

Abstract
Classical opioid analgesics, including morphine, mediate all of their desired and undesired effects by specific activation of the μ‐opioid receptor (μ receptor). The use of morphine for treating chronic pain, however, is limited by the development of constipation, respiratory depression, tolerance and dependence. Analgesic effects can also be mediated through other members of the opioid receptor family such as the κ‐opioid receptor (κ receptor), δ‐opioid receptor (δ receptor) and the nociceptin/orphanin FQ peptide receptor (NOP receptor). Currently, a new generation of opioid analgesics is being developed that can simultaneously bind with high affinity to multiple opioid receptors. With this new action profile, it is hoped that additional analgesic effects and fewer side effects can be achieved. Recent research is mainly focused on the development of bifunctional μ/NOP receptor agonists, which has already led to novel lead structures such as the spiroindole‐based cebranopadol and a compound class with a piperidin‐4‐yl‐1,3‐dihydroindol‐2‐one backbone (SR16835/AT‐202 and SR14150/AT‐200). In addition, the ornivol BU08028 is an analogue of the clinically well‐established buprenorphine. Moreover, the morphinan‐based nalfurafine exerts its effect with a dominant κ receptor‐component and is therefore utilized in the treatment of pruritus. The very potent dihydroetorphine is a true multi‐receptor opioid ligand in that it binds to μ, κ and δ receptors. The main focus of this review is to assess the paradigm of opioid ligands targeting multiple receptors with a single chemical entity. We reflect on this rationale by discussing the biological actions of particular multi‐opioid receptor ligands, but not on their medicinal chemistry and design.
Linked Articles
This article is part of a themed section on Emerging Areas of Opioid Pharmacology. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.14/issuetoc
Abbreviations
- DRG
dorsal root ganglion
- N/OFQ
nociceptin/orphanin FQ peptide
- NOP receptor
nociceptin/orphanin FQ peptide receptor
- PAG
periaqueductal grey
- RVM
rostral ventral medulla
Introduction
Opioids binding to the μ‐opioid receptor (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319&familyId=50&familyType=GPCR) remain the mainstay in the management of moderate to severe pain conditions (Smith and Peppin, 2014). However, their long‐term use is limited by various side effects, such as constipation, potentially life‐threatening respiratory depression, nausea, development of tolerance and physical dependence (Bailey and Connor, 2005). This situation has led to different approaches to develop drugs with a more favourable side effect profile. However, as yet, there is no single compound known in the vast repertoire of orthosteric μ receptor‐selective ligands that has analgesic action that is not also associated with side effects. In μ receptor knock out mice, both the antinociception and side effects of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627 are clearly mediated by μ receptor (Kieffer and Gaveriaux‐Ruff, 2002).
The design and clinical development of new analgesics has to consider several intracacies: (i) the multiplicity of neurochemical transmitters and receptors involved in pain pathways; (ii) the variety of physiological functions mediated by a single transmitter or receptor target; (iii) the different intrinsic activity (e.g. full vs. partial agonist vs. antagonist) or mechanism (orthosteric vs. allosteric) of a drug compound; (iv) different signalling mechanisms mediated by a single receptor species (e.g. ‘biased’ intracellular coupling); (v) receptor oligo‐ and/or heteromerization; (vi) anatomy, functional hierarchy and redundancy of pain pathways (e.g. central vs. spinal vs. peripheral; nociceptive vs. antinociceptive); (vii) species differences in translational animal models and clinical efficacy in humans; (viii) limited solubility due to high lipophilicity; and (ix) low bioavailability. Obviously, these complexities will multiply when more than a single drug target is therapeutically addressed at the same time. Despite these challenges, many recent efforts are trying to harness the powerful analgesic effects of μ receptor agonists with new strategies to circumvent typical μ receptor agonist‐associated side effects and thus demonstrate the insistent need for improved μ receptor‐directed analgesics. As one possible strategy to overcome this dilemma, recent studies have proposed the development of so‐called ‘biased’ μ receptor agonists, which are able to induce receptor conformations that preferentially trigger Gi‐dependent signalling without activating β‐arrestin pathways (Manglik et al., 2016). It is hypothesized that compounds promoting Gi‐signalling will produce analgesia while avoiding β‐arrestin‐dependent effects on respiration or reinforcement. Despite promising preclinical and early clinical data (DeWire et al., 2013; Viscusi et al., 2016), it is currently too early to validate this concept as, so far, only two biased μ receptor agonists have been investigated more closely.
While the μ receptor is the predominant target of morphine‐like compounds, analgesia can be mediated by activation of any of the four members of the opioid receptor family, as has been shown in human and non‐human settings for the κ‐opioid receptor (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=318&familyId=50&familyType=GPCR) (Pande et al., 1996; Schepers et al., 2008), δ‐opioid receptor (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=317) (Petrillo et al., 2003) and nociceptin/orphanin FQ peptide receptor (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=320&familyId=50&familyType=GPCR) (Xu et al., 1996; Yamamoto et al., 1997). However, thus far, the clinical development of selective δ or κ receptor compounds has failed due to limited analgesic potency and/or dysphoric effects in rodents (Eguchi, 2004; Bodnar, 2010) respectively. Although selective κ and δ receptor agonists lack the typical μ receptor‐mediated side effects, such as respiratory depression and constipation, they possess their own distinct profile of side effects, including dysphoria, sedation, diuresis and constipation for κ receptor agonists (Land et al., 2008; White et al., 2015), and convulsive effects have been observed with many δ receptor agonists (Jutkiewicz et al., 2006). Activation of the δ receptor has been proved to be effective in chronic pain conditions, a state that can only be insufficiently controlled by μ receptor agonists (Nadal et al., 2006; Gaveriaux‐Ruff et al., 2008). As an additional benefit, and in contrast to μ receptor agonists, δ receptor activation causes no physical dependence (Brandt et al., 2001). Furthermore, activation of δ receptors has low abuse liability, as δ receptor agonists are not self‐administered (Negus et al., 2009). Other important features of δ receptor agonists are their anxiolytic and anti‐depressant effects that are desirable in chronic pain conditions (Perrine et al., 2006), as indicated in Table 1.
Table 1.
Clinically reported analgesic effects and side effects mediated by each member of the opioid receptor family
| μ receptor | κ receptor | δ receptor | NOP receptor | ||
|---|---|---|---|---|---|
| Analgesic effects | Acute pain | +++ | + | + | − |
| Chronic pain | ++ | + | +++ | ++ | |
| Neuropathic pain | + | + | + | + | |
| Inflammatory pain | + | + | +++ | ++ | |
| Migraine | + | n.a. | + | n.a. | |
| Visceral pain | n.a. | + | n.a. | + | |
| Anti‐allodynic | + | + | −/+ | + | |
| Anti‐depressant | + | + | + | − | |
| Side effects | Rewarding | + | − | + | − |
| Respiratory depression, cough reflex | +++ | − | + | − | |
| Constipation, gastric motility | +++ | + | + | − | |
| Euphoria | + | n.a. | n.a. | n.a. | |
| Dysphoria | n.a. | + | n.a. | n.a. | |
| Epilepsy | + | − | + | − | |
| Nausea, vomiting tolerance | + | − | − | − | |
| Dependence | +++ | − | + | − | |
| Itch | +++ | − | n.a. | − | |
| Renal function | + | + | + | + |
The severity of the indicated effect is expressed as high (+++), moderate (++), low (+) or absent (−). No data available (n.a.).
