

Abstract
Recent genomic studies have identified subtypes of uterine leiomyoma (LM) with distinctive genetic alterations. Here we report the elucidation of the biological characteristics of the two most prevalent LM subtypes, MED12 mutant (MED12-LM) and HMGA2-overexpressing (HMGA2-LM) LM. Since each tumor carries only one genetic alteration, both subtypes are considered to be monoclonal. Approximately 90% of cells in HMGA2-LM were smooth muscle cells (SMC) with HMGA2 overexpression. In contrast, MED12-LM consisted of similar numbers of SMC and non-SMC, which were mostly tumor-associated fibroblasts (TAF). Paradoxically, TAF carried no mutations in MED12, suggesting an interaction between SMC and TAF to coordinate their growth. The higher amount of ECM in MED12-LM than HMGA2-LM was partially due to the high concentration of collagen-producing TAF. SMC growth in a xenograft assay was driven by progesterone in both LM subtypes. In contrast, TAF in MED12-LM proliferated in response to estradiol, whereas progesterone had no effect. The high concentration of estrogen-responsive TAF in MED12-LM explains the inconsistent discoveries between in vivo and in vitro studies on the mitogenic effect of estrogen and raises questions regarding the accuracy of previous studies utilizing MED12-LM cell culture. In addition, the differential effects of estradiol and progesterone on these LM subtypes emphasize the importance of subtypes and genotypes in designing non-surgical therapeutic strategies for LM.
Introduction
Uterine leiomyomata (LMs) or fibroids are benign tumors of myometrium (MM), which occur in women of reproductive age with the cumulative incidence of approximately 70% (1–3). Recent comprehensive genome analyses identified at least 4 major LM subtypes with unique genetic alterations (4,5). The most common subtype is MED12 mutant LMs (MED12-LMs), present in approximately 70% of all cases (6). The second major subtype of LMs overexpresses HMGA2, a non-histone chromosomal high-mobility group (HMG) protein (HMGA2-LMs) (4,7). The HMGA2-LMs account for approximately half of non-MED12-LMs (5,8,9).
Researchers of LM have presumed MED12 mutations or translocations of HMGA2 are the causal genetic alterations that promote the unregulated proliferation of the mutant myometrial cell, leading to the formation of tumors. In MED12-LMs, missense mutations in exon 2, in-frame-deletions in exon 2, and mutations in splice acceptor site for exon 2 resulting in aberrant splicing in MED12 are frequent genetic alterations (6). Additionally, a transgenic mouse study has established that the expression of missense-mutant MED12 can be the sole cause of LMs (10). While MED12 mutations in LMs are diverse, each MED12-LM contains only one type of MED12 mutation. Similarly, the t(12;14) translocation is the most common genetic alteration in HMGA2-LMs (11,12), and only one type of chromosome rearrangement is found in a single HMGA2-LM (13). These observations establish the monoclonal origin of MED12-LMs and HMGA2-LMs.
The ovarian hormone dependency of LM growth has been long accepted, and 17β-estradiol (E2) was considered to be the mitogen of LMs for decades (14). However, utilizing the human patient derived tumor xenograft (PDTX) model for LMs, we established that progesterone (P4) is actually the driver for LM growth. While E2 itself is not a mitogen, it plays an essential role in the growth of LMs by sensitizing LM cells to P4 through the up-regulation of progesterone receptor (PGR) (15). However, it is not known if there is a differential response to E2 and P4 across LM subtypes. Given the clinical phenotypes of MED12-LMs and HMGA2-LMs have not been fully addressed, we conducted an investigation to determine the hormonal response and characteristics of two major LM subtypes, MED12-LM and HMGA2-LM, in a PDTX model.
Materials and Methods
Collection of human LM and MM tissues
The acquisition and research use of surgical specimens were approved by the Institutional Review Board of the Ohio State University and Northwestern University, and were conducted in accordance with the Belmont Report. LM and MM samples were obtained from hysterectomy or myomectomy patients aged 28–52 years with prior written informed consent and delivered to research personnel within 5 hours of surgical removal. Should the tumor exceed 5 cm in diameter, only the tissues from the peripheral areas of the tumor were used, given the center was more necrotic and calcified. Tissues were digested into single cells as described in the following section. Portions of undigested MM and LM tissues were processed into paraffin blocks or stored at −80 °C for histological and genetic analyses.
Immunostaining and histochemistry
Immunohistochemistry with DAB (3,3′-diaminobenzidine, Sigma-Aldrich) (IHC) and immunofluorescence (IF) were performed as previously described (16). The following primary antibodies were used at indicated dilutions: anti-HMGA2 (1:800, #8179) and anti-vimentin (VIM) (1:200, #9856) antibodies from Cell Signaling Technologies; anti-MKI67 (1:100, ab92742), anti-calponin1 (CNN1) (1:100, ab197639), anti-αSMA (ACTA2) (1:500, ab7817), anti-S100A9 + Calprotectin (MCF387)(17) (1:100, ab22506), anti-caldesmon (CALD1) (1:200, ab32330), anti-desmin (DES) (1:2000, ab32362) and anti-procollagen I (1:50, ab64409) antibodies from Abcam; anti-collagen I (1:200, LS-B342) and anti-collagen III (1:200, LS-B693) antibodies from Lifespan Biosciences; and anti-MED12 (1:50, HPA003184, Sigma-Aldrich), anti-VWF (1:200, ECM590-21540, Millipore), anti-CD31 (PECAM1) (1:200, 90214, Chemicon), anti-ESR1 (1:100 RM9101-S, Lab Vision) and anti-PGR (1:200, A0098, Agilent Technologies) antibodies. The following donkey polyclonal secondary antibodies (Jackson ImmunoResearch) were used at 1:800 – 1:1,000 dilutions: Alexa-Fluor594 anti-mouse IgG (H+L) (715-586-150), Alexa-Fluor488 anti-rabbit IgG (H+L) (711-546-152), Alexa-Fluor594 anti-rabbit IgG (H+L) (711-586-152), biotinylated anti-rabbit IgG (H+L) (711-066-152) and biotinylated anti-rat IgG (H+L) (712-066-153). Biotinylated antibodies were used in conjunction with streptavidin-horseradish peroxidase (016-030-084, Jackson ImmunoResearch) and Alexa-Fluor488 conjugated streptavidin (1:500, S1123, Thermo Fisher Scientific). Bisbenzimide H 33258 (Hoechst 33258, 1:10,000, Sigma-Aldrich) was used for nuclear staining in IF assay. Images were captured on a fluorescence microscope (BZ-9000, Keyence). Masson’s trichrome staining was performed following a standard protocol (18).
HMGA2 expression and MED12 mutation analyses
LM cases were classified as HMGA2-LMs when >80% of cells showed intense nuclear staining in HMGA2 IHC (Supplemental Figure S1). We screened 230 cases and identified 11 HMGA2-LMs. For MED12 genotyping, genomic DNA was isolated from tissues and cells utilizing DNeasy Blood & Tissue Kit (Qiagen) or from formalin-fixed paraffin-embedded tissues utilizing QIAamp DNA FFPE Tissue Kit (Qiagen). The 391 bp sequence between intron 1 and intron 2 for NM_005120.2 was amplified by PCR utilizing Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific) with following primers: 5′-ggtggctgggaatcctagtg-3′ (forward) and 5′- ccctataagtcttcccaaccca-3′ (reverse) (9). The PCR products were Sanger sequenced at the Genomics Shared Resource Core and the Plant Microbe Genomics Facility at The Ohio State University using the same primers. None of MMs overexpressed HMGA2 (n = 20) nor carried mutations in MED12 (n = 31).
