

Abstract
Dietary flavonoids are used in treatment of multiple diseases, and their antiinflammatory effects in the intestine are due, in part, to interactions with gut microflora and possibly due to modulation of aryl hydrocarbon receptor (AhR) signaling. In this study, we investigated the structure-dependent AhR activity of 14 flavonoids in Caco2 colon cancer cells using induction of CYP1A1 and UGT1A1 gene expression as endpoints. A major structural determinant for AhR activation was the number of hydroxyl groups where pentahydroxyflavonoids (with the exception of morin) > hexahydroxyflavonoids > tetra-/trihydroxyflavonoids, and some of the latter compounds such as apigenin exhibited AhR antagonist activity for induction of CYP1A1. Simulations suggest that while quercetin and apigenin interact primarily with the same residues, the strength of interactions between specific AhR residues with CYP1A1 agonist, quercetin, in comparison with CYP1A1 antagonist, apigenin, is different; thus, such interactions are presumably indicative of potential switches for modulating CYP1A1 activity. The structure-dependent effects of the hydroxyl flavonoids on induction of UGT1A1 were similar to that observed for induction of CYP1A1 except that luteolin and apigenin induced UGT1A1 levels similar to that observed for TCDD, whereas both compounds were AhR antagonists for CYP1A1. Thus, the effects of the flavonoids in Caco2 cells on Ah-responsiveness and interactions with butyrate were both ligand structure- and response-dependent and these activities are consistent with hydroxyflavonoids being selective AhR modulators.
Keywords: structure-activity, flavonoids, Ah receptor ligands, Caco2 cells, CYP1A1, UGT1A1
Flavonoids are a class of polyphenolic secondary metabolites that are produced primarily in fruits and vegetables, and these compounds are generally recognized as health promoting and are components of traditional medicines and commercially-available nutraceuticals (Aoki et al., 2000; Havsteen, 2002; Panche et al., 2016; Ross and Kasum, 2002). A review of the literature shows that flavonoids are used in the treatment of multiple health problems including viral and bacterial infections, inflammatory conditions, cardiovascular disease, and cancer (Aoki et al., 2000; Havsteen, 2002; Panche et al., 2016; Ross and Kasum, 2002). Flavonoids contain a common 4-H-chromen-4-one (chromone) ring structure with a phenyl substituent at C-2 or C-3 and hydroxyl substituents in both the chromone and phenyl rings and include flavones, flavonols, and isoflavones. Anthocyanins which are substituted flavylium cations (2-phenylbenzopyrilium) are often classified as flavonoids. The complexity of flavonoid structures is due not only to their degree of hydroxylation and position of the hydroxyl substituents, but also various degrees of hydroxyl group methylation and glycosylation. The health-promoting properties of flavonoids and polyphenolics are due to multiple activities including their antioxidant and antiinflammatory effects, direct or indirect inhibition of key enzymes and pathways, and interactions with other sub-cellular targets including receptors such as the estrogen receptor and aryl hydrocarbon receptor (AhR) (Hertog et al., 1997; Hugel et al., 2016; Kim et al., 2004a; Kumar and Pandey, 2013; LeJeune et al., 2015; Panche et al., 2015; Pietta, 2000; Salaritabar et al., 2017; Vezza et al., 2016; Xue et al., 2017).
There is growing evidence in laboratory animal studies that flavonoids protect against inflammation of the gastrointestinal tract (Salaritabar et al., 2017; Vezza et al., 2016) and this includes flavonoids such as quercetin, morin, kaempferol, and luteolin (Galvez et al., 2001; Guazelli et al., 2013; Kwon et al., 2005; Nishitani et al., 2013; Ocete et al., 1998; Park et al., 2012). The direct use of individual flavonoids and polyphenolics for treating inflammatory intestinal conditions in humans has been limited (Algieri et al., 2015; Dryden et al., 2013) but warranted based on the successful studies in laboratory animals (Salaritabar et al., 2017; Vezza et al., 2016). The effects of flavonoids on the gut and other organs/tissues are also modulated by their interactions with gut microbiota in which these compounds not only undergo microbial metabolism, but they also change the composition of the microbial population (Cassidy and Minihane, 2017; Espin et al., 2017). Metabolism of flavonoids is dependent on multiple factors including the composition of gut microbiomes; however, deglycosylation is one of the initial steps and results in formation of flavonoids and polyphenolics with only their base structure and fewer glycoside derivatives (Cassidy and Minihane, 2017). Polyphenolics can also modulate gut microbiota composition resulting in an enhanced diversity (Espin et al., 2017).
Research in our laboratories is focused on the role of the AhR in protecting the gut against inflammation and cancer and this includes characterization of the AhR agonist and antagonist activities of microbiota-derived AhR ligands (Cheng et al., 2015, 2017; Jin et al., 2014, 2017). Flavones and flavonols are dietary components and several hydroxyflavonoids including apigenin, luteolin, galangin, quercetin, kaempferol, and naringenin competitively bind to the AhR (Ashida et al., 2000). Hydroxyflavonoids have previously been investigated as AhR agonists or antagonists (Allen et al., 2001; Ashida et al., 2000; Ciolino et al., 1998, 1999; Kim et al., 2004b; Puppala et al., 2007; Van der Heiden et al., 2009; Xue et al., 2017; Yannai et al., 1998; Zhang et al., 2003); however, some reports have been contradictory and this may be due, in part, to the assay system used and cell context. Although flavonoids have been characterized as AhR ligands, their structure-dependent activities as AhR agonists or antagonists have not been investigated and most of these studies have only focused on CYP1A1-related response readout. In this study, we have investigated the structure-activity relationships among a series of 14 flavones/flavonols as AhR agonists or antagonists for 2 Ah-responsive genes (CYP1A1 and UGT1A1) in Caco2 colon cancer cells and the results show that major determining factors are the number and position of the hydroxyl groups and the specific response. Moreover, we also show that designation of a compound as an AhR agonist or antagonist is dependent on which Ah-responsive gene is used as an assay endpoint. Modeling studies show interactions of quercetin and apigenin in the AhR binding pocket. Both quercetin and apigenin interact with the binding pocket residues of AhR. Similarities and differences in the strength of interactions of quercetin, apigenin, and additionally TCDD (Jin et al., 2017) to AhR residues can shed light on interactions that may act as switches inducing agonist activity.
MATERIALS AND METHODS
Cell lines, antibodies, and reagents
The Caco2 human colon cancer cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) nutrient mixture supplemented with 20% fetal bovine serum (FBS), 1× MEM non-essential amino acid solution (Gibco), and 1× antibiotic/antimycotic reagent (Sigma-Aldrich) at 37°C in the presence of 5% CO2. Human CYP1A1 and AhR antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and β-actin was purchased from Sigma-Aldrich (St. Louis, MO). GAPDH, Acetyl-H3K27, and Acetyl-H3K8 antibodies were purchased from Cell Signaling (Beverly, MA). All flavonoid compounds were purchased from INDOFINE Chemical Company (Hillsborough, NJ). For single dose experiments with the flavonoids, 100 μM concentrations were used except for quercetin (10 μM; maximal CYP1A1 induction), gossypetin (10 μM; higher concentrations were toxic), and luteolin, kaempferol, and apigenin (10 μM; higher concentrations were poorly soluble).