Analgesic effects mediated by the NOP receptor are the most complex of any other member of the opioid receptor family. Depending on the exposure to exogenous opioid agonists, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1681 either blocks the analgesic effect of opioids, or mediates an analgesic effect by reducing hyperalgesia during opioid withdrawal conditions (Pan et al., 2000). Overall, N/OFQ appears to act as a ‘functional opioid antagonist’ in the CNS. Additionally, the N/OFQ system has different effects on spinal compared to supraspinal nociceptive circuits (Heinricher, 2005), and these pain circuits may differ between rodents and non‐human primates (Ko et al., 2009b). Due to the similarities and interactions with the classical opioid peptides and their receptors, the N/OFQ‐NOP receptor system was studied early on as a potential drug target on its own and in combination with opioid analgesics. For instance, the joint application of the NOP receptor‐selective agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=1684 and morphine in subthreshold analgesic doses reduced pain sensitivity in the hot plate assay in an additive manner (Reiss et al., 2008), although it showed hyperalgesic effects in the tail flick assay when administered alone. Interestingly, Ro64‐6198 inhibits pain in rhesus monkeys without mediating opioid‐like side effects (Ko and Naughton, 2009a) and displays clinically desirable antiallodynic actions in rats (Obara et al., 2005). However, Ro64‐6198 never reached clinical trials, most likely due to low oral bioavailability and substance‐specific side effects such as impairment of motor activity, learning and memory (Shoblock, 2007). Other selective NOP receptor agonists like MCOPPB and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8868 probably do not pass through the blood brain barrier, although some studies report anxiolytic effects of MCOPPB, which is indicative of central activity (Hirao et al., 2008). The role of N/OFQ in the development of opioid tolerance is also controversial. While one report showed attenuation of morphine tolerance after co‐administration of the natural peptide ligand N/OFQ (Lutfy et al., 2001), numerous studies suggested N/OFQ antagonists are useful as a co‐treatment to prevent the development of morphine tolerance (Ueda et al., 2000; Chung et al., 2006). Consistent with its function as an ‘anti‐opioid’ agent, endogenous N/OFQ may also be involved in precipitating side effects during opioid therapy (Zaveri, 2011). Due to the intimate functional interaction between the classical opioids and N/OFQ, recent attention has shifted to compounds that target both systems simultaneously.
This brief overview demonstrates that simultaneous targeting of multiple opioid receptors with compounds producing a mixture of agonistic and/or antagonistic effects may be a promising strategy to overcome the existing intrinsic limitations of the current opioid drugs.
Indeed, in the clinic, polypharmaceutical interventions are being employed to control opioid‐mediated side effects. One strategy used to alleviate side effects and to lower the abuse potential of opioids is to combine the analgesically active opioid with an antagonist that reduces constipation. This concept, known as PAMORA (peripherally acting μ receptor antagonist) is already clinically established in a fixed‐dose combination scheme of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1670 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1638 (Suboxone™) or http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7093 and naloxone (Targin™). However, in this case the management of one side effect requires intervention with a second drug, which alleviates only one symptomatic condition, that is, constipation, while other centrally mediated side effects persist. Thus, the aim for development of multi‐opioid receptor ligands should be a single chemical entity with improved analgesic activity and reduced side effects.
At first glance, the concept of addressing multiple opioid receptors seems rather old fashioned. Since the 1950s, a plethora of opioid compounds have been developed even though their pharmacology had not been elucidated due to a lack of molecular tools. Before the four members of the opioid receptor family were cloned in the early 1990s, the rationale in drug design was the creation of ever‐more selective ligands, commonly based on natural alkaloids such as morphine, and optimization of their pharmacokinetic profile. When pure opioid receptor preparations in cells transfected with cloned receptor cDNAs became available, reexamination of opioid agonists revealed mixed pharmacological profiles of several clinically successful drugs, such as buprenorphine, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8286 and naltrexamine (Sadee et al., 1982; Frink et al., 1996; Cami‐Kobeci et al., 2009) Together with earlier in vivo work, it became clear that one alternative strategy for developing more effective http://topics.sciencedirect.com/topics/page/Analgesic could be to use either mixtures of selective opioid receptor http://topics.sciencedirect.com/topics/page/Agonist or single compounds with mixed pharmacological profiles, in order to produce synergistic analgesic effects, combined with hopefully fewer side effects. Recent progress in medicinal chemistry has suggested the possible design, synthesis and testing of single molecule entities with mixed opioid receptor agonist or combined agonist/antagonist activities.
What options are there to address opioid receptors?
All marketed opioids are orthosteric agonists with different intrinsic activity (full, partial or inverse agonists, or neutral antagonists) that target either one or multiple opioid receptors. Clinically used opioids such as morphine require the expression of μ receptors to exert an effect, as shown in μ receptor knock out mice (Matthes et al., 1996). In contrast to orthosteric ligands, the design of allosteric modulators could help to control spatial and temporal receptor signalling in the presence or absence of the endogenous peptide opioid ligands. While less conserved than orthosteric sites, the allosteric agonist‐binding sites, when targeted, could produce higher ligand selectivity and therefore fewer off‐target effects. A proof‐of‐concept approach generated positive allosteric modulators for the μ receptor, namely http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9157/http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9156 (Burford et al., 2013), and BMS‐986187 for the δ receptor (Burford et al., 2015), as shown in Figure 1. Another approach used to overcome the limited efficacy of classical orthosteric ligands is the development of compounds with biased signalling towards G‐proteins or β‐arrestins. The μ receptor agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7334 was the first example of a G‐protein‐biased substance to mediate analgesia with fewer side effects compared to morphine (DeWire et al., 2013). The concept of ‘biased’ μ receptor agonists was recently extended by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9286, which shows an even higher selectivity for Gi‐activation and a favourable dissociation of analgesia from side effects in rodent models (Manglik et al., 2016). The complex pharmacology of opioids, contrasted by the very limited number of opioid receptors, might be explained by the existence of receptor subtypes and the formation of heterodimers. Targeting heterodimerized opioid receptors has led to the recent development of CYM51010, a μ‐δ receptor‐heteromer‐biased agonist (Gomes et al., 2013). MDAN‐21 was designed as a spacer‐linked bivalent (heterobivalent) compound with an http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7094‐derived μ receptor‐binding moiety and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1641‐related δ receptor‐binding component, which suppressed morphine‐dependent withdrawal signs in rhesus monkeys (Aceto et al., 2012). These findings suggest the simultaneous occupancy of δ receptors with an antagonist and μ receptors with an agonist as a promising strategy to dissociate antinociception from side effects. While there is no bitopic (dualsteric) compound so far for opioid receptors, this has been made for other class A GPCRs, for example, the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=13&familyId=2&familyType=GPCR agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=290 (Valant et al., 2008) or the dopamine http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=215 agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7694 (Mistry et al., 2015).
Figure 1.
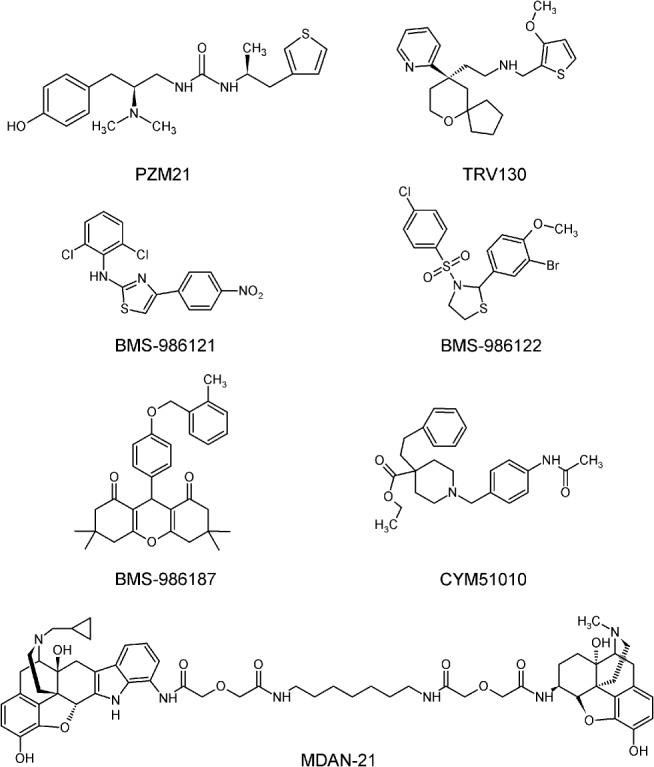
Structures of proof‐of‐concept opioid compounds.
