

Abstract
The 356 nt noncoding satellite RNA C (satC) of Turnip crinkle virus (TCV) is composed of 5′ sequences from a second TCV satRNA (satD) and 3′ sequences derived from TCV. SHAPE structure mapping revealed that 76 nt in the poorly-characterized satD-derived region form an extended hairpin (H2). Pools of satC in which H2 was replaced with 76, 38, or 19 random nt were co-inoculated with TCV helper virus onto plants and satC fitness assessed using in vivo functional selection (SELEX). The most functional progeny satCs, including one as fit as wild-type, contained a 38-39 nt H2 region that adopted a hairpin structure and exhibited an increased ratio of dimeric to monomeric molecules. Some progeny of satC with H2 deleted featured a duplication of 38 nt, partially rebuilding the deletion. Therefore, H2 can be replaced by a 38-39 nt hairpin, sufficient for overall structural stability of the 5′ end of satC.
Keywords: Turnip crinkle virus, satellite RNA, viral RNA evolution, SELEX
Introduction
The past fifteen years have led to a growing understanding of the structure and function of noncoding RNAs. This group of RNAs includes cellular microRNAs and also entities such as viroids and a subset of plant virus-associated satellite (sat) RNAs (Tsagris et al., 2008; Simon et al., 2004). Viroids replicate and traverse the plant independent of any accompanying virus, whereas satRNAs require a helper virus for replication and movement. Since noncoding satRNAs can intensify or attenuate the symptoms of their helper virus (Simon et al., 2004), these properties must be mediated by their primary sequence and/or higher-order structure.
Viral satRNAs typically do not share sequence similarity with the genomic (g)RNA of their helper virus, though exceptions have been found. For example, the 356 nt satC of Turnip crinkle virus (TCV) is a recombinant RNA composed of the nearly full-length sequence from a second TCV satRNA (satD) in its 5′ half, followed by two regions derived from the 3′ end of TCV gRNA in its 3′ half (Fig. 1A; Simon and Howell, 1986). When satC (or other recombinant molecules) was first generated, alterations likely occurred over time that allowed the original satD molecule to adapt to its newly acquired 3′ sequences. For example, since the 3′ region of TCV that is shared with satC contains elements required for translation, untranslated satC when first generated would have been free to modify these elements to support satRNA-specific functions, such as replication and mutualistic association with its helper virus.
FIG. 1.
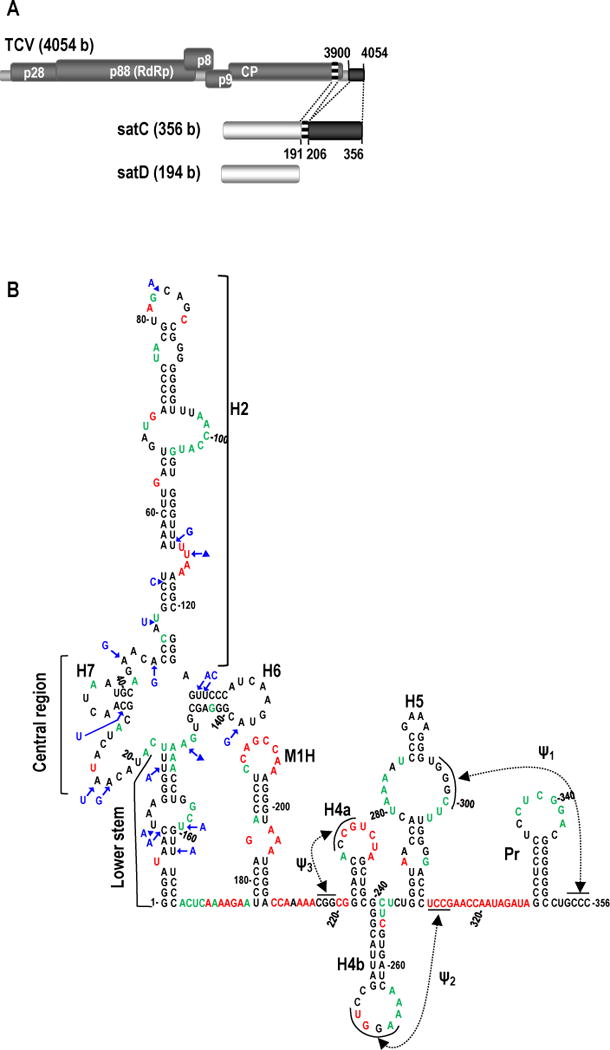
Origin and structure of TCV satC. (A) Top, TCV gRNA is 4054 bases (b) and contains five open reading frames. Replication is mediated by p28 and the RdRp readthrough protein, p88. Cell-to-cell movement is controlled by p8 and p9 (translated from a subgenomic RNA, not shown). CP (translated from a second subgenomic RNA, not shown) encodes the coat protein. Middle, the 356 b satC is composed of nearly all of the 194 b satD RNA (bottom) and two regions from the TCV 3′ end as illustrated by similar shading/patterns and the dotted lines between TCV and satC (Simon and Howell, 1986). (B) Model of wt satC structure as determined by SHAPE and mFold computational predictions. Nucleotides in red are strongly flexible; nucleotides in green are moderately/weakly flexible. Location of the lower stem (nt positions 1-17 and 150-166), central region (positions 18-47 and 124-149), and H2 region (positions 48-123) in the 5′ portion of satC are indicated. Blue residues denote how satD differs from satC in the extended hairpin region.
To gain an understanding of the evolution of a recombinant molecule such as satC, a detailed understanding of parental and progeny RNA structure and function is required. Using a combination of methods including computational analysis, solution structure mapping, compensatory mutagenesis, and in vivo functional selection, the satC sequences derived from TCV have been structurally and functionally mapped. All results were consistent with both satC and TCV 3′ regions having four hairpins (from 3′ to 5′: Pr, H5, H4b, and H4a) and three pseudoknots (for a review, see Simon, in press). In TCV, the Pr is a weak promoter and a key element in ribosomal-readthrough that generates the RNA-dependent RNA polymerase (Song and Simon, 1995; Sun and Simon, 2006; Cimino et al., 2011). In satC, this hairpin has been significantly changed by both base deletions and alterations and serves as the core promoter for satRNA minus-strand synthesis. Hairpin H5 is a critical hairpin for replication of both TCV and satC, and part of its internal symmetrical loop forms a pseudoknot (Ψ1) with four 3′-end residues (Zhang et al., 2004a; Zhang and Simon, 2005; Zhang et al., 2006c). In TCV, H5 also may be a chaperone for the viral RdRp (McCormack and Simon, 2004). In TCV but not satC, H5 along with H4a, H4b, Ψ2 (a pseudoknot formed from sequences immediately 3′ of H5 and in the loop of H4b) and Ψ3 (a pseudoknot between the loop of H4a and upstream adjacent sequence) fold into a T-shaped structure (TSS; McCormack et al., 2008) that binds to ribosomes and serves as a cap-independent translation enhancer (Stupina et al., 2008). This region in satC is not predicted to form a TSS and no longer binds efficiently to ribosomes due to six positional differences with the TCV parental sequence (Guo et al., 2011). However, hairpins H5, H4a, H4b and pseudoknot Ψ2 are important for satC accumulation (Zhang et al., 2006c), and Ψ3 appears to be important for satC fitness (Guo et al., 2011).
Hairpin exchanges with the TCV-related Cardamine chlorotic fleck virus (CCFV) revealed that H4a and H4b are a functional unit in satC (Zhang et al., 2006c). Using in vivo SELEX (systematic evolution of ligands by exponential enrichment [Ellington and Szostak, 1990; Tuerk and Gold, 1990]) to select for functional satC from an initial population with random sequences in the 45 nt H4a+H4b region, satC progeny were recovered that either retained two stem-loops or contained a single hairpin in the region, with both configurations maintaining Ψ2 (Guo et al., 2009). This plasticity in satC H4a+H4b likely is possible because satC lacks the translational function of this region that is critical for TCV.
A small portion of the satD 3′ terminal sequence in the central region of satC and two regions from TCV gRNA altogether form the M1H stem-loop (nucleotide [nt] positions 178-209). In satC (-)-strands, M1H functions as a replication enhancer (Nagy et al., 2001). In satC (+)-strands, M1H acts as a spacer element to bridge flanking sequences, which regulates fitness of satC in plants by repressing accumulation of TCV virions in an unknown manner (Sun and Simon, 2003; Zhang and Simon, 2003b). The resulting free coat protein, the TCV silencing suppressor (Qu et al., 2003; Thomas et al., 2003), therefore can better suppress RNA silencing, enhancing helper virus movement in infected plants (Zhang and Simon, 2003a).