Morphometric analysis
The proportion of smooth muscle cells (SMCs) and non-SMC in MMs and LMs was determined by counting the nuclei of SMCs (CNN1-positive) and non-SMCs (CNN1-negative) in IF-stained sections for CNN1 (green) with Bisbenzimide H 33258 nuclear staining. Although LM tissues were collected from peripheral regions of tumors, the distribution of SMCs showed substantial heterogeneity within tissue sections. To avoid selection bias towards the areas with CNN1-staining, the entire area of CNN1-stained sections (~25–100 mm2) were scanned, and the areas with different cellularities were imaged proportionally to the original tissue. The morphometric analyses were blindly performed by designated researchers: the vascular cells were manually excluded from each image, and the areas for morphometric analyses were manually selected to include SMCs or non-SMCs only. In SMC concentration analysis, at least 615 total cells from 3–6 view fields at 40× magnification were counted blindly for each LM or MM. The nuclear size (area) was measured in the blue channel (nuclear staining) of the same field using “Analyze particle” function of Image J (NIH). The MKI67 labeling indices of SMCs and non-SMCs in PDTXs were determined by counting positive and negative cells in double IF-stained sections for CNN1 and MKI67.
Isolation of LM cells
The isolation of LM cells was performed as previously described (18). Briefly, LM tissues were cut into pieces (< ~9 mm3), washed twice with Dulbecco’s PBS containing 1× Gibco® Antibiotic-Antimycotic (Thermofisher Scientific), and digested in Hanks’s Balanced Salt solution containing 1.5 mg/ml collagenase Type I (Sigma-Aldrich), 83.3 g/mL DNase I (Sigma-Aldrich), and 1 × Gibco® Antibiotic-Antimycotic at 37°C for 5 hours. After filtration through a 100 μm Falcon® cell strainer (BD Falcon), residual erythrocytes in cell suspensions were lysed with Red Blood Cell Lysis Buffer (00-4300-54, eBioscience) for 10 – 15 min at room temperature.
Fluorescence activated cell sorting (FACS) analysis
Freshly isolated LM cells were further dispersed into single cells by incubating with 0.1 % trypsin for 15 min at 37°C. Singly dispersed cells were stained with Live/Dead fixable Near-IR Dead Cell Stain kit (Thermo Fisher Scientific), fixed with 4% paraformaldehyde for 10 min and incubated with Flow Cytometry Permeabilization/Wash Buffer I (R&D system) for 10 min. The permeabilized LM cells were then incubated with anti-collagen I (1:100 LS-B342) and anti-collagen III (1:100 LS-B693) IgGs together or with normal rabbit IgG (negative control, 1:100, sc-2027, Santa Cruz Biotechnology) for 30 min on ice, followed by incubation with PE-conjugated F(ab’)2-donkey anti-rabbit IgG secondary antibody (1:500, 12-4739-81, eBioscience) for 45 min on ice. Then, cells were incubated with anti-CNN1 IgG-Alexa488 (1:25, ab197639, Abcam) on ice for 45 min (Supplemental Fig. S2), washed with Flow Cytometry Permeabilization/Wash Buffer I twice, sieved through 60 μm nylon mesh (Spectrum Laboratories) and re-suspended in sorting buffer. Cell population count was performed with a LSRII Flow Cytometer (Becton Dickinson), and cell sorting was performed with a BD FACSAria III cell sorter (Becton Dickinson) at the Analytical Cytometry Shared Resources Core. Forward-scatter height versus forward-scatter width (FSC-H versus FSC-W) and side-scatter area versus side-scatter width (SSC-A versus SSC-W) were used to eliminate cell aggregates and ensure single cell sorting. Dead cells were excluded from analysis as they display high background. Each experiment included one no-staining, three single-color and two fluorescence-minus-one (FMO) controls. The distribution of cell populations was analyzed by FlowJo software (FlowJo, LLC).
Patient Derived Tumor Xenograft (PDTX) model
All animal procedures were approved by the Institutional Animal Care and Use Committees of the Ohio State University and Northwestern University. The procedure for the preparation of cell grafts and subrenal grafting were described previously (15,18). Briefly, at the time of grafting, host NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, Jackson Laboratory) were ovariectomized and subcutaneously implanted with 70 mg slow-releasing hormone pellets. When the tumor volume of E2+P4 group was less than 1.0 mm3 at the endpoint (6 or 8 weeks after grafting), the LM was categorized as growth negative and removed from the analyses. Images of PTDXs on the kidney were captured from 3 directions (x, y and z axes) using a dissecting microscope connected to a computer with Leica Application Suite version 3.8 software (Leica Microsystems). The tumor volume was calculated by π/6× length × width × height. The average value of 3 to 9 xenografts per group in each experiment was considered as a single measurement. To study the growth control of SMCs and non-SMCs in MED12-LMs, PDTXs were prepared from a LM carrying a c.131G>A mutation in MED12, and the value of each xenograft (n ≥3) were considered as a single measurement.
Statistical analyses
The experimental data were presented as mean values with standard deviation (SD), except in a box plot graph. The methods of statistical analyses are specified for each figure in the legend.
Results
Cellular composition of MED12-LMs and HMGA2-LMs is distinctive
Using HMGA2 IHC in 230 LM samples, we identified 11 HMGA2-LMs (4.8%) (Fig. 1A and Supplementary Fig. S1). This incidence of HMGA overexpression was lower than that previously reported (5,8,9). One of the factors contributing to the lower incidence of HMGA2-LMs was our selection of smaller LMs, which have greater cell viability for PDTX studies than larger LMs; however, karyotype abnormalities, including those leading to rearrangements of HMGA2 are associated with larger tumor size (19).
Figure 1. Two most prevalent subtypes of human LMs show distinctive cellular compositions.