Rationale for the development of multi‐targeting opioid ligands
Structure and design of multi‐targeting opioid ligands
In principle, the simultaneous activation of two receptors can be achieved with two different ligands, each binding the receptor with its cognate pharmacophore. In contrast to this approach, which requires two drugs, a more refined way is the design of a single compound containing the pharmacophores for activation of both receptors, thus designing ligands with a more predictable pharmacokinetic and pharmacodynamic profile (Morphy and Rankovic, 2009). The goal of targeting multiple opioid receptors with a single chemical entity can be realized by designing either bivalent or bifunctional (monovalent) compounds. While for bivalent ligands, two distinct pharmacophores have to be fused with a linker (Portoghese, 1989), bifunctional ligands are non‐selective compounds that combine two or more receptor‐specific binding properties (Li et al., 2007). The recent development of multi‐opioid receptor ligands is mainly centred on the development of bifunctional and bivalent ligands simultaneously addressing μ/NOP receptor or μ/δ receptor as shown in Figure 2.
Figure 2.
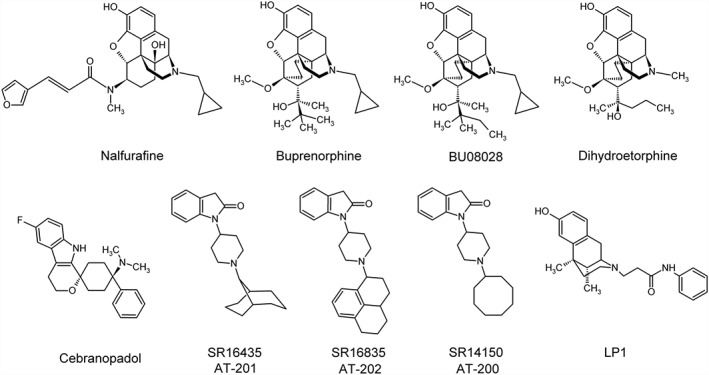
Structures of multi‐opioid receptor ligands.
μ/κ receptor mixed ligands
κ receptor agonists alone produce potent analgesia, especially in visceral pain models, but are often accompanied by many unwanted effects such as dysphoria and diuresis. However, they were found to reduce the rewarding effects of a variety of addictive drugs (Shippenberg et al., 2007) and are, therefore, potential analgesics to overcome opioid dependence. The recent development of selective κ receptor agonists has focused on selective peripherally acting analgesics due to their dysphoric central effects. For example, the co‐application of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1626 with the κ receptor agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1653 potentiates antinociception in rats (Briggs et al., 1998). In addition to the μ receptor, the κ receptor is also expressed in the spinal cord, dorsal root ganglia (DRGs), peripheral sensory neurons and supraspinal regions like the rostral ventral medulla (RVM) or the periaqueductal grey (PAG). The evidence for whether μ and κ receptors form heterodimers is ambivalent (Jordan and Devi, 1999; Wang et al., 2005). When applied individually, κ receptor agonists mediate antinociceptive effects. However, μ receptor activation can be partially attenuated by administration of the κ receptor agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1655 (Ackley et al., 2001), suggesting the existence of heterodimers in vivo, but not excluding a functionally antagonistic circuitry. The development of monofunctional κ receptor agonists is focused on the concept of targeting κ receptors in the periphery, with an emphasis on managing visceral pain (Black and Trevethick, 1998). The κ receptor agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1651 is used clinically in Japan as an antipruritic for the treatment of intractable pruritus in haemodialysis patients (Kumagai et al., 2010; Nakao et al., 2016) and also exhibits high‐antinociceptive activities (Endoh et al., 1999, 2000). Receptor binding and functional studies of recombinant human and rat opioid receptors have shown nalfurafine to be a potent and selective agonist at the κ receptor, but also to display weak and partial agonist activity at μ and δ receptors (Seki et al., 1999). μ receptor agonists carry a high risk of abuse potential, whereas selective κ receptor agonists generally lack a reinforcing effect (Negus et al., 2008). Accordingly, nalfurafine produced no reinforcing effects in rhesus monkeys in an intravenous self‐administration paradigm. These data indicated that nalfurafine can be used for long‐term treatment of intractable pruritus with minimal concern regarding the risk of dependence (Nakao et al., 2016). All other mixed μ/κ receptor agonists in clinical use, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7591, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1663 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1606, alleviate morphine‐induced pruritus; however, only butorphanol is used to treat chronic pruritus (Du et al., 2013). Dihydroetorphine is one of the most potent analgesic opioids known, with up to 12‐fold higher potency than morphine. Several in vitro and in vivo studies have shown that dihydroetorphine is a μ receptor agonist that also binds and activates κ and δ receptors (Katsumata et al., 1995). The onset of its analgesic action is rapid, its duration of action after systemic administration to rodents is rather short and it is largely ineffective after oral administration (Ohmori et al., 2000; Ohmori and Morimoto, 2002). Continuous exposure and repeated administration of dihydroetorphine results in analgesic tolerance, physical dependence, and a rewarding effect in normal animals, but not in rats with formalin‐induced inflammation (Ohmori et al., 2000). Clinical tests in China using sublingual application of dihydroetorphine showed a potent analgesic effect with only mild side effects, which included dizziness, somnolence, nausea, vomiting, constipation and shortness of breath (Ohmori et al., 2000; Ohmori and Morimoto, 2002). Transdermal delivery of dihydroetorphine via a patch produces continuous analgesic effects with minimal physical dependence and rewarding effects in rats suffering from chronic pain (Ohmori et al., 2000). While κ receptor agonists frequently precipitate dysphoric mood changes in humans and depression‐like symptoms in experimental animals (Ranganathan et al., 2012), several κ receptor antagonists were found to exert anti‐depressant effects (Lutz and Kieffer, 2013) (Table 2). A combined pharmacological profile of a μ receptor agonist and κ receptor antagonist could thus not only produce analgesia but also treat depressive symptoms, a frequent comorbidity in chronic pain patients (Taylor and Manzella, 2016). The compound best studied in this context is buprenorphine, which is a partial μ receptor agonist with a weak κ receptor antagonistic profile. Buprenorphine was found to produce antidepressant and anxiolytic effects in mice, which were mediated through the κ receptor (Falcon et al., 2015), although clinical observations in chronic pain patients treated with buprenorphine failed to detect lasting mood effects (Stein et al., 2015). Overall, compounds simultaneously targeting μ and κ receptors, either as agonist/agonist or agonist/antagonist, have shown some clinical efficacy, although the main limitations of chronic μ receptor activation, such as abuse liability, constipation and respiratory depression, are still apparent.
Table 2.
Activity profile of selected multi‐opioid receptor ligands
| Substance | μ receptor | κ receptor | δ receptor | NOP receptor | Profile/activity | Source |
|---|---|---|---|---|---|---|
| Dihydroetorphine | +++ | +++ | +++ | n.a./− | analgesic for severe and cancer pain | Ohmori et al. 2000, Ohmori and Morimoto, 2002, Ranganathan et al., 2012 |
| Nalfurafine | +++ | +++ | + | + | antipruritic and antinociceptive activity; clinical trial for pruritus | Kumagai et al., 2010, Nakao et al., 2016, Endoh et al., 1999 |
| Buprenorphine | ++ | n.a/− | n.a/− | + | opioid substitution therapy; postoperative pain control; chronic pain; cancer pain; neuropathic pain in combination with full μ receptor agonists | Lutfy and Cowan, 2004, Huang et al., 2001, Khroyan et al., 2009a, 2009b, van Niel et al., 2016, Bonhomme et al., 2012 |
| BU08028 | +++ | − | + | ++ | antinociceptive, antihypersensitive and antiallodynic activity; absence of respiratory and cardiovascular activities; absence of physical dependence and pruritus | Ding et al., 2016, Khroyan et al., 2011 |
| SR14150 (AT‐200) | ++ | + | n.a. | +++ | antinociceptive | Spagnolo et al. 2008, Khroyan et al., 2007, Toll et al., 2009, Sukhtankar et al., 2013 |
| SR16435 (AT‐201) | +++ | + | − | ++ | antinociceptive and antiallodynic, induces CPP, slower development of tolerance compared to μ receptor agonists under neuropathic pain | |
| SR16835 (AT‐202) | ++ | + | n.a. | +++ | not antinociceptive, induces no CPP, attenuates morphine‐mediated CPP | |
| Cebranopadol | +++ | ++ | + | +++ | clinical trial for severe chronic and neuropathic pain | Linz et al. 2014 |
The grade of intrinsic activity is expressed as high (+++), moderate (++), low (+) or absense (−) of intrinsic activity determined by GTPγS assay. (n.a.) Data are either not available or were not determined.