Unlike the well characterized TCV-derived 3′ portion of satC, little is known about the 5′ half of satC that has maintained 90% sequence similarity with the parental satD sequence (Simon and Howell, 1986). As such, understanding how satC (as a representative recombinant molecule) uses or has modified its parental satD-derived sequence for its own requirements is needed to both develop an overall functional map for satC and to understand how recombinant molecules adapt to a new combination of sequences. The H4a+H4b in vivo SELEX revealed that functional satC progeny could be recovered following randomization of a large region (13%), if sequence specificity is not a major requirement for function (Guo et al., 2009). In the current study, a 76 nt hairpin (H2; 21% of satC) in the 5′ region was replaced with 76, 38, or 19 randomized nt to assess the importance of the wild-type (wt) sequence, sequence length, and a hairpin in this region. Results indicate that a substitute sequence consisting of 38 or 39 nt is the preferred outcome of all SELEX experiments as well as an in vivo passaging experiment in which H2 was fully deleted. One satC with a 39 nt H2 region that folds into a hairpin structure was at least as fit as wt satC for accumulation with TCV gRNA in plants. This implies that wt H2 functions as a spacer element, yet its current size appears to be an evolutionary remnant from satD.
Results and Discussion
SHAPE structure probing of wt satC
The structure of the TCV-derived 3′ 140-nt region of satC was previously determined by biochemical probing and genetic analyses (Zhang et al., 2006a; Zhang et al., 2006b). Both methods suggested that the structure exists in two conformations: i) a basal, pre-active conformation that lacks H5, Pr, and Ψ1 and forms in vitro when the RNA transcripts contain precise 5′ and 3′ ends; and ii) an “active structure” conformation that includes hairpins and pseudoknots known to exist in this TCV-derived region. The latter structure forms when the 3′ terminus is extended with non-template residues, or when 2 to 3 residues at the 5′ or 3′ termini are deleted. For the current study, selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE; Merino et al., 2005) was used to determine the structure of full-length satC including the 5′ portion derived from satD. Since SHAPE analysis requires primer extension to evaluate flexible (NMIA-reactive) and non-flexible residues, transcripts of satC contained additional non-template residues to allow for primer binding when assessing the 3′ region of the satRNA.
As shown in Fig. 1B, the structure of satC in the 3′ region was similar to the previously defined active conformation that also is present in the comparable region in TCV (McCormack et al., 2008). Differences included unpaired residues at the 3′ base of the H4b stem and apparently poor formation or detection of Ψ2. Although Ψ2 is present in, and stabilizes, the satC pre-active structure, its existence in the active structure has not been confirmed (Zhang et al., 2006b). The lack of flexibility of guanylates on the 3′ side of the large symmetrical loop in H5 suggests the formation of Ψ1. Hairpin M1H flanked by single stranded sequences also was clearly defined.
In the 5′ portion of satC (positions 1-166), most residues were non-reactive to NMIA suggesting a highly structured domain. The secondary structure that best fits the SHAPE data, which also is very similar to the lowest free energy conformation predicted by mFold (Zuker, 2003), is consistent with a long stem-loop structure consisting of a lower stem connected to a central region containing few flexible bases (but without significant possibilities for Watson-Crick base-pairing), topped by a large 76 nt hairpin designated H2 (positions 48-123). The inflexible central region was predicted by mFold to contain small hairpin H7 (positions 31-41) upstream of H2, and hairpin H6 (positions 125-145) downstream of H2. If these two hairpins exist, the lack of any significant flexibility in their terminal loops and in the sequences surrounding H7 suggest that tertiary interactions reduce the flexibility of these sequences. In contrast, the presence of SHAPE reactive residues in most of the loops and bulges in H2 suggest that any higher-order structures in H6 and H7 may not extend significantly into this upper hairpin. Although the existence of H6 and H7 has not been confirmed, preliminary data from additional unpublished SELEX experiments support their interaction. For example, most satC recovered after randomizing just the H6 loop were wt, suggesting that when H7 is wt, the H6 loop also needs to be wt (D.B.K., unpublished). In contrast, wt H6 loop sequences were not recovered after SELEX of the H6 loop, H7, and sequences surrounding H7 (D.B.K., unpublished). Future experiments using compensatory mutations in H6 and H7 will be needed to provide firm evidence for this interaction. For simplicity, the putative H6 and H7 hairpins will be shown in the structures presented in this report.
Figure 1B also depicts the evolution of the original satD sequence to wt satC. Only five changes occurred in H2 (three single nt deletions, one nt insertion, and one base change) with only two (the deleted C at position 56, and the U to G transversion at position 112) affecting canonical base-pairing in the stem. These apparently minor alterations suggest that satD also has hairpin H2 in this location. The central region contains eight single base alterations and one base insertion. Of these changes, three are in the putative stems of H6 and H7, all of which are predicted to reduce the stability of these hairpins in satD as compared to satC. Five alterations (one deleted base and four base changes) are in the lower stem, with at least two predicted to reduce the stability of the stem. These alterations make it less likely that satD also contains the lower stem.
satC with a deletion of H2 is functional but not stable in planta
Since satC is a recombinant molecule composed of satD and TCV sequence, the question arose as to how much of the satD sequence is present for “historical” reasons and how much is actually required for satC viability? To begin addressing this question, the 76 nt hairpin H2 (positions 48-123) was deleted (satCΔH2) to determine: i) how the absence of H2 affects the flexibility of residues in the central region and lower stem (i.e., if H2 represents a separate RNA domain); ii) if satC can accumulate in the absence of 21% of its sequence; and iii) if accumulation of satCΔH2 is limited, whether or not and how quickly satC can adapt and recover from the loss of this sequence.
SHAPE of satCΔH2 transcripts revealed no significant changes to the lower stem compared to wt satC, with the exception of the loss of flexibility of the uracil residue in position 5 (Fig. 2C, left). In contrast with wt satC, satCΔH2 exhibited substantial flexibility in its H6 and H7 loop residues, suggesting that, at least in this truncated molecule, these hairpins exist and tertiary interactions are abolished. In addition, the residues upstream of H7 (positions 19-30) became nearly uniformly flexible. This result indicates that the proposed tertiary interactions in the central region of the satD-derived domain are dependent on the presence of H2.
FIG. 2.
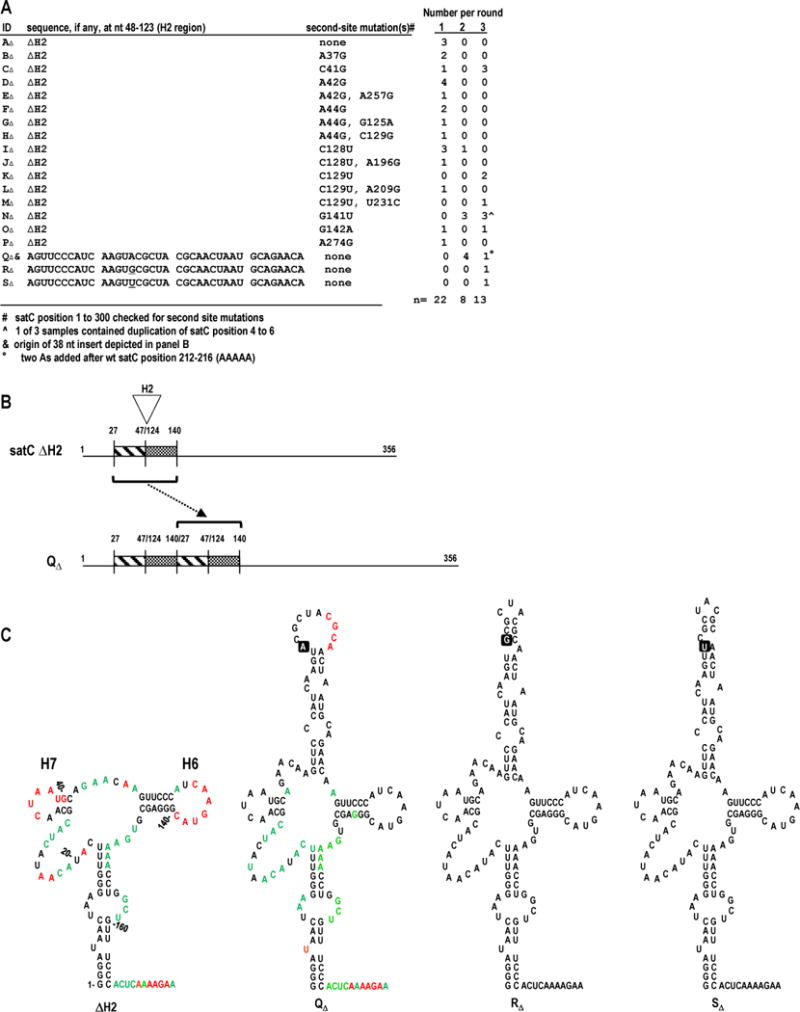
Progeny generated during in planta passaging of satC with H2 deleted (ΔH2) recover 38 nt in the H2 region. (A) satCΔH2 transcripts were mixed with TCV genomic RNA and passaged through 30 plants (Round 1). RNA was extracted, pooled, and passaged through 6 plants two sequential times (Rounds 2 and 3). satC progeny were cloned following each round. Samples AΔ through PΔ retained the deletion. Second-site mutations in samples BΔ through PΔ are noted. (B) Model depicting duplication leading to QΔ. Duplication (bracket) of nt 27-47 and 124-140 (H2-flanking sequences) from satCΔH2 resulted in a 38 nt H2 region. (C) Secondary structure predictions for the 5′ region of samples satCΔH2, QΔ, RΔ, and SΔ. SHAPE results are shown on the structures of satCΔH2 and QΔ (see Fig. 1B for nucleotide coloring). The single-nucleotide difference among QΔ, RΔ, and SΔ is boxed. Numbering is according to wt satC.