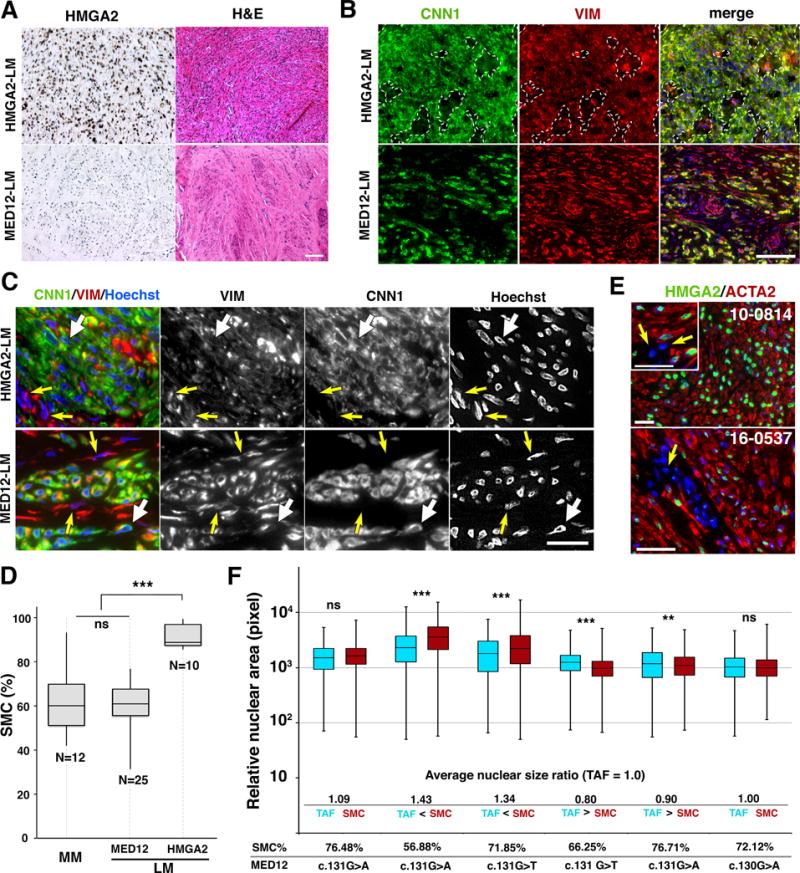
A. Histological characteristics of LM subtypes. Representative images for HMGAs and H&E of HMGA2-LM and MED12-LM subtypes. Bar = 100 μm B. Tissue distribution of SMCs and fibroblasts in HMGA2-LM and MED12-LM tissues. CNN1 (green) stains SMCs, whereas VIM (red) stains both SMCs and fibroblasts. In the HMGA2-LM tissue, perivascular connective tissues devoid of tumor SMCs were marked with dotted lines. In MED12-LMs, fibroblasts are indicated by white arrows. Bar = 100 μm C. Cellular and nuclear appearance of SMCs and tumor associated fibroblasts (TAFs). The nuclei of SMCs (white arrows) and fibroblasts (yellow arrows) are indicated by arrows. Bar = 25 μm. D. Box-plot for the concentration of SMCs in MMs and LMs. The MED12 mutations are listed in Table S1. The statistical significance by ANOVA was indicated as *** p< 0.001 and ns (not significant, P > 0.05). E. HMGA2 is overexpressed in SMCs but not in the nuclei of tumor-associated fibroblasts (arrow). Bar = 50 μm. F. Relative nuclear area of TAFs (blue box) and SMCs (red box) in MED12-LMs. The ratio between average nuclear sizes of SMCs and TAFs (SMC/TFA) is indicated below the boxplot. There was not clear association between nuclear size-ratio, mutation type and SMC concentration. The statistical significance by ANOVA was indicated as *** P< 0.001, ** P< 0.01 and ns (not significant, P > 0.05).
As assessed by the expression of calponin1/CNN1, both HMGA2-LMs and MED12-LMs contained smooth muscle cells (SMCs) and non-SMCs, which were mostly fibroblasts as stained for vimentin/VIM (Fig. 1 B and C). The distribution of fibroblasts was distinctive between HMGA2-LMs and MED12-LMs: in HMGA2-LMs, fibroblasts were concentrated in perivascular connective tissues (Fig. 1B, white dotted lines), whereas in MED12-LMs small clusters of SMCs were embedded in fibroblast-rich ECM (Fig. 1 B and C). As the cellular compositions of MED12-LMs and HMGA2-LMs appeared to be different, the percentage of SMCs and non-SMCs, excluding vascular cells, was determined in different LM subtypes. While MM consisted of 61.2 ± 14.9% of CNN1-positive SMCs, HMGA2-LMs consisted of 91.5 ± 5.6 % SMCs (Fig. 1D), suggesting that the overexpression of HMGA2 drives the overgrowth of SMCs leading to LMs. Indeed, immunostaining confirmed that HMGA2 was overexpressed only in SMCs (Fig. 1E). On the other hand, the SMC concentration of MED12-LMs (60.1 ± 10.6%) was not significantly different from that of MM (Fig. 1D). The SMC concentration in MED12-LMs widely ranged from 31.4 to 76.7%. Since the analysis of SMCs concentration can be biased by the size and morphology of nuclei, we assessed the correlation between SMC concentration and nuclear size ratio between SMCs and fibroblasts in 6 MED12-LMs (Fig. 1F). While the ratio of nuclear sizes in SMCs and fibroblasts was also variable among MED12-LMs, there was no correlation with SMC concentration. Thus the diverse SMC concentrations in MED12-LMs are not an artifact due to different nuclear sizes.
The similar concentration of SMCs and fibroblasts in MED12-LMs implied that these two populations arose from the same progenitor population. Indeed, Holdsworth-Carson et al. previously proposed that fibroblasts and SMCs in LMs arose from a same clone, based on a human androgen receptor assay (20). However, the sensitivity and accuracy of X-chromosome inactivation assay is limited, as it only determine the presence of 1 or 2 gene silencing patterns in the tissue. Furthermore, because silencing of the X-chromosome occurs during embryogenesis, the pattern must be shared by all cells in tissues derived from a progenitor cell (21). Hence, although fibroblasts and SMCs in LMs may be derived from the same Müllerian duct mesenchymal cell, MED12 mutations may occur exclusively in one population after their lineages are segregated. We therefore determined MED12 genotypes in SMCs and non-SMCs of MED12-LMs. MED12-LMs with a missense mutation in exon 2 of MED12 were digested into single cells, and 3 fractions were collected, based on the expression levels of CNN1 (CNN1−, CNN1+ and CNN1++). CNN1 preferentially stains tumor SMCs over vascular SMCs (v. 2A). Hence, CNN1− and CNN1++ fractions should mostly consist of fibroblasts and LM SMCs respectively, whereas CNN1+ should be a mixture of fibroblasts, vascular cells and LM SMCs. Each MED12-LM demonstrated a unique population distribution for the CNN1 signal levels (Fig.2B). Nevertheless, MED12 mutations were totally absent in non-SMCs, whereas CNN1+ and CNN1++ fractions contained MED12 mutant cells.
Figure 2. MED12 mutations are present in SMCs but not in TAFs.
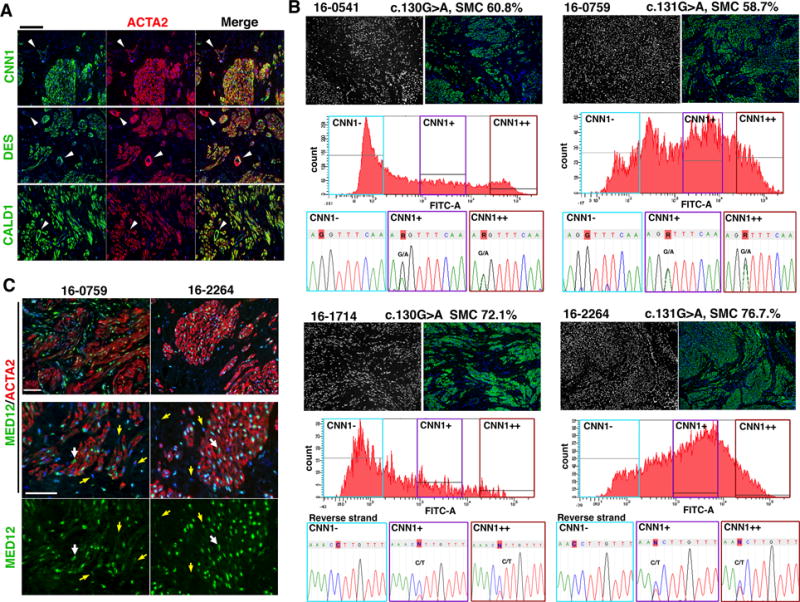
A. Expression patterns of SM markers in MED12-LMs. ACTA2, CNN1, DES and CALD1 were uniformly expressed in the SMCs of MED12-LMS. Vascular SMCs (arrowheads) showed significantly lower expression of CNN1 and DES compared to tumor SMCs. B. MED12 mutation analysis of SMC and non-SMC fraction in MED12-LM cases. The case ID number, MED12 missense mutation and SMC concentration by morphometric analysis in the original LM are indicated on top of the panels. IF images demonstrate the distribution of nuclei (black and white) and ATCA2 (green) in the original LM tissues. The FACS charts indicate the distributions of CNN1-signal intensity among isolated cells, and 3 fractions are separated by CNN1 expression level. In all four cases, the missense mutation of the original LM was absent in the CNN1-negative faction, whereas CNN1+ and CNN1++ fractions display peaks for the mutation. The base at the position of point mutation is highlighted with red. C. Expression patterns of MED12 (green) and ACTA2 (red) in MED12-LMs. The nuclei of fibroblasts (yellow arrows) contained lower levels of MED12 compared to that of ACTA2-positive SMCs (white arrows). Bar = 50 μM.