μ/δ receptor mixed ligands
The main rationale behind simultaneously targeting μ/δ receptors is to develop drugs with attenuated tolerance profiles suitable for the management of persistent pain conditions (Abdelhamid et al., 1991). The concomitant application of the δ receptor agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1611 with a μ receptor agonist such as fentanyl potentiated the antinociceptive actions of the δ receptor agonist (Stevenson et al., 2003). The mixed μ/δ receptor antagonist LP1 was reported as a valuable substance for the treatment of persistent pain conditions (Parenti et al., 2013). Compared to the pain relief triggered upon μ receptor activation in acute pain states, activation of the δ receptor alone is relatively ineffective (Gallantine and Meert, 2005). However, δ receptor activation can be therapeutically beneficial in the treatment of chronic pain states, especially inflammatory pain (Gaveriaux‐Ruff et al., 2008), and co‐administration of a δ receptor agonist with a μ receptor agonist increases the overall analgesic effects (Kovelowski et al., 1999). There is less abuse potential for δ receptor agonists compared to μ receptor agonists (Carmo et al., 2009), fewer gastro intestinal‐related problems (Tavani et al., 1990) and less respiratory depression (Szeto et al., 1999). Another reason for simultaneously targeting μ and δ receptors is their co‐localization in nociceptive sensory neurons (Wang et al., 2010) and receptor crosstalk (Zhang and Pan, 2010). The existence of μ/δ receptor heterodimers with a unique binding pocket, signalling and trafficking (Gomes et al., 2004) has led to the development of bivalent ligands. In contrast to other opioid receptors, the δ receptor was mainly found to be located intracellularly in basal conditions. Different stimuli, such as stress (Commons, 2003), chronic morphine exposure (Hack et al., 2005), neuropathic and inflammatory pain (Cahill et al., 2003; Kabli and Cahill, 2007) were demonstrated to enhance δ receptor‐mediated analgesia as a function of membrane trafficking. In a proof‐of‐concept study for a mixed μ/δ receptor agonist, a bifunctional enkephalin‐like peptide exhibited antiallodynic and antihyperalgesic effects in rats (Lee et al., 2011). Conjointly targeting μ and δ receptors is a therapeutic option to potentially lower opioid tolerance in neuropathic pain, compared to classical opioids (Balboni et al., 2010). As a general observation from these studies, it appears that δ receptor agonists require μ receptors for their activity. A special incentive for the development of dual μ/δ receptor agonists is the anti‐depressant like activity induced by δ receptor agonists. Based on this concept, the fentanyl‐like dual μ/δ receptor agonist RV‐Jim‐C3 was designed, which acts as an effective antinociceptive compound in neuropathic pain models with limited motor‐activating effects (Podolsky et al., 2013). The benzomorphan‐based LP1 was found to have potent supraspinal antinociceptive activity in the tail flick test with longer lasting antinociception compared to morphine (Parenti et al., 2012). However, further studies on the possible mood‐elevating properties of these compounds have not yet been published. The evaluation of a novel class of 6β‐acylaminomorphinans revealed that 6β‐cinnamoylmorphinamine is a potent analgesic and that its effects can be attenuated by selective antagonists of μ and δ receptors. Interestingly, no alterations in respiratory rate were found in mice (Varadi et al., 2013).
μ/NOP receptor mixed ligands
The μ and NOP receptor share common signalling pathways and are both functionally expressed in pain pathways such as the spinal cord, PAG and RVM, although mostly in distinctly separate neurons. There is evidence of only limited co‐localization of μ and NOP receptors in DRGs as recently shown in NOP receptor‐eGFP knock‐in mice (Ozawa et al., 2015). Also the heterodimerization of μ and NOP receptors, as reported in heterologous expression systems, is still controversial (Pan et al., 2002). Probably most important for drug development was the discovery that NOP receptor agonists show better potential for the treatment of neuropathic pain than classical opioids (Schroder et al., 2014). While the results from rodent studies produced conflicting views on the role of N/OFQ and the NOP receptor in analgesia, in rhesus monkey studies the NOP receptor has been shown to be a valid target for the treatment of pain (Ko et al., 2009b). These effects appear to be mediated at the spinal level and peripheral nociceptive sensory neurons. In addition, early studies indicated the potential of mixed μ/NOP receptor agonists to alleviate some of the side effects of pure μ receptor agonists, including the development of tolerance and dependence. Counterintuitively, a potential mechanism for this desirable effect might be chronic desensitization of NOP receptor signalling in reward and tolerance circuits (Lutfy et al., 2001), since similar effects were also reported for a co‐treatment regimen with μ receptor agonists and NOP receptor antagonists (Ueda et al., 2000; Chung et al., 2006). Based on these findings, a clear rationale for the design of bivalent μ/NOP receptor agonists was provided (Zaveri et al., 2013), which was supported by observations from opioid compounds that are already in clinical use, such as buprenorphine. Buprenorphine, is a unique opioid compound with mixed agonist–antagonist activity at classical opioid receptors that has been approved for the treatment of opioid dependency and is also used as an analgesic (Lutfy and Cowan, 2004). Buprenorphine has partial intrinsic agonistic activities at μ and NOP receptors and very low intrinsic activity at κ and δ receptors (Huang et al., 2001). The buprenorphine dose–response curve is sometimes submaximal, or even bell‐shaped, in nociceptive assays (Lutfy and Cowan, 2004). Partial agonism at μ receptors and, in some cases, antagonism at κ or δ receptors have been considered as possible underlying mechanisms for the ceiling effect and bell‐shaped dose–response curve of buprenorphine. While ceiling effects can be explained by the partial agonist activity of buprenorphine, the bell‐shaped dose–response curve has been postulated to be due to the suppression of μ receptor efficacy at higher doses by its NOP receptor partial agonist activity, at least in acute pain assays (Khroyan et al., 2009a). Buprenorphine's analgesic activity has been characterized in several acute and chronic pain models and it is a very potent long‐lasting analgesic that is useful after surgery in humans (Khroyan et al., 2009a). In the clinical setting, analgesia induced by buprenorphine is indiscernible from that of a full μ receptor agonist, without a ceiling effect (van Niel et al., 2016). However, there is a ceiling effect for respiratory depression, reducing the likelihood of this potentially fatal adverse event occurring (Dahan et al., 2006). The ceiling effect may also be responsible for the well‐known reduced abuse liability of buprenorphine, which is therefore often used as a substitute opioid in heroin replacement programmes (Bonhomme et al., 2012). Overall, these properties make it useful for opioid maintenance therapy (Khroyan et al., 2009a; Mello and Mendelson, 1980). It is also reported that buprenorphine does not diminish morphine‐induced antinociception, and even interacts in an additive to supra‐additive manner with morphine in preclinical rodent models (van Niel et al., 2016).