Transcripts of satCΔH2 were inoculated with TCV gRNA onto thirty turnip plants and allowed to establish an infection for three weeks. At this point, total RNA was harvested from new leaves, pooled, and inoculated onto six new seedlings; this procedure then was repeated for a third round of passaging through six plants. After each round, satCΔH2 progeny molecules were cloned and sequenced.
After the first round, 22 satCΔH2 clones contained 13 different sequences (clones AΔ through JΔ, LΔ, OΔ, and PΔ; Fig. 2A). Twelve of these 13 sequences contained either one or two second-site mutations between positions 37 and 274. After round 1, all recovered satCΔH2 progeny contained second-site mutations. Six of the fifteen unique second-site changes were in between positions 125 and 145, and all are predicted to reduce the stability of the putative H6 stem. Four of the second-site changes were in, or just downstream of, putative hairpin H7. Most of these alterations were predicted to improve possible base-pairing between this region and sequences within H6. For example, A37G resulted in the sequence 37-GAUG-40 which can pair with 130-CAUC-133; C41G resulted in the sequence 38-AUGG-41 which can pair with 129-CCAU-132; and A42G resulted in the sequence 42-GGAAC-46 which can pair with 125-GUUCC-129. These second-site mutations likely were acquired in response to the absence of H2 since previous sequencing of 25 progeny of wt satC transcripts revealed no sequence alterations after three weeks in planta (Zhang et al., 2004b). The location of the second-site changes suggests a benefit from alterations near the H2 deletion to adjust for the absence of the 76 nt hairpin.
In round 2, several satCΔH2 progeny contained a 38 nt duplication of sequence between positions 27 and 140 (clone QΔ; position numbers reflect their original locations in wt satC; Fig. 2B). SHAPE structure mapping of QΔ (Fig. 2C) revealed that the 38 nt duplication folds into a hairpin that maintains H6 and H7, and that unlike satCΔH2, nearly all of H6 and H7 now are inflexible, similar to what was seen for H6 and H7 in wt satC (Fig. 1B). In addition, no major structural variations from wt satC were found in the lower stem. Recovery of the proposed tertiary interactions in the H6/H7 region in QΔ strongly suggest that they are dependent on a hairpin in the H2 location, but independent of the sequence/structure of the replaced hairpin. In contrast, the unpaired sequence upstream of H7 (positions 19-30) continued to exhibit enhanced flexibility compared with wt satC, similar to satCΔH2. This suggests that the structure of these residues is affected by the sequence/structure in the H2 location.
Two QΔ-derived progeny (RΔ and SΔ) had different single base alterations at the same position within the duplicated sequence. Both base alterations were predicted to stabilize the recovered hairpin by increasing base-pairing in the upper stem (Fig. 2C). Taken together, these data suggest that while satCΔH2 is capable of accumulation and movement through plants, stability likely requires second-site mutations or duplicated sequences that generate a new stem-loop in the H2 location.
In vivo evolution of satC with 76 random nt in place of the H2 region results in satC progeny with a truncated 38 nt H2-replacement sequence
The results from Fig. 2 indicate that a hairpin in the H2 region does not need to be wt length (76 nt) for satC accumulation in plants. Therefore, we investigated the fitness of satC containing H2 of different lengths, compositions, and structures by replacing H2 with different lengths of random sequences. In vivo functional selection was first performed on a pool of satC transcripts containing 76 randomized nt in place of H2 (satC76). It is important to note that the pool is not saturating (the pool does not possess a satC for every possible 76 nt permutation in H2) and severe bottlenecks occur when the RNAs are first inoculated into plants (the satRNA must be present in the same cells that receive the helper virus). However, the goal is to start with a large number of combinations for H2 and only the very few functional satC within the pool will move through plants and ultimately be cloned. These satC likely will possess critical sequence and/or structural features, allowing for improved understanding of the role of H2 for satC movement in plants and replication. As such, satC76 transcripts were mixed with TCV gRNA and inoculated onto 30 turnip plants. Progeny accumulating in new leaves at three weeks post-inoculation were cloned. Six new seedlings were inoculated with pooled RNA extracted from the 30 round 1 plants. Subsequent cloning of progeny and inoculation with pooled RNA was repeated for a total of five rounds.
As depicted in Fig. 3A, 18 of 30 round 1 satC76 progeny (clones A, C through R, and AA) either retained a 76 nt H2 replacement sequence (A, and C through M), or contained a reduced-length H2 replacement sequence (N through R, and AA). For example, N contained 75 nt, O had 57 nt, closely-related clones P through R contained 38 nt, and AA was 31 nt. Notably, after all progeny extracted from the 30 round 1 plants were pooled and inoculated onto 6 new seedlings, 9 of 10 round 2 progeny contained a 38 nt H2, and only one (clone B) retained a full-length 76 nt H2 region similar to clone A. By round 3, all recovered satC76 progeny contained a 38 nt H2 region, and all of these satCs were derived from a single selected sequence. At the end of round 5, closely related samples S and Z were the only satC76 progeny recovered and therefore were termed satC76 SELEX winners.
FIG. 3.
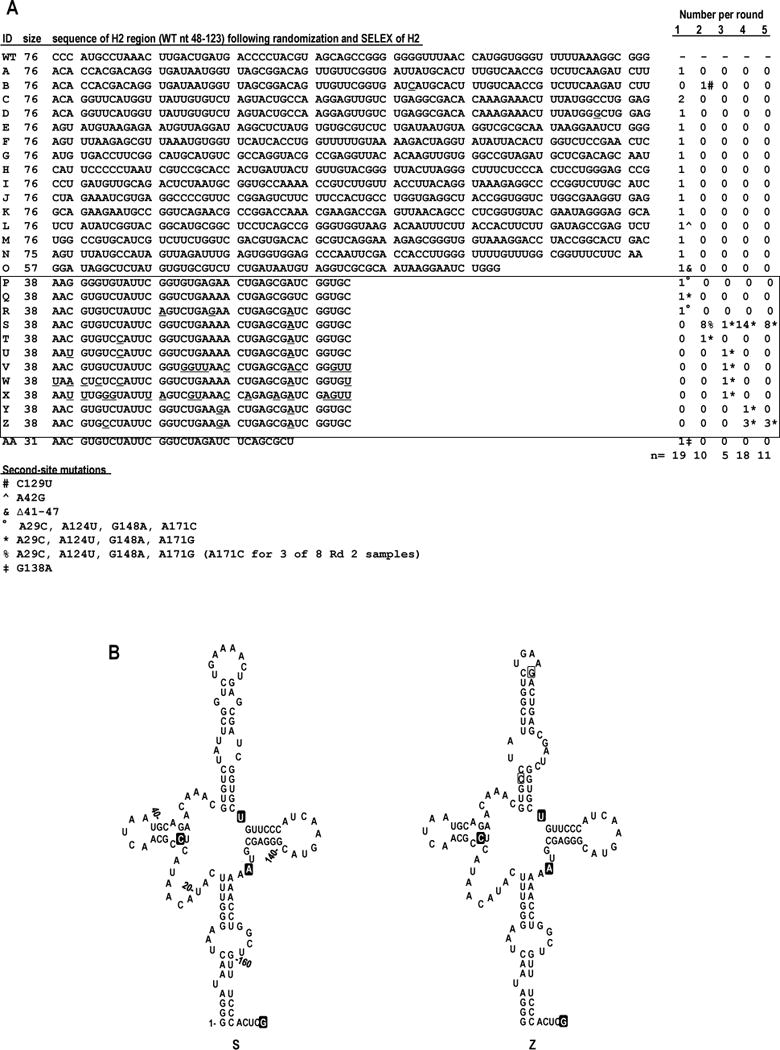
In vivo SELEX of satC76. (A) Results of five rounds of passaging satC with 76 random bases replacing H2 (satC76). Sequences related to the two nearly identical, most functional (“winning”) satCs (samples S and Z) are boxed. Underlined nt denote differences between related satC clones. Second-site mutations are identified by symbols and defined at the bottom of the figure. For reference, WT sequence for satC nt 48-123 is shown above sample A. (B) Secondary structure models for the 5′ region of SELEX winners S (left) and Z (right). Nucleotides within a filled box denote second-site mutations. Open boxed nucleotides in clone Z denote differences between S and Z.
