

Conspectus
The mystery associated with catalysis by what were once regarded as protein black boxes, diminished with the X-ray crystallographic determination of the three-dimensional structures of enzyme–substrate complexes. The report that several high-resolution X-ray crystal structures of orotidine 5′-monophosphate decarboxylase (OMPDC) failed to provide a consensus mechanism for enzyme-catalyzed decarboxylation of OMP to form uridine 5′-monophosphate, therefore, provoked a flurry of controversy. This controversy was fueled by the enormous 1023-fold rate acceleration for this enzyme, which had “jolted many biochemists’ assumptions about the catalytic potential of enzymes.” Our studies on the mechanism of action of OMPDC provide strong evidence that catalysis by this enzyme is not fundamentally different from less proficient catalysts, while highlighting important architectural elements that enable a peak level of performance. Many enzymes undergo substrate-induced protein conformational changes that trap their substrates in solvent occluded protein cages, but the conformational change induced by ligand binding to OMPDC is incredibly complex, as required to enable the development of 22 kcal/mol of stabilizing binding interactions with the phosphodianion and ribosyl substrate fragments of OMP. The binding energy from these fragments is utilized to activate OMPDC for catalysis of decarboxylation at the orotate fragment of OMP, through the creation of a tight, catalytically active, protein cage from the floppy, open, unliganded form of OMPDC. Such utilization of binding energy for ligand-driven conformational changes provides a general mechanism to obtain specificity in transition state binding. The rate enhancement that results from the binding of carbon acid substrates to enzymes is partly due to a reduction in the carbon acid pKa that is associated with ligand binding. The binding of UMP to OMPDC results in an unusually large >12 unit decrease in the pKa = 29 for abstraction of the C-6 substrate hydrogen, due to stabilization of an enzyme-bound vinyl carbanion, which is also an intermediate of OMPDC-catalyzed decarboxylation. The protein–ligand interactions operate to stabilize the vinyl carbanion at the enzyme active site compared to aqueous solution, rather than to stabilize the transition state for the concerted electrophilic displacement of CO2 by H+ that avoids formation of this reaction intermediate. There is evidence that OMPDC induces strain into the bound substrate. The interaction between the amide side chain of Gln-215 from the phosphodianion gripper loop and the hydroxymethylene side chain of Ser-154 from the pyrimidine umbrella of ScOMPDC position the amide side chain to interact with the phosphodianion of OMP. There are no direct stabilizing interactions between dianion gripper protein side chains Gln-215, Tyr-217, and Arg-235 and the pyrimidine ring at the decarboxylation transition state. Rather these side chains function solely to hold OMPDC in the catalytically active closed conformation. The hydrophobic side chains that line the active site of OMPDC in the region of the departing CO2 product may function to stabilize the decarboxylation transition state by providing hydrophobic solvation of this product.
I. Introduction
Enzymes operate at different levels of performance;1 and one of the challenges for mechanistic enzymologists is to define protein architectural elements that enable enzymes to perform at the highest level.2 Amyes and Richard recognized this challenge, after spending years investigating the mechanism for nonenzymatic reactions.3,4 The obvious target for study was the decarboxylation reaction catalyzed by orotidine 5′-monophosphate decarboxylase (OMPDC), because the 1023-fold rate acceleration for this enzyme corresponds to an enormous 31 kcal/mol stabilization of the transition state for the chemically difficult decarboxylation of orotidine 5′-monophosphate (OMP) to give uridine 5′-monophosphate (UMP, Scheme 1) represented an extraordinary rate enhancement for a unimolecular reaction.5
Scheme 1. OMDPC-Catalyzed Decarboxylation of OMP to form UMP.

High-resolution X-ray crystal structures of OMPDC resulted in a flurry of controversy and activity, because of their failure to lead to a consensus decarboxylation reaction mechanism.6 This gave rise to provocative statements. For example, it was claimed that “This proficient enzyme jolted many biochemists’ assumptions about the catalytic potential of enzymes...”7 A second group claimed that “the 1023M–1proficiency of ODC must arise from covalent catalysis, since only 1011M–1proficiency is possible by noncovalent binding,”8 even though there was no solid experimental evidence to support covalent catalysis by OMPDC. We eagerly joined in the quest to determine how OMPDC works, while viewing these statements with skepticism. In fact, the rate acceleration by OMPDC is only incrementally greater than that for other enzymes.1 We have therefore embraced, and worked to build on, the extraordinary work of mechanistic enzymologists over the past 50 years, in defining the mechanism of action for OMPDC.
II. The Pauling Principle
Catalysis by OMPDC is due to stabilization of the decarboxylation transition state by interactions with the protein catalyst.9,10 These must be 31 kcal/mol to account for the 1023-fold rate acceleration. The falloff in the second-order rate constants for OMPDC-catalyzed decarboxylation observed upon the truncation of the phosphodianion and the ribosyl phosphate from the substrate OMP, provide intrinsic binding energies of 11.8 and 10.6 kcal/mol for these substrate fragments, and of 8.6 kcal/mol for the pyrimidine ring (Scheme 2).11,12
Scheme 2. Partitioning of the Transition State Binding Energy for Yeast OMPDC (ScOMPDC)-Catalyzed Decarboxylation of OMP.
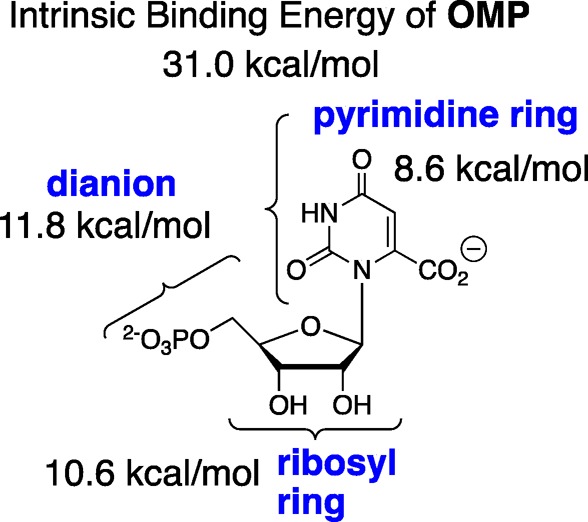
This pedantic division of the substrate intrinsic binding energy provides limited insight into reaction mechanism, but is the first step toward the more enlightening characterization of the specificity of these substrate fragments for binding to the transition state for OMPDC-catalyzed decarboxylation. For example, phosphite dianion shows barely detectable binding to free ScOMPDC (Kd > 0.1 M). However, an analysis of strong phosphite dianion activation of ScOMPDC-catalyzed decarboxylation of the truncated substrate 1-(β-d-erythrofuranosyl)orotic acid (EO) gives Kact‡ = 2.2 × 10–6 M for dianion binding to the transition state (eq 1, derived for Scheme 3), where (kcat/Km)0 = 0.026 M–1 s–1 and (kcat/Km)HPi/Kd = 12 000 M–2 s–1 are the experimental rate constants for unactivated and phosphite dianion activated ScOMPDC-catalyzed decarboxylation of EO.13,14 These data show that 8 of the 12 kcal/mol intrinsic phosphodianion binding energy of OMP is recovered in HPi, as activation of ScOMPDC for catalysis of decarboxylation of EO. The remaining 4 kcal/mol of binding energy represents the largely entropic advantage of unimolecular enzyme-catalyzed decarboxylation of the whole substrate compared with the bimolecular reaction of the substrate pieces.15
| 1 |
Scheme 3. Kinetic Scheme That Describes the Activation of ScOMPDC-Catalyzed Decarboxylation of EO and FEO by Phosphite Dianion (eq 1).