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation (ChIP) assay was performed using ChIP-IT Express Magnetic Chromatin Immunoprecipitation kit (Active Motif, Carlsbad, CA) according to the manufacturer's protocol. Caco2 cells were treated with sodium butyrate overnight, and TCDD was subsequently added into the media for 2 h prior to cell harvest. Cells were then fixed with 1% formaldehyde, and the cross-linking reaction was stopped by addition of 0.125 M glycine. After washing twice with phosphate-buffered saline, cells were scraped and pelleted. Collected cells were hypotonically lysed, and nuclei were collected. Nuclei were then sonicated to desired chromatin length (∼200–1500 bp). The sonicated chromatin (25 μg) was immunoprecipitated with primary antibodies (25 μg) and protein A-conjugated magnetic beads at 4°C for 12 h. After the magnetic beads were extensively washed, protein-DNA crosslinks were reversed and eluted. DNA was prepared by proteinase K digestion followed by PCR amplification. The human CYP1A1 primers were 5′-TCA ATC AAG AGG CGC GAA CCT C-3′ (sense), and 5′-CTA CAG CCT ACC AGG ACT CG-3′ (antisense), and then amplified by targeting a 203-bp region of human CYP1A1 promoter which contained the AhR binding sequences. The human UGT1A1 primers were 5′-GTG TTA TCT CAC CAG AAC AAA-3′ (sense) and 5′-TAC CCT CTA GCC ATT CTG-3′ (antisense), and subsequently amplified by targeting a 190-bp region of human UGT1A1 promoter, which contained the AhR-binding sequences. PCR products were resolved on a 2% agarose gel in the presence of ETBR.
Quantitative real-time reverse transcriptase PCR
cDNA was prepared from the total RNA of cells using High Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA). Each PCR was carried out in triplicate using Bio-Rad SYBR Universal premix for 1 min at 95°C for initial denaturing, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min in the Bio-Rad iCycler (MyiQ™2) real-time PCR System. The comparative CT method was used for relative quantitation of samples. Values for each gene were normalized to expression levels of TATA-binding protein (TBP). The sequences of the primers used for real-time PCR were as follows: CYP1A1 sense 5′-GAC CAC AAC CAC CAA GAA C-3′, antisense 5′-AGC GAA GAA TAG GGA TGA AG-3′; UGT1A1 sense 5′-GAA TCA ACT GCC TTC ACC AAA AT-3′, antisense 5′-AGA GAA AAC CAC AAT TCC ATG TTC T-3′; TBP sense 5′-GAT CAG AAC AAC AGC CTG CC-3′, antisense 5′-TTC TGA ATA GGC TGT GGG GT-3′.
Western blot analysis
Cells (3×105) were plated in 6-well plates in DMEM media containing 2.5% FBS for 24 h and then treated with different concentrations of the compounds. Cellular lysates were prepared in lysis buffer containing 50 mM HEPES, 0.5 M NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, and 1% Triton-X-100, each 1× protease and phosphatase inhibitor cocktail (GenDEPOT), and 1% NP-40. The cells were disrupted and extracted at 4°C for 30 min. After centrifugation, the supernatant was obtained as the cell lysate. Protein concentrations were measured using the Bio-Rad protein assay. Aliquots of cellular proteins were electrophoresed on 10% SDS–polyacrylamide gel electrophoresis (PAGE) and transferred to a PVDF membrane (Bio-Rad, Hercules, CA). The membrane was allowed to react with a specific antibody, and detection of specific proteins was carried out by enhanced chemiluminescence. Loading differences were normalized using a polyclonal β-actin or GAPDH antibody.
Generation of AhR-deficient Caco2 cells
Two AhR CRISPR guide RNAs, in a Cas9 vector (pSpCas9 BB-2A-GFP PX458) which also expresses GFP, were purchased from GenScript (Piscataway, NJ). Sequences of the guide RNAs were 5′-AAG TCG GTC TCT ATG CCG CT-3′ and 5′-AGA CCG ACT TAA TAC AGA GT-3′. Caco2 cells were transfected with each plasmid and 48 h later, cells were sorted by flow cytometry to collect the 5% highest GFP expressing cells. Clonal cells were grown and tested for knock-out of AhR protein by Western blots and CYP1A1 mRNA induction following treatment with TCDD and analysis by RT-PCR.
Ethoxyresorufin-O-deethylase assay
Cells (1.5×105) were plated in 12-well plates for 24 h and then treated with solvent control (0.2% v/v DMSO) or compounds dissolved in DMEM medium (no phenol red) containing 2.5% charcoal stripped FBS. After 24 h, the treatment medium was replaced with DMEM medium (no phenol red) containing 10 μM ethoxyresorufin for incubation. After 3 h of incubation, 100 μl medium (without cells) was transferred to a 96-well opaque black-wall solid-bottom plate. Resorufin-associated fluorescence was measured using the Synergy Mx (BioTek) microplate reader with excitation at 535 nm and emission at 590 nm (9 nm bandwidth for both). Media containing 10 μM ethoxyresorufin or 10 μM resorufin were used as negative (background) and positive controls, respectively.
Statistics
All of the experiments were repeated a minimum of three times. The data are expressed as the means ± SD. Statistical significance was analyzed using either Unpaired-Student’s t test (two-tailed) or analysis of variance (ANOVA) test. A p value of <.05 was considered statistically significant.
Modeling of the human AhR protein
Residues 247 through 406 (sequence HGQKKKGKDG-SILPPQLALF-AIATPLQPPS-ILEIRTKNFI-FRTKHKLDFT-PIGCDAKGRI-VLGYTEAELC-TRGSGYQFIH-AADMLYCAES-HIRMIKTGES-GMIVFRLLTK-NNRWTWVQSN-ARLLYKNGRP-DYIIVTQRPL-TDEEGTEHLR-KRNTKLPFMF) of the human AhR were investigated in complex with both quercetin and apigenin. The human AhR protein domain investigated in this study is analogous to the mouse AhR (mAhR) protein domain we investigated in complex with TCDD and DHNA (Jin et al., 2017). The investigated human AhR residues were extracted from the crystal structure of human AhR in complex with (5S, 7R)-5,7-bis(3-bromophenyl)-4,5,6,7-tetrahydrotetrazolo[1,5-a]pyrimidine, a tetrazole-containing antagonist [PDB ID: 4XT2 (Scheuermann et al., 2015)], and residues missing from the crystal structure were modeled using I-TASSER (Yang et al., 2015). The N- and C-terminal ends of the modeled protein were acetylated and amidated to avoid errors due to the artificial placement of positively and negatively charged groups at the backbone termini of the truncated ends of the modeled systems under investigation.
Docking quercetin and apigenin to AhR
To investigate the docking of quercetin and apigenin to human receptor AhR, we employed our in-house developed docking protocol which was previously used to determine interactions within the binding pocket of AhR (Jin et al., 2017). In brief, this protocol can be considered as a tool to refine and elucidate the structure of a docked ligand into a receptor’s binding pocket by combining the following features: (1) the use of short MD docking simulations to nearly exhaustively search conformations of the ligand within the receptor binding pocket; (2) the use of both harmonic and quartic spherical potentials to constrain the ligand within the binding pocket during the multiple short docking simulations; and (3) the use of all-atom, explicit water MD simulations and free energy calculations are used elucidate the most energetically favorable binding mode of the ligand in complex with the receptor. As a result, the most energetically favorable binding modes of quercetin and apigenin independently in complex with AhR were identified, analogously to our previous study elucidating the most energetically favorable binding mode of TCDD and DHNA in complex with mAhR (Yang et al., 2015). The docking procedure followed is described below.
Quercetin and apigenin were independently positioned into the binding site of AhR by structural superposition to the experimentally resolved (5S, 7R)-5,7-bis(3-bromophenyl)-4,5,6,7-tetrahydrotetrazolo[1,5-a]pyrimidine via the ShaEP algorithm (Vainio et al., 2009). The structures for both quercetin and apigenin were obtained from the ZINC database (Irwin and Shoichet, 2005) and were parameterized using CGenFF (Vanommeslaeghe and MacKerell, 2012). Six separate docking simulation systems were introduced independently to investigate the binding of quercetin and apigenin to AhR. In the docking simulations, each ligand independently was constrained using harmonic or quartic potential energy functions to the docked binding site through the MMFP module of CHARMM (Brooks et al., 2009).
