fractions from HPLC were analyzed using LC/MS (Scripps Center for Metabolomics and Mass Spectrometry). The fraction showing the correct mass was dried using an evaporator (SpeedVac SVC-100, Savant).
AF594 labeling and final product verification
Full-length α-synuclein with confirmed mass was dissolved in pro-NCL buffer with 20 mM DTT. Then it was buffer exchanged into pro-NCL buffer without DTT using a spin filter while concentrating the sample to 100μl. The concentration of AF488 on the peptide was measured using Absλ=495nm, and a 10-fold molar excess of Alexa Fluor 594 C5 maleimide (Thermo Fisher Scientific) was used for labeling. The sample was incubated at 4°C for overnight (15 hours). Excess free dye was removed by buffer exchange into pro-NCL buffer using a spin filter. The fully labeled sample’s mass was confirmed by LC/MS (Figure S1). The final product concentration was measured using absorbance on a NanoDrop uv-vis spectrophotometer. The final yield was ~2% (1.2 nmole) based on N-terminal fragment as the starting material (~60 nmoles). The sample was aliquoted and stored at −80°C until smFRET experiments.
Choice of labeling positions
The labeling positions for the proof-of-principle construct described here were chosen to provide information about both N- and C-terminal regions of the protein. A general consideration for the types of single-molecule studies to be carried out with constructs labeled using the described method is that labeling positions should be chosen such that labeling with dyes will have a minimal impact on the protein properties. For the current case, we note that we have successfully used similar labeling with negatively charged Alexa dyes for previous 2-color smFRET studies on the N-terminal region of this protein, and the results showed no significant effect on the folding-binding properties of the protein. Furthermore, the C-terminal region has a large number (14) of negatively charged residues, making the addition of a single negatively-charged dye of minimal impact. Comparison of the presented results to data from our previous study (Ferreon, et al., 2009) indeed confirms that we do not observe significant perturbation. In addition, the A85C mutation provides a preceding glycine, which has been shown to increase the efficiency of the intein cleavage reaction used in the strategy (New England BioLabs, 2015).
Collection of three-color smFRET data
smFRET experiments were carried out using a confocal smFRET detection setup as explained previously (Ferreon, et al., 2009), with an altered optical setup for three-color signal collection as described below (Figure S2). The light source used was a 488 nm argonion laser (543-AP-A01, Melles Griot), with a dichroic beamsplitter (Di01-R405/488/561/635) in the microscope to separate fluorescence from excitation light. The emission was further separated by two additional dichroic beamsplitters, first by DC560LPXR (Chroma) to separate out donor (AF488) emission from the acceptors, and then by FF677-Di0 (Semrock) to separate the acceptor 1 (AF594) fluorescence from acceptor 2 (AF680) fluorescence. The emission signals were further refined by additional filters, HQ525/50M bandpass filter (donor, Chroma), 590LPV2 long pass filter (acceptor 1, Chroma), and D725/60M bandpass filter (acceptor 2, Chroma). For the two-color FRET control experiment (Figure S3), a 561nm diode-pumped solid-state laser (CL561-025-O, CrystaLaser) was used for excitation of AF594 dye, and the same optical setup was used for the data collection. For all smFRET experiments, the emission photons were detected by the avalanche photo diodes (APDs; Figure S2) and the total numbers of photons counted by each APD per bin time (0.5 ms) were recorded.
Samples were placed in a Tween-20 coated chambered borosilicate glass coverslip (Thermo Scientific). The measurements were carried in 50 mM Tris-Cl, 150 mM NaCl, pH 7.5 buffer. To avoid issues from aggregation, samples were diluted (to ~100 pM) in measurement buffer, then immediately used for smFRET measurements. In addition to the buffer, propyl gallate (PG) prepared in acetonitrile was added to reduce photo-bleaching, where all experiments were carried out with 0.5% (v/v) acetonitrile and 0.5 mM PG.