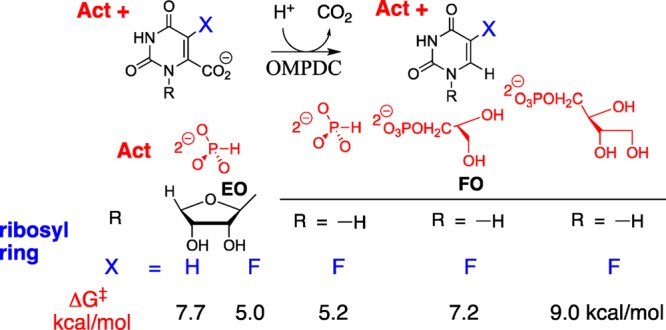
The activation of ScOMPDC for catalysis of decarboxylation of EO observed for a wide range of dianions, with intrinsic dianion binding energies ΔG‡ shown in Chart 1, is consistent with a binding locus at ScOMPDC where dianion binding energy is utilized to enhance catalysis at a distant pyrimidine binding locus.14,16ScOMPDC also catalyzes decarboxylation of 1-(β-d-erythrofuranosyl)-5-fluoroorotate (FEO) and of 5-fluoroorotate (FO).11 Each enzymatic reaction is activated by phosphite dianion (see Scheme 3), and a similar 5 kcal/mol stabilization of the respective reaction transition states by 1.0 M dianion is observed (Scheme 4).11 The transition state for ScOMPDC-catalyzed decarboxylation of FO is stabilized by 5.2, 7.2, and 9.0 kcal/mol, respectively, by 1.0 M phosphite dianion, d-glycerol 3-phosphate and d-erythritol 4-phosphate, so that binding interactions between both the substrate phosphodianion and the ribosyl hydroxyls are utilized to activate ScOMPDC for catalysis.(11)
Chart 1. Binding Energies ΔG‡ for Stabilization of the Transition State for ScOMPDC-Catalyzed Decarboxylation of EO by Different Dianions, Calculated from the Dissociation Constants for Breakdown of Dianion Transition State Complexes14,16.

Scheme 4. Activation of ScOMPDC-Catalyzed Decarboxylation, Where ΔG‡ is for Activator Binding to the Transition State for OMDC-Catalyzed Decarboxylation of a Truncated Substrates EO or FO (Scheme 3).
III. Enzyme-Activating Conformational Changes
Scheme 5 shows the model that rationalizes the activation of OMPDC for catalysis by interactions with dianions (Chart 1) and ribosyl-type hydroxyl groups (Scheme 4).10,17,18 OMPDC undergoes a large thermodynamically unfavorable (KC ≪ 1, Scheme 5) conformational change that transforms this enzyme from an inactive, floppy and disordered open form (EO) to an active, tight structured substrate cage (EC).2,19,20 Activation of OMPDC and other enzymes results when ligand binding gives rise to an increase in the fraction of protein present as EC.21Scheme 5 shows a substrate-induced fit that was referred to by Koshland, in another context, as an “induced-fit”.22 Koshland’s induced fit model was criticized because it did not provide a rational for the evolution of enzymes that exist mainly as inactive EO.23 However, we expect that there should normally be a barrier to conversion of a loose, open, enzyme EO to the tight, closed, caged complexe EC, and have proposed that this represents the barriers to extrusion of protein bound waters to bulk solvent; and, to freezing motions at protein loops and catalytic side chains at the structured cage.17
Scheme 5. Enzyme-Activating, Ligand Driven Conformational Change.

X-ray Crystal Structures of EO and EC
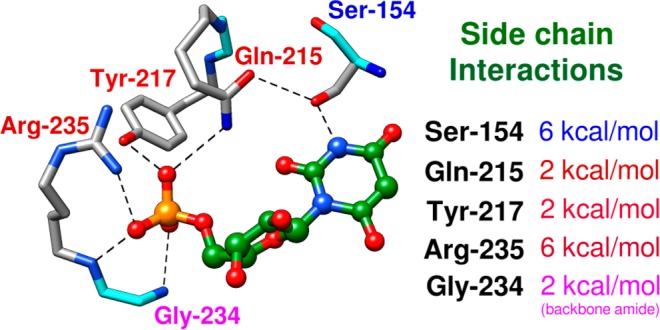
Figure 1 (top structures) shows space filling representations of the unliganded and the 6-hydroxyuridine 5′-monophosphate (BMP) complex to OMPDC from yeast (ScOMPDC). The solvent exposed active site of EO lies between open phosphodianion (Pro-202 to Val-220) and pyrimidine gripper (Ala-151 to Thr-165) loops that are shaded blue. These loops close over BMP at EC. The side chain of Arg-235 (shaded green) at the surface of ScOMPDC forms an ion pair to the ligand phosphodianion. The R235A mutation of ScOMPDC results in a 5.8 kcal/mol destabilization of the transition state for ScOMPDC-catalyzed decarboxylation; and, 3.0 kcal/mol of this effect is “rescued” by guanidine cation, which mimics the excised side chain.24
Figure 1.
