

Abstract
Following the recent emergence of Zika virus (ZIKV), many murine and human neutralizing anti-ZIKV antibodies have been reported. Given the risk of virus escape mutants, engineering antibodies that target mutationally constrained epitopes with therapeutically relevant potencies can be valuable for combating future outbreaks. Here, we applied computational methods to engineer an antibody, ZAb_FLEP, that targets a highly networked and therefore mutationally constrained surface formed by the envelope protein dimer. ZAb_FLEP neutralized a breadth of ZIKV strains and protected mice in distinct in vivo models, including resolving vertical transmission and fetal mortality in infected pregnant mice. Serial passaging of ZIKV in the presence of ZAb_FLEP failed to generate viral escape mutants, suggesting that its epitope is indeed mutationally constrained. A single-particle cryo-EM reconstruction of the Fab-ZIKV complex validated the structural model and revealed insights into ZAb_FLEP’s neutralization mechanism. ZAb_FLEP has potential as a therapeutic in future outbreaks.
Keywords: Antibody, Therapeutic, Zika virus (ZIKV), Flavivirus, Structure, Quaternary Epitope, Neutralization, Efficacy, Cryo-EM
INTRODUCTION
Zika virus (ZIKV) has recently emerged as a global arboviral threat that has caused an estimated 30,000 human infections worldwide in the last 1 year alone (Lessler et al., 2016; Musso, 2015; Zhang et al., 2017). The rapid spread of the virus, aided in part by its cellular tropism, diverse transmission modes, and ability to bypass the human type I interferon response, calls for the rapid development of effective countermeasures (Rodriguez-Morales et al., 2016). While an effective vaccine against ZIKV is still many years away (Barouch et al., 2017), monoclonal antibodies have shown promise in controlling infection in animal models (Sapparapu et al., 2016; Wang et al., 2016; Zhao et al., 2016), indicating that they could serve as a potential immunotherapy option for infected or at-risk individuals.
Before the time of ZIKV emergence in humans, an increasing number of studies were reporting highly potent broad-spectrum neutralizing monoclonal antibodies against Dengue virus (DENV, a related flavivirus) that were either engineered or isolated from infected patients (Beltramello et al., 2010; de Alwis et al., 2011; Lai et al., 2013). The progress in the DENV field has led to a rapid acceleration in the identification of neutralizing antibodies for ZIKV. Like DENV, the surface envelope (E) protein of ZIKV is responsible for viral entry and fusion, which makes it the main target of neutralizing antibodies (Heinz et al., 1994). Examples of anti-ZIKV antibodies include DENV antibodies that crossreact with ZIKV and ZIKV-specific antibodies (generated in murine models or isolated from infected human patients) that target different regions of the E-protein with neutralization potencies (PRNT50) ranging from 0.014 to 6.560 μg/mL (Barba-Spaeth et al., 2016; Sapparapu et al., 2016; Wang et al., 2016; Zhao et al., 2016). Despite these developments, no prior approaches have described antibody engineering from the standpoint of targeting specific epitope surfaces on the whole ZIKV spherical assembly. Such an approach may be highly desired, for instance, in order to target highly conserved regions of the E-protein assembly and therefore minimize the chance that the virus will escape the neutralizing antibody response. Indeed, a previous study isolated escape viruses to anti-ZIKV monoclonal antibody therapy by using an in vivo animal model and by means of serially passaging the virus in cell culture (Wang et al., 2017). Additionally, since these outbreaks are sporadic and short-lived, obtaining affinity matured antibodies from the peripheral blood mononuclear cells of ZIKV-infected individuals can be challenging. Consequently, a rational approach for engineering antibodies against mutationally constrained epitopes with potencies similar to previously reported monoclonal antibodies will likely be valuable for combating future outbreaks.
Herein, we first defined potential epitope surface residues of the E-protein based on residue mutability constraints in the context of the entire spherical assembly of ZIKV. Toward this goal, we built a 3D homology model of E-protein ZIKV assembly to correlate the structural and network properties of the epitope surfaces with in vitro neutralization data obtained using a panel of ZIKV-specific and cross-reactive antibodies. Based on this, we demonstrated that antibodies that target highly networked, solvent accessible, inter-chain epitope surfaces are associated with more potent neutralization than antibodies that target intra-domain epitopes. Importantly, we identified a quaternary epitope surface proximal to the fusion loop (FLEP) that is the most structurally constrained among the various epitope surfaces. Driven by this careful epitope selection procedure, we then applied a computational approach to engineer an antibody (ZAb_FLEP) that specifically targets the FLEP quaternary epitope surface in ZIKV. Based on the computational analyses, we postulated that this antibody would show potent neutralization and that the evolution of ZIKV escape mutants that could evade neutralization would be unlikely.
We first validated our hypothesis by performing biochemical binding studies and PRNT-based in vitro neutralization assays. Notably, ZAb_FLEP demonstrated neutralization of ZIKV strains from different lineages and circulation periods. Second, passive transfer of ZAb_FLEP in an in vivo lethal mouse model of ZIKV infection conferred 100% survival. Third, administration of ZAb_FLEP to pregnant mice in a post-exposure challenge resolved maternal-fetal placental transmission and fetal demise, highlighting ZAb_FLEP’s potential for treating or preventing ZIKV fetal disease in humans. Fourth, serial passaging of ZIKV in the presence of ZAb_FLEP failed to generate viral escape mutants, which is consistent with our prediction that FLEP epitope is mutationally constrained. Finally, determination of the cryoelectron microscopy (cryo-EM) structure of ZIKV in complex with the antibody-binding fragment (Fab) of ZAb_FLEP validated the theoretical structural model and helped explain the neutralization mechanism of the engineered antibody.
RESULTS
Structure-guided analysis of potential E-protein quaternary epitopes
Optimal epitope selection is a critical first step in the engineering of a neutralizing antibody. Examples of important factors to consider during epitope selection include sequence conservation of the epitope across strains, accessibility of the epitope (in the context of the viral assembly), and the epitope’s conformational flexibility. To gain a systematic understanding of how these epitope characteristics affect the neutralization ability of antibodies that target them, we examined the accessibility, conservation, mode of binding (e.g., occupancy) and binding affinity of a panel of different DENV antibodies and correlated these factors with the reported PRNT50 values (STAR Methods). The analysis revealed that the inter-chain locking potential (i.e., the ability of the antigen binding fragment to engage two or more chains), solvent accessibility, and network score are dominant factors that correlate with PRNT50 (Figure S1). In other words, antibodies that target quaternary epitopes spanning more than one chain are generally the most potent. Among the quaternary epitope targeting antibodies, those that target intra-dimer epitopes also exhibit a relatively high degree of epitope sequence conservation and limited conformational flexibility, highlighting their potential as promising therapeutic candidates. However, given the limited viral structural information for ZIKV (during the start of this work), we built the 3D model of E-protein ZIKV assembly starting from the homology model of soluble E-protein in order to understand the various neutralizing antibody epitope surfaces in the context of an assembled virus (STAR Methods). Simultaneously, we performed a PRNT-based neutralization of ZIKV for a panel of chosen antibodies (Figure S2). This analysis showed that the inter-chain locking potential is the most dominant factor that correlates with PRNT50, similar to what was observed with DENV (Table S1). Further, the analysis permitted us to define thresholds for network score and accessibility based on matching the corresponding values of the different antibodies (STAR Methods).
Taking advantage of this information, we employed a computational method that uses a residue interaction network based on graph theory in the context of the modeled E-protein ZIKV assembly to identify epitope surfaces that exhibit inter-chain locking potential, a high degree of residue interconnectivity, and epitope accessibility (STAR Methods). Accordingly, each residue in the assembly is assigned three variables: (1) a network score that reflects the constraints imposed on it by its environment; (2) its location relative to the 5-fold, 3-fold and 2-fold symmetry axes; and (3) solvent and antibody accessibility in the viral assembly. The geometrical parameters are especially important when identifying potential quaternary epitopes that span or fall near E-protein dimer interfaces, which are extensively influenced by the membrane curvature and inter-chain interface orientations about symmetry axes of the assembly (STAR Methods). A potential epitope surface is defined as a set of spatially clustered, surface-exposed residues that are highly inter-connected by non-covalent interactions (STAR Methods).
Our analyses of the modeled E-protein ZIKV assembly revealed that the region involving the fusion loop (E-DII) of one monomer and a small portion of E-DI and E-DIII of the adjacent monomer is highly networked and accessible compared with other neutralizing antibody epitopes (Figure 1A-B). Consistent with the observed extensive inter-residue interactions in the fusion loop epitope-proximal region, only seven amino acid mutations were observed in the FLEP epitope in the 254 ZIKV strains found in GenBank (Figure S3). Notably, the fusion loop epitope-proximal epitope (FLEP) we define is enclosed within a dimer region, which indicates that the entire assembly presents 180 sites available for binding (at full occupancy) (Figure 1B). Furthermore, FLEP appears to be structurally invariant in the different pH-sensitive viral forms, indicating that the antibody is unlikely to dissociate from the antigen even in the low pH compartments of the endosome (Figure 1C). The quaternary nature of the FLEP epitope led us to postulate that antibodies that target this region would be capable of neutralizing intracellularly by blocking the fusion process and therefore would lower any risk of antibody-dependent enhancement or ADE (Chan et al., 2011).
Figure 1. Computational Identification of Highly Constrained E-protein Epitopes.