Quantification and Statistical Analysis
Visualization of smFRET data
The acquired three-color FRET data were processed and visualized using Matlab (Mathworks) using a procedure adapted from standard 2-color smFRET analysis. The raw data were background-corrected using average signal from each channel, and points that satisfied the threshold condition (total number of photons after background correction from all three channels added together fall between 40 to 200) were used to calculate PC1 and PC2 (See Figure 2 for equations) for the 2D-histogram. The size of bins for the 2D-histogram was 0.017 for each axis.
Supplementary Material
SIGNIFICANCE.
Cellular proteins are often conformationally plastic, a feature important in their function. A noteworthy case is that of proteins that encode intrinsic disorder (IDPs), which frequently have complex patterns of interactions and conformational distributions. This complexity is linked to important cellular function and misfunction, including regulation, signaling and protein misfolding, but can be challenging to study using structural and biophysical methods. In this context, smFRET has begun to provide significant capabilities to decipher complex conformational distributions and dynamics. However, although smFRET has been used successfully, it has been limited with respect to direct measurements of structural relationships between multiple regions of a protein. In this study, we have developed and demonstrated a method for labeling proteins fully site-specifically with three chemical fluorophores for single-molecule FRET studies. The described approach uses relatively simple chemistry, widely available reactive dyes, and can use but does not require chemically synthesized peptides. The method can be used to study how conformational complexity of IDPs and multidomain proteins links with and regulates allosteric and other functional aspects of the global biophysics of IDPs.
Highlights.
Site-specific labeling of a single polypeptide is presented for three-color smFRET.
Convergent method combines regiospecific protection and native chemical ligation.
Full-length protein is synthesized from expressed and labeled fragments.
Modular and expandable method enables more colors and combined biophysical study.
Acknowledgments
Funding is gratefully acknowledged from NIGMS/NIH (RO1 GM06833 to AAD) and NSF (grant MCB 1121959 to AAD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes additional experimental details regarding DNA oligo designs and three supplemental figures.
AUTHOR CONTRIBUTIONS
Developing concept and methodology, C.R.M., T.C.L., P.E.D., and A.A.D.; Experiments, data acquisition, analysis, T.C.L., C.R.M., P.A.C.; Manuscript preparation, T.C.L., C.R.M., P.A.C., P.E.D., and A.A.D.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee PR, Deniz AA. Shedding light on protein folding landscapes by single-molecule fluorescence. Chem Soc Rev. 2014;43:1172–1188. doi: 10.1039/c3cs60311c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Tompa P, Pappu RV. Polymer physics of intracellular phase transitions. Nat Phys. 2015;11:899–904. [Google Scholar]
- Brucale M, Schuler B, Samori B. Single-molecule studies of intrinsically disordered proteins. Chemical reviews. 2014;114:3281–3317. doi: 10.1021/cr400297g. [DOI] [PubMed] [Google Scholar]
- Clamme JP, Deniz AA. Three-color single-molecule fluorescence resonance energy transfer. Chemphyschem : a European journal of chemical physics and physical chemistry. 2005;6:74–77. doi: 10.1002/cphc.200400261. [DOI] [PubMed] [Google Scholar]
- Csizmok V, Follis AV, Kriwacki RW, Forman-Kay JD. Dynamic Protein Interaction Networks and New Structural Paradigms in Signaling. Chemical reviews. 2016;116:6424–6462. doi: 10.1021/acs.chemrev.5b00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz AA, Laurence TA, Beligere GS, Dahan M, Martin AB, Chemla DS, Dawson PE, Schultz PG, Weiss S. Single-molecule protein folding: diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5179–5184. doi: 10.1073/pnas.090104997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst S, Duser MG, Zarrabi N, Borsch M. Three-color Forster resonance energy transfer within single F(0)F(1)-ATP synthases: monitoring elastic deformations of the rotary double motor in real time. J Biomed Opt. 2012;17:011004. doi: 10.1117/1.JBO.17.1.011004. [DOI] [PubMed] [Google Scholar]