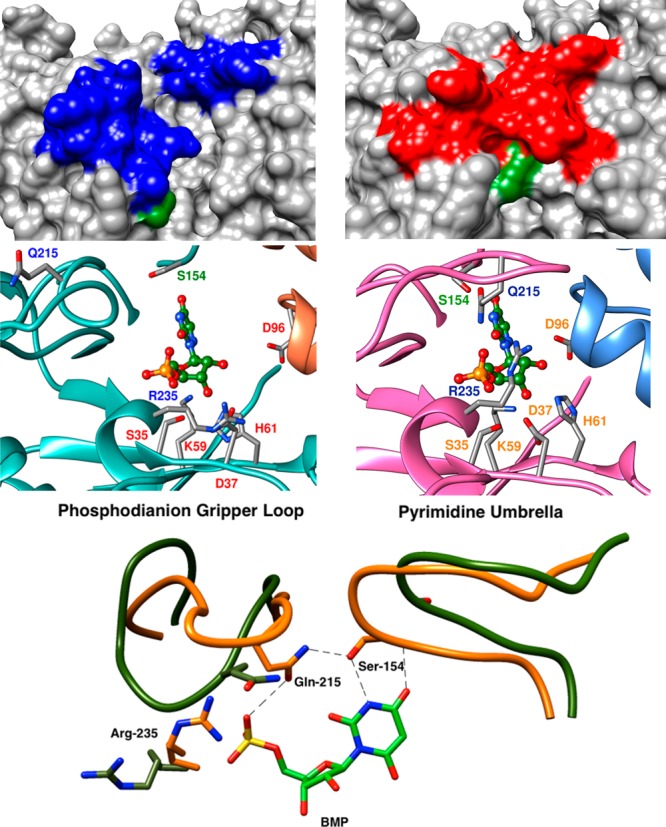
Representations of the open (EO) and the closed (EC) forms of ScOMPDC. Top: Space filling models of open unliganded ScOMPDC (left, PDB entry 1DQW) and the caged complex to BMP (right, 1DQX). The Arg-235 guanidine side chain is shaded green. Middle: Representations of the open (EO, left, 3GDK) and closed (EC, right, 3GDL) forms of ScOMPDC.25 The azaUMP ligand is placed at EO at the position determined for EC. Bottom: Image that superimposes partial X-ray crystal structures of the OMPDC·BMP complex (PDB entry 1DQX) over the structure for unliganded ScOMPDC (1DQW). The movement of the phosphodianion gripper loop (Pro-202 to Val-220) toward the pyrimidine umbrella (Ala-151 to Thr-165) is shown. Reprinted with permission from ref (39). Copyright 2013 American Chemical Society.
The middle structures from Figure 1 show the complex between ScOMPDC and 6-aza uridine 5′-monophosphate (azaUMP) on the right, and the structure of unliganded ScOMPDC on the left, in which a hypothetical 6-aza uridine 5′-monophosphate (azaUMP) ligand has been inserted at the position determined for the ScOMPDC·azaUMP complex.25 The middle structures from Figure 1 show that ligand binding is driven by the development of stabilizing interactions between the phosphodianion and the side chains of Arg-235 and Gln-215, and between the ribosyl hydroxyls and the side chains of Asp-96, His-61, Asp-37, and Lys-59. All of these interactions act to stabilize EC relative to EO, and activate ScOMPDC for catalysis of decarboxylation of truncated substrates (Scheme 4).
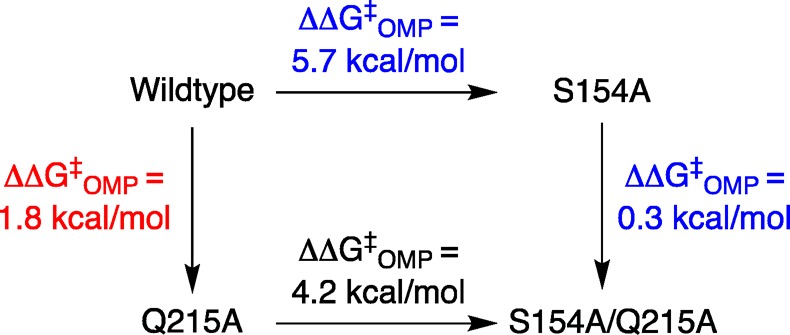
The bottom structures from Figure 1 show interactions, that arise from the ligand driven conformational change, of the amide side chain of Gln-215 (phosphodianion gripper loop) with the phosphodianion and with the hydroxymethylene side chain of Ser-154 (pyrimidine umbrella). The role of this interloop interaction in catalysis was probed by determining the effects of S154A, Q215A and S154A/Q215A mutations on ΔG‡ for ScOMPDC-catalyzed decarboxylation of OMP (Scheme 6).26 The Q215A mutation results in 0.5 and 1.8 kcal/mol increases, respectively, in the barrier for ScOMPDC-catalyzed decarboxylation of EO and OMP that are consistent with an interaction between the amide side chain and the phosphodianion of OMP. The Q215A mutation of wild type ScOMPDC results in a 1.8 kcal/mol destabilization of the decarboxylation transition state, while the same mutation at the S154A mutant results in only a 0.3 kcal/mol change,27 because the interloop interaction between the side chains of Ser-154 and Gln-215 wild type ScOMPDC (Figure 1, bottom) is required to hold the amide side chain in a position to interact with the substrate phosphodianion.
Scheme 6. Effect S154A and Q215A Mutations on the Activation Barrier G‡ (kcat/Km) for Wild Type ScOMPDC-Catalyzed Decarboxylation of OMP.
Steric Effects on Cage Formation
Small changes in substrate bulk result in dramatic changes in the barrier to ScOMPDC-catalyzed decarboxylation (Chart 2). For example, adding a −CH2OH group to EO or an −OH group to phosphite dianion results in 8.3 and 4.7 kcal/mol increases, respectively, in the barrier to the dianion-activated decarboxylation; the combined effect of these two substitutions is ca. 13 kcal/mol.13,28 These data define an estimated rate constant of 3.4 × 10–6 M–1 s–1 for phosphate dianion activation of ScOMPDC-catalyzed decarboxylation orotidine, which was too small to detect by experiment.29 Addition of a −CH2OH group to EO results in a 3.0 kcal/mol increase in the barrier to unactivated ScOMPDC-catalyzed decarboxylation, while addition of a −PO3–2 group of orotidine results in a 14.7 kcal/mol reduction in the barrier to decarboxylation that is even larger than the 11.7 kcal/mol effect of addition of a −CH2OPO32 group to EO (Chart 2).13,28
Chart 2. Rate Constants and Changes in Activation Barriers ΔΔG‡ for ScOMPDC-Catalyzed Decarboxylation of the Substrate Pieces (Top Row) Or the Whole Substrate OMP (Bottom Row)a.

a ΔΔG‡ is the effect of substitutions on the stability of the transition state for either dianion activated ScOMPDC-catalyzed decarboxylation of EO or direct decarboxylation of OMP.