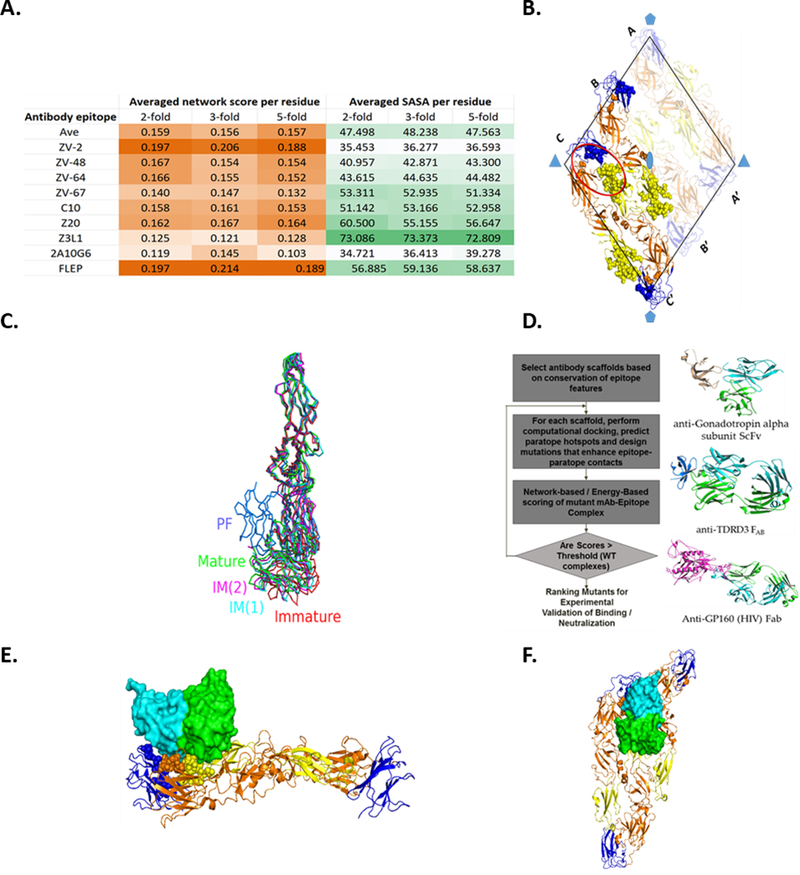
(A) Average (per residue) network score and SASA of Zika antibody epitopes in the 2-fold, 3-fold and 5-fold axes of the ZIKV cryo-EM assembly. Values in each column are colored into lighter shades (lower value) and darker shades (higher value). (B) Putative FLEP epitope depicted on an asymmetric unit in a raft structure. The raft contains three E-protein dimers forming two asymmetric units. The three E-chains that are oriented parallel to each other in the raft are labeled A, B, and C, and those oriented in the opposite direction, A′, B′, and C′, respectively. The rhombus that encloses the six chains is represented by black line. The domains are color coded as follows: orange, domain I; yellow, domain II; blue, domain. The 532 axes of symmetry are highlighted by blue polygons. Residues predicted to form the epitope are represented by spheres in one of the asymmetric units (chains labeled B-C-C'). The FLEP epitope proximal to the 3-fold axis of symmetry is highlighted by an oval. Note the proximity of the epitope residues to the adjacent dimer, which greatly influences the epitope’s surface geometry. (C) Flexibility and conformational changes of E-protein. PDB structures corresponding to the different E-protein conformations were selected (PDB: 1K4R, mature; PDB: 1UZG, IM(1); PDB: 1TG8, IM(2); PDB: 1OK8, PF; PDB: 5U4W, immature) and aligned, where the alignment is based on the DI-DII domain. The N- and C- terminal residues are represented by spheres and colored in brown and yellow, respectively. While DI-DII domains (e.g., FLEP epitope) appear to be largely invariant between the structures, significant conformational differences were observed in the DI-DIII region (viz a viz ZV67 epitope), particularly in the hinge region connecting the two domains. IM:Intermediate; PF:Postfusion. (D) Illustration of rational engineering of antibodies targeting FLEP-like surface. Overview of antibody scaffold selection and paratope engineering (left); representative scaffolds targeting FLEP-like surface (right). (E and F) Computationally docked model of Fv interacting with the E-protein asymmetric unit. Views perpendicular (E) and along (F) the 2-fold axes are shown. Select residues that make contact with Fv are show as spheres.
While some antibody epitopes possess higher network score or solvent accessible surface area (SASA) than the FLEP region, none of the known epitopes possess inter-chain locking potential as well as higher network score and SASA values (Figure 1A). For instance, Z3L1 (Wang et al., 2016) targets a highly accessible region but its epitope residues are poorly networked and, hence, susceptible to viral escape. On the other hand, ZV-2 (Zhao et al., 2016) targets a highly networked epitope, but the region is partially hidden on the viral assembly, thereby increasing the risk of ADE (Figure 1A). Based on FLEP’s promising characteristics taken together with the repertoire of antibodies that engage this region, we chose this epitope as the target for our antibody engineering effort as discussed below.
Rational engineering of neutralizing antibodies against the FLEP epitope
Herein, we took a two-pronged approach for antibody engineering, which included (1) scaffold selection and (2) paratope engineering. First, scaffolds targeting fusion-loop-like structures were used as anchoring points to develop the initial ZIKV antigen:antibody structural models (STAR Methods). In paratope engineering, we investigated complementarity-determining region (CDR) substitutions that provided optimal contacts with the epitope surface, using the approach described in the DENV studies (STAR Methods). The engineering approach took into consideration important differences in the accessibility and network scores of epitope residues, as these parameters vary across different icosahedral vertices on the spherical assembly. The engineered antibodies were tested for binding to three different strains, MR766 (1947/Uganda virus), H/PF/2013, and PF13 (both French Polynesia, 2013), via ELISA (STAR Methods). In parallel, we also evaluated the engineered antibodies for their ability to inhibit ZIKV infection using a PRNT-based neutralization assay (STAR Methods). A significant number of the engineered antibodies failed to show binding, but the analyses yielded a few ZIKV binding antibodies with varying affinities against the three strains. After multiple iterations of testing (STAR Methods), ZAb_FLEP was obtained and showed neutralization of ZIKVs at PRNT50 values of = 0.022–5.098 μg/ml (Table 1).
Table 1. Binding and neutralization characteristics of antibody scaffolds.
ZAb_FLEP binds and neutralizes ZIKV from all three lineages tested. Reflecting its binding affinities, ZAb_FLEP most strongly neutralizes African strain (MR766) followed by the French Polynesian (H/PF/2013) and Brazilian (Paraiba) strains. In contrast, the widely used 4G2 antibody showed no neutralization of ZIKV, despite binding. We also examined the neutralization potency of ZAb_FLEP against clinical isolates representative of DENV serotypes 1–4. ZAb_FLEP was able to potently neutralize DENV-1 and −2 and to a lesser extent DENV-3 and −4.
| Antibody | DENV-1 (EDEN2402) | DENV-2 (ST) | DENV-3 (EDEN863) | DENV-4 (EDEN2270) | ZIKV (H/PF/2013) | ZIKV (PF13) | ZIKV (MR766) | |
|---|---|---|---|---|---|---|---|---|
| Affinity (Kd - μg/ml) | 4G2 | 0.192 | 0.149 | 2.482 | 0.814 | 10.290 | 6.405 | 0.042 |
| PGT124 | No/weak binding | No/weak binding | No/weak binding | No/weak binding | No/weak binding | 388.200 | No/weak binding | |
| ZAb_FLEP | 0.604 | 2.665 | 36.740 | 0.925 | 22.990 | 19.320 | 0.029 | |
| Neutralization PRNT50 (μg/ml) | 4G2 | No neutralization | No neutralization | No neutralization | No neutralization | >500 | No neutralization | No neutralization |
| PGT124 | No neutralization | No neutralization | No neutralization | No neutralization | >500 | No neutralization | No neutralization | |
| ZAb_FLEP | < 3.900 | < 3.900 | 15.600 | 15.600 | 3.579 | 5.092 | 0.022 |
Based on the theoretical structural model of antibody binding at the 2-fold and 3-fold vertices, the variable region displayed potential to make contacts with the adjacent E-dimer, thereby effectively locking two adjacent dimers (Figures 1E and 1F). On the other hand, the theoretical model predicted that binding near the 5-fold vertices would be prevented due to steric hindrance from the adjacent dimer (STAR Methods). The in vitro neutralization potential of ZAb_FLEP approaches the potency of select Zika antibodies (e.g. PRNT50 of DIII binding antibody ZV67 against H/PF/2013 measured in-house is 1.966 μg/ml; data not shown). Interestingly, ZAb_FLEP showed better binding to ZIKV (MR766) relative to the more recent ZIKV (H/PF/2013) and ZIKV (PF13) strains (Table 1). This could be due to the absence of N-glycosylation at Asn154 of MR766 (Musso and Gubler, 2016), which may promote better accessibility to the fusion loop epitope. Despite the lower binding activity, ZAb_FLEP showed useful ZIKV neutralization activity against all strains tested (Figure 2). Additionally, ZAb_FLEP cross-reacted and neutralized the four serotypes of DENV (Low et al., 2006), albeit weakly (Table 1). We opted to focus on ZAb_FLEP for further studies given its ability to neutralize a wide variety of genetically diverse strains (Table 1; Figure 2).
Figure 2. In Vitro Neutralization of Zika Strains by Illustrative Antibodies.