In Systems 1 through 4, the harmonic spherical potential was enacted on the ligands. The harmonic spherical potential is defined by Equation (1):
| (1) |
where is the potential and is the difference between the ligands' initial and new center of mass, and is a user defined offset such that the potential is zero if is less than .
In Systems 5 and 6, the quartic spherical potential enacted on the ligands. The quartic spherical potential is defined by Equation (2):
| (2) |
where is the potential, is the difference between the ligands' initial and new center of mass, and is a defined offset distance such that the potential is zero if is less than . The quartic spherical potential simultaneously constrains the ligand to the binding site while energetically encouraging the ligand to explore docking poses at which the center of mass will not coincide with the center of mass of its initial placement.
In System 1, is set to 0.5 Å; in System 2, is set to 1.5 Å; in System 3, is set to 2.5 Å; in System 4, is set to 3.5 Å; in System 5, set to 0.5 Å; in System 6, is set to 1.5 Å. In each of the six separate docking simulation systems, 20 independent runs comprising 200 short 2 ps simulations were performed. In each step, prior to the short MD simulation run, the ligands were independently rotated about a randomly generated axis. After the short MD simulation run, an energy minimization was performed on the complex conformation and the structure was saved for evaluation. This procedure resulted in the generation of 4000 binding conformations for each ligand-AhR complex per docking simulation system. As an initial screening, out of the 4000 complex structures produced per system, we extracted the 3 complex structures with the lowest interaction energy for further analysis. This resulted in 18 docking conformations of quercetin-AhR complex and 18 docking conformations of apigenin-AhR complex extracted for further investigation using MD simulations and free energy calculations. The 18 binding modes for both the quercetin-AhR and apigenin-AhR complexes were named based on which docking system they originated from. For example, binding mode 05q1 originated from System 5, which uses a quartic spherical potential with an of 0.5 Å, and was the binding mode with the lowest interaction energy value within System 5; binding mode 25h3 originated from System 3, which uses a harmonic spherical potential with an of 2.5 Å, and was the binding mode with the second lowest interaction energy value within System 3.
MD simulations were introduced in order to refine the extracted ligand-AhR structures, optimize intermolecular interactions, determine the structural stability of the selected binding modes, and assess-identify the most energetically favorable binding modes of the quercetin-AhR and apigenin-AhR complexes. The structures corresponding to the 18 selected quercetin-AhR docking conformations and the 18 selected apigenin-AhR docking conformations were used as initial structures for the 36 independent MD simulations. All MD simulations of the 36 systems under investigation were performed in explicit solvent using CHARMM (Carney et al., 2008) and CHARMM36 topology and parameters (Vanommeslaeghe et al., 2010) with periodic boundary conditions.
Subsequently, the Molecular Mechanics Generalized Born Surface Area (MM-GBSA) approximation was introduced to identify the most energetically favorable conformation of quercetin and apigenin in complex with AhR (Carney et al., 2008; Sambuy et al., 2005; Tamamis et al., 2012; Yang et al., 2015). We calculated the association-free energy of the 18 complexes per ligand over the 10 ns production runs using snapshots extracted from the simulations every 20 ps. The simulations of the quercetin-AhR and apigenin-AhR binding modes with the most favorable MM-GBSA association-free energies (eg, lowest MM-GBSA association-free energies) were selected as the ones representing the most likely naturally occurring binding conformations of the 2 ligands in complex with AhR.
Per AhR residue interaction free energy analysis
To determine the key interactions occurring in the lowest association free energy simulated binding modes, the average per AhR residue interaction-free energies between the AhR protein and each ligand of the structures with the lowest MM-GBSA binding-free energies were calculated for the 50 ns production runs in intervals of 20 ps (Tamamis et al., 2012; Tamamis et al., 2010; Yang et al., 2015).
RESULTS
Ah-Responsiveness of Caco2 Cells to Flavonoids: Induction of CYP1A1 and UGT1A1
Initial screening of 14 flavonoids (Figure 1A) (10–100 μM) as inducers of CYP1A1 in CaCo2 cells showed that the most obvious structure-dependency was the number of hydroxyl groups. The hexahydroxy analogs myrcetin and gossypetin induced <20% of the maximal response observed for 10 nM TCDD (Figure 1B). In contrast, 4 of the 5 pentahydroxyflavonoids (quercetin, taxifolin, 5,7,3′,4′,5-pentahydroxyflavone and robinetin) induced >50% of the maximal response observed for TCDD (Figure 1C). Morin was the only outlier among the pentahydroxy compounds, and the results suggest that maximal induction activity required at least 2 of 3′-, 4′, and 5′-hydroxyl groups in the phenyl ring, whereas the 2′,4′-dihydroxyl substituents in morin resulted in decreased induction of CYP1A1. The tri- and tetrahydroxyflavonoids (10–100 μM) only minimally induced CYP1A1 mRNA (Figure 1D). Among the 7 tetra- and trihydroxyflavones, only the 3,6,2′,4-substituted compound induced CYP1A1 protein, whereas induction was observed for all pentahydroxy flavonoids (Figure 1E). TCDD induced CYP1A1 protein and downregulated AhR protein expression; however, among the flavonoids, only the pentahydroxy compounds decreased AhR expression but the decrease was much less than observed for TCDD. Figure 1F summarizes the induction of ethoxyresorufin-O-deethylase (EROD) activity by 3,6,2′,4-tetrahydroxy flavone, quercetin, morin, robinetin, and myrcetin, and the remainder of the compounds were inactive (data not shown). The induction of CYP1A1 protein and EROD activity by morin was relatively low compared to TCDD but higher than observed for CYP1A1 mRNA (Figure 1C) and this is currently being investigated.
Figure 1.

Flavonoids as inducers of CYP1A1 in Caco2 cells. (A) Structure of flavonoids used in this study. Induction of CYP1A1 by hexahydroxy (B), pentahydroxy- (C) and tetra-/trihydroxy flavonoids (D). Cells were treated with TCDD and hydroxyflavonoids for 18 h, and mRNA expression was determined by real-time PCR. Results are expressed as means ± SD for 3 replicate determinations, and significant (p < .05) induction is indicated (*). (E) Caco2 cells were treated with TCDD and hydroxyflavonoids for 24 h, and whole cell lysates were analyzed by Western blots. EROD activity (F) was determined for all compounds and only those that significantly (p < .05) induced activity are shown. The rationale for the hydroxyflavonoid concentrations are outlined in the Materials and Methods.
TCDD (10 nM) induced expression of another Ah-responsive gene, UGT1A1, by >3-fold (Figure 2A) and this was significantly lower than the >250-fold induction of CYP1A1 mRNA (Figure 2B). However, the structure-activity relationships among the hexa- and pentahydroxyflavonoids for induction of CYP1A1 and UGT1A1 were similar except that compounds weakly activating CYP1A1 (myrcetin, gossypetin, and morin) did not significantly induce UGT1A1 expression (Figs. 2A and 2B). The fold induction of UGT1A1 observed for 10–100 μM quercetin and robinetin were equal to or greater than the induction observed for TCDD. The most dramatic differences in the induction of UGT1A1 compared to CYP1A1 were observed for the tetra- and trihydroxyflavonoids (luteolin and apigenin, respectively) where their fold induction response was similar to that observed TCDD (Figure 2C). Using CRISPR/Cas9 technology, we knocked down the AhR in CaCo2 cells (Figure 2D) and the 2 cell lines (KO1 and KO2) expressed low background levels of the AhR. The KO2 cell line was used as a model and we observed that TCDD, quercetin, taxifolin, morin, robinetin, luteolin, and apigenin did not induce CYP1A1 (Figure 2E) or UGT1A1 (Figure 2F), and similar results were observed in K01 cells (data not shown), demonstrating that the hydroxyflavonoid-induced responses were AhR-dependent.