Since CNN1 and ACTA2 can be detected in uterine soft-tissue tumors of non-SMC origin, we assessed the expression patterns of two additional SMC markers, desmin (DES) and caldesmon (CALD1), which are more specific for SM tumors (22). Vascular SMCs expressed DES and CNN1 at noticeably lower levels compared to non-vascular SMCs. Meanwhile, tumor SMCs in MED12-LMs expressed all four SMC markers (N = 10) (Fig. 2A), indicating that the CNN1-positive population in MED12-LMs represents homogenous SMCs. Although MED12 was expressed in both SMCs and fibroblasts, the expression level was higher in the SMCs than fibroblasts (Fig. 2C), suggesting the critical roles of MED12 in SMCs.
In MED12-LMs, cells positive for MC387 (granulocytes, monocytes and macrophages) (Supplemental Figure S3) and endothelial cell markers (VWF and PECAM1) (Supplemental Figure S4) did not contribute significantly to the tissue mass, indicating that fibroblasts constitutes the major fraction of non-SMCs. Our results establish that MED12-LMs consist of approximately equal number of MED12 mutant SMCs and MED12 wild type non-SMCs, which are mostly fibroblasts.
Tumor associated fibroblasts (TAFs) contribute ECM to the MED12-LMs
The accumulation of excess ECM is a characteristic of LMs (15,18). However, HMGA2-LMs and MED12-LMs displayed striking differences in the distribution of ECM, as revealed by Masson’s trichrome staining and IHC for Type I collagen (Fig. 3A). Particularly, large pools of ECM, which contain fibroblasts but not SMCs, were frequently observed in MED12-LMs. Hence, we hypothesized that the high number of fibroblasts contributes to the accumulation of excess ECM in MED12-LMs. Indeed, the cytoplasm of both SMCs and TAFs in MED12-LMs was stained for Type I collagen (Fig. 3B). We further analyzed the intracellular contents of collagens in SMCs and non-SMCs by FACS. FACS analysis for Type I + Type III collagens versus CNN1 indicated that both SMC and non-SMC population produce ECM in MED12-LMs (Fig. 3C). Additionally, we assessed the regulation of procollagen I by E2 and P4 in PDTXs of MED12-LMs. Procollagen I was detected in both SMCs and TAFs in PDTXs of MED12-LMs irrespective of hormone treatment (Fig. 3D). Interestingly, procollagen signal in SMCs increased with E2+P4-treament, whereas procollagen I levels in fibroblasts seemed unchanged between E2 versus E2+P4 groups. These observations indicate that ECM in MED12-LMs are produced by both MED12 mutant SMCs and non-mutant TAFs. However, the regulation of collagen synthesis by E2 and P4 appears to be different in SMCs and TFAs.
Figure 3. Tumor-associated fibroblasts contribute collagens to MED12-LMs.
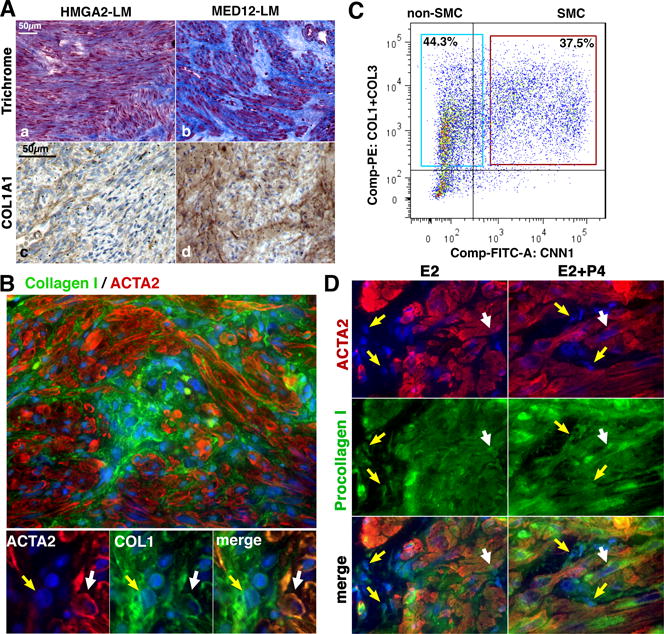
A. Distribution of ECM in HMGA2-LMs and MED12-LMs. Representative images of Masons Trichrome staining (a and b) and IHC for collagen type I (c and d) in HMGA2-LMs and MED12-LMs. B. IF staining of a MED12-LM for ACTA2 (red) and collagen type I (green). The cytoplasm of fibroblasts (yellow arrow) and SMCs (white arrow) showed signals for collagen. C. FACS analysis of intracellular collagen contents. Single cells isolated from a MED12-LM were stained with anti-collagen antibodies (anti-Type I + anti-Type III collagens, PE-labeled) and anti-CNN1 IgG (FITC-labeled). Both SMCs and non-SMCs contained detectable levels of collagens. D. IF for pro-collagen type I (green) and ACTA2 (red) in MED12-LM PDTXs. Type I procollagen was detected in both SMCs and TAFs in MED12-LM PDTXs treated with E2 alone or E2+P4. The treatment protocol is presented in Figure 5A.
The growth regulation of LM subtypes
The distinctive cellular composition and ECM contents implied fundamental differences in the growth control of MED12-LMs and HMGA2-LMs. Hence, we compared growth regulation of these LM subtypes by E2 and P4 utilizing PDTX model (15). In PDTXs of both LM subtypes, tumor volume significantly increased only when the hosts were treated with E2+P4 (Fig.4 A and B). Likewise, SMCs had a significantly increased proliferation rate (MKI67 labeling index) in response to E2+P4 but not to E2 or P4 only (Fig. 4 C and D). Therefore, our previous findings are confirmed in both MED12-LMs and HMGA2-LMs (15,18,23).
Figure 4. Growth control of HMGA2 and non-HMGA2 LMs.