Bases corresponding to wt positions 1-171 in the 5′ region of S and Z were subjected to mFold analysis. S, first detected in round 2, contained H2 selected sequence that was predicted to fold into a hairpin, and also contained three single second-site mutations in the central region, with one (A29C) predicted to extend the H7 stem (Fig. 3B, left). Z, first detected in round 4, contained two additional base changes (compared with S) that were predicted to stabilize the H2 replacement stem-loop (Fig. 3B, right). In addition to S and Z, all satC76 progeny that contained an H2 region shorter than 75 or 76 nt (clones O through AA) possessed second-site mutations. As described above, prominent second-site mutation A29C, found in both SELEX winners S and Z, was predicted to lengthen the stem of hairpin H7 (Fig. 3B). These results suggest that usage of shortened H2 hairpins is assisted by second-site mutations that compensate for the reduced length of the hairpin in the H2 region.
In vivo evolution of satC with 38 random nt in place of the H2 region results in functional satC progeny that maintain the size of the 38 nt replacement
Since 3 of the 13 samples cloned following round 3 of the satCΔH2 experiment (Fig. 2A) and the two SELEX winners from the satC76 experiment (Fig. 3A) each possessed evolved 38 nt H2 regions, this suggested that satC might prefer a half-sized (38 nt) H2 region. Therefore, satC with 38 random nt replacing wt H2 (satC38) was subjected to in vivo SELEX. Pools of satC38 were passaged with TCV gRNA through plants for a total of 5 rounds. Nineteen of 21 satC38 progeny isolated from round 1, and all satC38 progeny isolated in subsequent rounds, contained a 38 nt H2 region (Fig. 4A; exceptions in round 1 were apparent wt satC cloning contaminant Ah, and clone Vh that had a 37 nt H2 region). Of the three satC38 SELEX winners in round 5, Zh had no second-site mutations and Wh and DDh had only single second-site mutations. Zh and DDh contained sequences in the H2 region that were predicted to form a hairpin similar to wt satC and to satC76 winners S and Z (Fig. 4B, center and right). The A29C second-site change in DDh was the same putative H7-extending second-site mutation in satC76 SELEX winners S and Z (Fig. 3B), which suggested that this second-site mutation arises for reasons independent of the composition of the H2 replacement sequence. SHAPE RNA structure probing of Wh transcripts suggested that the H2 replacement sequence in Wh folds into a three-way branched structure with two hairpins of differing lengths (Fig. 4B, left). The C32U second-site mutation in Wh was predicted to alter and extend the length of hairpin H7 (Fig. 4B), similar to the suggested consequence of A29C in DDh. Interestingly, this change in H7 was accompanied by increased flexibility of both a guanylate at the base of hairpin H6 (G125) and the adjacent upstream adenylate (A124). The flexibility of sequence downstream of these residues was unchanged compared to wt satC. The new structure that replaced H2 also was accompanied by enhanced flexibility of residues in positions 21-26, supporting the hypothesis that the structure of these residues in wt satC is dependent on wt H2. Recovery of different H2 replacement sequences in Wh, Zh, and DDh, as well as in S, Z, and QΔ, suggests that satC can support a half-sized hairpin of variable sequence and structure in the H2 region.
FIG. 4.
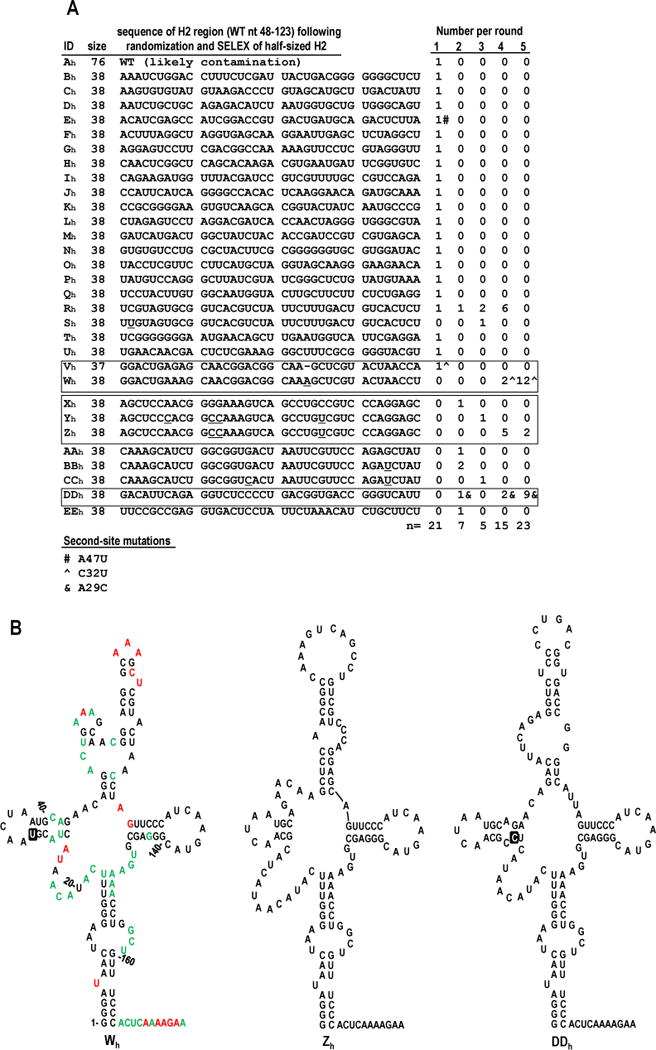
In vivo SELEX of satC38. (A) Results of five rounds of passaging satC with 38 random bases replacing H2 (satC38). Sequences related to the three distinct “winning” satCs (samples Wh, Zh, and DDh) are boxed. Underlined nt denote differences between related satC clones. Second-site mutations are identified by symbols and defined at the bottom of the figure. (B) Secondary structure models for the 5′ region of SELEX winners Wh, Zh, and DDh. Results of SHAPE structure probing are shown on the model of Wh (see Fig. 1B for nucleotide coloring). Boxed nucleotides in Wh and DDh denote second-site mutations.
In vivo evolution of satC with 19 random nt in place of the H2 region results in functional satC progeny with an expanded sequence in the H2 region
To determine if a 19 nt H2 region is stable in satC, in vivo evolution of satC with 19 random nt replacing wt H2 (satC19) was performed. Pools of satC19 were passaged with TCV gRNA through plants for 5 rounds, and progeny were cloned and sequenced as described above. After the first round, 21 progeny clones were recovered (Fig. 5A; clones Aq through Uq), each retaining 19 nt in the H2 region. Similar results were found for 8 of the 10 clones after round 2 (clones Vq through AAq). The two exceptions, clones BBq and DDq, respectively contained 38 nt and 58 nt in the H2 region. The expanded H2 region in clone DDq, which arose from sample CCq with a 19 nt insert, contained a duplication of the region from positions 37 through 132 (positions according to wt satC), including the 19 nt CCq SELEX insert (Fig. 5B). Although DDq was cloned (round 2) before CCq (round 3; Fig. 5A), it still is likely that DDq (58 nt H2 region) arose from CCq (19 nt H2 region) because (i) all clones from round 1 were 19 nt (not larger) so it is unlikely that a satC with a 58 nt H2 region with a direct repeat (DDq) was present prior to a satC with the single copy of the region (CCq), and (ii) the small number of clones obtained is not saturating. Clone IIq (and related JJq), recovered in rounds 3 through 5, had a 45 nt H2 region consisting of an apparent duplication of the 19 nt SELEX sequence and extending through position 131 (Fig. 5C). Clone HHq contained a perfect duplication of the 19 nt SELEX sequence from round 5 winner GGq, with one adenylate of unknown origin connecting the duplicated sequences (Fig. 5D).
FIG. 5.


In vivo SELEX of satC19. (A) Results of five rounds of passaging satC with 19 random bases replacing H2 (satC19). Sequences related to the four distinct “winning” satCs (samples FFq, GGq, HHq, and IIq) are boxed. Underlined nt denote differences between related satC clones. Second-site mutations are identified by symbols and defined at the bottom of the figure. (B-D) Models for sequence expansion in DDq, IIq, and HHq. (E) Secondary structure models for the 5′ region of SELEX winners FFq, GGq, HHq and IIq. Results of SHAPE structure probing are shown on the model of HHq (see Fig. 1B for nucleotide coloring). Boxed nucleotides in GGq and HHq denote second-site mutations.
Nearly all of the round 1 through 5 winners contained selected sequences that were predicted to form a hairpin structure (Fig. 5A, 5E). The hairpin predicted for GGq, the most prevalent sequence cloned in round 5, required upstream sequence from H7 to form the hairpin. Recovery of clones BBq, DDq, EEq, HHq, IIq, and JJq (all with enlargements of the 19 nt H2 replacement sequence), and especially the late evolution of GGq to HHq in which the 19 nt selected sequence in GGq was duplicated, suggests that a shorter hairpin in the H2 region might limit the fitness of satC. The result from an in vivo competition experiment between equal amounts of HHq, GGq, and FFq (described below) supports this idea.