The buprenorphine analogue http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9294 with mixed μ/NOP receptor agonist activity was designed to have increased affinity and efficacy at NOP receptors. BU08028 is the first reported ‘universal opioid ligand’ that binds with high affinity to each member of the opioid receptor family, with a higher functional efficacy at the NOP receptor compared with buprenorphine in vitro (Ding et al., 2016; Khroyan et al., 2011). BU08028 produces long‐lasting antinociceptive and antihypersensitive actions mediated by both receptors, without reinforcing effects. In contrast to other μ receptor agonists, BU08028 possesses a higher safety profile and does not either inhibit respiratory functions, impair cardiovascular activities or produce acute physical dependence (Ding et al., 2016). In recent years, a series of modestly selective small‐molecule NOP receptor agonists has been developed, such as SR14150 (AT‐200) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8867 (AT‐202), and a non‐selective μ/NOP receptor agonist SR16435 (AT‐201). These bifunctional ligands, which can simultaneously activate both NOP and μ receptors, have a wider therapeutic window and the ability to alleviate severe pain conditions. Both SR14150 and SR16835 have higher selectivity and efficacy at NOP receptors compared to SR16435, and lower affinity and efficacy at μ receptors (Toll et al., 2009). SR14150 has antinociceptive activity in the tail‐flick assay that seems to be caused by activation of the μ receptor, because it was naloxone‐reversible. This suggests that the partial agonist efficacy of SR14150 at the μ receptor is sufficient to produce an acute antinociceptive effect without rewarding effects. SR16835 has slightly lower NOP receptor affinity than SR14150 and does not produce μ receptor‐mediated behavioural effects, such as conditioned place preference (CPP) (Toll et al., 2009). In a chronic pain model (sciatic nerve ligation in mice), both SR14150 and SR16835 display potent antiallodynic activity, with SR14150 being more potent than SR16835. In contrast to thermal nociception, this effect was completely blocked by the NOP receptor antagonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1693 but was not altered by naloxone, indicating that both compounds have NOP receptor‐mediated and not μ receptor‐mediated antiallodynic activity (Khroyan et al., 2011). In contrast, SR16435, a partial agonist at μ and NOP receptors, is a potent analgesic with reduced and slower development of tolerance to its antiallodynic effects compared to buprenorphine, but produces CPP to the same extent as morphine (Khroyan et al., 2007; Sukhtankar et al., 2013). The NOP receptor antagonist SR14148 was ineffective on its own but found to potentiate the antiallodynic activity of morphine and the mixed NOP/μ receptor agonist SR16435. Therefore, NOP receptor antagonists may provide a favourable therapeutic combination for neuropathic pain treatment that would allow for a reduction in opioid dosage. Furthermore, modulation of μ receptor‐mediated analgesia by NOP receptors using dual ligands with a profile of MOR agonist and NOPR antagonist activity may be particularly useful for the treatment of chronic pain (Khroyan et al., 2009b). In contrast, NOP receptor mediated antiallodynic activity may be more effective than that induced by an opiate medication and both NOP receptor agonists and antagonists could potentially become useful treatments for neuropathic pain (Khroyan et al., 2011). Importantly, compounds with affinity for both NOP and μ receptors may have a useful profile as analgesics with slower tolerance development and reduced addiction liability (Toll et al., 2009; Cremeans et al., 2012). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8866, a novel mixed μ/NOP receptor ligand, is currently in clinical development (phase 3 clinical trial) for the treatment of severe chronic nociceptive and neuropathic pain that targets the NOP receptor and other members of the opioid receptor family, in particular the μ receptor (Linz et al., 2014). Cebranopadol has full agonist activity at μ receptors, near‐full activity at the κ and NOP receptor, and partial activity at δ receptors. In various pain states, cebranopadol displays broad activity and is highly potent and efficacious in animal models of acute nociceptive, inflammatory, cancer and, especially, chronic neuropathic pain. Furthermore, even when administered at doses higher than those required to produce analgesia, cebranopadol does not affect either motor coordination or respiratory function and thus displays a better tolerability profile than opioids. In contrast to morphine, cebranopadol displays higher analgesic potency in chronic pain, especially of neuropathic origin, than in acute nociceptive pain (Linz et al., 2014). Both BU08028 and cebranopadol are proof‐of‐concept compounds to show that ligands with a multi‐opioid receptor pharmacology can produce a clinically useful therapeutic spectrum with a reduced side effect profile.
Conclusions
The search for improved opioid analgesics has clearly changed direction during the past two decades. When the first molecular structures of the four members of the opioid receptor family became available in the early 1990s, the initial hope was that ever‐more selective compounds targeting a single opioid receptor may eventually lead to more potent analgesics with fewer or no side effects. Subsequently, a deeper understanding of the anatomy, physiological functions and interactions between the four opioid systems, and last but not least detailed insights into the molecular basis of distinct intracellular signal transduction pathways of all members of the opioid receptor family have changed this view. Consequently, compounds that were until recently denounced as ‘dirty drugs’, because they interacted with multiple opioid receptors, now receive new attention. New research has demonstrated the value of simultaneously targeting two or even more members of the opioid receptor family, thus applying these new insights into the complex functional interactions of these pain‐modulating systems for devising new strategies in drug development.
The goal has always remained the same: creating a ‘better morphine’, with analgesic effects equal to morphine but none of the very serious liabilities, such as dependence, or possibility of fatal overdoses. Most progress has been made in the development of μ/NOP receptor bifunctional agonists for which the scientific rationale is probably strongest at this moment. But similar cases can be made for μ/δ receptor and μ/κ receptor mixed compounds, especially when taking into account a more narrow therapeutic application for specific pain states. The mixed μ/κ receptor agonist nalfurafine and its successful development for treatment of pruritus is a good example for the therapeutic benefits of such novel opioids with a mixed pharmacological profile that are highly effective in a limited therapeutic spectrum. The two μ/NOP receptor mixed agonists BU08028 and cebranopadol independently showed their potential for treatment of chronic inflammatory and neuropathic pain, which are largely unmet therapeutic needs.
It appears that balancing efficacies at multiple opioid receptors with a single compound may be a promising avenue to develop new opioid analgesics that are able to address specific pain conditions, which are not adequately managed with the drugs currently available. This strategy is not in competition, but hopefully complementary, to the recently developed concept of biased μ receptor agonists, both aim to reveal novel opioid analgesics with improved pharmacological profiles.
Outstanding issues
Do opioid receptors have to be compartmentalized when they are addressed with one ligand?
Is targeting multi‐opioid receptors one step back in the emerging search for selective ligands?
Is the peripheral restriction of the targeted opioid receptors the key to overcome opioid‐mediated side effects?
What is the perfect balance of ligands with a preferential μ/NOP receptor profile?
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Conflict of interest
S.S. received research grants from Grünenthal GmbH, Aachen, Germany and Mundipharma GmbH, Limburg, Germany. T.G., P.D., A.M., E.M., A.K., S.F. and R.S. have nothing to disclose.
Acknowledgement
This work was supported by the Deutsche Forschungsgemeinschaft grants SCHU924/11‐3, SCHU924/14‐1 and SCHU924/15‐1.
Günther T., Dasgupta P., Mann A., Miess E., Kliewer A., Fritzwanker S., Steinborn R., and Schulz S. (2018) Targeting multiple opioid receptors – improved analgesics with reduced side effects?, British Journal of Pharmacology, 175:2857–2868, https://doi.org/10.1111/bph.13809.