Figure 2.

Flavonoids as inducers of UGT1A1 and role of the AhR. Caco2 cells were treated with hexahydroxy- (A), pentahydroxy- (B), and tetra-/trihydroxy (C) flavonoids for 18 h, and induction of UGT1A1 mRNA levels were determined by real-time PCR. Results are expressed as means ± SD for 3 replicate determinations, and significant (p < .05) induction is indicated (*). (E) Caco2 KO1 and KO2 AhR knockout cells were obtained after CRISPR/Cas9 knockout of the AhR, and whole cell lysates were analyzed by Western blots. Caco2-AhR-KO2 cells were treated with TCDD and selected hydroxyflavonoids, and induction of CYP1A1 (E) and UGT1A1 (F) mRNA levels were determined by real-time PCR. Data determination and analysis were obtained as outlined for (A–C). Significantly (p < .05) decreased responses in (E) and (F) are indicated (*).
Modeling of Quercetin and Apigenin in Complex With AhR to Identify the Most Energetically Favorable Binding Conformations
The quercetin-AhR and apigenin-AhR binding modes acquiring the lowest MM-GBSA association free energy across all 18 simulated quercetin-AhR and apigenin-AhR binding modes, respectively, were selected for further analysis. The quercetin-AhR binding mode 15q3 and the apigenin-AhR binding mode 05h3 were selected for further investigation. Apigenin-AhR binding modes 15h1 and 25h3 have the second and third lowest association-free energy values of the 18 binding modes investigated and have similar binding modes within the AhR binding site as the lowest association free energy binding mode 05h3. Thus, apigenin-AhR binding modes 15h1 and 25h3 were not analyzed further. The average association free energies of the simulated quercetin-AhR and apigenin-AhR binding modes are shown in Figures 3A and B. The binding modes were named based on which docking system they originated from. For example, binding mode 15q3 originated from System 6, which uses a quartic spherical potential with an of 1.5 Å, and was the binding mode with the third lowest interaction energy value within System 6.
Figure 3.
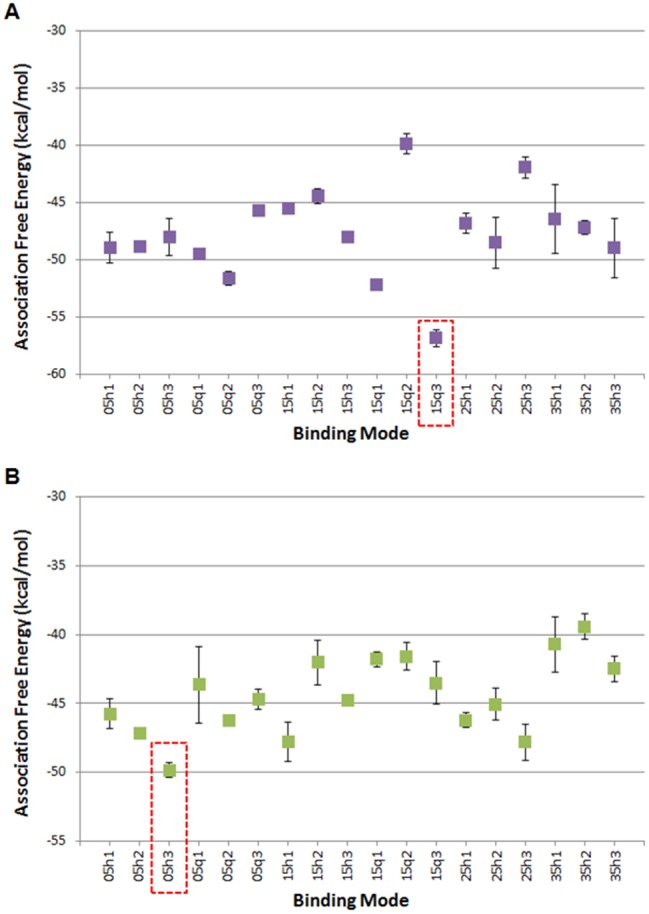
Average association-free energies (in kcal/mol) of the MD simulated quercetin (A) and apigenin (B) binding modes. Binding mode names correspond to each of the 6 docking systems from which the binding mode originated. The binding mode with the lowest average association free energy, which is selected for further analysis, is boxed using dotted lines. The average and standard deviation values for the association-free energies were calculated over 4 measurements, of which the first, second, third, and fourth measurements correspond to the individual average association-free energy of the first, second, third, and fourth 2.5 ns segment of the 10 ns MD simulation production run.
Interactions Between Quercetin and AhR
The average per-residue interaction-free energy between AhR residues and quercetin are decomposed into polar and non-polar contributions, and interactions with average interaction-free energy values <−1.0 kcal/mol are presented in Figure 4A. In the simulation of the selected quercetin-AhR complex structure, the hydroxyl group at position 3 of quercetin forms a hydrogen bond with the ND group of His337; the hydroxyl group at position 5 of quercetin forms a hydrogen bond with the backbone N of Gly321; the hydroxyl group at position 7 of quercetin forms a hydrogen bond with the NE group of Gln383; the hydroxyl group at position 3′ of quercetin forms a hydrogen bond with the hydroxyl group of Ser365; and the hydroxyl group at position 4′ of quercetin forms a hydrogen bond with the hydroxyl groups of Ser336 and Ser346. These hydrogen bonds are indicated with black dotted lines in Figure 4B. As the binding site of AhR is highly hydrophobic, the binding of quercetin in AhR is primarily stabilized by non-polar interactions. A π-π interaction is formed between the aromatic rings of quercetin and Tyr322. In addition, Van der Waals interactions are formed between quercetin and the side-chain atoms of residues His291, Phe295, Ile325, Cys333, His337, Met340, Phe351, Ala367, Ile379, and Val381 as well as the backbone atoms of residues Gly347 and Asn366 due to their close proximity to the bound quercetin molecule.
Figure 4.

A, Average interaction-free energies (kcal/mol) decomposed into polar (orange or red) and non-polar (purple or blue) components for AhR interacting residues in complex with quercetin (first bar per residue), and in complex with apigenin (second bar per residue). The sum of the polar and non-polar components corresponds to the total average per AhR residue interaction-free energy. Only per residue interactions with total interaction free energies less than −1.0 kcal/mol are considered in the plot. The average polar and non-polar interaction-free energy component values and their standard deviation values were calculated over 4 measurements in which the first, second, third, and fourth measurement correspond to the individual average interaction-free energy component of the first, second, third, and fourth 12.5 ns segment of the 50 ns MD simulation production run. Molecular graphics images of (B) the lowest association-free energy binding mode of the quercetin: AhR complex and (C) the lowest association-free energy binding mode of apigenin: AhR complex. For both panels, the ligand is shown in licorice representation, AhR is shown in transparent cyan new cartoon representation, and key residues are shown in thin licorice representation. D, 2D diagram of hydroxyl group positions of quercetin and apigenin. The quercetin molecule has hydroxyl groups at positions 3, 5, 7, 3′, and 4′. The apigenin molecule has hydroxyl groups at positions 5, 7, and 4′. Dotted lines indicate hydrogen bonds to AhR residues. Residue names and dotted lines colored orange indicate interactions to quercetin. Residue names and dotted lines colored cyan indicate interactions to apigenin.