SHAPE structure probing conducted on winner HHq revealed strong similarities with QΔ and Wh, and also several unique features. As with QΔ and Wh, the structure in the lower stem was unchanged from wt satC and the residues within H6 and H7 were mainly inflexible (Fig. 5E). However, in the unpaired sequence upstream of H7, which was mainly flexible in QΔ and Wh, only positions 21-23 had enhanced flexibility compared with wt satC. In addition, the sequence just upstream from the H2 replacement sequence (positions 46-47), which was inflexible in wt satC, QΔ, and Wh, exhibited major new flexibility in HHq. Altogether, the structures of HHq, QΔ, and Wh revealed: i) the lower stem is independent of sequence and/or structural alterations in the H2 and central regions; ii) base inflexibility in the H6 and H7 regions is dependent on a hairpin in the H2 region but independent of the sequence/structure of the hairpin; and iii) base flexibility in the sequence between positions 18-30 is dependent on specific sequence and/or structure in the H2 region.
In planta competition experiments reveal that clone HHq (39 nt H2) is at least as fit as wt satC
Direct competition experiments were conducted to determine which of the selected satC winners was the most fit to engage in a mutualistic association with the helper virus in plants (Table 1). Competition A compared the three winners from the satC38 SELEX (Wh, DDh, and Zh). Three weeks after inoculation of three plants with equal amounts of transcripts of the three winners and TCV gRNA, 10 of the 12 cloned progeny were Wh and the remaining two were DDh. Three of the winners from the satC19 SELEX (HHq, GGq, and FFq) were assessed in competition B, which resulted in a clear winner in 39 nt HHq (19/21 clones). Since GGq and FFq each only had a 19 nt H2 region, the finding that HHq won this competition (despite GGq having been cloned more often in round 5) is consistent with the importance of enhanced structural stability via the rapid expansion of H2 observed during the course of the satC19 SELEX. Competition C paired HHq against fellow satC19 SELEX winner IIq, with recovery of only HHq (36/36). Competition D, between HHq, Wh, and satC76 winner S, resulted in recovery of only HHq (16/16). These competition experiments thus clearly identified HHq as the overall SELEX winner among the clones tested.
Table 1.
Results of in planta competition experiments
| Competition A: satC38 SELEX winners | n | ||||
|
| |||||
| ID | Size | Sequence at positions 48-123 | |||
| Wh | 38 | GGACUGAAAGCAACGGACGGCAAAGCUCGUACUAACCU | 10 | ||
| DDh | 38 | GACAUUCAGAGGUCUCCCCUGACGGUGACCGGGUCAUU | 2 | ||
| Zh | 38 | AGCUCCAACGGCCAAAGUCAGCCUGUCGUCCCAGGAGC | 0 | ||
|
| |||||
| Competition B: satC19 SELEX winners | n | ||||
|
| |||||
| ID | Size | Sequence at positions 48-123 | |||
| HHq | 39 | GAGAUCCGGCGUGAAUCAUAGAGAUCCGGCGUGAAUCAU | 19 | ||
| GGq | 19 | GAGAUCCGGCGUGAAUCAU | 1 | ||
| FFq | 19 | UGCGGUAUCAUUCAACUGC | 1 | ||
|
| |||||
| Competition C: satC19 SELEX winners | n | ||||
|
| |||||
| ID | Size | Sequence at positions 48-123 | |||
| HHq | 39 | GAGAUCCGGCGUGAAUCAUAGAGAUCCGGCGUGAAUCAU | 36 | ||
| IIq | 45 | UCUGGAAAGUCUUUUUUUGGUAGUUCCCAGAAAGUCUUUUUUGGU | 0 | ||
|
| |||||
| Competition D: satC76 winner S vs. Wh and HHq | n | ||||
|
| |||||
| ID | Size | Sequence at positions 48-123 | |||
| HHq | 39 | GAGAUCCGGCGUGAAUCAUAGAGAUCCGGCGUGAAUCAU | 16 | ||
| Wh | 38 | GGACUGAAAGCAACGGACGGCAAAGCUCGUACUAACCU | 0 | ||
| S | 38 | AACGUGUCUAUUCGGUCUGAAAACUGAGCGAUCGGUGC | 0 | ||
|
| |||||
| Competition E: Wh, DDh, Zh, S | n | ||||
|
| |||||
| ID | Size | Sequence at positions 48-123 | |||
| Wh | 38 | GGACUGAAAGCAACGGACGGCAAAGCUCGUACUAACCU | 5 | ||
| DDh | 38 | GACAUUCAGAGGUCUCCCCUGACGGUGACCGGGUCAUU | 0 | ||
| Zh | 38 | AGCUCCAACGGCCAAAGUCAGCCUGUCGUCCCAGGAGC | 0 | ||
| S | 38 | AACGUGUCUAUUCGGUCUGAAAACUGAGCGAUCGGUGC | 0 | ||
|
| |||||
| Competition F: satCΔH2 progeny winners | n | ||||
|
| |||||
| ID | Size | Sequence at positions 48-123 | |||
| QΔ | 38 | AGUUCCCAUCAAGUACGCUACGCAACUAAUGCAGAACA | 19 | ||
| AΔ | 0 | ΔH2 | 0 | ||
|
| |||||
| Competition G: Wh and QΔ | n | ||||
|
| |||||
| ID | Size | Sequence at positions 48-123 | |||
| Wh | 38 | GGACUGAAAGCAACGGACGGCAAAGCUCGUACUAACCU | 8 | ||
| QΔ | 38 | AGUUCCCAUCAAGUACGCUACGCAACUAAUGCAGAACA | 0 | ||
|
| |||||
| Competition H: wt satC and Wh | Comp 1 | Comp 2 | sum | ||
|
| |||||
| ID | Size | Sequence at positions 48-123 | |||
| WT | 76 | WT | 10 | 5 | 15 |
| Wh | 38 | GGACUGAAAGCAACGGACGGCAAAGCUCGUACUAACCU | 0 | 3 | 3 |
|
| |||||
| Competition I: wt satC and HHq | Comp 1 | Comp 2 | sum | ||
|
| |||||
| ID | Size | Sequence at positions 48-123 | |||
| HHq | 39 | GAGAUCCGGCGUGAAUCAUAGAGAUCCGGCGUGAAUCAU | 4 | 11 | 15 |
| WT | 76 | WT | 0 | 6 | 6 |
To determine the next most competitive satC, competition E paired three satC38 winners previously tested in competition A (Wh, DDh, and Zh) with the satC76 winner S. Although only a limited number of clones were evaluated, all five were Wh, consistent with the results from competition A. Competition F compared satC lacking H2 (AΔ) and a winner of the satCΔH2 infection (QΔ); QΔ, with its 38 nt regenerated H2 sequence, was the more fit satRNA (19/19), consistent with the rapid expansion of H2 from 0 nt observed during in planta passaging (Fig. 2). Since competition between QΔ and Wh resulted in only Wh being recovered (8/8; competition G), this suggests that HHq was the most fit satC, followed by Wh.
To determine how the fitness of wt satC compared with HHq and Wh, competition experiments were conducted between wt satC and each of the two SELEX winners. Two independent assays (competition H) indicated that wt satC was more fit than Wh (15/18 clones). In direct competition between wt satC and HHq, 15/21 clones recovered were HHq, suggesting that HHq was as least as fit as wt satC for mutualistic association with TCV gRNA in plants. This was unexpected, as wt satC has always been significantly more competitive than winners from SELEXing various regions within the 3′ portion of satC, unless the winning sequence was virtually identical with wt satC (Carpenter and Simon, 1998; Guan et al., 2000a; Guan et al., 2000b; Zhang et al., 2004b; Sun et al., 2005; Guo et al., 2009).
To determine if HHq and/or Wh would incorporate additional alterations if allowed to continue accumulating in infected plants, each of these satC winners was independently passaged through three plants with TCV gRNA for a total of six additional rounds (three weeks per passage). Neither HHq nor Wh (12 or 10 clones sequenced after round 6, respectively) acquired any additional mutations (data not shown). This suggests that these satC SELEX winners are now relatively stable.
Since HHq and other winning progeny of these experiments have H2 regions of 38-39 nt (instead of the wt H2 length of 76 nt), the question arises as to why wt satC has not reduced the length of its H2 to 38 nt during routine passaging in plants? One possibility is that the wt 76 nt H2 region is already structurally stable and produces a highly functional and fit satRNA (Fig. 1B). While the viral RdRp is error prone (Drake and Holland, 1999), the enzyme would have to generate a 38 nt deletion within H2 (unlikely, because as noted above, no nt changes were observed in 25 progeny of wt satC transcripts cloned after three weeks in planta [Zhang et al., 2004b]), and after its generation this truncated satC would have to be sufficiently fit to compete with the vast majority of molecules that remain wt satC, which also is unlikely. SELEX, however, provides an opportunity for satCs with different sequence lengths in the H2 region to arise through positive selection (Domingo et al., 2012) since the starting pool of molecules is so diverse and poorly functional.