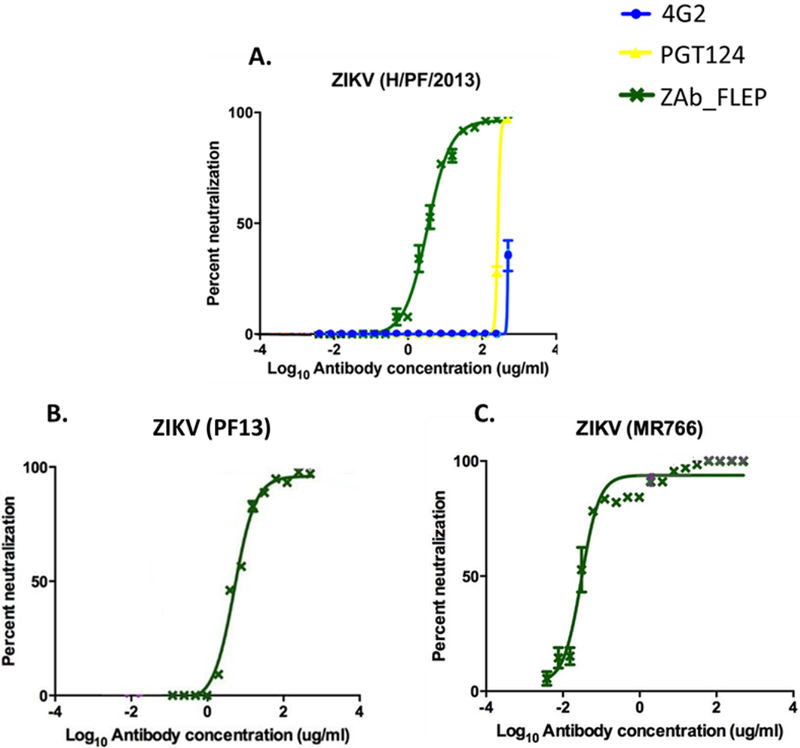
While 4G2 and PGT124 did not neutralize Zika virus efficiently (A), ZAb_FLEP neutralized all three strains with dose response (PRNT50 = 0.022 – 5.092 μg/ml) (A-C). Data points represent averages of n=3 experiments with error bars representing SD. See also Figure S5.
In vivo efficacy of ZAb_FLEP in A129 mice
To assess the prophylactic and therapeutic efficacy of ZAb_FLEP, A129 mice (8–11 weeks old, N=4), which lack the receptor for type I interferon (IFN α/β), were given 50μg (~1.7mg/kg) of antibody or control immunoglobulin (Ig)G and infected intraperitoneally on −1 (day post-infection [d.p.i.]) and +1 (d.p.i) with H/PF/2013 strain at 103 plaque-forming units (PFU) (STAR Methods). Analysis of this ZIKV infection model shows that virus infection causes 100% mortality by day 10 post-infection, accompanied with signs of neurological deficits, as well as high viral loads in brain, spinal cord, serum, spleen, testes (data not shown). Efficacy of the antibody was monitored by assessing mortality, weight loss, and viremia reduction. While mice administered control IgG died by day 11, 100% of ZAb_FLEP-administered mice survived until day 30 (Figure 3). It must be noted that in the antibody protection studies published thus far, survival is monitored only up to 15–20 days (Sapparapu et al., 2016; Wang et al., 2016; Zhao et al., 2016). Furthermore, ZAb_FLEP prevented weight loss and reduced viremia by 2–3 logs when compared with the control antibody, explaining the reason behind the survival rate (Figures 3A-D). In light of these data, we monitored weight loss and viral load reduction in A129 mice with 10mg/kg (300μg), 2mg/kg (60μg) and 0.2mg/kg (6μg) doses of ZAb_FLEP under a therapeutic challenge (+1 d.p.i) with H/PF/2013 strain at 103 PFU (Figures 4A-C). Remarkably, higher doses of ZAb_FLEP protected mice without resulting in cytotoxicity (Figures 4A-C). Importantly, we did not see differences in viremia or accelerated death even when significantly lower doses (0.2mg/kg or 6μg) of ZAb_FLEP were administered. This contrasts with the findings of dengue antibody studies in A129/AG129 mice that showed that lower doses can result in enhanced disease progression resulting in increased or accelerated death due to ADE (Balsitis et al., 2010).
Figure 3. In Vivo Efficacy of ZAb_FLEP ZAb_FLEP.
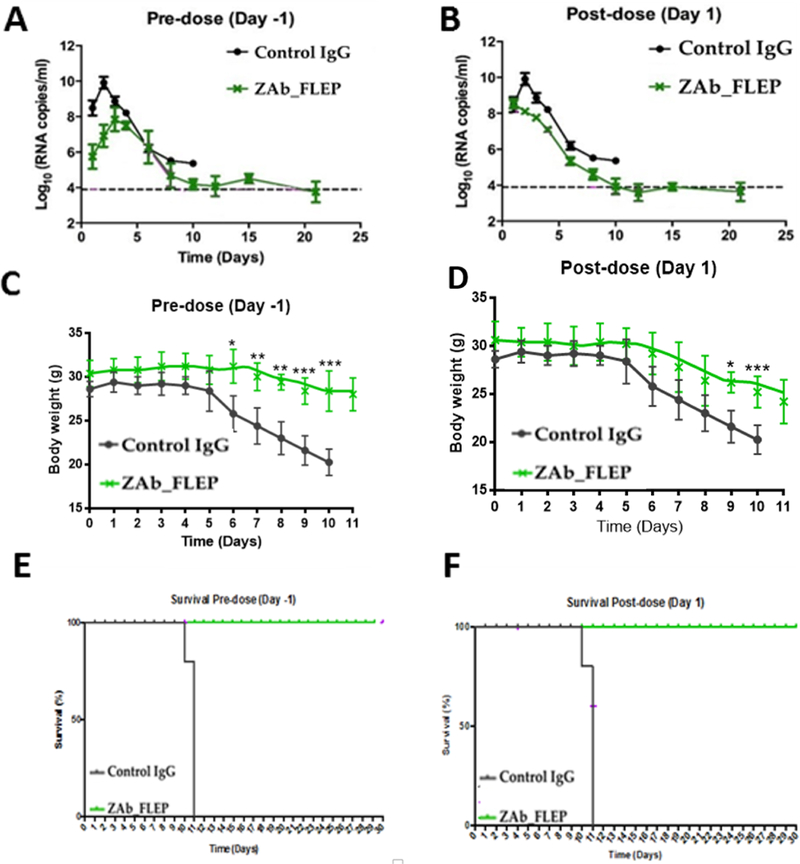
ZAb_FLEP (50μg) reduces viremia (A and B) and protects A129 mice against weight loss (C and D) and death (E and F) when administered −1 or +1 day post infection. n=5 for each condition, experiment performed twice, error bars indicate SD of the data, *p<0.05, **p<0.01, ***p<0.001. See also Table S2.
Figure 4. Examining the Effects of ZAb_FLEP Dosing on In Vivo Efficacy.
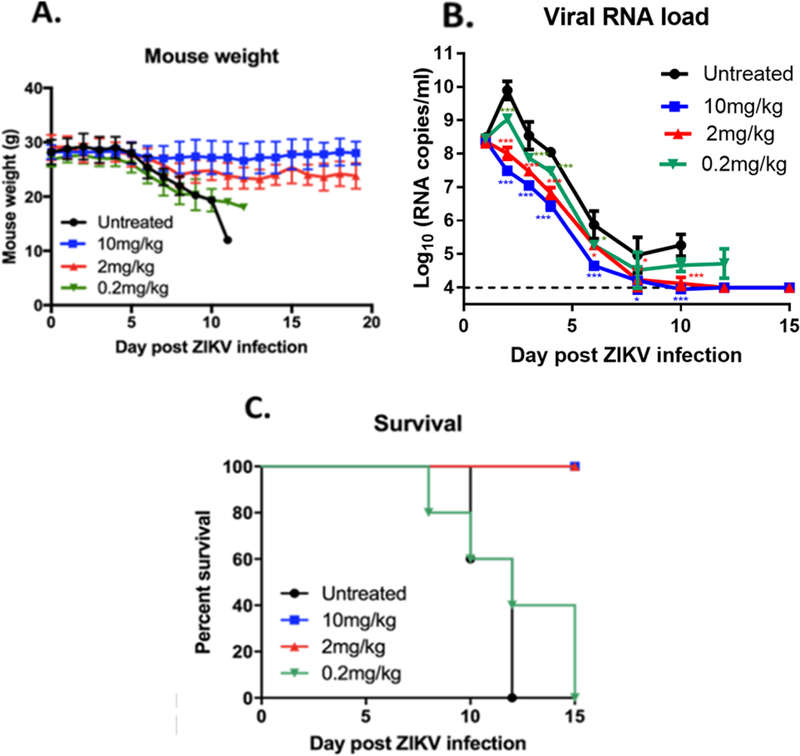
Evaluating the effect of ZAb_FLEP dosing on weight loss (A), viremia (B), and survival (C) in A129 mice. A total of 2mg/kg (60μg) ZAb_FLEP reduces viremia and provides complete protection against weight loss and survival. Remarkably, there were no observable differences in viremia or accelerated death even when significantly lower doses (0.2mg/kg or 6μg) of ZAb_FLEP were administered. N=5 for each condition, experiment performed twice, error bars indicate standard deviation of the data, *P<0.05, ***P<0.001.
ZAb_FLEP therapy prevents vertical transmission and protects fetuses in pregnant mice
Next, we evaluated the potential of ZAb_FLEP to prevent vertical infection and fetal mortality in pregnant A129 mice. Others and we have observed placental and fetal infection and fetal mortality in pregnant A129 mice that are infected with ZIKV (Sapparapu et al., 2016). A total of N=18 pregnant A129 mice that were mated to male mice for 4 days (starting on day 0 evening and separated on day 4 morning) were injected 103 PFU of ZIKV (H/PF/2013 strain of Asian lineage) intravenously on day 10 (corresponding to embryo day 7–10; i.e. E7–10). These mice were treated with 50 μg of ZAb_FLEP (N=9) or an isotype control IgG (N=9) 24 hours (E8-E11) after infection. The mice were sacrificed on day 17 (E14–17) and viral RNA levels were analyzed on day 12 (blood) and day 17 (blood, placenta and fetal compartments) (Figure 5A) (STAR Methods). When compared with isotype control IgG treated mice, the ZAb_FLEP treated mice had substantially lower levels of viral RNA in blood on day 12 (p-value < 0.0001; two-sided t-test) and day 17 (p-value = 0.0018; two-sided t-test) (Figures 5B-C). Significantly, ZAb_FLEP treatment reduced placental (p-value < 0.0001; two-sided t-test) and fetal (p-value < 0.0001; two-sided t-test) infection and provided protection against fetal mortality. Notably, fetuses harvested from dams in the control group had 100% lethality (Figures 5D-F). Similarly, treatment with the isotype control led to significant levels of placental and fetal infection, with concomitant fetal mortality by day 17 (Figure 5D). Fetuses harvested from dams that were treated with ZAb_FLEP showed signs of normal embryo development without any developmental impairment; on the other hand, fetuses from isotype control IgG treated dams were dead before harvesting, revealing a stark morphological difference compared with normal fetuses (Figures 5G-H).