- Evans TC, Jr, Benner J, Xu MQ. Semisynthesis of cytotoxic proteins using a modified protein splicing element. Protein Sci. 1998;7:2256–2264. doi: 10.1002/pro.5560071103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon AC, Ferreon JC, Wright PE, Deniz AA. Modulation of allostery by protein intrinsic disorder. Nature. 2013;498:390–394. doi: 10.1038/nature12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrie JJ, Ieda N, Haney CM, Walters CR, Sungwienwong I, Yoon J, Petersson EJ. Multicolor protein FRET with tryptophan, selective coumarincysteine labeling, and genetic acridonylalanine encoding. Chem Commun (Camb) 2017;53:11072–11075. doi: 10.1039/c7cc05492k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambin Y, Deniz AA. Multicolor single-molecule FRET to explore protein folding and binding. Molecular bioSystems. 2010;6:1540–1547. doi: 10.1039/c003024d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackeng TM, Griffin JH, Dawson PE. Protein synthesis by native chemical ligation: expanded scope by using straightforward methodology. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:10068–10073. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejjaoui M, Haj-Yahya M, Kumar KS, Brik A, Lashuel HA. Towards elucidation of the role of ubiquitination in the pathogenesis of Parkinson’s disease with semisynthetic ubiquitinated alpha-synuclein. Angew Chem Int Ed Engl. 2011;50:405–409. doi: 10.1002/anie.201005546. [DOI] [PubMed] [Google Scholar]
- Hohng S, Joo C, Ha T. Single-molecule three-color FRET. Biophysical journal. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohng S, Lee S, Lee J, Jo MH. Maximizing information content of single-molecule FRET experiments: multi-color FRET and FRET combined with force or torque. Chem Soc Rev. 2014;43:1007–1013. doi: 10.1039/c3cs60184f. [DOI] [PubMed] [Google Scholar]
- Hohng S, Zhou R, Nahas MK, Yu J, Schulten K, Lilley DM, Ha T. Fluorescence-force spectroscopy maps two-dimensional reaction landscape of the holliday junction. Science. 2007;318:279–283. doi: 10.1126/science.1146113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo C, Balci H, Ishitsuka Y, Buranachai C, Ha T. Advances in single-molecule fluorescence methods for molecular biology. Annual review of biochemistry. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- Juette MF, Terry DS, Wasserman MR, Zhou Z, Altman RB, Zheng Q, Blanchard SC. The bright future of single-molecule fluorescence imaging. Current opinion in chemical biology. 2014;20:103–111. doi: 10.1016/j.cbpa.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastantin M, Faulon Marruecos D, Grover N, Yu McLoughlin S, Schwartz DK, Kaar JL. Connecting Protein Conformation and Dynamics with Ligand-Receptor Binding Using Three-Color Forster Resonance Energy Transfer Tracking. Journal of the American Chemical Society. 2017 doi: 10.1021/jacs.7b03978. [DOI] [PubMed] [Google Scholar]
- Kim E, Lee S, Jeon A, Choi JM, Lee HS, Hohng S, Kim HS. A single-molecule dissection of ligand binding to a protein with intrinsic dynamics. Nature chemical biology. 2013;9:313–318. doi: 10.1038/nchembio.1213. [DOI] [PubMed] [Google Scholar]
- Kim H, Abeysirigunawarden SC, Chen K, Mayerle M, Ragunathan K, Luthey-Schulten Z, Ha T, Woodson SA. Protein-guided RNA dynamics during early ribosome assembly. Nature. 2014;506:334–338. doi: 10.1038/nature13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Ha T. Single-molecule nanometry for biological physics. Reports on progress in physics. Physical Society. 2013;76:016601. doi: 10.1088/0034-4885/76/1/016601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochendoerfer GG, Chen SY, Mao F, Cressman S, Traviglia S, Shao H, Hunter CL, Low DW, Cagle EN, Carnevali M, et al. Design and chemical synthesis of a homogeneous polymer-modified erythropoiesis protein. Science. 2003;299:884–887. doi: 10.1126/science.1079085. [DOI] [PubMed] [Google Scholar]