No significant electronic effects are expected by groups distant from the reacting pyrimidine ring; and, only small steric effects are expected by these groups for ligand binding to the open enzyme (EO). The results from Chart 2 are consistent with a reaction that proceeds through an extraordinarily crowded and reactive preorganized closed protein cage EC, where small disruptions of the cage structure result in large increases in the activation barrier for ScOMPDC-catalyzed decarboxylation.30,31 The 3.0 kcal/mol larger barrier for unactivated ScOMPDC-catalyzed decarboxylation of orotidine compared to EO is a surprising −CH2OH substituent effect, because the larger −CH2OPO32– group results in an 11.9 reduction in the reaction barrier. Apparently, the binding energy for OMP, orotidine, and EO is utilized for the stabilization of a similar caged protein complexes EC, whose stability is perturbed in some way by the −OH group of orotidine.
IV. Reaction Mechanism
The simplest mechanism for OMPDC, direct decarboxylation of OMP to form a vinyl carbanion reaction intermediate, was viewed with skepticism,32,33 in part because of the failure to detect OMPDC-catalyzed exchange of deuterium from solvent D2O with the C-6 H of UMP to form d-UMP (Scheme 7).34 While the cut of Occam’s razor is sometimes ragged, we suspected that the experiments to detect the OMPDC-catalyzed deuterium exchange reaction were inconclusive, due to the use of a low concentration of OMPDC, a too-short reaction time, and a less than optimal pD.34 This skepticism led to experiments which detected (a) ScOMPDC-catalyzed (0.10–0.30 mM enzyme) exchange for deuterium of the C-6 protons of uridine 5′-monophosphate (UMP) using 1H NMR spectroscopy (Scheme 7);35,36 (b) the faster ScOMPDC deuterium exchange reaction of 5-fluorouridine 5′-monophosphate (F-UMP); and (c) a sharp increase in the reaction velocity as the pD was increased from 7.0 to 9.4.
Scheme 7. Decarboxylation and Deuterium Exchange Reactions Catalyzed by ScOMPDC.

The following observations provide strong evidence that the OMPDC-catalyzed decarboxylation and deuterium exchange reactions proceed through a common vinyl carbanion intermediate (Scheme 7).
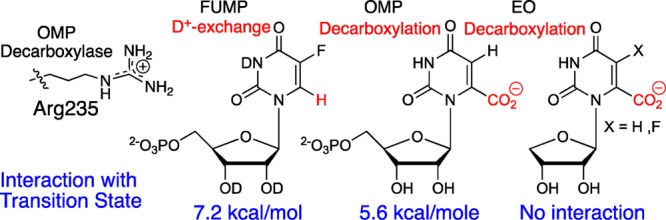
(1) The electron-withdrawing C-5 F pyrimidine substituent shows substantial 4.8 and 3.2 kcal/mol stabilizing interactions, respectively, with the transition states for the wild type ScOMPDC-catalyzed deuterium exchange reaction of FUMP and the Q215A/R235A mutant ScOMPDC-catalyzed decarboxylation reaction of OMP,35,37 where chemistry is strongly rate-determining for the decarboxylation reaction. This is consistent with a large buildup of negative charge at C-6 for the two transition states.
(2) The transition states for ScOMPDC-catalyzed decarboxylation reactions of EO and FEO (Scheme 3) and for the ScOMPDC-catalyzed deuterium exchange reaction of FEU (Scheme 8) are stabilized by 7.8, 5.0, and 5.8 kcal/mol, respectively, by the binding of 1.0 M phosphite dianion.13,28,38 The similar dianion activation of ScOMPDC-catalyzed decarboxylation and deuterium exchange reactions of FEO and FEU, respectively, provides strong evidence that activation is due to the stabilization of a common vinyl carbanion reaction intermediate.
Scheme 8. Phosphite Dianion Activation of the ScOMPDC-Catalyzed Deuterium Exchange Reaction of h-FEU.

(3) The guanidine side chain of Arg-235 forms an ion pair with the C-5′ phosphodianion of substrates bound to ScOMPDC (Figure 1, top panel). The R235A mutation of ScOMPDC results in a 5.6 kcal/mol destabilization of the transition states for enzyme-catalyzed decarboxylation of OMP, and a 7.2 kcal/mol destabilization of the transition state for enzyme-catalyzed exchange of the C-6 proton of FUMP (Scheme 9).39 Guanidine cation, an analog of the arginine side chain, likewise shows a high efficiency for rescue of the lost activity for ScOMPDC-catalyzed decarboxylation and deuterium exchange reactions that result from an R235A mutation.24,39
Scheme 9. Stabilizing Interaction between the R235 Side Chain and Transition States for ScOMPDC-Catalyzed Reactions.
Stepwise or Concerted Decarboxylation?
Loss of CO2 and the addition of a proton to carbon-6 of OMP may proceed by a stepwise (Scheme 7) or a concerted reaction mechanism, where there is electrophilic push to the loss of CO2.33 The 50:50 yield of h-UMP and d-UMP products determined for ScOMPDC-catalyzed decarboxylation of OMP in 50:50 (v/v) H2O/D2O (eq 2 for Scheme 10) gives a PIE = 1.0 for the product deuterium isotope effect, where the PIE is equal to the 1°DKIE on the barrier to formation of the decarboxylation transition state from solvent, OMPDC and OMP.40 This result provides strong evidence for stepwise decarboxylation, because the increase in bonding to the transferred hydron that occurs on proceeding to the transition state for a concerted reaction mechanism requires the observation of a 1°DKIE of >1.41,42
| 2 |
Scheme 10. Decarboxylation of OMP in 50:50 (v/v) H2O/D2O To Form h-UMP and d-UMP.

We proposed that the statistical yield of h-UMP and d-UMP is enforced because the vinyl carbanion intermediate cannot select between protonation by −H and −D from the −CH2–NL3+ group of Lys-93 [the catalytic acid], due to restriction of C–N bond rotation by hydrogen bonds to Asp-91 and Asp-96 carboxylate groups, so that krot ≪ kp (Scheme 11).20 A rate constant of krot ≈ 104 s–1 was estimated from the approximate value of krot = 1011 s–1 for unhindered bond rotation,43 and barriers of ca. 5 kcal/mol to cleavage of each of the hydrogen bonds.41 Values of PIE = 1.0, also consistent with krot ≪ kp (Scheme 11), were determined for ScOMPDC-catalyzed decarboxylation of OMP and FOMP catalyzed by R235A, Y217A, Q215A, S154A, and S154A/Q215A mutant enzymes.41
Scheme 11. Mechanism for ScOMPDC-Catalyzed Decarboxylation, Which Shows Partitioning of the Vinyl Carbanion Intermediate between Direct Protonation (k–p), and C–N Bond Rotation of the −CH2–NL3+ Group (krot) Followed by Transfer of a Second Hydron (k′–p).

