

Abstract
Biosynthesis of the gibberellin plant hormones evolved independently in plants and microbes, but the pathways proceed via similar transformations. The combined demethylation and γ-lactone ring forming transformation is of significant mechanistic interest, yet remains opaque. The relevant CYP112 from bacteria was probed via activity assays and 18O2 labeling experiments. Notably, the ability of tert-butyl hydroperoxide to drive this transformation indicates use of the ferryl-oxo (Compound I) from the CYP catalytic cycle for this reaction. Together with the confirmed loss of C-20 as CO2, this necessitates two catalytic cycles for carbon-carbon bond scission and γ-lactone formation. The ability of CYP112 to hydroxylate the δ-lactone form of GA15 shown by the labeling studies is consistent with the implied use of a further oxygenated heterocycle in the final conversion of GA24 to GA9, with the partial labeling of GA9 demonstrating that CYP112 partitions its reactants between two diverging mechanisms.
Keywords: enzyme mechanism, oxygenase, lactol, decarboxylation, anhydride
Graphical Abstract
Gibberellins are important phytohormones, requiring complex biosynthetic processes that independently evolved in bacteria and fungi as well as plants. The requisite coupled demethylation and γ-lactone ring formation, catalyzed by the bacterial cytochrome P450 CYP112, was mechanistically probed, providing insight into this complex transformation.
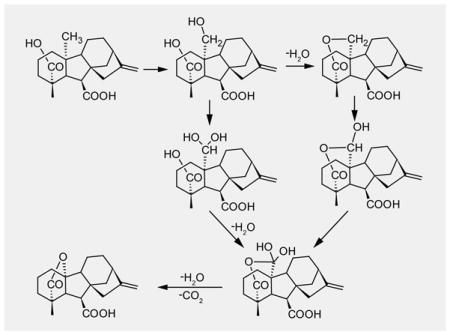
Gibberellins (GAs) are essential hormones in plants, which has led some plant-associated fungi and bacteria to produce GA to manipulate their host plants. The biosynthetic steps of the individual pathways are essentially identical in plants and bacteria, and differ from the fungal pathway only in the order of occurrence of one peripheral (C-3β) hydroxylation step. Yet on the enzyme level it is evident from the low sequence identity that GA biosynthesis evolved separately in plants, fungi and bacteria.[1] One hallmark transformation in GA biosynthesis is the conversion of 20-carbon GAs to the 19-carbon GAs, involving the combined loss of a methyl group and formation of an intramolecular γ-lactone bridge. In plants this complex multi-step reaction is catalyzed by iron/α-ketoglutarate-dependent dioxygenases termed GA 20-oxidase (GA20ox), while it is catalyzed by cytochrome P450 (CYP) monooxygenases in fungi (CYP68B) and bacteria (CYP112).[1a] Although fungi and bacteria both use CYPs, these fall into different families that share less than 15% sequence identity. Nonetheless the reactions catalyzed by the three individual enzymes appear to be identical, and involve the stepwise oxidation of C-20 from a methyl group in GA12 in plants and bacteria, and GA12 as well as the 3β-hydroxy derivative GA14 in fungi, to the corresponding alcohols GA15 or GA37 and further oxidation to the aldehyde function, yielding GA24 or GA36, respectively. The final step is combined loss of the oxidized methyl group and formation of a γ-lactone bridge between carbon C-19 and C-10, producing GA9 or GA4, respectively (Scheme 1). While the fungal enzyme does not react with the intermediates GA15/GA37 and GA24/GA36, the bacterial and plant enzymes can use GA15 and GA24 as substrates to produce GA9.[1a, 1b] Plants and fungi also produce the C-20 carboxylic acids GA25 or GA13 as side products, since neither the plant GA20ox nor the fungal CYP68B further convert GA25/GA13 to GA9/GA4 (Scheme 1).[2] For the fungal CYP68B very little is known about the catalytic mechanism, only that C-20 appears to be lost as CO2.[3] With one plant GA20ox catalyzing this multi-step reaction some artificial substrates have been tested, and it has been demonstrated that C-20 also is lost as CO2.[4] The combination of the apparently straightforward first two reactions with the complex last reaction, and the known differences between plants and fungi, makes the catalytic mechanism of CYP112 of significant interest, which was investigated here.
Scheme 1.