SELEX winners have increased satC dimer:monomer ratios in protoplasts
To determine if fitness to accumulate in plants correlates with replication efficiency in protoplasts, the fittest SELEX winners were inoculated with TCV gRNA into Arabidopsis thaliana protoplasts and RNA levels were assayed 40 hours later. In addition, control satCs with either 76 (ctrl76), 38 (ctrl38), or 19 (ctrl19) nt of randomly selected sequence replacing H2 were assayed at the same time for accumulation in protoplasts. Like most satRNAs, wt satC normally accumulates in both monomeric and dimeric forms, with monomers accumulating at a significantly higher percentage compared with dimers (often 20:1; Fig. 6A). Monomers of HHq, the most fit SELEX winner, accumulated to 50% of wt satC levels and dimers accumulated to levels 6-fold higher than wt satC (Fig. 6A-C). Monomers and dimers of IIq accumulated slightly more than those of HHq, despite HHq being more competitive for accumulation in plants (Table 1). Monomers of the second-most fit SELEX winner, Wh, accumulated to about 25% of wt satC levels and dimers accumulated at levels similar to wt satC. Monomers of all other SELEX winners accumulated to about 25% of wt satC or lower. Interestingly, GGq, which like FFq retained the 19 nt length in its H2, generated almost no detectable monomers (similar to ctrl19 levels) and mainly accumulated dimers, resulting in the greatest dimer:monomer ratio (>8; Fig. 6D). GGq also contained the only H2 replacement sequence predicted to not contain putative hairpin H7 (Fig. 5E). Interestingly, all satC19 SELEX winners, including two that retained the 19 nt size replacement sequence, had significantly higher levels of dimers compared with wt satC, whereas other SELEX winners had dimer levels comparable to wt satC (Fig. 6C).
FIG. 6.
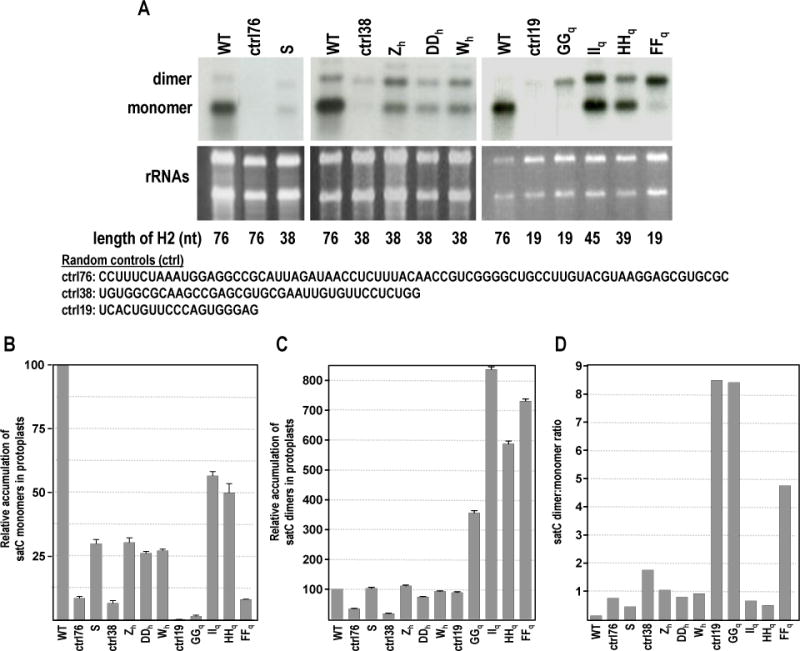
Replication of SELEX winners in protoplasts. (A) Northern blot analysis of RNA extracted from protoplasts at 40 hpi. Protoplasts were inoculated with wt satC, negative control (unSELEXed) satCs with various length H2 regions (sequences listed below the blots), or satC SELEX winners. A set of three representative blots from one of three independent experiments is shown. Each blot contained a wt satC control for normalization purposes; normalization to the rRNA levels in the EtBr stained gel also was performed. The positions of satC monomers and dimers, as well as the length of the H2 region, are indicated to the left and below the figure, respectively. (B) Quantitation of monomers and (C) dimers from the replicate experiments. Standard deviations are indicated. (D) Dimer:monomer ratio from one set of blots.
Taken together, these data suggest a correlation between the length of H2 and the dimer:monomer ratio, with shorter H2 having high levels of dimers and very low levels of monomers. This is consistent with prior work, in which deletion of nt 93-100 within H2 was sufficient to drive accumulation to 50% dimers (Carpenter et al., 1991). A larger deletion of nt 79-100 caused a 16-fold reduction in (+)-strand momomer accumulation and a 2.3-fold increase in accumulation of dimers, increasing the dimer:monomer ratio from 0.10 for wt to 3.80 for satC (Zhang, 2006). This suggests that the size of the H2 region (either in the [+]-strand, or in the [–]-strand replication intermediate), and not any specific sequence, controls the ratio of dimers to monomers, in a manner that remains unknown. Interestingly, agroinfiltration of satC monomers in the absence of TCV gRNA results in dimer detection (R. Guo and A.E. Simon, unpublished results); dimerization in the absence of helper virus also has been shown for a satRNA of Cucumber mosaic virus (Choi et al., 2012). Further studies will need to be performed to elucidate the mechanism of satC dimerization and the connection to H2 length.
Conclusions
As previously observed with the SELEX of H4a and H4b (45 nt; Guo et al., 2009), it is possible to evolve large regions of a noncoding satRNA (76 nt; 21% of the molecule in the current report) when a large, diverse pool of molecules is provided during SELEX. The requirement in the H2 region (for a fit satRNA) appears to be a stem-loop of flexible sequence and length, with an apparent preference for 38-39 nt. When H2 is too short (0 or 19 nt), it rapidly expands (as quickly as round 2 [6 weeks]), restoring much of the wt residue flexibility pattern in the central region of the 5′ portion of satC (Fig. 1B, 2C, 4B, 5E). As a recombinant noncoding RNA, the rapid selection and/or duplication of sequences capable of folding into a hairpin in this region may be revealing that the central region tertiary interactions that are restored by the hairpin are important. Achieving a 38-39 nt hairpin in the H2 region for QΔ, DDq, and IIq/JJq required duplication of SELEXed sequences and/or sequences surrounding the SELEXed region. Interestingly, this expansion was specific to the H2 location and did not occur elsewhere in the region. The use of self-sequence to expand H2 when it was less than 38 nt not only illustrates evolutionary selection pressure for a minimal functional length for H2 but also raises questions about the mechanism of this repair. Similar, head-to-tail concatemerization of whole viral RNAs has been reported during passaging of dsRNA reoviruses in tissue culture (e.g. Anthony et al., 2011), but the nucleotide additions seen here are much smaller in length and occur within the viral RNA. Our observations are similar to those reported by Olsthoorn and van Duin (Olsthoorn and van Duin, 1996) when they deleted 19 nt from the bacteriophage MS2 RNA genome between the maturation and coat protein ORFs. This deletion was predicted to destroy two stem-loops necessary for function. One revertant isolated from infected bacteria was a 14 nt insertion that was predicted to restore the two stem-loops. Further passaging of this revertant resulted in addition of 4 more nt to restore the spacing between the stem-loops. Although the source of these 18 nt was unclear, both examples illustrate rapid RNA repair to restore secondary structure, possibly via RdRp-mediated RNA recombination (reviewed in Barr and Fearns, 2010). Further study in this area could help understand the mechanism of this repair in satC and possibly other recombinant or modified viral RNAs undergoing similar evolution.
Materials and Methods
In vivo SELEX
In vivo genetic selection was performed as previously described (Carpenter and Simon, 1998; Guo et al., 2009) with a few exceptions. To generate the template for in vitro transcription of satC with random sequence in place of the 76 nt H2 region (positions 48-123), two fragments were generated by separate PCRs with pC(+) (pUC19 containing full-length satC cDNA) as a template. The 5′ fragment was produced by using primers T7C5′ (5′-GTAATACGACTCACTATAGGGATAACTAAGGGTTTCA-3′; T7 promoter sequence is in italics) and 28-47rev (5′-TGTTCTGCATTAGTTGCGTA-3′; MwoI site is underlined). The 3′ fragment containing either 19, 38, or 76 random nt was generated by using respective forward primer 28-135randomize (5′-TACGCAACTAATGCAGAACA[N19 or N38 or N76]AGTTCCCATCAA-3′) and reverse primer oligo 7 (5′-GGGCAGGCCCCCCGTCCGA-3′; complementary to 19 nt at the 3′ end of satC). PCR products were subjected to electrophoresis, purified using Wizard® SV Gel and PCR clean-up system columns (Promega, Madison, WI), digested with MwoI (all enzymes procured from New England Biolabs, Ipswich, MA, except where noted), phenol/chloroform extracted, and respective 5′ and 3′ fragments ligated together to produce full-length satC cDNA. These satC cDNAs with randomized wt or reduced length H2 were directly in vitro transcribed using T7 RNA polymerase. TCV gRNA was in vitro transcribed from SmaI-linearized pT7TCVms (Oh et al., 1995). Both satC and TCV gRNA transcripts contain precise 5′ and 3′ ends. For the first round of selection, 2 μg of wt TCV gRNA transcripts and 5 μg of satC transcripts with specific randomized sequences were inoculated onto each of 30 turnip seedlings (Turnip Hybrid Just Right, Gurney’s, Greendale, IN or Wammock Farm Service, Sylvania, GA). Total RNA was extracted from uninoculated leaves after 21 days, pooled, and inoculated onto six new turnip seedlings for an additional 21-day infection. This procedure was repeated for a total of 5 rounds. After each round, satC RNA was subjected to reverse transcription using M-MuLV-RT and oligo 7 as primer, then amplified by PCR (GoTaq® Flexi DNA polymerase, Promega, Madison, WI) using primers T7C5′ and oligo 7. PCR products were treated with 5 U Klenow fragment for 15 minutes at room temperature then extracted using phenol/chloroform. After gel purification of the Klenow-treated PCR products, the DNA was incubated with 5 U of T4 polynucleotide kinase for 25 minutes at 37°C, ligated at 16°C into SmaI-linearized pUC19, and transformed into competent E. coli DH5αF’. Blue-white screening in the presence of X-gal was used to check for inserts, and plasmid DNA with cloned satC was sequenced (Clemson University Genomics Institute, Clemson, SC).