Figure 5. In Vivo Efficacy of ZAb_FLEP in a Maternal Transfer Mouse Model.
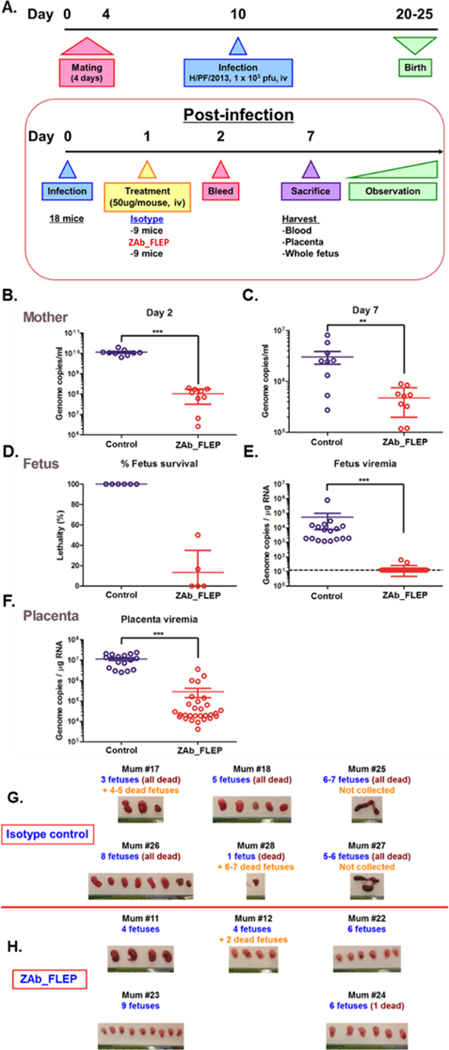
(A) The overall study design. (B) Viral load reduction in the blood collected from pregnant A129 mice on day 12 and day 17. In each case, significance was established by comparing viral RNA copies from the ZAb_FLEP treated (n=9) and isotype control IgG treated (n=9) mice. (C) Fetal survival observed by day 17, (D and E) Viral RNA found in fetal (D) and placental (E) compartments by day 17 in ZAb_treated (n=29 samples) and isotype control IgG treated (n=17 samples) mice; (F) Difference in placental viremia observed between ZAb_FLEP-treated and control mice; (G and H) Fetuses harvested on day 17 from ZAb_FLEP treated (H) and isotype control IgG (G) mice. Error bars indicate SD of the data, **p<0.01, ***P<0.001.
Analysis of the mechanism of neutralization of ZAb_FLEP
To investigate the mechanism of neutralization and to validate the structural model of the ZAb_FLEP:E-protein interaction, we determined the 9.7 Å structure of Fab ZAb_FLEP in complex with ZIKV using cryo-EM (STAR Methods; Figure S4). Interestingly, we observed a total of 120 Fab fragments bound to the viral particle around the icosahedral 2-fold and 3-fold axes (Figure 6A). Remarkably, there were no Fab fragments bound around the twelve 5-fold axes, which accounts for the absence of 60 Fab molecules (Figure 6A). The reconstruction process performed with a Fab-free model converged to the same solution with 120 Fab molecules, confirming very low to zero occupancy of Fab around the 5-fold axis. Thus, ZAb_FLEP is able to efficiently neutralize ZIKV infection even without completely covering the viral particle (Figure 6B). We fit 180 copies of E-monomer atomic structure onto the density map and docked the ZAb_FLEP binding fragment (Fv) against the E-protein using the predicted docked model as the reference (STAR Methods). Although the exact orientation of heavy and light chains could not be discerned from the density map, the Fv fitted into the electron density with good correlation, thus validating the structural modeling procedure. Modeling of a complex with 180 Fv bound gave rise to some exciting observations: (1) ZAb_FLEP makes inter-dimer contacts, as predicted by the computational modeling, and (2) while the Fab is nicely accommodated at the 2-fold and 3-fold vertices, binding of the Fab near the 5-fold vertex is sterically hindered by the adjacent E-protein in the 5-fold assembly (Figure 6C). The recently described envelope dimer epitope human monoclonal antibodies (C8, C10, A11, B7) target overlapping regions (Dejnirattisai et al., 2016) (Barba-Spaeth et al., 2016), suggesting that the region is in fact immunogenic in humans. However, comparison between the cryo-EM complex structures of C10, which binds to all 180 sites on the viral assembly, and ZAb_FLEP shows differences in the orientation of ZAb_FLEP with respect to the E asymmetric unit (Figure 6D). Additionally, ZAb_FLEP makes more contacts with the adjacent dimer residues than C10 (Figure 6E), although the contributions of these residues toward the binding energetics may still be marginal relative to the intra-dimer epitope residues. In summary, the topological nature of ZAb_FLEP epitope combined with the differences in the arrangement of the E-protein near the different icosahedral vertices potentially restricts the occupancy sites to 120. The spacing between the bound Fab fragments suggests the possibility of a bivalent binding pattern of ZAb_FLEP antibody, wherein the two arms (Fab) of a single IgG contact the epitopes near the 2-fold and 3-fold icosahedral axes, respectively (Figure 6B). The binding mode of the Fabs and the distances between the heavy-chain C terminus of adjacent Fab fragments have been used by others to evaluate such a bivalent mode of engagement (Ye et al., 2016). The epitope specificity of ZAb_FLEP mapping to the fusion loop peptide combined with its ability to lock the intra-dimer and inter-dimer interfaces suggests that the monoclonal antibody (mAb) neutralizes by preventing the necessary conformational changes required for the fusion event.
Figure. 6. CryoEM Structure of 9.7 Å ZIKV:ZAb_FLEP Fab Complex.
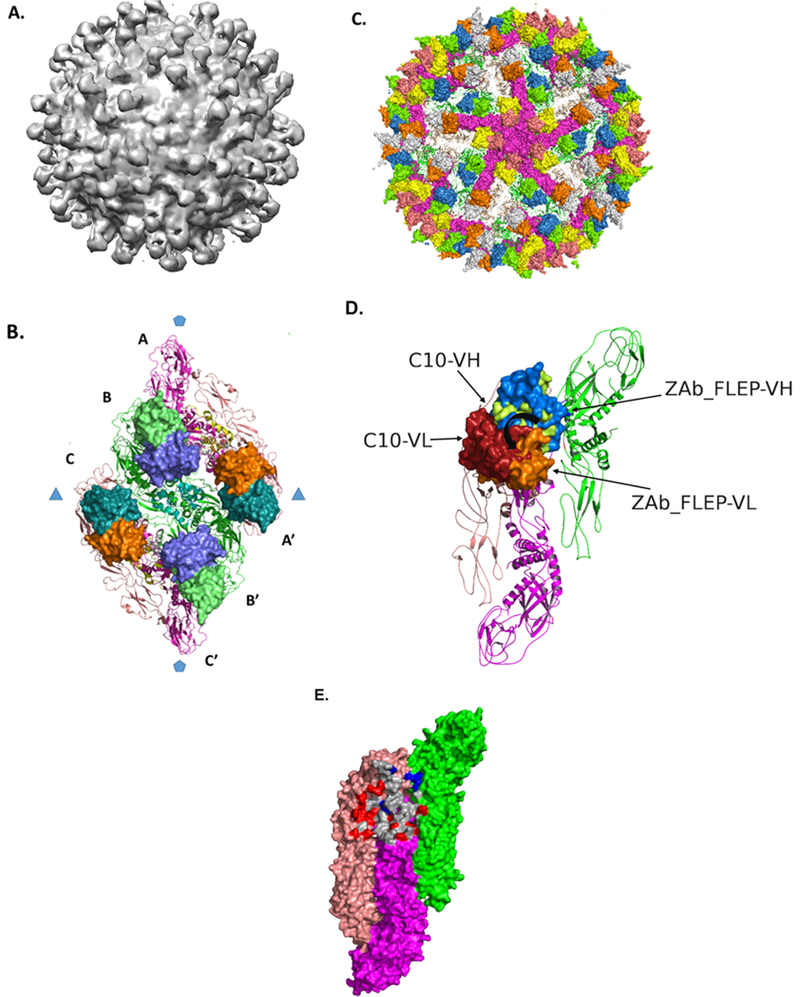
(A) Electron density of ZIKV:Fab complex is shown where the view is along a 5-fold axis. The density map highlights the lack of occupancy around the 5-fold axis. (B) ZAb_FLEP epitope on E-protein raft. The ZAb_FLEP Fv is shown in surface format. The spacing between the C-termini of the Fvs suggests bivalent binding pattern where one arm engages the 2-fold epitope and the other engages the 3-fold epitope. (C) Modeling of a complex with 180 Fv bound indicates steric hindrance as a reason for the lack of binding around the 5-fold axes. Shown in the figure is a view of the complex along the 5-fold where the E-monomers that form the pentamer are in surface format (magenta), and the Fv molecules that occupy the sites around the 5-fold axes are in orange and yellow. Notice that the footprint of the 5-fold Fv overlaps with the E protein monomers in the adjacent raft. (D) Comparison between the orientations of C10 and ZAb_FLEP Fv with respect to the E surface. The heavy and light chains of the two Fvs are represented in surface format and labeled. When viewed down the 2-fold symmetry axis, ZAb_FLEP Fv is oriented 30° counterclockwise relative to C10 Fv, which brings the vestigial loops of its VH nearer to the adjacent dimer. (E) Comparison between the epitopes recognized by ZAb_FLEP and C10. Overlapping residues are shaded in gray, whereas residues unique to C10 and ZAb_FLEP epitopes are shaded in red and blue, respectively. View is looking down the 2-fold axis of the E-dimer. See also Figure S4.