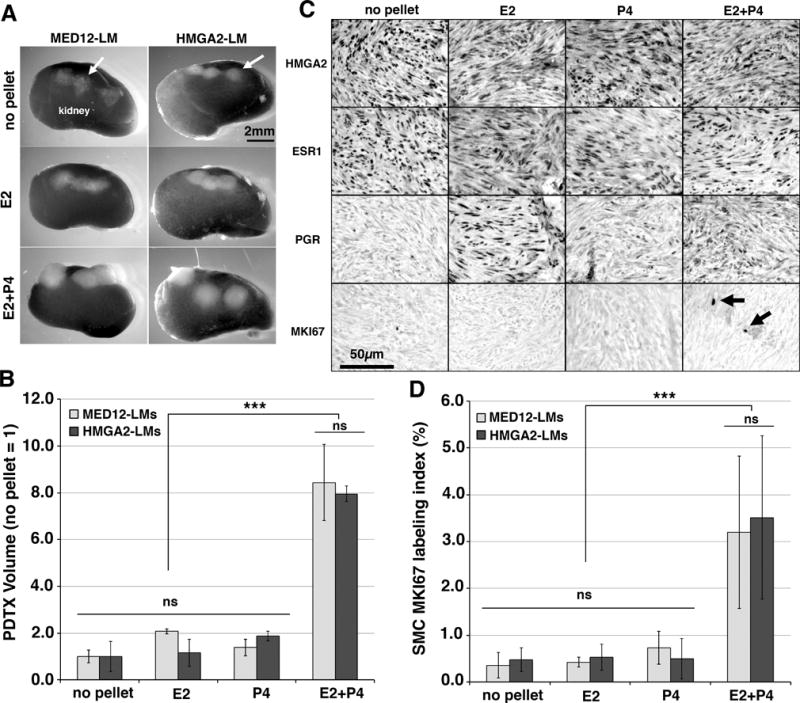
PDTXs were prepared with MED12-LMs (MED12 c.130G>C, c.131G>T and c.131G>C) and HMGA2-LMs (no MED12 mutations)(n = 3 each) and grown in NSG mice supplemented with different hormones for 8 weeks.
A. Gross appearance of PTDXs on the host kidney. B. Regulation of HMGA2, ESR1, PGR and MKI67 by E2 and P4 in HMGA2-LM PDTXs. HMGA2 expression did not change by hormone treatment. Expression of PGR in HMGA2-LMs was E2 dependent. C. Proliferation rate (MKI67 labeling index) of SMCs in PDTXs of HMGA2-LMs and MED12-LMs. D. PDTX volumes of MED12-LMs and HMGA2-LMs. Data are presented as mean values with SD. Statistical significance by Analysis of Variance was indicated as *** P < 0.001 and ns (not significant, P > 0.05).
In HMGA2-LM PDTXs, HMGA2 was constitutively expressed in the entire tumor tissue, and the expression pattern did not change by hormone treatments. Meanwhile, expression of PGR in HMGA2-LMs was E2 dependent (Fig 4B).
Growth characteristics of TAFs in MED12-LMs
If MED12 mutation drives growth of SMC cell-autonomously, SMCs is expected to be the predominant cell type, as found in HMGA2-LMs. However, MED12-LMs contain a similar number of MED12-mutant SMCs and non-mutant TAFs, thus raising a question about the growth control of these two cell populations. Accordingly, we assessed the growth response of SMCs and TAFs to E2 and P4 in PDTXs of MED12-LMs. PDTXs were generated from a MED12-LM with c.131G>A missense mutation in MED12. MED12-LM PDTXs were grown in hosts supplemented with E2+P4 for 4 weeks, and then subjected to no hormone, E2, or E2+P4 treatment (Fig. 5A). The volume increase from 4 to 6 weeks in PDTXs of E2+P4 groups confirmed that the LM cells were actively growing (Fig. 5B). Interestingly, E2 stimulated the proliferation of SMCs and TAFs differentially: while the MKI67 labeling index of SMCs was significantly elevated by E2+P4 but not E2, E2 and E2+P4 equally increased MKI67 labeling index in TAFs (Fig. 5 C and D). Nevertheless, MKI67 labeling index was significantly higher in SMCs than TAFs in E2+P4 treated MED12-LMs. Our results suggest that the diverse ratio of SMCs versus TAFs among MED12-LM cases reflects the history of endocrine milieu in the patient, given E2 exclusively stimulates TAFs, whereas P4 preferentially promotes growth of SMCs. In addition, the removal of P4 significantly reduced the ratio of SMCs to TAFs (Fig. 5E). This is likely due to a reduction of cellular/nuclear volume in SMCs. While both SMCs and TAFs expressed ESR1, the signal intensity per nucleus was always higher in SMCs than TAFs. When E2 induced PGR in PDTXs, the expression levels of PGR were also higher in SMCs than TAFs (Fig. 5F), suggesting that TAFs in MED12-LMs are less sensitive to E2 and P4 compared to MED12 mutant SMCs.
Figure 5. Growth control of SMCs and TAFs in MED12-LM.
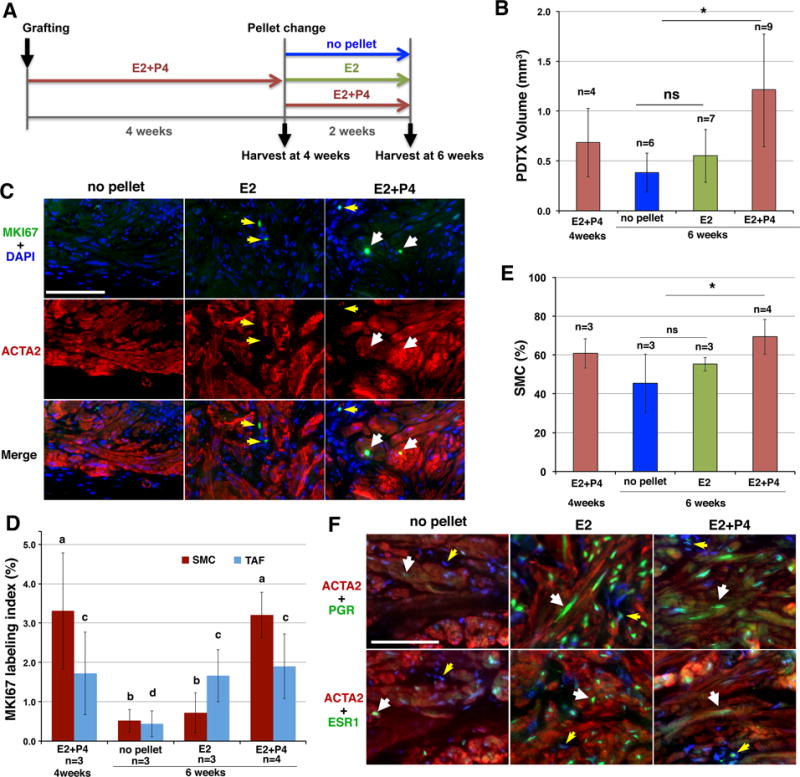
PDTXs were prepared from a MED12-LM (MED12 c.131 G>A). Statistical significance was determined by Analysis of Variance. A. Timeline. PDTXs were grown for 4 weeks with E2+P4, and then subjected to one of three different treatments: no hormone (no pellet), E2 or E2+P4 treatment for 2 weeks, at which point the PDTXs were harvested for analyses. B. The volume of PDTXs. *P < 0.05 and ns (not significant) C. Effect of E2 and P4 on MKI67 expression in SMCs (white arrows) and TAFs (yellow arrows) within MED12-LM PDTXs. D. Proliferation rate (MKI67 labeling index) of SMCs (dark red) and TAFs (light blue). The MKI67 labeling indices were significantly higher in groups marker with “a” than “b” (P < 0.01), and in groups marked with “c” than “d” (P < 0.05). E. Effect of E2 and P4 on SMC concentrations (%) in MED12-LM PDTXs. *P < 0.05. F. Regulation of ESR1 and PGR by E2 and P4 in SMC (white arrows) and TAFs (yellow arrows) in MED12-LMs.