In vivo self-evolution
In the presence of TCV gRNA, satC with a deletion of H2 (satCΔH2) was passaged through plants for 3 rounds. To prepare satCΔH2, a similar strategy was used as described above. The 5′ fragment was generated using primers T7C5′ and 28-47rev. The 3′ fragment was generated using primers ΔH2 (5′-ACTACGCAACTAATGCAGAACAAGTTCCCATCAAGTACGGGAGC-3′ (MwoI site is underlined and deletion-flanking wt bases 47 and 124 italicized) and oligo 7. In addition, selected Round 5 SELEX winner satC sequences were transcribed from their pUC19-based plasmids following linearization with SmaI, and in the presence of TCV gRNA, each was independently passaged through three plants. Total RNA was extracted and pooled and inoculated onto three new plants; passaging was repeated for a total of 6 rounds. After each round, extracted RNA was reverse transcribed and progeny satC cloned and sequenced.
In vivo competitions between wt satC and/or SELEX winners
Competition experiments between equal amounts of T7 polymerase-generated transcripts of SELEX winners, in vivo satCΔH2 passaging winners, and/or wt satC were performed as previously described (Zhang and Simon, 2005; Guo et al., 2009). Wt satC transcription template was generated by PCR using pC(+) as template and primers T7C5′ and oligo 7. The PCR product was gel purified and phenol/chloroform extracted before use in transcription reactions with T7 RNA polymerase. For all competitions, control experiments were performed simultaneously in which individual plants were co-infected with TCV gRNA and RNA of each satC SELEX winner, to verify that each set of satC transcripts was functional in planta and able to be cloned from extracted RNA.
Accumulation of viral RNAs in protoplasts
TCV gRNA and satC transcripts were in vitro transcribed with T7 RNA polymerase using plasmids pT7TCVms and pT7C(+) after linearization with SmaI (Song and Simon, 1994) or directly from PCR products. Protoplasts (5 × 106) prepared from callus cultures of Arabidopsis thaliana ecotype Col-0 were inoculated with 20 μg of TCV gRNA transcripts with or without 2 μg of satC RNA transcripts using polyethylene glycol-CaCl2, as previously described (Zhang et al., 2006b). Total RNA isolated from protoplasts at 40 hours post inoculation (hpi) was subjected to RNA gel blot analysis. A [γ-32P]ATP-labeled, satC-specific oligonucleotide (oligo 7) was used to probe the RNA. To obtain clones of satC with randomized H2 (76 nt, 38 nt, or 19 nt) for use as negative controls in protoplast experiments, ligated satC cDNAs (described above) were directly cloned into the SmaI site of pUC19.
SHAPE RNA structure probing
Six pmoles of in vitro transcribed WT and mutant (HHq, Wh, and QΔ) satC transcripts each were heated at 65°C for 5 min, snap-cooled on ice for 2 min, and then incubated at 37°C for 20 min in SHAPE Folding Buffer-2 (80 mM Tris-HCl pH 8, 11 mM Mg(CH3COO)2, 160 mM NH4Cl). Three pmoles of the folded RNA was combined with either N-methylisatoic anhydride (NMIA) or DMSO at a final concentration of 15 mM. RNA reaction mixtures were incubated at 37°C for 35 min (5 half-lives of NMIA) followed by ethanol precipitation. RNA was resuspended in 8 μl of 0.5× TE buffer. Primer extension reactions were performed using 32P-labeled oligonucleotides and Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA) as previously described (Wilkinson et al., 2006). For WT satC structural analysis, oligonucleotides used were complementary to positions 334-313, 235-221 or 128-114. To probe the 3′ terminal region of satC, satC transcripts were extended with 67 nt of plasmid-derived sequence, and an oligonucleotide was used that was complementary to the plasmid sequence. For mutant satC, oligonucleotides annealed to positions 235-221 or 152-131 to probe M1H and the region of satC upstream of H2. Radioactively-labeled products of reverse transcription were resolved on 8% denaturing polyacrylamide gels and visualized using a phosphorimager.
Acknowledgments
The authors thank Matthew Beamer, Alice Duchon, Maxwell James, Kristen Kocher, Caitlin Mehalick, Noah Morgenstein, Nicole Myers, Minh Nguyen, Anita Robin, and Lauren Saunders, who along with A.M.M., J.L.N., H.F.T., and N.B.J.-J. performed the first two rounds of the in vivo SELEXes during the laboratory portion of BIOL 419 at Dickinson College. Dickinson students Jessica Sinchi and Ashley Young generated the ctrl76 and ctrl38 templates during prematriculation research supported by a National Science Foundation STEP grant (DUE-0856704). This research was supported by a grant from the National Science Foundation (MCB-0918624), and funds from the Research and Development Committee of Dickinson College, to D.B.K. and National Science Foundation grant (MCB-1157906) to A.E.S.
References
- Anthony SJ, Darpel KE, Belaganahalli MN, Maan N, Nomikou K, Sutton G, Attoui H, Maan S, Mertens PPC. RNA segment 9 exists as a duplex concatemer in an Australian strain of epizootic haemorrhagic disease virus (EHDV): genetic analysis and evidence for the presence of concatemers as a normal feature of orbivirus replication. Virology. 2011;420:164–171. doi: 10.1016/j.virol.2011.09.009. http://dx.doi.org/10.1016/j.virol.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Barr JN, Fearns R. How RNA viruses maintain their genome integrity. J Gen Virol. 2010;91:1373–1387. doi: 10.1099/vir.0.020818-0. http://dx.doi.org/10.1099/vir.0.020818-0. [DOI] [PubMed] [Google Scholar]
- Carpenter CD, Cascone PJ, Simon AE. Formation of multimers of linear satellite RNAs. Virology. 1991;183:586–594. doi: 10.1016/0042-6822(91)90987-m. http://dx.doi.org/10.1016/0042-6822(91)90987-M. [DOI] [PubMed] [Google Scholar]
- Carpenter CD, Simon AE. Analysis of sequences and predicted structures required for viral satellite RNA accumulation by in vivo genetic selection. Nuc Acids Res. 1998;26:2426–2432. doi: 10.1093/nar/26.10.2426. http://dx.doi.org/10.1093/nar/26.10.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimino PA, Nicholson BL, Wu B, Xu W, White KA. Multifaceted regulation of translational readthrough by RNA replication elements in a tombusvirus. PLoS Pathog. 2011;7:e1002423. doi: 10.1371/journal.ppat.1002423. http://dx.doi.org/10.1371/journal.ppat.1002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Seo JK, Kwon SJ, Rao ALN. Helper virus-independent transcription and multimerization of a satellite RNA associated with Cucumber mosaic virus. J Virol. 2012;86:4823–4832. doi: 10.1128/JVI.00018-12. http://dx.doi.org/10.1128/JVI.00018-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E, Sheldon J, Perales C. Viral quasispecies evolution. Microbiol Mol Biol Rev. 2012;76:159–216. doi: 10.1128/MMBR.05023-11. http://dx.doi.org/10.1128/MMBR.05023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Holland JJ. Mutation rates among RNA viruses. Proc Natl Acad Sci USA. 1999;96:13910–13913. doi: 10.1073/pnas.96.24.13910. http://dx.doi.org/10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. http://dx.doi.org/10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Guan H, Carpenter CD, Simon AE. Analysis of cis-acting sequences involved in plus-strand synthesis of a Turnip crinkle virus-associated satellite RNA identifies a new Carmovirus replication element. Virology. 2000a;268:345–354. doi: 10.1006/viro.1999.0153. http://dx.doi.org/10.1006/viro.1999.0153. [DOI] [PubMed] [Google Scholar]
- Guan H, Carpenter CD, Simon AE. Requirement of a 5´-proximal linear sequence on minus strands for plus-strand synthesis of a satellite RNA associated with Turnip crinkle virus. Virology. 2000b;268:355–363. doi: 10.1006/viro.1999.0154. http://dx.doi.org/10.1006/viro.1999.0154. [DOI] [PubMed] [Google Scholar]