(Keq)enz for Deprotonation of Enzyme-Bound UMP and FUMP
The equilibrium constants (Keq)enz = (k–p/kp) for formation of vinyl carbanions generated by deprotonation of UMP or FUMP at the active site of ScOMPDC were estimated from the values of (kex)max for ScOMPDC-catalyzed deuterium exchange (eq 3, krot ≪ kp, Scheme 12), and using the above value of krot ≈ 104 s–1 (eq 4). The values of (kex)max = 1.2 × 10–5 and 0.041 s–1 that were determined for deuterium exchange into enzyme-bound UMP and FUMP, respectively, were substituted into eq 4 to give equilibrium constants for proton transfer of (Keq)enz = 1.2 × 10–9 and 4.1 × 10–6, respectively. By comparison, the equilibrium constants for proton transfer from C-6 of UMP and F-UMP to a primary amine base of pKRNH3 = 7 in water were estimated to be (Keq)aq = 2 × 10–22 and 8 × 10–19, respectively, for UMP and FUMP. We concluded that the binding of UMP or FUMP to ScOMPDC results in a ca. 17 kcal/mol stabilization of the enzyme bound vinyl carbanions by interaction with the protein catalyst, which corresponds to a 12 unit decrease in the pKa for the enzyme-bound carbon acid. This is a substantial fraction of the total 31 kcal/mol stabilization of the transition state for ScOMPDC-catalyzed decarboxylation, but falls short of accounting for the whole rate acceleration.35
| 3 |
| 4 |
Scheme 12. Reversible ScOMPDC-Catalyzed Deprotonation of UMP or FUMP Followed C–N Bond Rotation of the −CH2–NL3+ Group That Leads to Exchange of the C-6 Hydrogen for Deuterium.

V. Utilization of Intrinsic Dianion Binding Energy
We examined the activation of ScOMPDC from protein-dianion interactions by preparing all combinations of single, double, and triple mutations of the protein side chains that interact with the phosphodianion [Figure 2; Arg-235 (R235A), Gln-215 (Q215A), and Tyr-217 (Y217F)], and characterizing their effect on enzyme activity.21 The results of these studies show the following.
| 5 |
Figure 2.
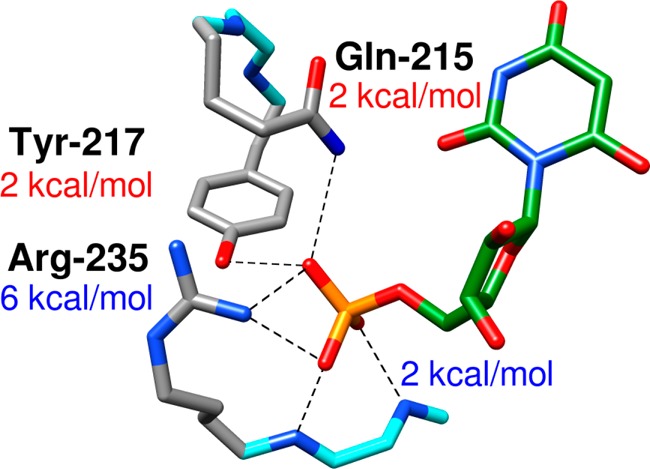
Contribution of ScOMPDC gripper side chains to the intrinsic phosphodianion binding energy (eq 5) utilized for stabilization of the transition state for OMPDC-catalyzed decarboxylation.12
(1) The effect of single Q215A, Y217F, and R235A mutations on the intrinsic dianion binding energy (ΔG‡)Pi was calculated from their effect on the ratio of the second-order rate constants for ScOMPDC-catalyzed reactions of whole and truncated substrates (eq 5). Figure 2 shows that these interactions account for (6 + 2 + 2) = 10 of the total 12 kcal/mol dianion binding energy. The remaining 2 kcal/mol of the dianion binding was attributed to hydrogen bonds to the backbone amides of Gly-234 and Arg-235.
(2) The mutant cube (Figure 3) summarizes the effects of mutations of Q215, Y217, and R235 on (ΔG‡)OMP for ScOMPDC-catalyzed decarboxylation of OMP. This cube shows that the effect of a point mutation of residue X on (ΔΔGX)OMP is approximately the same when the mutation is carried out at wild type ScOMPDC, singly mutated or doubly mutated forms of ScOMPDC.21 We concluded that mutations of these phosphodianion gripper side chains do not severely affect the functioning of the remaining side chains, and that there are only small interactions between the individual side chains.
Figure 3.
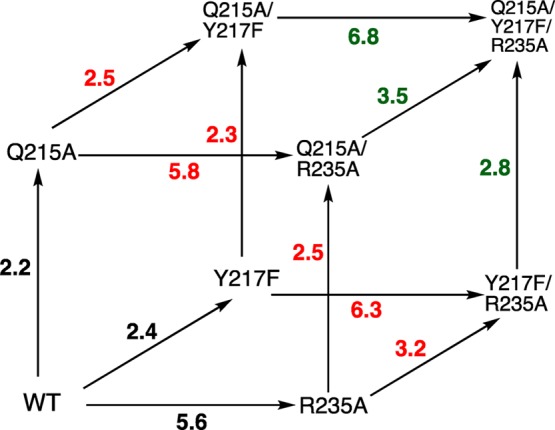
Triple mutant cube that summarizes the effects (ΔΔG‡)OMP, in kcal/mol, of single amino acid mutations on (ΔG‡)OMP for the decarboxylation of OMP catalyzed by wild type ScOMPDC (black values), by single mutants of ScOMPDC (red values) and by double mutants of ScOMPDC (green values).
(3) The Q215A, Y217F, and R235A (Scheme 9) mutations result in ≤2.4-fold decreases in kcat/Km for decarboxylation of (EO), while the Q215A/Y217F/R235A triple mutation results in only a 9-fold decrease in this kinetic parameter. Essentially the entire effect of these mutations is expressed as a decrease in the third-order rate constant for activation of the decarboxylation of EO by phosphite dianion, and this falloff in activity was so sharp that no dianion activation was detected for the reactions catalyzed by the Q215A/R235A, Y217F/R235A, and Q215A/Y217F/R235A mutants.27 These results show that the dianion binding interactions with Gln-215, Tyr-217, and Arg-235 serve the exclusive function of activating OMPDC for catalysis at the pyrimidine binding site.