Interactions Between Apigenin and AhR
The average per residue interaction-free energy between AhR residues and apigenin are decomposed into polar and non-polar contributions, and interactions with interaction-free energy values <−1.0 kcal/mol are presented in Figure 4A. Residues with the largest associated interaction-free energy contributions are presented in Figure 4C. According to the simulation of the selected apigenin-AhR complex structure, the carboxyl O of apigenin forms a hydrogen bond with the backbone N of Gly321; the hydroxyl group at position 7 (according to the position labels in Figure 4D) of apigenin forms a hydrogen bond with the hydroxyl group of Ser365; and the hydroxyl group at position 4′ of apigenin forms a hydrogen bond with the backbone O of Leu293. These hydrogen bonds are indicated using black dotted lines in Figure 4C as well as cyan dotted lines in Figure 4D. Similarly to quercetin, apigenin also primarily forms hydrophobic interactions with AhR residues. A π-π interaction is formed between the aromatic rings of apigenin and Tyr322. Hydrophobic interactions are also formed between apigenin and side chain atoms of His 291, Phe295, His337, Met340, Met348, Ala367, Ile379, Val381, and Gln383 as well as backbone atoms of Lys292 and Asn366.
Interaction of Flavonoids With TCDD and Butyrate
Previous studies show that some flavonoids exhibit cell-specific AhR agonist and antagonist activities (Allen et al., 2001; Ashida et al., 2000; Ciolino et al., 1999; Ciolino et al., 1998; Kim et al., 2004b; Puppala et al., 2007; Van der Heiden et al., 2009; Xue et al., 2017; Yannai et al., 1998; Zhang et al., 2003), and results in Figure 5A show that 100 μM myricetin and gossypetin do not inhibit TCDD-induced CYP1A1 or UGT1A1 mRNA levels in Caco2 cells. The pentahydroxy flavonoids did not inhibit TCDD-induced CYP1A1 or UGT1A1 mRNA levels and, for some of these compounds, the responses were additive (Figure 5B). With the exception of 3,6,2′,3′-flavone and naringenin, all of the flavonoids with 4 or 3 hydroxyl substituents inhibited TCDD-induced CYP1A1 gene expression (Figure 5C), whereas naringenin, apigenin, and kaempferol inhibited induction of UGT1A1 by TCDD. A recent study showed that the short chain fatty acid butyrate, propionate, and acetate enhanced the activity of TCDD and other AhR agonist in Caco2 cells (Jin et al., 2017) and in this study, we also investigated interactions of butyrate with TCDD and the hydroxyflavonoids as inducers of CYP1A1 and UGT1A1 in Caco2 cells (Figure 6). Results obtained for the hexa-, penta-, and tetra-/tri-hydroxyflavonoids showed that cotreatment with butyrate enhanced flavonoid-induced CYP1A1 gene expression 2–4-fold and similar results were observed for TCDD (Figs. 6A–C). In contrast, with the exception of 5,7,3′,4′,5′-flavone, butyrate enhancement of UGT1A1 induction by the flavonoids was <2-fold and, for some compounds, no enhancement was observed. We also examined the effects of butyrate on induction of UGT1A1 by microbiota-derived AhR ligands (Cheng et al., 2015, 2017; Jin et al., 2014, 2017) and showed that DHNA-induced (but not tryptamine- or indole-induced) UGT1A1 mRNA levels were enhanced (Figure 6D). Thus, the effect of the histone deacetylase (HDAC) inhibitor butyrate on flavonoid-induced Ah-responsive genes in Caco2 cells was both gene- (ie, CYP1A1 vs UGT1A1) and structure-dependent. Using quercetin as a model, we investigated the effects of 1, 2.5, 5, and 10 μM butyrate on induction of CYP1A1, CYP1B1, and UGT1A1 and 5 or 10 μM quercetin in Caco2 cells. Both concentrations of quercetin exhibited similar inducibility and were significantly enhanced by cotreatment with 1–10 μM butyrate (Supplementary Figure 1).
Figure 5.

Hydroxyflavonoids as AhR antagonists. Caco2 cells were treated with TCDD and hexahydroxy- (A), pentahydroxy- (B), and tetra-/trihydroxy (C) flavonoids alone or in combination, and inhibition of TCDD-induced CYP1A1 or UGT1A1 gene expression was determined by real-time PCR. Results are expressed as means ± SD for 3 replicate determinations, and significant (p < .05) inhibition of TCDD-induced gene expression is indicated (*).
Figure 6.

Butyrate effects on hydroxyflavonoid Ah-responsiveness. Caco2 cells were treated with TCDD and hexahydroxy- (A), pentahydroxy- (B), and tetra-/trihydroxy (C) flavonoids alone or in combination with 5 mM butyrate, and butyrate enhanced expression of CYP1A1 or UGT1A1 gene expression was determined by real-time PCR. (D) The effects of butyrate on microbiota-derived AhR ligands on UGT1A1 expression in Caco2 cells was also determined by real-time PCR. Results are expressed as means ± SD for 3 replicate determinations, and significant (p < .05) butyrate-enhanced gene expression is indicated (*).
We also examined ligand-induced interactions of the AhR on the CYP1A1 and UGT1A1 gene promoters containing cis-acting dioxin responsive elements using a ChIP assay. TCDD induced AhR binding to the CYP1A1 promoter in Caco2 cells, whereas only minimal AhR interaction was observed after treatment with quercetin and binding induced by kaempferol and apigenin was non-detectable (Figure 7A). TCDD also enhanced H3K27ac and H4K8ac and this has previously been observed for TCDD (Jin et al., 2017) and is consistent with a more ‘open’ chromatin structure and activation of transcription. We also observed that 2 of the AhR antagonists, kaempferol and apigenin, (Figure 7B) inhibited TCDD-induced recruitment of the AhR to the CYP1A1 promoter and this is consistent with their AhR antagonist activities. Recruitment of the AhR to the Ah-responsive region of the UGT1A1 gene promoter and enhanced acetylation of H3K27 and H4K8 was also observed in Caco2 cells after treatment with TCDD (Figure 7C), whereas apigenin but not kaempferol induced AhR-DNA interactions. Both apigenin and kaempferol inhibited TCDD-induced recruitment of the AhR to the UGT1A1 promoter (Figure 7D) and this was consistent with their inhibition of induction of UGT1A1 by TCDD in Caco2 cells (Figure 6C). These studies confirm the structure-dependent AhR agonist and antagonist activities of flavonoids in Caco2 cells, and ongoing studies are focused on their in vivo activities and interactions with microbiota-derived AhR ligands.
Figure 7.

ChIP analysis of hydroxyflavonoid-induced AhR recruitment to CYP1A1/UGT1A1 gene promoters. The effects of TCDD and selected hydroxyflavonoids alone (A) and in combination with TCDD (B) on recruitment of the AhR to the Ah-responsive region of the CYP1A1 gene promoter was determined in a ChIP assay. The effects of TCDD and selected hydroxyflavonoids alone (C) and in combination with TCDD (D) on recruitment of the AhR to the Ah-responsive region of the UGT1A1 gene promoter was determined in a ChIP assay.
DISCUSSION
The health promoting effects of flavonoids and other botanical extracts have been well-documented and there is evidence that their interactions with the microbiome contribute to the overall health impacts of these compounds (Cassidy and Minihane, 2017; Espin et al., 2017; Galvez et al., 2001; Guazelli et al., 2013; Kwon et al., 2005; Nishitani et al., 2013; Ocete et al., 1998; Park et al., 2012). For example, gut microorganisms metabolically activate flavonoid and other polyphenolic conjugates by hydrolysis and these same compounds can also affect the gut microbial population (Cassidy and Minihane, 2017; Espin et al., 2017). There is also evidence that intestinal AhR and its interactions with gut microbiota-derived AhR-active metabolites such as tryptophan and its metabolites and 1,4-dihydroxy-2-naphthoic acid (DHNA) are important for protection against gut inflammatory diseases (Fukumoto et al., 2014; Hubbard et al., 2015; Lamas et al., 2016; Zelante et al., 2013). Thus, diets enriched in AhR-active compounds such as the flavonoids could also significantly alter the overall intestinal exposure to AhR ligands and modulate their potency.