Discussion
This study demonstrated for the first time that SMCs and TAFs are independent populations in both HMGA2-LMs and MED12-LMs, and the causal mutations are present only in the SMCs. The cellular composition of HMGA2-LMs and MED12-LMs significantly differs as HMGA2-LMs mostly consist of SMC population, whereas MED12-LMs contain equal number of SMCs and TAFs.
The concentration of SMCs in LMs is likely lower than the estimate in this study. We had avoided collecting LM tissues from the central region of the tumor, due to necrosis and calcification, however, the central region is low in tumor cell number and high in ECM (24). In addition, we had excluded vascular cells from the SMC concentration analysis.
The high concentration of TAFs explains why MED12 mutant cells are quickly lost from the primary culture of LMs (25,26); fibroblasts attach the culture dish better and grow faster than SMCs in standard culture conditions, conditions originally developed to optimize fibroblast growth (27). Overgrowth of fibroblasts is a classic problem for the primary culturing of SMCs including MM, and many techniques to repress the growth of fibroblasts in SMC culture have been proposed (20,28–32). However, this common problem has been often overlooked in LM and MM cell cultures because cell types are usually assessed in whole cell lysates, in which SMC markers can be detected even when SMCs are a minor population (33,34). In addition, ACTA2, the most commonly used SMC marker, cannot accurately detect the disappearance of SMCs from cell cultures, as ACTA2 can be induced in non-SMCs of the human uterus (35–39). Given the high initial concentration of TAFs with superior in vitro growth potential, freshly prepared primary cultures of MED12-LMs would contain more fibroblasts than SMCs, and the concentration of SMCs should further decrease, as TAFs grow faster to reach confluence. Indeed, Bloch et al. recently demonstrated that the concentration of MED12 mutant cells gradually decreases in the primary culture of MED12-LMs even without passaging (26). The study by Bloch et al. also suggested that the loss of MED12 mutant cells is partially due to the detachment of mutant cells from the culture flask, which likely reflects the weaker adherence of SMCs to the plastic surface compared to fibroblasts (40). Then, the subsequent passaging further dilutes SMCs in MED12-LM cell culture (25). Conversely, the low fibroblast concentration in HMGA2-LMs explains why HMGA2 mutant cells can be maintained in vitro for multiple passages (25).
Our current study questions the validity of previous studies that utilized LM cell culture as the primary research model: these studies likely analyzed the cell cultures that mostly consisted of TAFs (26). Indeed, LM and MM cells rapidly change their gene expression profiles in vitro (41). While the change in gene expression was attributed to the effects of artificial culture conditions, our result indicate that the overgrowth of TAFs is the primary factor that alters the gene expression of LM and MM cells in culture. Furthermore, the rapid loss of ESR1 and PGR expression in primary LM cell culture (42) is undoubtedly due to the overgrowth of TAFs, which express ESR1 and PGR at lower levels compared to LM SMCs. Several studies have demonstrated the growth-promoting effect of estrogens on primary LM cell cultures (43,44), contradicting with results from our PDTX studies, in which E2 does not stimulate the growth of LM SMCs. These conflicting observations can also be explained by the overgrowth of TAFs in LM cell culture as their growth is stimulated by E2 alone.
As assessed in histology sections, MED12 mutant SMCs are severalfold larger than TAFs, particularly when they are stimulated by E2 and P4. Hence, even when LMs contain an equal number of fibroblasts and SMCs, SMCs should contribute severalfold higher amount of proteins and RNAs to the tumor. Accordingly, gene/protein expression profiles of the original LM tissues are expected to represent the characteristics of LM SMCs. On the other hand, the amount of genetic materials is equal in each cell irrespective of the type and size. Hence, epigenetic analysis of MED12-LMs requires extra caution, as tumors contain a similar number of SMCs and non-SMCs including TAFs. For example, epigenetic signatures of MED12-LMs, such as DNA methylation patterns or histone modification patterns, should equally reflect that of SMCs and TAFs. Hence, we predict the epigenome of original MED12-LMs as whole should show more fibroblast-like patterns compared to HMGA2-LMs. As the concentrations of TAFs vary between tumor cases as well as intra-tumor locations, the epigenetics of MED12-LMs would require additional refining to normalize the concentrations of cell types in the sample.
We have previously shown that the level of miR-29b in LMs was significantly lower compared to MM and that the down-regulation of miR-29b is essential for the accumulation of ECM proteins in LMs (18). Since miR-29 family members are enriched in vascular SMCs compared to fibroblasts (45), it is possible that the lower levels of miR-29b in LMs reflect the lower ratio of SMCs to fibroblasts, which generally express lower levels of miR-29b compared to SMCs. In our previous study, collagen mRNAs and miR-29b were moderately up- and down- regulated respectively in PDTXs with 2 weeks of E2 treatment (18). This may be due to the increase in fibroblast concentration in PDTXs by E2 treatment. Our previous study did not delineate the effects of miR-29b levels on SMCs and TAFs, given the primary cultures of LMs contained both cell types. In order to elucidate the role of miR-29b in the pathogenesis of LMs, the expression patterns of miR-29b in LM SMCs versus TAFs should be further determined.
Due to the requirement of both E2 and P4 for the proliferation of tumor SMCs, selective progesterone receptor modulators (SPRMs) should be an effective treatment for both MED12-LMs and HMGA2-LMs. On the other hand, SPRMs may cause fibrosis of ECM-prominent MED12-LMs, as TAFs are stimulated by E2. Hence, to design non-surgical therapeutic strategies for LMs, it is critical to consider the LM subtype.
The cell-autonomous growth-promoting effect of HMGA2 in SMCs explains the pathogenesis of HMGA2-LMs. On the other hand, the coordinated growth of SMCs and TAFs in MED12-LMs implies the presence of paracrine interactions between these two cell types. We hypothesize that E2 stimulates MED12-mutant SMCs to secrete paracrine factors that promote the growth of TAFs, and TAFs in turn support the growth of LM SMCs. Several studies detected mRNAs for growth factors that are differentially expressed in paired LMs and MMs (46–49). The regulation of these growth factors should be revisited in light of potential paracrine interactions between SMCs and TAFs. Our PDTX model would be an ideal platform to explore the nature of paracrine interactions and mediators.
Supplementary Material
Acknowledgments
We gratefully thank Sheila Yasin Martinez for her assistance in patient consent. Tissue samples were provided by the Tissue Procurement Shared Resource at The Ohio State University Comprehensive Cancer Center, Columbus, Ohio. We also thank Jason Bice and Daphne Bryant (Histology Core) and the OSUCCC-Analytic Cytometry and OSUCCC-Genomics cores for technical support.
Grant Support
This work was funded in part by the Eunice Kennedy Shriver National Institute of Child Health and Human Development grant R01 HD064402 (T. Kurita) and the National Cancer Institute grants R01 CA154358 (T. Kurita) and P30 CA016058 (T. Kurita).