- Lee J, Lee S, Ragunathan K, Joo C, Ha T, Hohng S. Single-molecule four-color FRET. Angew Chem Int Ed Engl. 2010;49:9922–9925. doi: 10.1002/anie.201005402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NK, Kapanidis AN, Koh HR, Korlann Y, Ho SO, Kim Y, Gassman N, Kim SK, Weiss S. Three-color alternating-laser excitation of single molecules: monitoring multiple interactions and distances. Biophysical journal. 2007;92:303–312. doi: 10.1529/biophysj.106.093211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon S, Rospigliosi CC, Eliezer D. Charge neutralization and collapse of the C-terminal tail of alpha-synuclein at low pH. Protein Sci. 2009;18:1531–1540. doi: 10.1002/pro.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milles S, Koehler C, Gambin Y, Deniz AA, Lemke EA. Intramolecular three-colour single pair FRET of intrinsically disordered proteins with increased dynamic range. Molecular bioSystems. 2012;8:2531–2534. doi: 10.1039/c2mb25135c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrea DM, Cika JA, Guy CS, Ban D, Banerjee PR, Stanley CB, Nourse A, Deniz AA, Kriwacki RW. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. Elife. 2016;5 doi: 10.7554/eLife.13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir TW. Semisynthesis of proteins by expressed protein ligation. Annual review of biochemistry. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- Muir TW, Dawson PE, Kent SB. Protein synthesis by chemical ligation of unprotected peptides in aqueous solution. Methods Enzymol. 1997;289:266–298. doi: 10.1016/s0076-6879(97)89052-0. [DOI] [PubMed] [Google Scholar]
- Munro JB, Altman RB, Tung CS, Cate JH, Sanbonmatsu KY, Blanchard SC. Spontaneous formation of the unlocked state of the ribosome is a multistep process. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:709–714. doi: 10.1073/pnas.0908597107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New England BioLabs. IMPACT Kit Instruction Manual. New England BioLabs: Ipswich; 2015. [Google Scholar]
- Roy R, Kozlov AG, Lohman TM, Ha T. SSB protein diffusion on single-stranded DNA stimulates RecA filament formation. Nature. 2009;461:1092–1097. doi: 10.1038/nature08442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler B, Hofmann H. Single-molecule spectroscopy of protein folding dynamics–expanding scope and timescales. Curr Opin Struct Biol. 2013;23:36–47. doi: 10.1016/j.sbi.2012.10.008. [DOI] [PubMed] [Google Scholar]
- Sevcsik E, Trexler AJ, Dunn JM, Rhoades E. Allostery in a disordered protein: oxidative modifications to alpha-synuclein act distally to regulate membrane binding. Journal of the American Chemical Society. 2011;133:7152–7158. doi: 10.1021/ja2009554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci. 2012;37:509–516. doi: 10.1016/j.tibs.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Trexler AJ, Rhoades E. Single molecule characterization of alpha-synuclein in aggregation-prone states. Biophysical journal. 2010;99:3048–3055. doi: 10.1016/j.bpj.2010.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi S, Lemke EA. Single-molecule FRET and crosslinking studies in structural biology enabled by noncanonical amino acids. Curr Opin Struct Biol. 2015;32:66–73. doi: 10.1016/j.sbi.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. The Journal of biological chemistry. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- Villain M, Vizzavona J, Rose K. Covalent capture: a new tool for the purification of synthetic and recombinant polypeptides. Chem Biol. 2001;8:673–679. doi: 10.1016/s1074-5521(01)00044-8. [DOI] [PubMed] [Google Scholar]
- Voss S, Zhao L, Chen X, Gerhard F, Wu YW. Generation of an intramolecular three-color fluorescence resonance energy transfer probe by site-specific protein labeling. J Pept Sci. 2014;20:115–120. doi: 10.1002/psc.2590. [DOI] [PubMed] [Google Scholar]
- Weiss S. Fluorescence spectroscopy of single biomolecules. Science. 1999;283:1676–1683. doi: 10.1126/science.283.5408.1676. [DOI] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KP, Weinstock DS, Narayanan C, Levy RM, Baum J. Structural reorganization of alpha-synuclein at low pH observed by NMR and REMD simulations. Journal of molecular biology. 2009;391:784–796. doi: 10.1016/j.jmb.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.