Bioflavonoids have been extensively investigated as AhR ligands which exhibit both AhR agonist and antagonist activities and these compounds vary with respect to structure and also the cell context and assay used (rev. in Xue et al., 2017). One study reported the quercetin was a weak agonist/partial antagonist in human Hep G2 liver and T47D breast cancer cells expressing an Ah-responsive luciferase reporter gene (Van der Heiden et al., 2009). In contrast, quercetin exhibited maximal (equivalent to TCDD) induction of CYP1A1 mRNA and reporter gene activity in MCF7 breast cancer cells, and induced gene expression was maximal after 12 h and decreased significantly 24 h after treatment (Ciolino et al., 1999). In this study, we investigated a series of structurally-related hydroxyflavones and flavanones using Caco2 cells as a model for determining their AhR agonist/antagonist activities and impact on the gut. Surprisingly, after completion of the initial concentration-dependent induction of CYP1A1 mRNA in Caco2 cells by 14 different compounds, the most apparent structure-dependent effect was the number of hydroxyl substituents. The order of potency for induction of CYP1A1 in Caco2 cells was pentahydroxyflavones > hexahydroxyflavones > tetra-/tri-hydroxyflavones (Figure 1), and the major exception was morin, a pentahydroxyflavone, which was weakly active. The only structural difference between quercetin (highly active) and morin was the shift of the 3′-hydroxyl group (quercetin) to the 2′- position (morin), indicating the importance of hydroxyl substitution at this position for induction of CYP1A1 gene expression. SARs for induction of UGT1A1 by the pentahydroxyflavones was similar to that observed for CYP1A1, and the hexahydroxyflavones were inactive. In contrast, major differences were observed among the tetra-/tri-hydroxyflavones; kaempferol, fisetin, 3,6,2,3′- and 3,6,2,4-flavone, and naringenin did not induce UGT1A1 or CYP1A1 (Figure 2), whereas luteolin (5,7,3′,4′-) and apigenin (5,7,4′-) maximally induced UGT1A1 (compared to TCDD) but did not affect CYP1A1 expression. There are some reports showing that luteolin and apigenin can bind and activate PXR (Dong et al., 2010; Walle and Walle, 2002; Xu et al., 2015) which regulates expression of some UGTs; however, this does not explain their effects on UGT1A1 expression in wild-type Caco2 cells since neither compound induced UGT1A1 in Caco2-AhR-KO cells (Figure 5).
Previous studies show that some flavonoids including kaempferol exhibit AhR antagonist activities and inhibit TCDD-induced CYP1A1 (Xue et al., 2017) and, among the flavonoids used in this study, we observed that most of the tetra-/trihydroxyflavones inhibited TCDD-induced CYP1A1 expression in Caco2 cells (Figure 4C). 3,6,2′,3′-Tetrahydroxy-, the penta-, and hexahydroxyflavones exhibited minimal AhR antagonist activity for induction of CYP1A1 by TCDD, and similar results were observed for induction of UGT1A1. However, there were some differences between the tetra-/tri-hydroxyflavones that inhibited TCDD-induced CYP1A1 (luteolin, kaempferol, fisetin, 3,6,2′,4′-hydroxyflavone, naringenin, and apigenin) or TCDD-induced UGT1A1 (kaempferol, naringenin, and apigenin). The reason for these response-specific differences in the AhR antagonist activity of the hydroxyflavones is unclear. A recent study also reported that HDAC inhibitors such as butyrate enhanced (2–4-fold) the CYP1A1 inducibility of TCDD and several microbial-derived AhR ligands (Jin et al., 2017) and this was also observed for the hydroxyflavonoids (Figure 6). In contrast, with few exceptions (5,7,3′,4′,5′-pentahydroxyflavone and robinetin), butyrate did not enhance polyhydroxyflavone-induced UGT1A1 in Caco2 cells (Figure 6). In Caco2 cells, previous studies showed that butyrate enhanced the induction of CYP1A1, CYP1B1, TIPARP, and AhRR by microbiota metabolites; however, effects on UGT1A1 were not determined (Jin et al., 2017). Therefore, we re-examined interactions between butyrate and TCDD, DHNA, tryptamine, and indole and observed minimal enhancement of UGT1A1 induction (Figure 6D), and these results correlate with the effects observed for hydroxyflavonoids. Thus, butyrate-enhanced Ah-responsiveness in Caco2 cells treated with TCDD, microbial metabolites (Jin et al., 2017) and hydroxyflavonoids is gene-dependent and minimal for UGT1A1.
We previously used modeling studies to visualize interactions of TCDD and DHNA within the AhR binding pocket (Jin et al., 2017) and we employed a similar approach to examine AhR interactions with quercetin and apigenin which differ in AhR agonist potency (quercetin > apigenin), and apigenin is also a partial AhR antagonist. Despite the structural similarities of the 2 flavonoids, quercetin and apigenin bind to human AhR with different orientations within the binding site and accordingly, the binding of the 2 flavonoids to human AhR affect CYP1A1 transcription differently. While the binding of quercetin to AhR results in an increase in the inducibility of CYP1A1 activity, the binding of apigenin to AhR does not. The difference in which AhR residues interact strongly with quercetin and apigenin can provide insights into why these 2 compounds have different effects on CYP1A1 activities.
Quercetin interacts significantly more strongly to AhR residues Cys333, Ser336, His337, Ser346, Gly347, and Gln383 than apigenin. Hydrogen bonds are formed between the hydroxyl groups of quercetin and Ser336, His337, Ser346, and Gln383 of AhR. Previous studies suggest that Gln383 contributes to TCDD binding through hydrogen bonding (Bisson et al., 2009; Cheng et al., 2017). The orientation of quercetin within the binding site of AhR puts the compound in closer proximity to Cys333 and Gly347, resulting in a stronger Van der Waals interaction compared to apigenin. Apigenin interacts more strongly to AhR residues Lys292 and Leu293 than quercetin. A hydrogen bond is formed between apigenin and the backbone O of Leu293. In addition, the orientation of apigenin within the binding site of AhR results in its close proximity to the backbone atoms of Lys292. Because apigenin is known to have minimal CYP1A1 activity, Lys292 and Leu293 may not be critical residues for AhR mediated CYP1A1 transcription. Interestingly, previous mutational analysis and modeling of TCDD-mAhR and DHNA-mAhR interactions (Jin et al., 2017) show that TCDD and DHNA interact with many similar side chain residues within the AhR binding pocket and their major differences were primarily due to the negatively charged carboxyl group in DHNA. The TCDD-mAhR simulation corresponding to the most energetically favorable binding mode provided in our previous study was extended here to 50 ns, and was used to compare the interactions between AhR residues and TCDD to those formed between AhR residues and quercetin or apigenin (Jin et al., 2017). Prior to comparing the interactions of the 3 compounds to AhR, the sequence of the mAhR was aligned to hAhR using structural superposition. The comparative analysis suggests that stronger interactions formed between compounds and AhR (human/mouse) residues Ile325/319, Cys333/327, Ser336/330, Met348/342, Phe351/345, and Gln383/377 can potentially constitute AhR switches leading to CYP1A1 agonist activity.