References
- 1.Marshall LM, Spiegelman D, Barbieri RL, Goldman MB, Manson JE, Colditz GA, et al. Variation in the incidence of uterine leiomyoma among premenopausal women by age and race. Obstetrics and gynecology. 1997;90:967–73. doi: 10.1016/s0029-7844(97)00534-6. [DOI] [PubMed] [Google Scholar]
- 2.Parker WH. Etiology, symptomatology, and diagnosis of uterine myomas. Fertility and sterility. 2007;87:725–36. doi: 10.1016/j.fertnstert.2007.01.093. [DOI] [PubMed] [Google Scholar]
- 3.Stewart EA, Laughlin-Tommaso SK, Catherino WH, Lalitkumar S, Gupta D, Vollenhoven B. Uterine fibroids. Nat Rev Dis Primers. 2016;2:16043. doi: 10.1038/nrdp.2016.43. [DOI] [PubMed] [Google Scholar]
- 4.Mehine M, Kaasinen E, Mäkinen N, Katainen R, Kampjarvi K, Pitkanen E, et al. Characterization of uterine leiomyomas by whole-genome sequencing. The New England journal of medicine. 2013;369:43–53. doi: 10.1056/NEJMoa1302736. [DOI] [PubMed] [Google Scholar]
- 5.Mehine M, Kaasinen E, Heinonen HR, Mäkinen N, Kampjarvi K, Sarvilinna N, et al. Integrated data analysis reveals uterine leiomyoma subtypes with distinct driver pathways and biomarkers. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:1315–20. doi: 10.1073/pnas.1518752113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mäkinen N, Mehine M, Tolvanen J, Kaasinen E, Li Y, Lehtonen HJ, et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science (New York, NY. 2011;334:252–5. doi: 10.1126/science.1208930. [DOI] [PubMed] [Google Scholar]
- 7.Kazmierczak B, Hennig Y, Wanschura S, Rogalla P, Bartnitzke S, Van de Ven W, et al. Description of a novel fusion transcript between HMGI-C, a gene encoding for a member of the high mobility group proteins, and the mitochondrial aldehyde dehydrogenase gene. Cancer Res. 1995;55:6038–9. [PubMed] [Google Scholar]
- 8.Markowski DN, Bartnitzke S, Loning T, Drieschner N, Helmke BM, Bullerdiek J. MED12 mutations in uterine fibroids–their relationship to cytogenetic subgroups. International journal of cancer. 2012;131:1528–36. doi: 10.1002/ijc.27424. [DOI] [PubMed] [Google Scholar]
- 9.Bertsch E, Qiang W, Zhang Q, Espona-Fiedler M, Druschitz S, Liu Y, et al. MED12 and HMGA2 mutations: two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod Pathol. 2014;27:1144–53. doi: 10.1038/modpathol.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mittal P, Shin YH, Yatsenko SA, Castro CA, Surti U, Rajkovic A. Med12 gain-of-function mutation causes leiomyomas and genomic instability. J Clin Invest. 2015;125:3280–4. doi: 10.1172/JCI81534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandberg AA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: leiomyoma. Cancer genetics and cytogenetics. 2005;158:1–26. doi: 10.1016/j.cancergencyto.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 12.Quade BJ, Weremowicz S, Neskey DM, Vanni R, Ladd C, Dal Cin P, et al. Fusion transcripts involving HMGA2 are not a common molecular mechanism in uterine leiomyomata with rearrangements in 12q15. Cancer Res. 2003;63:1351–8. [PubMed] [Google Scholar]
- 13.Mehine M, Heinonen HR, Sarvilinna N, Pitkanen E, Mäkinen N, Katainen R, et al. Clonally related uterine leiomyomas are common and display branched tumor evolution. Human molecular genetics. 2015;24:4407–16. doi: 10.1093/hmg/ddv177. [DOI] [PubMed] [Google Scholar]
- 14.Yu L, Moore AB, Dixon D. Receptor tyrosine kinases and their hormonal regulation in uterine leiomyoma. Semin Reprod Med. 2010;28:250–9. doi: 10.1055/s-0030-1251482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishikawa H, Ishi K, Serna VA, Kakazu R, Bulun SE, Kurita T. Progesterone is essential for maintenance and growth of uterine leiomyoma. Endocrinology. 2010;151:2433–42. doi: 10.1210/en.2009-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terakawa J, Rocchi A, Serna VA, Bottinger EP, Graff JM, Kurita T. Molecular endocrinology. Vol. 30. Baltimore, Md: 2016. FGFR2IIIb-MAPK Activity Is Required for Epithelial Cell Fate Decision in the Lower Mullerian Duct; pp. 783–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandtzaeg P, Jones DB, Flavell DJ, Fagerhol MK. Mac 387 antibody and detection of formalin resistant myelomonocytic L1 antigen. J Clin Pathol. 1988;41:963–70. doi: 10.1136/jcp.41.9.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiang W, Liu Z, Serna VA, Druschitz SA, Liu Y, Espona-Fiedler M, et al. Down-Regulation of miR-29b Is Essential for Pathogenesis of Uterine Leiomyoma. Endocrinology. 2014;155:663–9. doi: 10.1210/en.2013-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rein MS, Powell WL, Walters FC, Weremowicz S, Cantor RM, Barbieri RL, et al. Cytogenetic abnormalities in uterine myomas are associated with myoma size. Molecular human reproduction. 1998;4:83–6. doi: 10.1093/molehr/4.1.83. [DOI] [PubMed] [Google Scholar]
- 20.Holdsworth-Carson SJ, Zaitseva M, Vollenhoven BJ, Rogers PA. Clonality of smooth muscle and fibroblast cell populations isolated from human fibroid and myometrial tissues. Molecular human reproduction. 2014;20:250–9. doi: 10.1093/molehr/gat083. [DOI] [PubMed] [Google Scholar]
- 21.Konishi I, Fujii S, Okamura H, Mori T. Development of smooth muscle in the human fetal uterus: an ultrastructural study. Journal of anatomy. 1984;139(Pt 2):239–52. [PMC free article] [PubMed] [Google Scholar]
- 22.Oliva E, Young RH, Amin MB, Clement PB. An immunohistochemical analysis of endometrial stromal and smooth muscle tumors of the uterus: a study of 54 cases emphasizing the importance of using a panel because of overlap in immunoreactivity for individual antibodies. The American journal of surgical pathology. 2002;26:403–12. doi: 10.1097/00000478-200204000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Wang G, Ishikawa H, Sone K, Kobayashi T, Kim JJ, Kurita T, et al. Nonobese diabetic/severe combined immunodeficient murine xenograft model for human uterine leiomyoma. Fertility and sterility. 2014;101:1485–92. doi: 10.1016/j.fertnstert.2014.01.054. [DOI] [PubMed] [Google Scholar]
- 24.Yang EJ, Mutter GL. Biomarker resolution of uterine smooth muscle tumor necrosis as benign vs malignant. Mod Pathol. 2015;28:830–5. doi: 10.1038/modpathol.2015.35. [DOI] [PubMed] [Google Scholar]
- 25.Markowski DN, Tadayyon M, Bartnitzke S, Belge G, Maria Helmke B, Bullerdiek J. Cell cultures in uterine leiomyomas: rapid disappearance of cells carrying MED12 mutations. Genes, chromosomes & cancer. 2014;53:317–23. doi: 10.1002/gcc.22142. [DOI] [PubMed] [Google Scholar]