To verify the mechanism of action of ZAb_FLEP, we evaluated the neutralization potential of ZAb_FLEP in FcγR-expressing THP-1.2S monocyte cells and used 4G2 antibody as a reference, as 4G2 has been well established as a fusion-inhibiting antibody (Watterson et al., 2016). Studies carried out in our laboratory have shown that antibodies that are able to inhibit virus fusion with phagosomal membranes are most likely to prevent infection of FcγR-expressing cells (Chan KR et al., 2011). Different concentrations of antibody were incubated with ZIKV (strain H/PF/2013) for 1 hr prior to infecting THP1.2S monocytes. Three days later, the virus replication in culture supernatants was measured by plaque assay on BHK21 cells. We observed complete neutralization following treatment with ZAb_FLEP and near-complete neutralization with 4G2, but not with a weak-affinity ZAb_FLEP variant (Kd > 50ug/ml) (Figure S5A). These results provide mechanistic support for the anti-fusion activity of ZAb_FLEP. We also noted that a weak affinity variant of ZAb_FLEP failed to neutralize the virus even at the highest concentration, resulting in 3-fold higher peak titers compared to ZAb_FLEP (Figure S5B). The potent neutralizing activity of ZAb_FLEP in FcγR-expressing cells combined with its epitope specificity (which, as observed by cryoEM, maps to a region spanning the fusion loop peptide) provides a basis and a rationale for the fusion blocking mechanism of ZAb_FLEP.
Since ZAb_FLEP targets a highly networked region, we hypothesized that there should be a limited opportunity for the emergence of viral escape mutants. To test this hypothesis, we attempted to select for neutralization escape mutants after serial passaging of viruses in cell culture (STAR Methods). Briefly, we propagated H/PF/2013 French Polynesian ZIKV strain in Vero cells at 37°C with 5% CO2 for 48–72 hours in the presence of neutralizing concentration of antibody (90% effective concentration [EC90]). EC90 was chosen to ensure that there was sufficient selection pressure for the generation of escape mutants. The choice of EC90 is consistent with previously published reports (Beltramello et al., 2010; de Alwis et al., 2011; Lai et al., 2013)). The combination of selection and propagation was repeated for a total of seven passages. At each passage, the viral RNA was extracted and the E gene was amplified and sequenced. Importantly, we did not identify any escape mutants against ZAb_FLEP. Although this does not guarantee that escape mutation will not arise, it does imply the existence of underlying structural and/or evolutionary constraints that confer resistance against the antibody selection pressure.
DISCUSSION
There are a few important features of our epitope-driven engineering approach that were critical to the overall effectiveness of ZAb_FLEP. First, a key challenge in developing effective antiviral therapies is the development of resistance due to viral evolution (Low et al., 2017; Morens and Fauci, 2013; Ter Meulen, 2011), so we chose to enforce low epitope mutability and an inter-chain locking mechanism as strict constraints during the initial epitope selection. Maintaining the desired epitope engagement is likely what conferred ZAb_FLEP with its neutralizing ability that approaches that of those isolated from convalescent Zika patients (Barba-Spaeth et al., 2016; Robbiani et al., 2017; Sapparapu et al., 2016; Wang et al., 2016; Zhao et al., 2016). It must be noted that conventional epitope scanning methods using sequence-based approaches or analysis of a single domain or E-protein dimer did not lead to the selection of the FLEP epitope. As mentioned earlier, there was limited structural information available at the start of this work, and as a consequence we had to develop a homology model of the E-protein ZIKV assembly, which is validated by several recent structural studies (Kostyuchenko et al., 2016; Sirohi et al., 2016; Zhang et al., 2016b). This is significant because it was important to capture geometrical parameters when identifying potential quaternary epitopes that span or fall near E-protein dimer interfaces, as these are enormously influenced by the membrane curvature and inter-chain interface orientations about symmetry axes of the assembly.
The recently published quaternary epitope targeting anti-Zika antibody ZIKV-117 showed in vivo efficacy in a similar maternal transfer model, albeit in a prophylaxis setting (Sapparapu et al., 2016). While both ZIKV-117 and ZAb_FLEP recognize different epitopes, they potentially neutralize the virus by locking the E-monomers in the raft (Hasan et al., 2017). Similar to ZAb_FLEP, binding of ZIKV-117 Fab to the 5-fold vertex is restricted due to steric hindrance between the Fab and the E-monomer from the adjacent raft. It seems that the potency of ZIKV-117 and ZAb_FLEP is partly due to their ability to lock multiple antigens on the surface. Dosing studies indicate that ZAb_FLEP has the potential to provide partial protection even at low concentrations without increasing viremia associated with disease progression. Aside from the quaternary epitope, we believe that the potency of ZAb_FLEP is due to a combination of factors including binding affinity, epitope accessibility, and stoichiometry. Indeed, the fact that ZAb_FLEP conferred 100% survival of mice for a significantly longer period of time than reported by previous reports strongly indicates its therapeutic potential (Table S2).
Finally, the emergence of infectious disease outbreaks such as ZIKV and DENV calls for the rapid development and clinical testing of vaccines or therapies. In addition to ZIKV and DENV, the ongoing threat of a Yellow Fever outbreak in Brazil (Goldani, 2017) and the re-emergence of Ebola in West Africa (Dudas et al., 2017) could lead to substantial worldwide morbidity and mortality. Because our epitope selection method does not rely on any characteristics intrinsic to ZIKV or flaviviruses but instead leverages well-defined structural metrics, we believe that it can be applied to any virus assembly, which can then guide appropriate antibody engineering efforts regardless of the platforms that might be used to actually generate the antibody.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Ram Sasisekharan (rams@mit.edu)
Experimental Model and Subject Details
Ethics Statement
Mouse studies were conducted at the DUKE‐NUS Medical School in Singapore following approval from the Institutional Animal Care and Use Committee (IACUC).
Vertebrate Animals
Sv/129 mice deficient in type I IFN receptor (A129), purchased from B&K Universal (UK), were housed in the BSL-2 animal facility at Duke-NUS, Singapore and all animal experiments (protocol 2016/SHS/1197) were approved by the Institutional Animal Care And Use Committee at Singapore Health Services. Eight to eleven week-old male A129 mice were used to assess prophylactic or therapeutic efficacy of antibodies. For this, Mice were injected intraperitoneally (ip) with antibodies one day before (prophylactic) or after (therapeutic) infection with 5 x 104 pfu of H/PF/2013 ZIKV strain (ip). Mouse blood was collected from facial vein on days 1–21 pi to measure serum viremia level by real-time PCR. Mouse weight and survival were monitored until 30 days post-infection (pi). To assess the protective efficacy of the antibody against fetal infection in pregnancy, 9–10 weeks old female A129 mice were housed with 9–10 weeks old male A129 mice in same cages (1 female with 1 male) for 4 days. Female mice were infected intravenously (iv) with 1 x 103 pfu of H/PF/2013 strain on day 10 (corresponding to embryo day 7–10). The mice were injected iv with 50μg of antibodies one day pi. Blood was collected from facial vein on day 2 pi to measure viremia. Mice were euthanized by CO2 inhalation on day 7 pi and blood was collected from the post cavalvein. Placenta and whole futuses were harvested and snap-frozen in liquid nitrogen. Frozen samples were then homogenized by TissueLyser (Qiagen) in the presence of 500–1000μl of PBS, and the supernatants after centrifugation were used to measure tissue viral load. For the viral load measurement, total RNA was extracted from the homogenates using the TRIzol extraction method. 80ng of RNA was used to measure the viral load by real-time PCR.
Method Details
Structural Analysis of DENV Antibody Complexes
Our goal was to define residues that constitute an epitope surface on the entire viral assembly by taking into account the distinct local environments of the residues. In the assembly of E-proteins in a closed viral particle surface (or in complexes of viral surface with antibodies), there are distinct interfaces. These include i) the interface of 2-fold, 3-fold, 5-fold axis along planar and curved regions of the viral surface, ii) location on outer or inner surface of the viral particle, and iii) location at a known paratope-epitope interface on the entire assembly. Thus, to expand the datasets related to antibody footprint on the virus assembly, we modeled the X-ray co-crystal structures of E-protein dimer antibody complexes on to the cryo-EM structures and resolved steric hindrance through energy minimization. Next, we computed the solvent accessible surface area (square Angstrom) of each E-protein residues in these various structures using DSSP software, which also permitted identification of the epitope footprint of the various antibodies on the entire virus assembly. We employed an approach that we used previously to compute a network score that captured the structural constraints imposed on a residue due to its non-bonded interactions with the neighboring residues in the local environment. The solvent accessibility score and network scores were normalized based on highest network score of a residue in the aforementioned interfaces. The higher the network score, the more the constraints imposed by the environment of a residue on its ability to mutate. Subsequently, in order to investigate the structural correlates of potent neutralization of DENV antibodies, we examined the accessibility, conservation, mode of binding (e.g. occupancy) and binding affinity of a panel of different DENV antibodies (including 1A1D-2 (PDB: 2R6P), HM14c10 (PDB: 4CAU), 1F4 (PDB: 4C2I), 5J7 (PDB: 3J6U), B7 (PDB: 4UT6), Ab513 (PDB: 5AAW), 2D22 (PDB: 5A1Z)) and compared them with their reported PRNT50 values (Figure S1).