In summary, our results show that the AhR agonist and antagonist activity in Caco2 cells is structure-dependent and some variability is also gene-specific (CYP1A1 vs UGT1A1). We also observed response-specific differences with respect to the effects of butyrate which preferentially enhances CYP1A1 versus UGT1A1 gene expression. Molecular modeling studies demonstrate that, compared to TCDD and DHNA, the flavonoids quercetin and apigenin interact with the same set of AhR residues within the binding pocket with few differences. The overall significance of differences in ligand-Ah receptor binding pocket interactions has previously been observed for other ligands (Flaveny et al., 2009; Soshilov and Denison, 2014; Xing et al., 2012) and this undoubtedly contributes to their activity as selective AhR modulators. Current studies are focused on flavonoid-induced responses and interactions in vivo and their impacts on intestinal inflammation.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
Texas A&M AgriLife Research, the Sid Kyle Chair Endowment; the Cancer Prevention Research Institute of Texas (RP160589); and the National Institutes of Health (R01-ES025713, R01-CA202697, R35-CA197707). A.A.O. acknowledges support from the Graduate Diversity Fellowship awarded by the Texas A&M OGAPS.
Supplementary Material
REFERENCES
- Algieri F., Rodriguez-Nogales A., Rodriguez-Cabezas M. E., Risco S., Ocete M. A., Galvez J. (2015). Botanical drugs as an emerging strategy in inflammatory bowel disease: A review. Mediators Inflamm. 2015, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen S. W., Mueller L., Williams S. N., Quattrochi L. C., Raucy J. (2001). The use of a high-volume screening procedure to assess the effects of dietary flavonoids on human cyp1a1 expression. Drug Metab. Dispos. 29, 1074–1079. [PubMed] [Google Scholar]
- Aoki T., Akashi T., Ayabe S. (2000). Flavonoids of leguminous plants: structure, biological activity, and biosynthesis. J. Plant Res. 113, 475–488. [Google Scholar]
- Ashida H., Fukuda I., Yamashita T., Kanazawa K. (2000). Flavones and flavonols at dietary levels inhibit a transformation of aryl hydrocarbon receptor induced by dioxin. FEBS Lett. 476, 213–217. [DOI] [PubMed] [Google Scholar]
- Bisson W. H., Koch D. C., O’Donnell E. F., Khalil S. M., Kerkvliet N. I., Tanguay R. L., Abagyan R., Kolluri S. K. (2009). Modeling of the aryl hydrocarbon receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands. J. Med. Chem. 52, 5635–5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R., Brooks C. L. 3rd, Mackerell A. D. Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S. et al. , . (2009). CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney M. W., Erwin K., Hardman R., Yuen B., Volz D. C., Hinton D. E., Kullman S. W. (2008). Differential developmental toxicity of naphthoic acid isomers in medaka (Oryzias latipes) embryos. Mar. Pollut. Bull. 57, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy A., Minihane A. M. (2017). The role of metabolism (and the microbiome) in defining the clinical efficacy of dietary flavonoids. Am. J. Clin. Nutr. 105, 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Jin U. H., Allred C. D., Jayaraman A., Chapkin R. S., Safe S. (2015). Aryl hydrocarbon receptor activity of tryptophan metabolites in young adult mouse colonocytes. Drug Metab. Dispos. 43, 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Jin U. H., Davidson L. A., Chapkin R. S., Jayaraman A., Tamamis P., Orr A., Allred C., Denison M. S., Soshilov A. et al. , . (2017). Editor's Highlight: Microbial-derived 1,4-dihydroxy-2-naphthoic acid and related compounds as aryl hydrocarbon receptor agonists/antagonists: Structure-activity relationships and receptor modeling. Toxicol. Sci. 155, 458–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciolino H. P., Daschner P. J., Yeh G. C. (1999). Dietary flavonols quercetin and kaempferol are ligands of the aryl hydrocarbon receptor that affect CYP1A1 transcription differentially. Biochem. J. 340, 715–722. [PMC free article] [PubMed] [Google Scholar]
- Ciolino H. P., Wang T. T., Yeh G. C. (1998). Diosmin and diosmetin are agonists of the aryl hydrocarbon receptor that differentially affect cytochrome P450 1A1 activity. Cancer Res. 58, 2754–2760. [PubMed] [Google Scholar]
- Dong H., Lin W., Wu J., Chen T. (2010). Flavonoids activate pregnane x receptor-mediated CYP3A4 gene expression by inhibiting cyclin-dependent kinases in HepG2 liver carcinoma cells. BMC Biochem. 11, 23.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryden G. W., Lam A., Beatty K., Qazzaz H. H., McClain C. J. (2013). A pilot study to evaluate the safety and efficacy of an oral dose of (-)-epigallocatechin-3-gallate-rich polyphenon E in patients with mild to moderate ulcerative colitis. Inflamm. Bowel Dis. 19, 1904–1912. [DOI] [PubMed] [Google Scholar]
- Espin J. C., Gonzalez-Sarrias A., Tomas-Barberan F. A. (2017). The gut microbiota: A key factor in the therapeutic effects of (poly)phenols. Biochem. Pharmacol. 139, 82–93. [DOI] [PubMed] [Google Scholar]
- Flaveny C. A., Murray I. A., Chiaro C. R., Perdew G. H. (2009). Ligand selectivity and gene regulation by the human aryl hydrocarbon receptor in transgenic mice. Mol. Pharmacol. 75, 1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto S., Toshimitsu T., Matsuoka S., Maruyama A., Oh-Oka K., Takamura T., Nakamura Y., Ishimaru K., Fujii-Kuriyama Y., Ikegami S. et al. , . (2014). Identification of a probiotic bacteria-derived activator of the aryl hydrocarbon receptor that inhibits colitis. Immunol. Cell Biol. 92, 460–465. [DOI] [PubMed] [Google Scholar]
- Galvez J., Coelho G., Crespo M. E., Cruz T., Rodriguez-Cabezas M. E., Concha A., Gonzalez M., Zarzuelo A. (2001). Intestinal anti-inflammatory activity of morin on chronic experimental colitis in the rat. Aliment. Pharmacol. Ther. 15, 2027–2039. [DOI] [PubMed] [Google Scholar]
- Guazelli C. F., Fattori V., Colombo B. B., Georgetti S. R., Vicentini F. T., Casagrande R., Baracat M. M., Verri W. A. Jr. (2013). Quercetin-loaded microcapsules ameliorate experimental colitis in mice by anti-inflammatory and antioxidant mechanisms. J. Nat. Prod. 76, 200–208. [DOI] [PubMed] [Google Scholar]
- Havsteen B. H. (2002). The biochemistry and medical significance of the flavonoids. Pharmacol. Ther. 96, 67–202. [DOI] [PubMed] [Google Scholar]
- Hertog M. G., Sweetnam P. M., Fehily A. M., Elwood P. C., Kromhout D. (1997). Antioxidant flavonols and ischemic heart disease in a Welsh population of men: The Caerphilly Study. Am. J. Clin. Nutr. 65, 1489–1494. [DOI] [PubMed] [Google Scholar]
- Hubbard T. D., Murray I. A., Perdew G. H. (2015). Indole and tryptophan metabolism: Endogenous and dietary routes to Ah receptor activation. Drug Metab. Dispos. 43, 1522–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugel H. M., Jackson N., May B., Zhang A. L., Xue C. C. (2016). Polyphenol protection and treatment of hypertension. Phytomedicine 23, 220–231. [DOI] [PubMed] [Google Scholar]
- Irwin J. J., Shoichet B. K. (2005). ZINC—a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin U. H., Cheng Y., Park H., Davidson L. A., Callaway E. S., Chapkin R. S., Jayaraman A., Asante A., Allred C., Weaver E. A. et al. , . (2017). Short chain fatty acids enhance aryl hydrocarbon (Ah) responsiveness in mouse colonocytes and Caco-2 human colon cancer cells. Sci. Rep. 7, 10163.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin U. H., Lee S. O., Sridharan G., Lee K., Davidson L. A., Jayaraman A., Chapkin R. S., Alaniz R., Safe S. (2014). Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol. Pharmacol. 85, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. P., Son K. H., Chang H. W., Kang S. S. (2004a). Anti-inflammatory plant flavonoids and cellular action mechanisms. J. Pharmacol. Sci. 96, 229–245. [DOI] [PubMed] [Google Scholar]