References
- Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE (1991). Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J Pharmacol Exp Ther 258: 299–303. [PubMed] [Google Scholar]
- Aceto MD, Harris LS, Negus SS, Banks ML, Hughes LD, Akgun E et al. (2012). MDAN‐21: a bivalent opioid ligand containing mu‐agonist and delta‐antagonist pharmacophores and its effects in rhesus monkeys. Int J Medicinal Chem 2012: 327257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackley MA, Hurley RW, Virnich DE, Hammond DL (2001). A cellular mechanism for the antinociceptive effect of a kappa opioid receptor agonist. Pain 91: 377–388. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Connor M (2005). Opioids: cellular mechanisms of tolerance and physical dependence. Curr Opin Pharmacol 5: 60–68. [DOI] [PubMed] [Google Scholar]
- Balboni G, Salvadori S, Trapella C, Knapp BI, Bidlack JM, Lazarus LH et al. (2010). Evolution of the bifunctional lead μ agonist/δ antagonist containing the 2′,6′‐dimethyl‐l‐tyrosine−1,2,3,4‐tetrahydroisoquinoline‐3‐carboxylic acid (Dmt−Tic) opioid pharmacophore. ACS Chem Nerosci 1: 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black D, Trevethick M (1998). The kappa opioid receptor is associated with the perception of visceral pain. Gut 43: 312–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar RJ (2010). Endogenous opiates and behavior: 2009. Peptides 31: 2325–2359. [DOI] [PubMed] [Google Scholar]
- Bonhomme J, Shim RS, Gooden R, Tyus D, Rust G (2012). Opioid addiction and abuse in primary care practice: a comparison of methadone and buprenorphine as treatment options. J Natl Med Assoc 104: 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt MR, Furness MS, Rice KC, Fischer BD, Negus SS (2001). Studies of tolerance and dependence with the delta‐opioid agonist SNC80 in rhesus monkeys responding under a schedule of food presentation. J Pharmacol Exp Ther 299: 629–637. [PubMed] [Google Scholar]
- Briggs SL, Rech RH, Sawyer DC (1998). Kappa antinociceptive activity of spiradoline in the cold‐water tail‐flick assay in rats. Pharmacol Biochem Behav 60: 467–472. [DOI] [PubMed] [Google Scholar]
- Burford NT, Clark MJ, Wehrman TS, Gerritz SW, Banks M, O'Connell J et al. (2013). Discovery of positive allosteric modulators and silent allosteric modulators of the μ‐opioid receptor. Proc Natl Acad Sci U S A 110: 10830–10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford NT, Livingston KE, Canals M, Ryan MR, Budenholzer LML, Han Y et al. (2015). Discovery, synthesis, and molecular pharmacology of selective positive allosteric modulators of the δ‐opioid receptor. J Med Chem 58: 4220–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Hoffert C, O'Donnell D, Beaudet A (2003). Up‐regulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain 101: 199–208. [DOI] [PubMed] [Google Scholar]
- Cami‐Kobeci G, Neal AP, Bradbury FA, Purington LC, Aceto MD, Harris LS et al. (2009). Mixed kappa/mu opioid receptor agonists: the 6 beta‐naltrexamines. J Med Chem 52: 1546–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo GPD, Folk JE, Rice KC, Chartoff E, Carlezon WA, Negus SS (2009). The selective non‐peptidic delta opioid agonist SNC80 does not facilitate intracranial self‐stimulation in rats. Eur J Pharmacol 604: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Pohl S, Zeng J, Civelli O, Reinscheid RK (2006). Endogenous orphanin FQ/nociceptin is involved in the development of morphine tolerance. J Pharmacol Exp Ther 318: 262–267. [DOI] [PubMed] [Google Scholar]
- Commons KG (2003). Translocation of presynaptic delta opioid receptors in the ventrolateral periaqueductal gray after swim stress. J Comp Neurol 464: 197–207. [DOI] [PubMed] [Google Scholar]
- Cremeans CM, Gruley E, Kyle DJ, Ko MC (2012). Roles of mu‐opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine‐induced physiological responses in primates. J Pharmacol Exp Ther 343: 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan A, Yassen A, Romberg R, Sarton E, Teppema L, Olofsen E et al. (2006). Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br J Anaesth 96: 627–632. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM et al. (2013). A G protein‐biased ligand at the mu‐opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther 344: 708–717. [DOI] [PubMed] [Google Scholar]
- Ding H, Czoty PW, Kiguchi N, Cami‐Kobeci G, Sukhtankar DD, Nader MA et al. (2016). A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proc Natl Acad Sci U S A 113: E5511–E5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du BX, Song ZM, Wang K, Zhang H, Xu FY, Zou Z et al. (2013). Butorphanol prevents morphine‐induced pruritus without increasing pain and other side effects: a systematic review of randomized controlled trials. Can J Anaesth = Journal canadien d'anesthesie 60: 907–917. [DOI] [PubMed] [Google Scholar]
- Eguchi M (2004). Recent advances in selective opioid receptor agonists and antagonists. Med Res Rev 24: 182–212. [DOI] [PubMed] [Google Scholar]
- Endoh T, Matsuura H, Tajima A, Izumimoto N, Tajima C, Suzuki T et al. (1999). Potent antinociceptive effects of TRK‐820, a novel kappa‐opioid receptor agonist. Life Sci 65: 1685–1694. [DOI] [PubMed] [Google Scholar]
- Endoh T, Tajima A, Suzuki T, Kamei J, Narita M, Tseng L et al. (2000). Characterization of the antinociceptive effects of TRK‐820 in the rat. Eur J Pharmacol 387: 133–140. [DOI] [PubMed] [Google Scholar]
- Falcon E, Maier K, Robinson SA, Hill‐Smith TE, Lucki I (2015). Effects of buprenorphine on behavioral tests for antidepressant and anxiolytic drugs in mice. Psychopharmacology (Berl) 232: 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frink MC, Hennies HH, Englberger W, Haurand M, Wilffert B (1996). Influence of tramadol on neurotransmitter systems of the rat brain. Arzneimittelforschung 46: 1029–1036. [PubMed] [Google Scholar]
- Gallantine EL, Meert TF (2005). A comparison of the antinociceptive and adverse effects of the mu‐opioid agonist morphine and the delta‐opioid agonist SNC80. Basic Clin Pharmacol Toxicol 97: 39–51. [DOI] [PubMed] [Google Scholar]
- Gaveriaux‐Ruff C, Karchewski LA, Hever X, Matifas A, Kieffer BL (2008). Inflammatory pain is enhanced in delta opioid receptor‐knockout mice. Eur J Neurosci 27: 2558–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Fujita W, Gupta A, Saldanha SA, Negri A, Pinello CE et al. (2013). Identification of a μ‐δ opioid receptor heteromer‐biased agonist with antinociceptive activity. Proc Natl Acad Sci U S A 110: 12072–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA (2004). A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci U S A 101: 5135–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack SP, Bagley EE, Chieng BC, Christie MJ (2005). Induction of delta‐opioid receptor function in the midbrain after chronic morphine treatment. J Neurosci 25: 3192–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinricher MM (2005). Nociceptin/orphanin FQ: pain, stress and neural circuits. Life Sci 77: 3127–3132. [DOI] [PubMed] [Google Scholar]
- Hirao A, Imai A, Sugie Y, Tamura T, Shimokawa H, Toide K (2008). Pharmacological properties of a novel nociceptin/orphanin FQ receptor agonist, 2‐(3,5‐dimethylpiperazin‐1‐yl)‐1‐[1‐(1‐methylcyclooctyl)piperidin‐4‐yl]‐1H‐benzim idazole, with anxiolytic potential. Eur J Pharmacol 579: 189–195. [DOI] [PubMed] [Google Scholar]
- Huang P, Kehner GB, Cowan A, Liu‐Chen LY (2001). Comparison of pharmacological activities of buprenorphine and norbuprenorphine: norbuprenorphine is a potent opioid agonist. J Pharmacol Exp Ther 297: 688–695. [PubMed] [Google Scholar]
- Jordan BA, Devi LA (1999). G‐protein‐coupled receptor heterodimerization modulates receptor function. Nature 399: 697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz EM, Baladi MG, Folk JE, Rice KC, Woods JH (2006). The convulsive and electroencephalographic changes produced by nonpeptidic delta‐opioid agonists in rats: comparison with pentylenetetrazol. J Pharmacol Exp Ther 317: 1337–1348. [DOI] [PubMed] [Google Scholar]