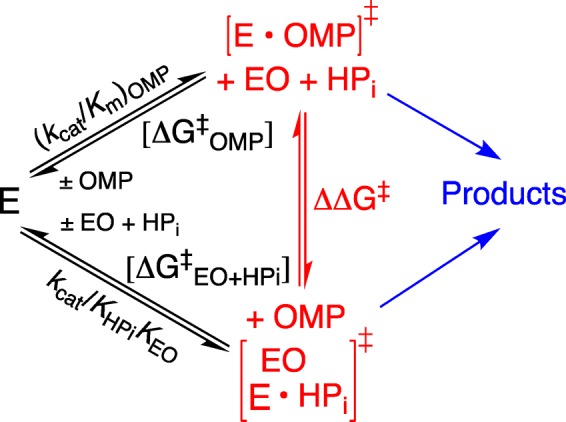
(4) There is a striking linear logarithmic correlation, with slope of 1.0, between the second and third order rate constants (kcat/Km)OMP and kcat/KHPiKEO, respectively, for ScOMPDC-catalyzed decarboxylation of OMP and for dianion activation of OMPDC-catalyzed decarboxylation of EO (Scheme 13).21 This correlation shows that these two OMPDC-catalyzed reactions proceed through similar transition states that show similar interactions with dianion gripper side chains. The results are simply rationalized by a model in which phosphodianion binding interactions serve the sole function of locking ScOMPDC into a reactive closed conformation (Scheme 5), so that these dianions are essentially spectators at the transition states for OMPDC-catalyzed decarboxylation.21
Scheme 13. OMPDC-Catalyzed Reactions of the Whole Substrate OMP and the Pieces EO + HPi.
VI. Other Issues
Structural Heterogeneity of Dianion Gripper Loops
OMPDCs from different organisms show an overall high degree of catalytic structural homology, except for their phosphate gripper loops.44 The loops for OMPDC from yeast (ScOMPDC) and from Escherichia coli (EcOMPDC) both extend 19 residues, from Pro-202 to Val-220 for ScOMPDC20 (Figure 1), and from Pro-189 to Pro-207 for EcOMPDC,45,46 while the corresponding loop of OMPDC from the thermophile Methanothermobacter thermautotrophicus (MtOMPDC) extends only 9 residues, from Pro-180 to Asp-188 (Figure 1).47 Each enzyme shows strong activation for catalysis of decarboxylation of EO by the binding of phosphite dianion. The activation parameters ΔH‡ and ΔS‡ determined for kcat for decarboxylation of OMP are 3.6 kcal/mol and 10 cal/K/mol more positive, respectively, for MtOMPDC than for ScOMPDC.44 This shows that ScOMPDC, which functions at relatively low temperatures, obtains the benefits of enthalpic transition state stabilization available from extensive loop–substrate interactions. By contrast, MtOMPDC functions at higher temperatures and has sacrificed enthalpic transition state stabilization from a large loop, for a smaller loop that shows a reduced entropic requirement for immobilization at the protein.44
Ground-State Effects
There is evidence that protein–ligand interactions introduce strain into the bound substrate, that is relieved at the decarboxylation transition state.47 (1) The binding of OMP to inactive mutants of OMPDC results in a ca. 40° movement of the carboxylate group at C-6 of OMP out of the plane of the pyrimidine ring.48 (2) The results from studies on the effect of a D70N mutation of MtOMPDC on the kinetic parameters for enzyme-catalyzed decarboxylation and deuterium exchange reactions are consistent with modest destabilization of the Michaelis complex by interactions between the substrate carboxylate and the carboxylate from Asp-70 of MtOMPDC.25
Large rate enhancements for decarboxylation are observed upon transfer of substrates, such as 1-methylorotate, from water to organic solvents.49 This has been attributed to the creation of electrostatic stress at the carboxylate in the organic solvent, that is relieved at the decarboxylation transition state. The pyrimidine ring of FUMP bound to ScOMPDC is “sandwiched” between the hydrophobic side chains of Pro-202 and Ile-318′ of the second subunit, while the fluorine of FUMP projects into a hydrophobic pocket lined by the side chains of Leu-150, Leu-153, and Ile-183 at ScOMPDC.35 It is not clear that these hydrophobic side-chains create electrostatic stress at OMP; and, we note that the substrate carboxylate lies close to the cationic side chain of Lys-93 (Scheme 12), so that the binding energy of OMP is ca. 3 kcal/mol larger than for UMP.50 We have proposed that the late transition state for ScOMPDC-catalyzed decarboxylation of OMP is stabilized by the development of stabilizing interactions between the departing CO2 and these hydrophobic protein side chains.35
VII. Summary and Conclusions
The controversy about the origin of the rate acceleration for OMPDC was partly due to the lack of protocol to evaluate the 31 kcal/mol intrinsic substrate binding energy.12 We have shown that interactions between ScOMPDC and the substrate ribosyl ring and phosphodianion are responsible for ca. 22 kcal/mol of this binding energy (Scheme 2). These interactions activate ScOMPDC for catalysis of decarboxylation, where the activation is accomplished through the utilization of substrate binding energy to drive an extensive change in protein conformation, from an open and loose structure that shows relatively weak interactions with the substrate, to a tight protein cage that shows strong interactions with the transition state. The utilization of binding interactions for the creation of tight and catalytically active protein cages from floppy open forms is a widespread phenomenon, that provides a general and underappreciated mechanism to obtain specificity in transition state binding.10,17
The correlation between the magnitude of rate enhancements for enzymes that catalyze deprotonation of carbon, and the complexity of conformational changes of the open enzymes that trap their substrates in a protein cage,2 extends to ScOMPDC-catalyzed decarboxylation and deuterium exchange reactions. These large rate accelerations are enabled by the expansive protein conformational change illustrated in Figure 1, which places the pyrimidine ring of OMP in a structured protein cage, where the barrier to decarboxylation is lower than in water. The mechanism for the reduction in activation barrier has been modeled in computational studies,47,51,52 but is still not entirely understood and will be the focus of our future work on OMPDC. Finally, we emphasize that this Account does not do justice to the complexity of the ligand-driven conformational change, in part because a full description of this change in protein structure is beyond the capabilities of the simple chemists in our group. We are not so simple as to fail to grasp the tremendous potential for protein engineers to contribute to our understanding of the mechanisms for protein catalysis, through modeling related conformational changes in designing catalytically active proteins.
Acknowledgments
This work was supported by grants from the United States National Institutes of Health: GM116921 and GM039754. We thank Prof. John Gerlt for insightful observations during our collaborative work on the mechanism of action of OMPDC.