- 26.Bloch J, Holzmann C, Koczan D, Helmke BM, Bullerdiek J. Factors affecting the loss of MED12-mutated leiomyoma cells during in vitro growth. Oncotarget. 2017;8:34762–72. doi: 10.18632/oncotarget.16711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao T, Asayama Y. Animal-cell culture media: History, characteristics, and current issues. Reproductive Medicine and Biology. 2017;16:99–117. doi: 10.1002/rmb2.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hongpaisan J. Inhibition of proliferation of contaminating fibroblasts by D-valine in cultures of smooth muscle cells from human myometrium. Cell Biol Int. 2000;24:1–7. doi: 10.1006/cbir.1999.0448. [DOI] [PubMed] [Google Scholar]
- 29.Terracio L, Ronnstrand L, Tingstrom A, Rubin K, Claesson-Welsh L, Funa K, et al. Induction of platelet-derived growth factor receptor expression in smooth muscle cells and fibroblasts upon tissue culturing. The Journal of cell biology. 1988;107:1947–57. doi: 10.1083/jcb.107.5.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frauli M, Ludwig H. Inhibition of fibroblast proliferation in a culture of human endometrial stromal cells using a medium containing D-valine. Archives of gynecology and obstetrics. 1987;241:87–96. doi: 10.1007/BF00931229. [DOI] [PubMed] [Google Scholar]
- 31.Xu S, Fu J, Chen J, Xiao P, Lan T, Le K, et al. Development of an optimized protocol for primary culture of smooth muscle cells from rat thoracic aortas. Cytotechnology. 2009;61:65–72. doi: 10.1007/s10616-009-9236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazzaro VA, Walker RJ, Duggin GG, Phippard A, Horvath JS, Tiller DJ. Inhibition of fibroblast proliferation in L-valine reduced selective media. Res Commun Chem Pathol Pharmacol. 1992;75:39–48. [PubMed] [Google Scholar]
- 33.Cavaillé F, Cabrol D, Ferre F. Human Myometrial Smooth Muscle Cells and Cervical Fibroblasts in Culture. In: Jones GE, editor. Human Cell Culture Protocols. Totowa, NJ: Humana Press; 1996. pp. 335–44. [DOI] [PubMed] [Google Scholar]
- 34.Mosher AA, Rainey KJ, Bolstad SS, Lye SJ, Mitchell BF, Olson DM, et al. Development and validation of primary human myometrial cell culture models to study pregnancy and labour. BMC Pregnancy Childbirth. 2013;13(Suppl 1):S7. doi: 10.1186/1471-2393-13-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oliver C, Montes MJ, Galindo JA, Ruiz C, Olivares EG. Human decidual stromal cells express alpha-smooth muscle actin and show ultrastructural similarities with myofibroblasts. Human reproduction (Oxford, England) 1999;14:1599–605. doi: 10.1093/humrep/14.6.1599. [DOI] [PubMed] [Google Scholar]
- 36.Czernobilsky B, Remadi S, Gabbiani G. Alpha-smooth muscle actin and other stromal markers in endometrial mucosa. Virchows Arch A Pathol Anat Histopathol. 1993;422:313–7. doi: 10.1007/BF01608341. [DOI] [PubMed] [Google Scholar]
- 37.Liu X, Shen M, Qi Q, Zhang H, Guo SW. Corroborating evidence for platelet-induced epithelial-mesenchymal transition and fibroblast-to-myofibroblast transdifferentiation in the development of adenomyosis. Human reproduction (Oxford, England) 2016;31:734–49. doi: 10.1093/humrep/dew018. [DOI] [PubMed] [Google Scholar]
- 38.Yen CF, Huang SJ, Lee CL, Wang HS, Liao SK. Reproductive sciences. Thousand Oaks, Calif: 2017. Molecular Characteristics of the Endometrium in Uterine Adenomyosis and Its Biochemical Microenvironment. 1933719117691141. [DOI] [PubMed] [Google Scholar]
- 39.Ibrahim MG, Sillem M, Plendl J, Chiantera V, Sehouli J, Mechsner S. Reproductive sciences. Thousand Oaks, Calif: 2017. Myofibroblasts Are Evidence of Chronic Tissue Microtrauma at the Endometrial-Myometrial Junctional Zone in Uteri With Adenomyosis. 1933719116687855. [DOI] [PubMed] [Google Scholar]
- 40.Nikolskaya A, Sharma V. Cell Culture Models and Methods. In: Sigg DC, Iaizzo PA, Xiao Y-F, He B, editors. Cardiac Electrophysiology Methods and Models. Boston, MA: Springer US; 2010. pp. 213–35. [Google Scholar]
- 41.Zaitseva M, Vollenhoven BJ, Rogers PA. In vitro culture significantly alters gene expression profiles and reduces differences between myometrial and fibroid smooth muscle cells. Molecular human reproduction. 2006;12:187–207. doi: 10.1093/molehr/gal018. [DOI] [PubMed] [Google Scholar]
- 42.Severino MF, Murray MJ, Brandon DD, Clinton GM, Burry KA, Novy MJ. Rapid loss of oestrogen and progesterone receptors in human leiomyoma and myometrial explant cultures. Molecular human reproduction. 1996;2:823–8. doi: 10.1093/molehr/2.11.823. [DOI] [PubMed] [Google Scholar]
- 43.Park SH, Ramachandran S, Kwon SH, Cha SD, Seo EW, Bae I, et al. Upregulation of ATP-sensitive potassium channels for estrogen-mediated cell proliferation in human uterine leiomyoma cells. Gynecol Endocrinol. 2008;24:250–6. doi: 10.1080/09513590801893315. [DOI] [PubMed] [Google Scholar]
- 44.Shimomura Y, Matsuo H, Samoto T, Maruo T. Up-regulation by progesterone of proliferating cell nuclear antigen and epidermal growth factor expression in human uterine leiomyoma. The Journal of clinical endocrinology and metabolism. 1998;83:2192–8. doi: 10.1210/jcem.83.6.4879. [DOI] [PubMed] [Google Scholar]
- 45.Cushing L, Costinean S, Xu W, Jiang Z, Madden L, Kuang P, et al. Disruption of miR-29 Leads to Aberrant Differentiation of Smooth Muscle Cells Selectively Associated with Distal Lung Vasculature. PLoS Genet. 2015;11:e1005238. doi: 10.1371/journal.pgen.1005238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arslan AA, Gold LI, Mittal K, Suen TC, Belitskaya-Levy I, Tang MS, et al. Gene expression studies provide clues to the pathogenesis of uterine leiomyoma: new evidence and a systematic review. Human reproduction (Oxford, England) 2005;20:852–63. doi: 10.1093/humrep/deh698. [DOI] [PubMed] [Google Scholar]
- 47.Litovkin KV, Ivanova OV, Bauer A, Hoheisel JD, Bubnov VV, Zaporozhan VN. Microarray study of gene expression in uterine leiomyoma. Exp Oncol. 2008;30:106–11. [PubMed] [Google Scholar]
- 48.Lee EJ, Kong G, Lee SH, Rho SB, Park CS, Kim BG, et al. Profiling of differentially expressed genes in human uterine leiomyomas. Int J Gynecol Cancer. 2005;15:146–54. doi: 10.1111/j.1048-891x.2005.15016.x. [DOI] [PubMed] [Google Scholar]
- 49.Skubitz KM, Skubitz AP. Differential gene expression in uterine leiomyoma. J Lab Clin Med. 2003;141:297–308. doi: 10.1016/S0022-2143(03)00007-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.