- Guo R, Lin W, Zhang J, Simon AE, Kushner DB. Structural plasticity and rapid evolution in a viral RNA revealed by in vivo genetic selection. J Virol. 2009;83:927–939. doi: 10.1128/JVI.02060-08. http://dx.doi.org/10.1128/JVI.02060-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Meskauskas A, Dinman JD, Simon AE. Evolution of a helper virus-derived, ribosome binding translational enhancer in an untranslated satellite RNA of Turnip crinkle virus. Virology. 2011;419:10–16. doi: 10.1016/j.virol.2011.07.019. http://dx.doi.org/10.1016/j.virol.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JC, Simon AE. Biased hypermutagenesis associated with mutations in an untranslated hairpin of an RNA virus. J Virol. 2004;78:7813–7817. doi: 10.1128/JVI.78.14.7813-7817.2004. http://dx.doi.org/10.1128/JVI.78.14.7813-7817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JC, Yuan X, Yingling YG, Kasprzak W, Zamora RE, Shapiro BA, Simon AE. Structural domains within the 3´ UTR of Turnip crinkle virus. J Virol. 2008;82:8706–8720. doi: 10.1128/JVI.00416-08. http://dx.doi.org/10.1128/JVI.00416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2´-hydroxyl acylation and primer extension (SHAPE) J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. http://dx.doi.org/10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- Nagy PD, Pogany J, Simon AE. In vivo and in vitro characterization of an RNA replication enhancer in a satellite RNA associated with Turnip crinkle virus. Virology. 2001;288:315–324. doi: 10.1006/viro.2001.1099. http://dx.doi.org/10.1006/viro.2001.1099. [DOI] [PubMed] [Google Scholar]
- Oh JW, Kong Q, Song C, Carpenter CD, Simon AE. Open reading frames of turnip crinkle virus involved in satellite symptom expression and incompatibility with Arabidopsis thaliana ecotype Dijon. Mol Plant Microbe Interact. 1995;8:979–987. doi: 10.1094/mpmi-8-0979. http://dx.doi.org/10.1094/MPMI-8-0979. [DOI] [PubMed] [Google Scholar]
- Olsthoorn RCL, van Duin J. Evolutionary reconstruction of a hairpin deleted from the genome of an RNA virus. Proc Natl Acad Sci USA. 1996;93:12256–12261. doi: 10.1073/pnas.93.22.12256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu F, Ren T, Morris TJ. The coat protein of Turnip crinkle virus suppresses posttranscriptional gene silencing at an early initiation step. J Virol. 2003;77:511–522. doi: 10.1128/JVI.77.1.511-522.2003. http://dx.doi.org/10.1128/JVI.77.1.511-522.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AE. 3´ UTRs of carmoviruses. Virus Res. 2015 doi: 10.1016/j.virusres.2015.01.023. in press. [DOI] [PubMed] [Google Scholar]
- Simon AE, Howell SH. The virulent satellite RNA of turnip crinkle virus has a major domain homologous to the 3´ end of the helper virus genome. EMBO J. 1986;5:3423–3428. doi: 10.1002/j.1460-2075.1986.tb04664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AE, Roossinck MJ, Havelda Z. Plant virus satellite and defective interfering RNAs: new paradigms for a new century. Annu Rev Phytopathol. 2004;42:415–437. doi: 10.1146/annurev.phyto.42.040803.140402. http://dx.doi.org/10.1146/annurev.phyto.42.040803.140402. [DOI] [PubMed] [Google Scholar]
- Song C, Simon AE. RNA-dependent RNA polymerase from plants infected with turnip crinkle virus can transcribe (+)- and (-)-strands of virus-associated RNAs. Proc Natl Acad Sci USA. 1994;91:8792–8796. doi: 10.1073/pnas.91.19.8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Simon AE. Requirement of a 3´-terminal stem-loop in in vitro transcription by an RNA-dependent RNA polymerase. J Mol Biol. 1995;254:6–14. doi: 10.1006/jmbi.1995.0594. http://dx.doi.org/10/1006.jmbi.1995.0594. [DOI] [PubMed] [Google Scholar]
- Stupina VA, Meskauskas A, McCormack JC, Yingling YG, Shapiro BA, Dinman JD, Simon AE. The 3´ proximal translational enhancer of turnip crinkle virus binds to 60S ribosomal subunits. RNA. 2008;14:2379–2393. doi: 10.1261/rna.1227808. http://dx.doi.org/10.1261/rna.1227808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Simon AE. Fitness of a Turnip crinkle virus satellite RNA correlates with a sequence-nonspecific hairpin and flanking sequences that enhance replication and repress the accumulation of virions. J Virol. 2003;77:7880–7889. doi: 10.1128/JVI.77.14.7880-7889.2003. http://dx.doi.org/10.1128/JVI.77.14.7880-7889.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Simon AE. A cis-replication element functions in both orientations to enhance replication of Turnip crinkle virus. Virology. 2006;352:39–51. doi: 10.1016/j.virol.2006.03.051. http://dx.doi.org/10.1016/j.virol.2006.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhang G, Simon AE. Short internal sequences involved in RNA replication and virion accumulation in a subviral RNA of Turnip crinkle virus. J Virol. 2005;79:512–524. doi: 10.1128/JVI.79.1.512-524.2005. http://dx.doi.org/10.1128/JVI.79.1.512-524.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CL, Leh V, Lederer C, Maule AJ. Turnip crinkle virus coat protein mediates suppression of RNA silencing in Nicotiana benthamiana. Virology. 2003;306:33–41. doi: 10.1016/s0042-6822(02)00018-1. http://dx.doi.org/10.1016/S0042-6822(02)00018-1. [DOI] [PubMed] [Google Scholar]
- Tsagris EM, Martinez de Alba AE, Gozmanova M, Kalantidis K. Viroids. Cell Microbiol. 2008;10:2168–2179. doi: 10.1111/j.1462-5822.2008.01231.x. http://dx.doi.org/10.1111/j.1462-5822.2008.01231.x. [DOI] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. http://dx.doi.org/10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Merino EJ, Weeks KM. Selective 2´-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc. 2006;1:1610–1616. doi: 10.1038/nprot.2006.249. http://dx.doi.org/10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]
- Zhang F, Simon AE. Enhanced viral pathogenesis associated with a virulent mutant virus or a virulent satellite RNA correlates with reduced virion accumulation and abundance of free coat protein. Virology. 2003a;312:8–13. doi: 10.1016/s0042-6822(03)00345-3. http://dx.doi.org/10.1016/S0042-6822(03)00345-3. [DOI] [PubMed] [Google Scholar]
- Zhang G, Simon AE. A multifunctional turnip crinkle virus replication enhancer revealed by in vivo functional SELEX. J Mol Biol. 2003b;326:35–48. doi: 10.1016/s0022-2836(02)01366-9. http://dx.doi.org/10.1016/S0022-2836(02)01366-9. [DOI] [PubMed] [Google Scholar]
- Zhang G, Zhang J, George AT, Baumstark T, Simon AE. Conformational changes involved in initiation of minus-strand synthesis of a virus-associated RNA. RNA. 2006a;12:147–162. doi: 10.1261/rna.2166706. http://dx.doi.org/10.1261/rna.2166706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Zhang J, Simon AE. Repression and derepression of minus-stand synthesis in a plus-strand RNA virus replicon. J Virol. 2004a;78:7619–7633. doi: 10.1128/JVI.78.14.7619-7633.2004. http://dx.doi.org/10.1128/JVI.78.14.7619-7633.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Ph D thesis. University of Maryland; College Park, MD: 2006. [Google Scholar]
- Zhang J, Simon AE. Importance of sequence and structural elements within a viral replication repressor. Virology. 2005;333:301–315. doi: 10.1016/j.virol.2004.12.015. http://dx.doi.org/10.1016/j.virol.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Zhang J, Stuntz RM, Simon AE. Analysis of a viral replication repressor: sequence requirements for a large symmetrical loop. Virology. 2004b;326:90–102. doi: 10.1016/j.virol.2004.05.006. http://dx.doi.org/10.1016/j.virol.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang G, Guo R, Shapiro BA, Simon AE. A pseudoknot in a preactive form of a viral RNA is part of a structural switch activating minus-strand synthesis. J Virol. 2006b;80:9181–9191. doi: 10.1128/JVI.00295-06. http://dx.doi.org/10.1128/JVI.00295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang G, McCormack JC, Simon AE. Evolution of virus-derived sequences for high-level replication of a subviral RNA. Virology. 2006c;351:476–488. doi: 10.1016/j.virol.2006.03.011. http://dx.doi.org/10.1016/j.virol.2006.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. http://dx.doi.org/10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]