Biographies
John P. Richard received a B.S. degree in biochemistry and his Ph.D. degree in chemistry (1979) from The Ohio State University. He is currently UB Distinguished Professor of Chemistry at the University at Buffalo, SUNY.
Tina L. Amyes received B.A. and M.A. degrees in natural sciences and her Ph.D. degree in chemistry (1986) from the University of Cambridge (England). She is currently Team Science Grant Coordinator at the Roswell Park Cancer Institute.
Archie C. Reyes received his B.S. degree in chemistry (2006) and M.S. degree in biochemistry (2010) from the University of the Philippines Los Baños, and his Ph.D. degree in Chemistry (2017) from the University at Buffalo SUNY, where he is currently employed as a Research Scientist.
The authors declare no competing financial interest.
References
- Wolfenden R.; Snider M. J. The Depth of Chemical Time and the Power of Enzymes as Catalysts. Acc. Chem. Res. 2001, 34, 938–945. 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. Enzyme architecture: on the importance of being in a protein cage. Curr. Opin. Chem. Biol. 2014, 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow J. R.; Amyes T. L.; Richard J. P. Phosphate Binding Energy and Catalysis by Small and Large Molecules. Acc. Chem. Res. 2008, 41, 539–548. 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Toteva M. M. Formation and Stability of Carbocations and Carbanions in Water and Intrinsic Barriers to Their Reactions. Acc. Chem. Res. 2001, 34, 981–988. 10.1021/ar0000556. [DOI] [PubMed] [Google Scholar]
- Radzicka A.; Wolfenden R. A proficient enzyme. Science 1995, 267, 90–93. 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Houk K. N.; Lee J. K.; Tantillo D. J.; Bahmanyar S.; Hietbrink B. N. Crystal structures of orotidine monophosphate decarboxylase: does the structure reveal the mechanism of nature’s most proficient enzyme?. ChemBioChem 2001, 2, 113–118. . [DOI] [PubMed] [Google Scholar]
- Callahan B. P.; Miller B. G. OMP decarboxylase - An enigma persists. Bioorg. Chem. 2007, 35, 465–469. 10.1016/j.bioorg.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Houk K. N. Why enzymes are proficient catalysts: beyond the Pauling paradigm. Acc. Chem. Res. 2005, 38, 379–385. 10.1021/ar040257s. [DOI] [PubMed] [Google Scholar]
- Pauling L. The nature of forces between large molecules of biological interest. Nature 1948, 161, 707–709. 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Specificity in transition state binding: The Pauling model revisited. Biochemistry 2013, 52, 2021–2035. 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. Enzyme Architecture: Erection of Active Orotidine 5′-Monophosphate Decarboxylase by Substrate-Induced Conformational Changes. J. Am. Chem. Soc. 2017, 139, 16048–16051. 10.1021/jacs.7b08897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 2006, 43, 219–410. 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P.; Tait J. J. Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J. Am. Chem. Soc. 2005, 127, 15708–15709. 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. The Activating Oxydianion Binding Domain for Enzyme-Catalyzed Proton Transfer, Hydride Transfer and Decarboxylation: Specificity and Enzyme Architecture. J. Am. Chem. Soc. 2015, 137, 1372–1382. 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 4046–4050. 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spong K.; Amyes T. L.; Richard J. P. Enzyme Architecture: The Activating Oxydianion Binding Domain for Orotidine 5′-Monophophate Decarboxylase. J. Am. Chem. Soc. 2013, 135, 18343–18346. 10.1021/ja4107513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Malabanan M. M.; Zhai X.; Reyes A. C.; Richard J. P. Enzyme activation through the utilization of intrinsic dianion binding energy. Protein Eng., Des. Sel. 2017, 30, 159–168. 10.1093/protein/gzw064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Amyes T. L.; Richard J. P. Hydron Transfer Catalyzed by Triosephosphate Isomerase. Products of the Direct and Phosphite-Activated Isomerization of [1-13C]-Glycolaldehyde in D2O. Biochemistry 2009, 48, 5769–5778. 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. A role for flexible loops in enzyme catalysis. Curr. Opin. Struct. Biol. 2010, 20, 702–710. 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. G.; Hassell A. M.; Wolfenden R.; Milburn M. V.; Short S. A. Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2011–2016. 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman L. M.; Amyes T. L.; Goryanova B.; Gerlt J. A.; Richard J. P. Enzyme Architecture: Deconstruction of the Enzyme-Activating Phosphodianion Interactions of Orotidine 5′-Monophosphate Decarboxylase. J. Am. Chem. Soc. 2014, 136, 10156–10165. 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland D. E. Jr. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. U. S. A. 1958, 44, 98–104. 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D. The role of induced fit and conformational changes of enzymes in specificity and catalysis. Bioorg. Chem. 1988, 16, 62–96. 10.1016/0045-2068(88)90038-7. [DOI] [Google Scholar]
- Barnett S. A.; Amyes T. L.; McKay Wood B.; Gerlt J. A.; Richard J. P. Activation of R235A Mutant Orotidine 5′-Monophosphate Decarboxylase by the Guanidinium Cation: Effective Molarity of the Cationic Side Chain of Arg-235. Biochemistry 2010, 49, 824–826. 10.1021/bi902174q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. K.; Wood B. M.; Fedorov A. A.; Fedorov E. V.; Imker H. J.; Amyes T. L.; Richard J. P.; Almo S. C.; Gerlt J. A. Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Evidence for Substrate Destabilization. Biochemistry 2009, 48, 5518–5531. 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; Wood B. M.; Gerlt J. A.; Richard J. P. Dissecting the Total Transition State Stabilization Provided by Amino Acid Side Chains at Orotidine 5′-Monophosphate Decarboxylase: A Two-Part Substrate Approach. Biochemistry 2008, 47, 7785–7787. 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Ming S. A.; Goldman L. M.; Wood B. M.; Desai B. J.; Gerlt J. A.; Richard J. P. Orotidine 5′-monophosphate decarboxylase: Transition state stabilization from remote protein-phosphodianion interactions. Biochemistry 2012, 51, 4630–4632. 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Spong K.; Amyes T. L.; Richard J. P. Catalysis by Orotidine 5′-Monophosphate Decarboxylase: Effect of 5-Fluoro and 4′-Substituents on the Decarboxylation of Two-Part Substrates. Biochemistry 2013, 52, 537–546. 