Predicting Quaternary Epitope Surface on Entire ZIKV Assembly
In order to investigate the neutralizing antibody epitope surfaces on ZIKV assembly, we built a 3D structure of ZIKV starting with the homology model of soluble E-protein. The procedure involved fitting the E-chain into the DENV density map (EMDB: EMD-5520). Specifically, the chains were fitted, one at a time, on the three E-protein molecules to form one-half of the raft. Then, the fitted chains were rotated and translated in a stepwise fashion using a steepest ascent optimization to orient the molecule within its local environment. After aligning the three chains in the density map, the remaining 59 copies in the symmetry-related orientations were generated using the 532 symmetry. Finally, the fitting of the 180 chains (60X3) within the density data was optimized using a global optimization program (Fit) in Chimera. Correlation coefficient was computed to determine the quality of fit of the atomic model in the EM map. The fitting of homology model of ZIKA E-protein on the EM map led to a correlation coefficient of 0.7432. Investigation of the rebuilt assembly showed a closed capsid without any major voids or steric overlaps. Interestingly, the rebuilt assembly contains far fewer (φ, ψ) outliers (0.61%) and steric overlaps (5 atoms per 1000) compared to published DENV cryoEM structures (Figure S6). As noted in Table S1, the homology model of ZIKV viral assembly was corroborated with the recently published ZIKV cryoEM structures (PDB structures 5H37, 5IRE and 5IZ7).
Next, we correlated the normalized network and solvent accessibility scores and other antibody-based structural features (computed in the context of the whole viral assembly) with experimental observations such as neutralization potency (PRNT50 values) of an antibody that was known to bind to a specific epitope region. Similar to DENV, this analysis suggested a trend that highly potent neutralizing antibodies target epitope surfaces that span multiple chains on the entire E-protein assembly (Table S1). Furthermore, this analysis taken together with the sequence conservation of a residue in a given position permitted us to define thresholds for normalized network score (0.15 +/− 0.02) and solvent accessible surface area (50 +/− 5 square angstrom) for the prediction of quaternary epitope surfaces across the entire E-protein assembly. By using these thresholds and searching for clusters of residues that could constitute an epitope surface, we selected an epitope surface that contained the largest quantity of highly networked residues and had the largest median solvent accessible surface area. This region was proximal to the fusion loop and so we designated this quaternary epitope surface as FLEP. Given that FLEP is defined in the context of the E-protein assembly on the viral particle surface, it cannot be viewed in the context of a single domain structure, which is typically provided as an input into established antibody engineering and docking software.
Antibody Engineering
Our antibody engineering approach comprised of two steps: (1) scaffold selection and (2) paratope engineering. To facilitate scaffold selection, we searched >450 antigen-antibody co-crystals in PDB database for Ig scaffolds that target homologous fusion-loop epitope like structures. The homology to the E-protein fusion-loop epitope was defined using an RMSD cutoff of 1.5A°. Each selected scaffold was docked against the ZIKV assembly, where the alignment was guided by the fusion-loop structure. To increase the chances of selecting suitable scaffolds, we incorporated CDR diversity from naturally occurring antibody sequences by considering all the VH and VL/K sequences having the same canonical groups as the chosen scaffolds. The matching antibody sequences were found by searching over 8,718 human VH, 8,513 human VK and 6,779 human VL sequences from the NCBI GenBank database. Antibody scaffolds that had high surface complementarity (Sc > 0.65) to the fusion-loop region and displayed potential to make energetically favorable interactions with the side chains of the fusion-loop residues were given preference. Interactions with E-protein residues that have accessible surface area > 20%, network score > 0.25 and B-factor (C-α) less than three standard deviations or less from the mean make were considered energetically favorable. In this overall structural context, Trp102 and Asn104 were considered critical from the epitope standpoint. In theory, the Fv of an antibody should lend itself to bind to an epitope on free E-protein in many different ways. However, binding to the envelope-embedded E-protein will be constrained by several steric factors: (1) spacing between the antigen, (2) spacing between the adjacently bound Fabs, and (3) multivalent interaction with E-protein. A combination of steric factors and epitope-paratope shape complementarity were employed to rank the antibody scaffolds. Scaffolds that had least constraints and were most permissive in terms of binding to E-protein without the steric factors described above were given higher preference. Three sets of scaffolds composed of different CDR canonical groups were included in the search: (1) L1:2/11A, L2:1/7A, L3:1/9A, H1:1/10A, H2:2/10A), (2) (L1:2/11A, L2:1/7A, L3: N/A, H1:1/10A, H2:1/9A), and (3) (L1:2/11A, L2:1/7A, L3:5/11A, H1:3/12A, H2:2/10A). Multiple antibody scaffolds (including mouse-derived pan-flavivirus 4G2, anti-EDE1 Dengue mAbs C8, C10 and anti-EDE2 Dengue mAb A11, anti-TDRD3 antibody and anti-HIV antibody PGT124) were used as starting templates for antibody engineering.
Having defined the antigen-antibody models, we explored favorable interactions between the CDR residues and remainder of the quaternary-epitope surface residues using amino acid interface (AIF) and evaluated them through experimentation to test their ability to bind to one or more ZIKV. Given the model of the antigen-antibody interaction, the contributions of the CDR residues to the binding were evaluated and ranked based on their interface buried surface area. Then, CDR residues were altered through AIF one at a time starting with the residue that is most buried going all the way to the residue that is least buried. Loop insertions and deletions were attempted if they led to an increase in surface complementarity. At each step, the structure was minimized using CHARMM minimization in Discovery Studio. During minimization, residues that were within 4.5 A° (in both antigen and antibody) from the position being modified were allowed to undergo both side chain and backbone changes whereas the remaining residues were kept fixed. The CDR change was accepted if the post-minimization CDR-antigen interaction energy was favorable when compared to the interaction energy before making the CDR change. Promising mutations from each interaction were used to remodel the antibody, dock against the antigen and generate a new antigen-antibody model using MLR as described before (Tharakaraman et al., 2013). The data from each iteration was used in remodeling the CDR loops, and subsequently to dock and refine the antigen-antibody model using the MLR metric (Tharakaraman et al., 2013). Certain CDR mutations had a detrimental effect on the interatomic network so different variants were tried or compensatory mutations were made in the spatially proximal CDR or framework regions. For example, the DENV E protein contains two N-linked glycosylation sites, at Asn-67 and Asn-153, with the former being in close proximity to the ZAb_FLEP binding domains. On the other hand, ZIKV does not possess the N67-linked glycosylation site. The engineering procedure included a framework mutation, S85K, to induce steric hindrance with the Asn-67 N-linked glycan of DENV E-protein and thereby impair binding to the DENV virus.
The engineered antibodies that satisfied the key epitope-paratope constraints while accounting for differences in accessibility and network scores of epitope residues (as these parameters vary across different icosahedral vertices on the spherical assembly) were taken forward and tested. The engineered ZAb_FLEP antibody buried a total of 1,089 Sq. A° in the epitope-paratope interface, out of which the VH and the VL domains contribute approximately 46% and 54% of the buried area, respectively. Based on the structural model, ZAb_FLEP's footprint crosses over into the adjacent dimer at the 2-fold and 3-fold vertices, where the adjacent dimer contributes 101 A° to the interface area at the 2-fold and 3-fold vertices. On the contrary, ZAb_FLEP showed restricted potential to interact at the 5-fold vertex due to probable steric hindrance from the adjacent dimer.
Expression and Purification of Antibodies
Antibodies were expressed in Freestyle 293 cells by transient transfection with Polyethyleneimine (PEI) and purified by protein A chromatography(Robinson et al., 2015). The purified antibodies were quantified by IgG ELISA. Briefly 96‐well plates were coated overnight at 4oC with appropriate antigen. The plates were washed and blocked with 1% blotto (Santa Cruz Biotechnologies). Serial dilution of antibodies were added to the plate and incubated for 2 hours at room temperature. Antigen bound IgG was detected using RbαHu IgG HRP Conjugated secondary antibody (Jackson ImmunoResearch) followed by TMB substrate (KPL) addition.
Antibody Affinity Measurements
Microtiter plates were coated with 0.05g of purified mouse 4G2 in carbonate buffer (pH 9.6) overnight at 4oC and blocked with 10% BSA for 2hrs at room temperature. Thereafter, 5x104 pfu of DENV-1 (EDEN2402), DENV-2 (ST), DENV-3 (EDEN863), DENV-4 (EDEN2270) or ZIKV were added. After 1hr incubation at room temperature, serial two‐fold dilutions of antibodies were added for 1hr, followed by goat anti‐human IgG Fc cross‐adsorbed HRP‐conjugated anti‐human IgG for 45min. Antibody binding was visualized by adding 3,3′,5,5′‐tetramethylbenzidene substrate and reaction was stopped after 10min with sulfuric acid. In between the different steps, plates were washed twice with PBST (PBS+0.05%Tween). Absorbance is read at 450nm using a plate reader (Robinson et al., 2015). The antibody affinity is obtained by measuring the mAb concentration that result in 50% maximal effective concentration of binding.
Plaque Reduction Neutralization Test
To assess if the monoclonal antibodies can neutralize DENV or ZIKV infection, we performed a plaque reduction neutralization test (PRNT), as described previously(Robinson et al., 2015). PRNT was performed on BHK‐21 cells. Serial two‐fold dilution of sera in RPMI maintenance media (MM) was incubated with 50pfu of DENV or ZIKV virus in equal volumes for 1hr before adding to BHK‐21. After 1hr incubation at 37°C, media was aspirated and cells were overlaid with 1% methyl cellulose in MM. After 5 days at 37°C, cells were fixed with 20% formaldehyde and stained with 1% crystal violet. At least 18 two-fold dilutions (starting from 500μg/ml) of the mAb were tested, and dose-response trend lines were calculated by fitting a variable slope sigmoidal equation to calculate the mAb concentration that neutralizes 50% (PRNT50).