- Kim J. Y., Han E. H., Shin D. W., Jeong T. C., Lee E. S., Woo E. R., Jeong H. G. (2004b). Suppression of CYP1A1 expression by naringenin in murine Hepa-1c1c7 cells. Arch. Pharmacal Res. 27, 857–862. [DOI] [PubMed] [Google Scholar]
- Kumar S., Pandey A. K. (2013). Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 162750.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon K. H., Murakami A., Tanaka T., Ohigashi H. (2005). Dietary rutin, but not its aglycone quercetin, ameliorates dextran sulfate sodium-induced experimental colitis in mice: Attenuation of pro-inflammatory gene expression. Biochem. Pharmacol. 69, 395–406. 10.1016/j.bcp.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Lamas B., Richard M. L., Leducq V., Pham H. P., Michel M. L., Da Costa G., Bridonneau C., Jegou S., Hoffmann T. W., Natividad J. M. et al. , . (2016). CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 22, 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeJeune T. M., Tsui H. Y., Parsons L. B., Miller G. E., Whitted C., Lynch K. E., Ramsauer R. E., Patel J. U., Wyatt J. E., Street D. S. et al. , . (2015). Mechanism of action of two flavone isomers targeting cancer cells with varying cell differentiation status. PLoS ONE 10, e0142928.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani Y., Yamamoto K., Yoshida M., Azuma T., Kanazawa K., Hashimoto T., Mizuno M. (2013). Intestinal anti-inflammatory activity of luteolin: Role of the aglycone in NF-kappaB inactivation in macrophages co-cultured with intestinal epithelial cells. Biofactors 39, 522–533. [DOI] [PubMed] [Google Scholar]
- Ocete M. A., Galvez J., Crespo M. E., Cruz T., Gonzalez M., Torres M. I., Zarzuelo A. (1998). Effects of morin on an experimental model of acute colitis in rats. Pharmacology 57, 261–270. [DOI] [PubMed] [Google Scholar]
- Panche A. N., Chandra S. R., Ad D., Harke S. (2015). Alzheimer's and current therapeutics: A review. Asian J. Pharm. Clin. Res. 8, 14–19. [Google Scholar]
- Panche A. N., Diwan A. D., Chandra S. R. (2016). Flavonoids: An overview. J. Nutr. Sci. 5, e47.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M. Y., Ji G. E., Sung M. K. (2012). Dietary kaempferol suppresses inflammation of dextran sulfate sodium-induced colitis in mice. Dig. Dis. Sci. 57, 355–363. [DOI] [PubMed] [Google Scholar]
- Pietta P. G. (2000). Flavonoids as antioxidants. J. Nat. Prod. 63, 1035–1042. [DOI] [PubMed] [Google Scholar]
- Puppala D., Gairola C. G., Swanson H. I. (2007). Identification of kaempferol as an inhibitor of cigarette smoke-induced activation of the aryl hydrocarbon receptor and cell transformation. Carcinogenesis 28, 639–647. [DOI] [PubMed] [Google Scholar]
- Ross J. A., Kasum C. M. (2002). Dietary flavonoids: Bioavailability, metabolic effects, and safety. Annu. Rev. Nutr. 22, 19–34. [DOI] [PubMed] [Google Scholar]
- Salaritabar A., Darvishi B., Hadjiakhoondi F., Manayi A., Sureda A., Nabavi S. F., Fitzpatrick L. R., Nabavi S. M., Bishayee A. (2017). Therapeutic potential of flavonoids in inflammatory bowel disease: A comprehensive review. World J. Gastroenterol. 23, 5097–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambuy Y., De Angelis I., Ranaldi G., Scarino M. L., Stammati A., Zucco F. (2005). The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 21, 1–26. [DOI] [PubMed] [Google Scholar]
- Scheuermann T. H., Stroud D., Sleet C. E., Bayeh L., Shokri C., Wang H., Caldwell C. G., Longgood J., MacMillan J. B., Bruick R. K. et al. , . (2015). Isoform-selective and stereoselective inhibition of hypoxia inducible factor-2. J. Med. Chem. 58, 5930–5941. [DOI] [PubMed] [Google Scholar]
- Soshilov A. A., Denison M. S. (2014). Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol. Cell. Biol. 34, 1707–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P., Lopez de Victoria A., Gorham R. D. Jr., Bellows-Peterson M. L., Pierou P., Floudas C. A., Morikis D., Archontis G. (2012). Molecular dynamics in drug design: New generations of compstatin analogs. Chem. Biol. Drug Des. 79, 703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P., Morikis D., Floudas C. A., Archontis G. (2010). Species specificity of the complement inhibitor compstatin investigated by all-atom molecular dynamics simulations. Proteins 78, 2655–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainio M. J., Puranen J. S., Johnson M. S. (2009). ShaEP: Molecular overlay based on shape and electrostatic potential. J. Chem. Inf. Model. 49, 492–502. [DOI] [PubMed] [Google Scholar]
- Van der Heiden E., Bechoux N., Muller M., Sergent T., Schneider Y. J., Larondelle Y., Maghuin-Rogister G., Scippo M. L. (2009). Food flavonoid aryl hydrocarbon receptor-mediated agonistic/antagonistic/synergic activities in human and rat reporter gene assays. Analytica Chimica Acta 637, 337–345. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I. et al. , . (2010). CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K., MacKerell A. D. Jr. (2012). Automation of the CHARMM general force field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezza T., Rodriguez-Nogales A., Algieri F., Utrilla M. P., Rodriguez-Cabezas M. E., Galvez J. (2016). Flavonoids in inflammatory bowel disease: A review. Nutrients 8, 211.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walle U. K., Walle T. (2002). Induction of human UDP-glucuronosyltransferase UGT1A1 by flavonoids-structural requirements. Drug Metab. Dispos. 30, 564–569. [DOI] [PubMed] [Google Scholar]
- Xing Y., Nukaya M., Satyshur K. A., Jiang L., Stanevich V., Korkmaz E. N., Burdette L., Kennedy G. D., Cui Q., Bradfield C. A. (2012). Identification of the Ah-receptor structural determinants for ligand preferences. Toxicol. Sci. 129, 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Luo M., Jiang H., Yu L., Zeng S. (2015). Involvement of CAR and PXR in the transcriptional regulation of CYP2B6 gene expression by ingredients from herbal medicines. Xenobiotica 45, 773–781. [DOI] [PubMed] [Google Scholar]
- Xue Z., Li D., Yu W., Zhang Q., Hou X., He Y., Kou X. (2017). Mechanisms and therapeutic prospects of polyphenols as modulators of the aryl hydrocarbon receptor. Food Funct. 8, 1414–1437. [DOI] [PubMed] [Google Scholar]
- Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y. (2015). The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yannai S., Day A. J., Williamson G., Rhodes M. J. (1998). Characterization of flavonoids as monofunctional or bifunctional inducers of quinone reductase in murine hepatoma cell lines. Food Chem. Toxicol. 36, 623–630. [DOI] [PubMed] [Google Scholar]
- Zelante T., Iannitti R. G., Cunha C., De Luca A., Giovannini G., Pieraccini G., Zecchi R., D’Angelo C., Massi-Benedetti C., Fallarino F. et al. , . (2013). Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385. [DOI] [PubMed] [Google Scholar]
- Zhang S., Qin C., Safe S. H. (2003). Flavonoids as aryl hydrocarbon receptor agonists/antagonists: Effects of structure and cell context. Environ. Health Perspect. 111, 1877–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.