10.1021/bi301650d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers A.; Wolfenden R. The effective molarity of the substrate phosphoryl group in the transition state for yeast OMP decarboxylase. Bioorg. Chem. 2005, 33, 45–52. 10.1016/j.bioorg.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Parson W. W. Modeling electrostatic effects in proteins. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 1647–1676. 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Warshel A. Electrostatic Origin of the Catalytic Power of Enzymes and the Role of Preorganized Active Sites. J. Biol. Chem. 1998, 273, 27035–27038. 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- Stanton C. L.; Kuo I. F.; Mundy C. J.; Laino T.; Houk K. N. QM/MM metadynamics study of the direct decarboxylation mechanism for orotidine-5′-monophosphate decarboxylase using two different QM regions: acceleration too small to explain rate of enzyme catalysis. J. Phys. Chem. B 2007, 111, 12573–12581. 10.1021/jp074858n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley T. P.; Ealick S. E. Enzymatic reactions involving novel mechanisms of carbanion stabilization. Curr. Opin. Chem. Biol. 2004, 8, 508–515. 10.1016/j.cbpa.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Smiley J. A.; DelFraino B. J.; Simpson B. A. Hydrogen isotope tracing in the reaction of orotidine-5′-monophosphate decarboxylase. Arch. Biochem. Biophys. 2003, 412, 267–271. 10.1016/S0003-9861(03)00062-6. [DOI] [PubMed] [Google Scholar]
- Tsang W.-Y.; Wood B. M.; Wong F. M.; Wu W.; Gerlt J. A.; Amyes T. L.; Richard J. P. Proton Transfer from C-6 of Uridine 5′-Monophosphate Catalyzed by Orotidine 5′-Monophosphate Decarboxylase: Formation and Stability of a Vinyl Carbanion Intermediate and the Effect of a 5-Fluoro Substituent. J. Am. Chem. Soc. 2012, 134, 14580–14594. 10.1021/ja3058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. Formation and Stability of a Vinyl Carbanion at the Active Site of Orotidine 5′-Monophosphate Decarboxylase: pKa of the C-6 Proton of Enzyme-Bound UMP. J. Am. Chem. Soc. 2008, 130, 1574–1575. 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Ming S.; Amyes T. L.; Gerlt J. A.; Richard J. P. Rate and Equilibrium Constants for an Enzyme Conformational Change during Catalysis by Orotidine 5′-Monophosphate Decarboxylase. Biochemistry 2015, 54, 4555–4564. 10.1021/acs.biochem.5b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Amyes T. L.; Gerlt J. A.; Richard J. P. OMP Decarboxylase: Phosphodianion Binding Energy Is Used To Stabilize a Vinyl Carbanion Intermediate. J. Am. Chem. Soc. 2011, 133, 6545–6548. 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Amyes T. L.; Gerlt J. A.; Richard J. P. Role of a Guanidinium Cation–Phosphodianion Pair in Stabilizing the Vinyl Carbanion Intermediate of Orotidine 5′-Phosphate Decarboxylase-Catalyzed Reactions. Biochemistry 2013, 52, 7500–7511. 10.1021/bi401117y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P.. Primary Deuterium Kinetic Isotope Effects From Product Yields: Rationale, Implementation, and Interpretation. In Methods in Enzymology; Michael E. H., Vernon E. A., Eds.; Academic Press, 2017; Vol. 596; pp 163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. Product Deuterium Isotope Effects for Orotidine 5′-Monophosphate Decarboxylase: Effect of Changing Substrate and Enzyme Structure on the Partitioning of the Vinyl Carbanion Reaction Intermediate. J. Am. Chem. Soc. 2010, 132, 7018–7024. 10.1021/ja102408k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. Product Deuterium Isotope Effect for Orotidine 5′-Monophosphate Decarboxylase: Evidence for the Existence of a Short-Lived Carbanion Intermediate. J. Am. Chem. Soc. 2007, 129, 12946–12947. 10.1021/ja076222f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Tsuji Y. Dynamics for Reaction of an Ion Pair in Aqueous Solution: The Rate Constant for Ion Pair Reorganization. J. Am. Chem. Soc. 2000, 122, 3963–3964. 10.1021/ja994453j. [DOI] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K. K.; Gerlt J. A.; Richard J. P. An Examination of the Relationship between Active Site Loop Size and Thermodynamic Activation Parameters for Orotidine 5′-Monophosphate Decarboxylase from Mesophilic and Thermophilic Organisms. Biochemistry 2009, 48, 8006–8013. 10.1021/bi901064k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris P.; Navarro Poulsen J.-C.; Jensen K. F.; Larsen S. Structural basis for the catalytic mechanism of a proficient enzyme: Orotidine 5′-monophosphate decarboxylase. Biochemistry 2000, 39, 4217–4224. 10.1021/bi992952r. [DOI] [PubMed] [Google Scholar]
- Harris P.; Poulsen J. C.; Jensen K. F.; Larsen S. Substrate binding induces domain movements in orotidine 5′-monophosphate decarboxylase. J. Mol. Biol. 2002, 318, 1019–1029. 10.1016/S0022-2836(02)00200-0. [DOI] [PubMed] [Google Scholar]
- Wu N.; Mo Y.; Gao J.; Pai E. F. Electrostatic stress in catalysis: structure and mechanism of the enzyme orotidine monophosphate decarboxylase. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2017–2022. 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujihashi M.; Ishida T.; Kuroda S.; Kotra L. P.; Pai E. F.; Miki K. Substrate distortion contributes to the catalysis of orotidine 5′-monophosphate decarboxylase. J. Am. Chem. Soc. 2013, 135, 17432–17443. 10.1021/ja408197k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis C. A.; Wolfenden R. Orotic Acid Decarboxylation in Water and Nonpolar Solvents: A Potential Role for Desolvation in the Action of OMP Decarboxylase. Biochemistry 2009, 48, 8738–8745. 10.1021/bi901085m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D. J. T.; Short S. A. Yeast Orotidine-5′-Phosphate Decarboxylase: Steady-State and Pre-Steady-State Analysis of the Kinetic Mechanism of Substrate Decarboxylation. Biochemistry 2000, 39, 11788–11800. 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- Vardi-Kilshtain A.; Doron D.; Major D. T. Quantum and Classical Simulations of Orotidine Monophosphate Decarboxylase: Support for a Direct Decarboxylation Mechanism. Biochemistry 2013, 52, 4382–4390. 10.1021/bi400190v. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Strajbl M.; Villa J.; Florian J. Remarkable Rate Enhancement of Orotidine 5′-Monophosphate Decarboxylase Is Due to Transition-State Stabilization Rather Than to Ground-State Destabilization. Biochemistry 2000, 39, 14728–14738. 10.1021/bi000987h. [DOI] [PubMed] [Google Scholar]