Protection Studies in A129 Mice
Mouse studies were conducted at the DUKE‐NUS Medical School in Singapore after the approval from the Institutional Animal Care and Use Committee (IACUC). The protective and therapeutic efficacy of monoclonal antibodies against ZIKV were tested in adult A129 mice (8‐11 weeks old). Mice were infected intraperitoneally (ip) with 5 x 104 pfu of H/PF/2013 strain (French Polynesia). To assess protective or prophylactic efficacy of antibodies, mice were injected ip with antibodies (50μg) one day prior to infection. To assess therapeutic efficacy, mice were injected ip with antibodies (50μg) one day post-infection (pi). Efficacy of antibody was monitored by assessing mortality, weight loss and viremia reduction. Briefly, mouse blood was collected from facial vein on days 1‐21 pi to measure serum viremia level by real‐time PCR while mouse weight and survival were monitored daily until 30 days pi. To assess the protective efficacy of the antibody against vertical infection in pregnancy, 9–10 weeks old female A129 mice were housed with adult male A129 mice in same cages (1 female with 1 male) for 4 days. Female mice were infected intravenously (iv) with 1 x 103 pfu of H/PF/2013 strain on day 10 (corresponding to embryo day 7–10). The mice were injected iv with 50μg of ZAb_FLEP or mouse isotype IgG one day pi. Blood was collected from facial vein on day 2 pi to measure viremia. Mice were euthanized by CO2 inhalation on day 7 pi and blood was collected from the postcaval vein. Placenta and whole fetuses were harvested and snap-frozen in liquid nitrogen. Frozen samples were then homogenized by TissueLyser (Qiagen) in the presence of 500–1000μl of PBS, and the supernatants after centrifugation were used to measure tissue viral load. For the viral load measurement, total RNA was extracted from the homogenates using the TRIzol extraction method and 80ng of RNA was used for real-time RT-PCR as described previously (Watanabe et al., 2015).
Virus Sample Preparation
Aedes albopictus C6/36 cells were used for amplification of ZIKA virus (ZIKV). The precipitation and purification were done following previously published virion preparation protocol(Tan and Lok, 2014). Briefly, virus was harvested from infected C6/36 cells 4 days post-infection by centrifuging virus-containing media at 9,000 g for 45 min. Virus was then precipitated with addition of 8% (w/v) polyethylene glycol (PEG) 8,000 in NTE buffer (10 mM Tris-HCl pH 8.0, 120 mM NaCl and 1 mM EDTA). Virus precipitate was pelleted by centrifugation at 14,000 g for 1 hr after overnight incubation. Pellet containing virus was then purified by 24% (w/v) sucrose cushion and 10–30% (w/v) potassium tartrate gradient. The virus band was visualized with a torch light in a linear potassium tartrate gradient and extracted with a syringe. It was then concentrated and buffer exchanged to NTE buffer by ultrafiltration using a 100 kDa molecular weight cut-off filter. The yield and purity of E protein were checked with SDS-PAGE using different amount of bovine serum albumin (BSA) as concentration standards.
Enzymatic Digestion of ZAb_FLEP
To obtain the Fab from ZAb_FLEP, papain digestion was carried out. Papain was firstly activated with L-cysteine. The activated papain was added to the digestion buffer (0.1 M Tris HCl pH 8.0, 2 mM EDTA) supplemented with L-cysteine. The reaction was incubated at 37°C for 7 hrs before it was stopped by addition of iodoacetamine to a final concentration of 20 mM. The Fab fragments were then purified with protein A affinity beads and gel filtration (Superdex 75 10/300 GL, GE Healthcare).
CryoEM Complex Preparation
To prepare ZIKV-Fab complexes for CryoEM, Fab and ZIKV virions were mixed with a E protein: Fab (ZAb_FLEP) molar ratio of 1:2 as described to ensure full occupancy (Zhang et al., 2016a). The immune complex was allowed to form by incubation at 37°C for 30 min. The virus-Fab complex was then kept on ice for about an hour prior applying to cryoEM grids.
Cryo-electron microscopy
2 μl of virus-Fab complex was applied onto glow-discharged 2-nm carbon coated holey grids (R1/2, Quantifoil GmbH, Germany) and incubated for 30 sec. Grids were manually blotted for 2 sec at RT and flash frozen in liquid N2-cooled liquid ethane. Grids were loaded to an FEI Tecnai Arctica cryo-transmission electron microscope, operated at 200 kV and equipped with a back-thinned Falcon III direct electron detection device. A total of 304 micrographs were recorded in a movie mode as a set of 31 frames (total dose of 36 electrons per Å2) at a calibrated magnification of 109,375 resulting in a pixel size of 1.28 Å on the object scale and at defocus values from 0.4–1.5 μm.
Image processing
287 micrographs were selected for data processing after removing micrographs with significant astigmatism. Whole-image drift correction was carried out using motioncorr(Li et al., 2013). CTF estimation was performed with CTFFIND3(Mindell and Grigorieff, 2003). Data was binned three times. Particles were picked from the micrographs using EMAN2(Bell et al., 2016). Data was processed with RELION 1.4(Scheres, 2012). In total, 4,256 particles were subjected to reference-free 2D classification to discard bad particles. 1,482 particles selected after 2D classifications were used for 3D classifications using a 60 Å filtered Zika EM map (EMDB: EMD-8116)(Sirohi et al., 2016). 925 particles selected after 3D refinement with icosahedral symmetry yielded an EM density map at 9.7Å resolution according to the gold-standard FSC = 0.143 criterion.
Assessment of predicted ZAb_FLEP:E-protein model using EM density map
The 3.8 Å resolution cryoEM structure of ZIKV (EMDB: EMD-8116)(Sirohi et al., 2016) was fitted into the density map using Chimera(Pettersen et al., 2004). Following this, the asymmetric unit structure of ZIKV (5IRE) were aligned on the three E-protein molecules to form one-half of the raft. After aligning the three chains in the density map, the remaining 59 copies in the symmetry-related orientations were generated using the 532 symmetry. Having generated the viral assembly structure, the Fv of ZAb_FLEP was docked onto each of the 120 sites (180 minus 60 5-fold sites) guided by the predicted docking model, and its orientation relative to the Fab density was assessed in Chimera.
Escape mutant analysis
To evaluate propensity of escape mutant generation against ZAb_FLEP, we premixed ZIKV strain H/PF/2013 with 22.50 μg/mL (EC90 of antibody that was predicted to neutralize the majority of the input virus), 500 μg/mL IgG1 isotype control antibody. Antibody was allowed to opsonize virus at 37°C for 1 hour in DMEM maintenance media, before being used to infect Vero cells for 48–72 hours to allow virus propagation to occur (1 passage). At the end of the 48–72 hour incubation, the supernatant containing virus was collected and half the volume was again pre-mixed with varying amounts of antibody, thus repeating the selection and propagation process. This propagation process was performed for a total of seven passages for each antibody combination. At each passage, aliquots of virus were frozen for plaque titer determination and subsequent sequencing of the virus by RT-PCR using primers designed to amplify the entire E gene, from the prM to NS1 gene. The resultant amplicon was run on an agarose gel, gel-extracted and sequenced. In the presence of control IgG1, wildtype virus was recovered in each passage. Sequencing of all detectable viruses was performed by first extracting viral RNA using the QIAamp Viral RNA Mini kit and then performing RT-PCR with primers designed to amplify the entire E gene (region between prM and NS1) using the One-Step Superscript III RT-PCR System with Platinum Taq. The resultant amplicon (~1,800 bp) was run on a 1% agarose gel, gel-extracted with the MinElute Gel Extraction Kit (Qiagen), and sequenced.
Quantification and Statistical Analysis
Statistical Methods
Statistical significance was determined by student’s t test between indicated groups using GraphPad Prism. Data presented represent the mean with error bars signifying the standard error of the mean.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Akshita Kumar for her help with sample preparation. We also thank Andrew Hatas, Drs. Rahul Raman and V. Sasisekharan for technical support and helpful discussions in support of the manuscript. This work was funded in part by National Institutes of Health Award (1R01AI111395; to RS) and National Research Foundation supported Interdisciplinary Research group in Infectious Diseases of SMART (Singapore MIT alliance for Research and Technology). Support from NMRC grant CBRG-0073–2014 to JL is also acknowledged.
Footnotes
Data and Software Availability
The informatics tool to determine the network scores from an X-ray crystal or cryoEM structure is made available online at glycomics2.mit.edu/matlab. Protein residue solvent accessibility is determined using the DSSP software available at http://swift.cmbi.ru.nl/gv/dssp/. The extent of sequence conservation in alignments of E-protein (DENV/ZIKV) was measured using Jalview, which is available at http://www.jalview.org/. Computational docking between antibody scaffolds and E-protein epitope was enabled by ZDOCK (http://zdock.umassmed.edu/). Methods for predicting native antigen-antibody-like structures and affinity enhancing mutations have been described before (Tharakaraman K et. al., PNAS 2013). Cryo-EM density map of the virion complexed with Fab ZAb_FLEP has been deposited with the Electron Microscopy Data Bank: EMD-7613.