- Kabli N, Cahill CM (2007). Anti‐allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain 127: 84–93. [DOI] [PubMed] [Google Scholar]
- Katsumata S, Minami M, Nakagawa T, Iwamura T, Satoh M (1995). Pharmacological study of dihydroetorphine in cloned mu‐, delta‐ and kappa‐opioid receptors. Eur J Pharmacol 291: 367–373. [DOI] [PubMed] [Google Scholar]
- Khroyan TV, Polgar WE, Cami‐Kobeci G, Husbands SM, Zaveri NT, Toll L (2011). The first universal opioid ligand, (2S)‐2‐[(5R,6R,7R,14S)‐N‐cyclopropylmethyl‐4,5‐epoxy‐6,14‐ethano‐3‐hydroxy‐6‐meth oxymorphinan‐7‐yl]‐3,3‐dimethylpentan‐2‐ol (BU08028): characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine‐induced reward. J Pharmacol Exp Ther 336: 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khroyan TV, Polgar WE, Jiang F, Zaveri NT, Toll L (2009a). Nociceptin/orphanin FQ receptor activation attenuates antinociception induced by mixed nociceptin/orphanin FQ/mu‐opioid receptor agonists. J Pharmacol Exp Ther 331: 946–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khroyan TV, Polgar WE, Orduna J, Jiang F, Olsen C, Toll L et al. (2009b). Activity of new NOP receptor ligands in a rat peripheral mononeuropathy model: potentiation of morphine anti‐allodynic activity by NOP receptor antagonists. Eur J Pharmacol 610: 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khroyan TV, Polgar WE, Orduna J, Zaveri NT, Judd AK, Tuttle DJ et al. (2007). Anti‐nociceptive and anti‐allodynic effects of a high affinity NOP hexapeptide [Ac‐RY(3‐Cl)YRWR‐NH2] (Syn 1020) in rodents. Eur J Pharmacol 560: 29–35. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Gaveriaux‐Ruff C (2002). Exploring the opioid system by gene knockout. Prog Neurobiol 66: 285–306. [DOI] [PubMed] [Google Scholar]
- Ko M‐C, Naughton NN (2009a). Antinociceptive effects of nociceptin/orphanin FQ administered intrathecally in monkeys. J Pain : official journal of the American Pain Society 10: 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Woods JH, Fantegrossi WE, Galuska CM, Wichmann J, Prinssen EP (2009b). Behavioral effects of a synthetic agonist selective for nociceptin/orphanin FQ peptide receptors in monkeys. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 34: 2088–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovelowski CJ, Bian D, Hruby VJ, Lai J, Ossipov MH, Porreca F (1999). Selective opioid delta agonists elicit antinociceptive supraspinal/spinal synergy in the rat. Brain Res 843: 12–17. [DOI] [PubMed] [Google Scholar]
- Kumagai H, Ebata T, Takamori K, Muramatsu T, Nakamoto H, Suzuki H (2010). Effect of a novel kappa‐receptor agonist, nalfurafine hydrochloride, on severe itch in 337 haemodialysis patients: a Phase III, randomized, double‐blind, placebo‐controlled study. Nephrol Dial Transplant 25: 1251–1257. [DOI] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C (2008). The dysphoric component of stress is encoded by activation of the dynorphin κ‐opioid system. J Neurosci: the official journal of the Society for Neuroscience 28: 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Kulkarani V, Cowell SM, Ma S‐w, Davis P, Hanlon KE et al. (2011). Development of potent μ and δ opioid agonists with high lipophilicity. J Med Chem 54: 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Shiotani K, Miyazaki A, Tsuda Y, Ambo A, Sasaki Y et al. (2007). Bifunctional [2′,6′‐dimethyl‐L‐tyrosine1]endomorphin‐2 analogues substituted at position 3 with alkylated phenylalanine derivatives yield potent mixed mu‐agonist/delta‐antagonist and dual mu‐agonist/delta‐agonist opioid ligands. J Med Chem 50: 2753–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linz K, Christoph T, Tzschentke TM, Koch T, Schiene K, Gautrois M et al. (2014). Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J Pharmacol Exp Ther 349: 535–548. [DOI] [PubMed] [Google Scholar]
- Lutfy K, Cowan A (2004). Buprenorphine: a unique drug with complex pharmacology. Curr Neuropharmacol 2: 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K, Hossain SM, Khaliq I, Maidment NT (2001). Orphanin FQ/nociceptin attenuates the development of morphine tolerance in rats. Br J Pharmacol 134: 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz PE, Kieffer BL (2013). Opioid receptors: distinct roles in mood disorders. Trends Neurosci 36: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G et al. (2016). Structure‐based discovery of opioid analgesics with reduced side effects. Nature 537: 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I et al. (1996). Loss of morphine‐induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu‐opioid‐receptor gene. Nature 383: 819–823. [DOI] [PubMed] [Google Scholar]
- Mello NK, Mendelson JH (1980). Buprenorphine suppresses heroin use by heroin addicts. Science 207: 657–659. [DOI] [PubMed] [Google Scholar]
- Mistry SN, Shonberg J, Draper‐Joyce CJ, Klein Herenbrink C, Michino M, Shi L et al. (2015). Discovery of a novel class of negative allosteric modulator of the dopamine D2 receptor through fragmentation of a bitopic ligand. J Med Chem 58: 6819–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morphy R, Rankovic Z (2009). Designing multiple ligands – medicinal chemistry strategies and challenges. Curr Pharm Des 15 (6): 587–600. [DOI] [PubMed] [Google Scholar]
- Nadal X, Banos JE, Kieffer BL, Maldonado R (2006). Neuropathic pain is enhanced in delta‐opioid receptor knockout mice. Eur J Neurosci 23: 830–834. [DOI] [PubMed] [Google Scholar]
- Nakao K, Hirakata M, Miyamoto Y, Kainoh M, Wakasa Y, Yanagita T (2016). Nalfurafine hydrochloride, a selective kappa opioid receptor agonist, has no reinforcing effect on intravenous self‐administration in rhesus monkeys. J Pharmacol Sci 130: 8–14. [DOI] [PubMed] [Google Scholar]
- Negus SS, Bear AE, Folk JE, Rice KC (2009). Role of delta opioid efficacy as a determinant of mu/delta opioid interactions in rhesus monkeys. Eur J Pharmacol 602: 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Schrode K, Stevenson GW (2008). Micro/kappa opioid interactions in rhesus monkeys: implications for analgesia and abuse liability. Exp Clin Psychopharmacol 16: 386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara I, Przewlocki R, Przewlocka B (2005). Spinal and local peripheral antiallodynic activity of Ro64‐6198 in neuropathic pain in the rat. Pain 116: 17–25. [DOI] [PubMed] [Google Scholar]
- Ohmori S, Hayashi T, Kawase M, Saito S, Sugibayashi K, Morimoto Y (2000). Transdermal delivery of the potent analgesic dihydroetorphine: kinetic analysis of skin permeation and analgesic effect in the hairless rat. J Pharm Pharmacol 52: 1437–1449. [DOI] [PubMed] [Google Scholar]
- Ohmori S, Morimoto Y (2002). Dihydroetorphine: a potent analgesic: pharmacology, toxicology, pharmacokinetics, and clinical effects. CNS Drug Rev 8: 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa A, Brunori G, Mercatelli D, Wu J, Cippitelli A, Zou B et al. (2015). Knock‐in mice with NOP‐eGFP receptors identify receptor cellular and regional localization. J Neurosci 35: 11682–11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YX, Bolan E, Pasternak GW (2002). Dimerization of morphine and orphanin FQ/nociceptin receptors: generation of a novel opioid receptor subtype. Biochem Biophys Res Commun 297: 659–663. [DOI] [PubMed] [Google Scholar]
- Pan Z, Hirakawa N, Fields HL (2000). A cellular mechanism for the bidirectional pain‐modulating actions of orphanin FQ/nociceptin. Neuron 26: 515–522. [DOI] [PubMed] [Google Scholar]
- Pande AC, Pyke RE, Greiner M, Cooper SA, Benjamin R, Pierce MW (1996). Analgesic efficacy of the kappa‐receptor agonist, enadoline, in dental surgery pain. Clin Neuropharmacol 19: 92–97. [DOI] [PubMed] [Google Scholar]
- Parenti C, Turnaturi R, Arico G, Gramowski‐Voss A, Schroeder OH, Marrazzo A et al. (2013). The multitarget opioid ligand LP1's effects in persistent pain and in primary cell neuronal cultures. Neuropharmacology 71: 70–82. [DOI] [PubMed] [Google Scholar]
- Parenti C, Turnaturi R, Arico G, Marrazzo A, Prezzavento O, Ronsisvalle S et al. (2012). Antinociceptive profile of LP1, a non‐peptide multitarget opioid ligand. Life Sci 90: 957–961. [DOI] [PubMed] [Google Scholar]