AUTHOR CONTRIBUTIONS
K.T., S.W., K.R.C., S.G.V., E.E.O., R.S. conceived and designed the study experiments. K.T., D.Q., A.R., R.S. conducted computational analyses, including epitope prediction and antibody engineering. K.R.C., V.S., Y.H.C, M.M, E.Z.O., E.S.G., H.C.T., E.E.O. performed the biochemical, in vitro and regular A129 in vivo experiments. S.W., K.R.C, E.Z.O., E.S.G., H.C.T. and S.G.V. designed and conducted the pregnant A129 mice experiments. J.H., A.T., S.B. and J.L. solved the cryo-electron microscopy structure of virus:Fab complex and completed structural analysis. K.T., A.R., S.G.V., J.L., E.E.O. and R.S. wrote the manuscript.
DECLARATION OF INTERESTS
MIT and Duke-NUS own intellectual property related to anti-Zika monoclonal antibodies that are described in part in this manuscript. Drs. Ooi and Sasisekharan are board members of Tychan Pte. Ltd. that is conducting clinical studies related to anti-Zika therapeutics.
DENV, Dengue virus; EDEN, Early Dengue Infection and Outcome Study; ZIKV, Zika virus.
REFERENCES
- Balsitis SJ, Williams KL, Lachica R, Flores D, Kyle JL, Mehlhop E, Johnson S, Diamond MS, Beatty PR, and Harris E (2010). Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog 6, e1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barba-Spaeth G, Dejnirattisai W, Rouvinski A, Vaney MC, Medits I, Sharma A, Simon-Loriere E, Sakuntabhai A, Cao-Lormeau VM, Haouz A, et al. (2016). Structural basis of potent Zika-dengue virus antibody cross-neutralization. Nature 536, 48-53. [DOI] [PubMed] [Google Scholar]
- Barouch DH, Thomas SJ, and Michael NL (2017). Prospects for a Zika Virus Vaccine. Immunity 46, 176-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JM, Chen M, Baldwin PR, and Ludtke SJ (2016). High resolution single particle refinement in EMAN2.1. Methods 100, 25-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramello M, Williams KL, Simmons CP, Macagno A, Simonelli L, Quyen NT, Sukupolvi-Petty S, Navarro-Sanchez E, Young PR, de Silva AM, et al. (2010). The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 8, 271-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KR, Zhang SL, Tan HC, Chan YK, Chow A, Lim AP, Vasudevan SG, Hanson BJ, and Ooi EE (2011). Ligation of Fc gamma receptor IIB inhibits antibody-dependent enhancement of dengue virus infection. Proc Natl Acad Sci U S A 108, 12479-12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alwis R, Beltramello M, Messer WB, Sukupolvi-Petty S, Wahala WM, Kraus A, Olivarez NP, Pham Q, Brien JD, Tsai WY, et al. (2011). In-depth analysis of the antibody response of individuals exposed to primary dengue virus infection. PLoS Negl Trop Dis 5, e1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba-Spaeth G, Duangchinda T, Sakuntabhai A, Cao-Lormeau VM, Malasit P, Rey FA, et al. (2016). Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat Immunol 17, 1102-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas G, Carvalho LM, Bedford T, Tatem AJ, Baele G, Faria NR, Park DJ, Ladner JT, Arias A, Asogun D, et al. (2017). Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 544, 309-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldani LZ (2017). Yellow fever outbreak in Brazil, 2017. Braz J Infect Dis 21, 123-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan SS, Miller A, Sapparapu G, Fernandez E, Klose T, Long F, Fokine A, Porta JC, Jiang W, Diamond MS, et al. (2017). A human antibody against Zika virus crosslinks the E protein to prevent infection. Nat Commun 8, 14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz FX, Auer G, Stiasny K, Holzmann H, Mandl C, Guirakhoo F, and Kunz C (1994). The interactions of the flavivirus envelope proteins: implications for virus entry and release. Arch Virol Suppl 9, 339-348. [DOI] [PubMed] [Google Scholar]
- Kostyuchenko VA, Lim EX, Zhang S, Fibriansah G, Ng TS, Ooi JS, Shi J, and Lok SM (2016). Structure of the thermally stable Zika virus. Nature 533, 425-428. [DOI] [PubMed] [Google Scholar]
- Lai CY, Williams KL, Wu YC, Knight S, Balmaseda A, Harris E, and Wang WK (2013). Analysis of cross-reactive antibodies recognizing the fusion loop of envelope protein and correlation with neutralizing antibody titers in Nicaraguan dengue cases. PLoS Negl Trop Dis 7, e2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessler J, Chaisson LH, Kucirka LM, Bi Q, Grantz K, Salje H, Carcelen AC, Ott CT, Sheffield JS, Ferguson NM, et al. (2016). Assessing the global threat from Zika virus. Science 353, aaf8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, and Cheng Y (2013). Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods 10, 584-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JG, Ooi EE, Tolfvenstam T, Leo YS, Hibberd ML, Ng LC, Lai YL, Yap GS, Li CS, Vasudevan SG, et al. (2006). Early Dengue infection and outcome study (EDEN) - study design and preliminary findings. Ann Acad Med Singapore 35, 783-789. [PubMed] [Google Scholar]
- Low JG, Ooi EE, and Vasudevan SG (2017). Current Status of Dengue Therapeutics Research and Development. J Infect Dis 215, S96-S102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, and Grigorieff N (2003). Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142, 334-347. [DOI] [PubMed] [Google Scholar]
- Morens DM, and Fauci AS (2013). Emerging infectious diseases: threats to human health and global stability. PLoS Pathog 9, e1003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso D (2015). Zika Virus Transmission from French Polynesia to Brazil. Emerg Infect Dis 21, 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso D, and Gubler DJ (2016). Zika Virus. Clin Microbiol Rev 29, 487-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605-1612. [DOI] [PubMed] [Google Scholar]
- Robbiani DF, Bozzacco L, Keeffe JR, Khouri R, Olsen PC, Gazumyan A, Schaefer-Babajew D, Avila-Rios S, Nogueira L, Patel R, et al. (2017). Recurrent Potent Human Neutralizing Antibodies to Zika Virus in Brazil and Mexico. Cell 169, 597-609 e511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson LN, Tharakaraman K, Rowley KJ, Costa VV, Chan KR, Wong YH, Ong LC, Tan HC, Koch T, Cain D, et al. (2015). Structure-Guided Design of an Anti-dengue Antibody Directed to a Non-immunodominant Epitope. Cell 162, 493-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Morales AJ, Bandeira AC, and Franco-Paredes C (2016). The expanding spectrum of modes of transmission of Zika virus: a global concern. Ann Clin Microbiol Antimicrob 15, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapparapu G, Fernandez E, Kose N, Bin C, Fox JM, Bombardi RG, Zhao H, Nelson CA, Bryan AL, Barnes T, et al. (2016). Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 540, 443-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, and Kuhn RJ (2016). The 3.8 A resolution cryo-EM structure of Zika virus. Science 352, 467-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JL, and Lok SM (2014). Dengue virus purification and sample preparation for cryo-electron microscopy. Dengue: Methods and Protocols, 41-52. [DOI] [PubMed] [Google Scholar]
- Ter Meulen J (2011). Monoclonal antibodies in infectious diseases: clinical pipeline in 2011. Infect Dis Clin North Am 25, 789-802. [DOI] [PubMed] [Google Scholar]
- Tharakaraman K, Robinson LN, Hatas A, Chen YL, Siyue L, Raguram S, Sasisekharan V, Wogan GN, and Sasisekharan R (2013). Redesign of a cross-reactive antibody to dengue virus with broad-spectrum activity and increased in vivo potency. Proc Natl Acad Sci U S A 110, E1555-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Bardelli M, Espinosa DA, Pedotti M, Ng TS, Bianchi S, Simonelli L, Lim EXY, Foglierini M, Zatta F, et al. (2017). A Human Bi-specific Antibody against Zika Virus with High Therapeutic Potential. Cell 171, 229-241 e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Yang H, Liu X, Dai L, Ma T, Qi J, Wong G, Peng R, Liu S, Li J, et al. (2016). Molecular determinants of human neutralizing antibodies isolated from a patient infected with Zika virus. Sci Transl Med 8, 369ra179. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Chan KW, Wang J, Rivino L, Lok SM, and Vasudevan SG (2015). Dengue Virus Infection with Highly Neutralizing Levels of Cross-Reactive Antibodies Causes Acute Lethal Small Intestinal Pathology without a High Level of Viremia in Mice. J Virol 89, 5847-5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson D, Robinson J, Chappell KJ, Butler MS, Edwards DJ, Fry SR, Bermingham IM, Cooper MA, and Young PR (2016). A generic screening platform for inhibitors of virus induced cell fusion using cellular electrical impedance. Sci Rep 6, 22791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Fan C, Ku Z, Zuo T, Kong L, Zhang C, Shi J, Liu Q, Chen T, Zhang Y, et al. (2016). Structural Basis for Recognition of Human Enterovirus 71 by a Bivalent Broadly Neutralizing Monoclonal Antibody. PLoS Pathog 12, e1005454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Sun K, Chinazzi M, Pastore YPA, Dean NE, Rojas DP, Merler S, Mistry D, Poletti P, Rossi L, et al. (2017). Spread of Zika virus in the Americas. Proc Natl Acad Sci U S A 114, E4334-E4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Kostyuchenko VA, Ng T-S, Lim X-N, Ooi JS, Lambert S, Tan TY, Widman DG, Shi J, and Baric RS (2016a). Neutralization mechanism of a highly potent antibody against Zika virus. Nature Communications 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Kostyuchenko VA, Ng TS, Lim XN, Ooi JS, Lambert S, Tan TY, Widman DG, Shi J, Baric RS, et al. (2016b). Neutralization mechanism of a highly potent antibody against Zika virus. Nat Commun 7, 13679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Fernandez E, Dowd KA, Speer SD, Platt DJ, Gorman MJ, Govero J, Nelson CA, Pierson TC, Diamond MS, et al. (2016). Structural Basis of Zika Virus-Specific Antibody Protection. Cell 166, 1016-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.