- Perrine SA, Hoshaw BA, Unterwald EM (2006). Delta opioid receptor ligands modulate anxiety‐like behaviors in the rat. Br J Pharmacol 147: 864–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrillo P, Angelici O, Bingham S, Ficalora G, Garnier M, Zaratin PF et al. (2003). Evidence for a selective role of the delta‐opioid agonist [8R‐(4bS*,8aalpha,8abeta, 12bbeta)]7,10‐Dimethyl‐1‐methoxy‐11‐(2‐methylpropyl)oxycarbonyl 5,6,7,8,12,12b‐hexahydro‐(9H)‐4,8‐methanobenzofuro[3,2‐e]pyrrolo[2,3‐g]isoquinoli ne hydrochloride (SB‐235863) in blocking hyperalgesia associated with inflammatory and neuropathic pain responses. J Pharmacol Exp Ther 307: 1079–1089. [DOI] [PubMed] [Google Scholar]
- Podolsky AT, Sandweiss A, Hu J, Bilsky EJ, Cain JP, Kumirov VK et al. (2013). Novel fentanyl‐based dual mu/delta‐opioid agonists for the treatment of acute and chronic pain. Life Sci 93: 1010–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portoghese PS (1989). Bivalent ligands and the message‐address concept in the design of selective opioid receptor antagonists. Trends Pharmacol Sci 10: 230–235. [DOI] [PubMed] [Google Scholar]
- Ranganathan M, Schnakenberg A, Skosnik PD, Cohen BM, Pittman B, Sewell RA et al. (2012). Dose‐related behavioral, subjective, endocrine, and psychophysiological effects of the kappa opioid agonist Salvinorin A in humans. Biol Psychiatry 72: 871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss D, Wichmann J, Tekeshima H, Kieffer BL, Ouagazzal AM (2008). Effects of nociceptin/orphanin FQ receptor (NOP) agonist, Ro64‐6198, on reactivity to acute pain in mice: comparison to morphine. Eur J Pharmacol 579: 141–148. [DOI] [PubMed] [Google Scholar]
- Sadee W, Rosenbaum JS, Herz A (1982). Buprenorphine: differential interaction with opiate receptor subtypes in vivo. J Pharmacol Exp Ther 223: 157–162. [PubMed] [Google Scholar]
- Schepers RJ, Mahoney JL, Shippenberg TS (2008). Inflammation‐induced changes in rostral ventromedial medulla mu and kappa opioid receptor mediated antinociception. Pain 136: 320–330. [DOI] [PubMed] [Google Scholar]
- Schroder W, Lambert DG, Ko MC, Koch T (2014). Functional plasticity of the N/OFQ‐NOP receptor system determines analgesic properties of NOP receptor agonists. Br J Pharmacol 171: 3777–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, Awamura S, Kimura C, Ide S, Sakano K, Minami M et al. (1999). Pharmacological properties of TRK‐820 on cloned mu‐, delta‐ and kappa‐opioid receptors and nociceptin receptor. Eur J Pharmacol 376: 159–167. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Zapata A, Chefer VI (2007). Dynorphin and the pathophysiology of drug addiction. Pharmacol Ther 116: 306–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoblock JR (2007). The pharmacology of Ro 64‐6198, a systemically active, nonpeptide NOP receptor (opiate receptor‐like 1, ORL‐1) agonist with diverse preclinical therapeutic activity. CNS Drug Rev 13: 107–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HS, Peppin JF (2014). Toward a systematic approach to opioid rotation. J Pain Res 7: 589–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolo B, Calo G, Polgar WE, Jiang F, Olsen CM, Berzetei‐Gurske I et al. (2008). Activities of mixed NOP and mu‐opioid receptor ligands. Br J Pharmacol 153: 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein MD, Herman DS, Bailey GL, Straus J, Anderson BJ, Uebelacker LA et al. (2015). Chronic pain and depression among primary care patients treated with buprenorphine. J Gen Intern Med 30: 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson GW, Folk JE, Linsenmayer DC, Rice KC, Negus SS (2003). Opioid interactions in rhesus monkeys: effects of delta + mu and delta + kappa agonists on schedule‐controlled responding and thermal nociception. J Pharmacol Exp Ther 307: 1054–1064. [DOI] [PubMed] [Google Scholar]
- Sukhtankar DD, Zaveri NT, Husbands SM, Ko MC (2013). Effects of spinally administered bifunctional nociceptin/orphanin FQ peptide receptor/mu‐opioid receptor ligands in mouse models of neuropathic and inflammatory pain. J Pharmacol Exp Ther 346: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH, Soong Y, Wu D, Olariu N, Kett A, Kim H et al. (1999). Respiratory depression after intravenous administration of delta‐selective opioid peptide analogs. Peptides 20: 101–105. [DOI] [PubMed] [Google Scholar]
- Tavani A, Petrillo P, La Regina A, Sbacchi M (1990). Role of peripheral mu, delta and kappa opioid receptors in opioid‐induced inhibition of gastrointestinal transit in rats. J Pharmacol Exp Ther 254: 91–97. [PubMed] [Google Scholar]
- Taylor GT, Manzella F (2016). Kappa opioids, salvinorin A and major depressive disorder. Curr Neuropharmacol 14: 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Khroyan TV, Polgar WE, Jiang F, Olsen C, Zaveri NT (2009). Comparison of the antinociceptive and antirewarding profiles of novel bifunctional nociceptin receptor/mu‐opioid receptor ligands: implications for therapeutic applications. J Pharmacol Exp Ther 331: 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Inoue M, Takeshima H, Iwasawa Y (2000). Enhanced spinal nociceptin receptor expression develops morphine tolerance and dependence. J Neurosci 20: 7640–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valant C, Gregory KJ, Hall NE, Scammells PJ, Lew MJ, Sexton PM et al. (2008). A novel mechanism of G protein‐coupled receptor functional selectivity. Muscarinic partial agonist McN‐A‐343 as a bitopic orthosteric/allosteric ligand. J Biol Chem 283: 29312–29321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel JC, Schneider J, Tzschentke TM (2016). Efficacy of full micro‐opioid receptor agonists is not impaired by concomitant buprenorphine or mixed opioid agonists/antagonists – preclinical and clinical evidence. Drug Res 66: 562–570. [DOI] [PubMed] [Google Scholar]
- Varadi A, Hosztafi S, Le Rouzic V, Toth G, Urai A, Noszal B et al. (2013). Novel 6beta‐acylaminomorphinans with analgesic activity. Eur J Med Chem 69: 786–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscusi ER, Webster L, Kuss M, Daniels S, Bolognese JA, Zuckerman S et al. (2016). A randomized, phase 2 study investigating TRV130, a biased ligand of the mu‐opioid receptor, for the intravenous treatment of acute pain. Pain 157: 264–272. [DOI] [PubMed] [Google Scholar]
- Wang D, Sun X, Bohn LM, Sadee W (2005). Opioid receptor homo‐ and heterodimerization in living cells by quantitative bioluminescence resonance energy transfer. Mol Pharmacol 67: 2173–2184. [DOI] [PubMed] [Google Scholar]
- Wang HB, Zhao B, Zhong YQ, Li KC, Li ZY, Wang Q et al. (2010). Coexpression of delta‐ and mu‐opioid receptors in nociceptive sensory neurons. Proc Natl Acad Sci U S A 107: 13117–13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK et al. (2015). The G protein‐biased kappa‐opioid receptor agonist RB‐64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther 352: 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XJ, Hao JX, Wiesenfeld‐Hallin Z (1996). Nociceptin or antinociceptin: potent spinal antinociceptive effect of orphanin FQ/nociceptin in the rat. Neuroreport 7: 2092–2094. [PubMed] [Google Scholar]
- Yamamoto T, Nozaki‐Taguchi N, Kimura S (1997). Analgesic effect of intrathecally administered nociceptin, an opioid receptor‐like1 receptor agonist, in the rat formalin test. Neuroscience 81: 249–254. [DOI] [PubMed] [Google Scholar]
- Zaveri N, Jiang F, Olsen C, Polgar W, Toll L (2013). Designing Bifunctional NOP receptor‐mu opioid receptor ligands from NOP receptor‐selective scaffolds. Part I. Bioorg Med Chem Lett 23: 3308–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri NT (2011). The nociceptin/orphanin FQ receptor (NOP) as a target for drug abuse medications. Curr Top Med Chem 11: 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Pan ZZ (2010). Synaptic mechanism for functional synergism between delta‐ and mu‐opioid receptors. J Neurosci 30: 4735–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]