Reactions catalyzed by CYP112, CYP68B and GA20ox in GA biosynthesis from bacteria, fungi and plants, respectively. Sequential oxidation and elimination of C-20, while the plant and fungal enzyme release CO2 upon elimination of C-20, this was unknown for the bacterial CYP112.
Previously, isoforms of CYP112 were characterized by addition of GA12 to recombinant Escherichia coli cultures.[1b, 1d] This however did not permit detailed investigation of the catalytic mechanism. Here the isoform from Erwinia tracheiphila (EtCYP112), chosen as it is in the same Enterobacteriaceae family as E. coli, was His-tagged, purified and found to exhibit full activity in a reconstituted system with purified ferredoxin reductase from E. tracheiphila (EtFdR) and spinach ferredoxin (Fd), converting GA12 to GA9 (Figure 1). Under optimal conditions (i.e., with a molar excess of NADPH), no intermediates were detected, even when turnover was not yet complete, indicating their retention in the active site. It was possible to observe the expected C-20 hydroxylated intermediate GA15 and aldehyde intermediate GA24 under NADPH-limited conditions. These assays also produced the C-20 carboxylate derivative GA25 (Figures 1 and S1). However, while GA15 and GA24 were readily converted to GA9, GA25 is not (Figure S2).
Figure 1.

In vitro enzyme activity of EtCYP112. GC-MS chromatograms of assays with purified EtCYP112, spinach Fd and EtFdR under limiting NADPH concentrations (A), or NADPH in excess stopped after 30 min (B), with GA12 as substrate.
The form in which C-20 is released by EtCYP112 was examined by 18O2 labeling, using the reconstituted enzymatic system with an excess of NADPH under an 18O2 atmosphere, with GC-MS analysis of the head-space. In the absence of substrate only unlabeled CO2 was found, while addition of GA12 resulted in production of CO2 with one or two 18O labels (Figure 2). The observed loss of label presumably originates from the interconversion of CO2 with bicarbonate during the incubation period of the enzyme assay.[5] Thus, EtCYP112 clearly releases C-20 as CO2. In addition, the resulting GA9 product was extracted, methylated and analyzed by GC-MS. Surprisingly, 44% had incorporated one 18O (Figures 3 and S2, and Table S1). This was found not only in the molecular ion, but also the fragments at m/z = 298/299 and 270, which retain the γ-lactone ring,[6] wherein the 18O label must have been inserted.
Figure 2.

Figure CO2 loss during γ-lactone formation. (A) GC-MS chromatograms of enzyme assays with purified EtCYP112, spinach Fd, EtFdR, NADPH in excess and either no substrate or GA12 as the substrates under an 18O2 atmosphere. (B) MS-spectra of the CO2 peak.
Figure 3.

In vitro enzyme activity of EtCYP112 under an 18O2 atmosphere. (A) GC-MS chromatograms of enzyme assays with purified EtCYP112, spinach Fd and EtFdR with an excess of NADPH and GA12 or under limiting NADPH concentrations with GA12, GA15 (closed lactone) or GA24 as the substrate under an 18O2 atmosphere. (B) MS-spectra of GA9 showing the molecular ion at m/z 330 or 332 and two larger fragments corresponding to the loss of the methylated acid at C-7.
18O incorporation during the transformation of GA12 to GA9 was further probed with limiting NADPH, enabling examination of the intermediates GA15 and GA24. While this reduced the amount of 18O incorporated into GA9 (to 14%), a substantial proportion of the observed GA24 accumulated two 18O (37%). The remainder of the GA24 contained a single 18O. GA15 is observed in the closed δ-lactone form, and was essentially fully labeled with a single 18O. This demonstrated that GA12 is converted to GA15 via hydroxylation rather than direct formation of the δ-lactone ring. Analogous labeling experiments were carried out with GA15 (open lactone), GA15 (closed δ-lactone) or GA24 as substrate (Figure 3 and Table S1). GA9 produced from these intermediates did not contain 18O. However, GA24 produced from the closed (δ-lactone) form of GA15 was fully labeled with 18O. Thus, EtCYP112 readily hydroxylates the δ-lactone form of GA15 to the lactol form of GA24. Given that GA24 is observed in its aldehyde form,[7] the incorporated 18O is retained by direct opening of the lactol. Starting from the open form of GA15, 67% of GA24 was labeled with 18O, indicating that EtCYP112 produces a geminal-diol, which upon dehydration loses ~50% of the label and forms the observed GA24 aldehyde. The greater amount of labeled species observed here originates from both a slight bias towards removal of the unlabeled hydroxyl-group,[8] but presumably largely from partial closure of the open GA15 to its δ-lactone form in the assay buffer before conversion by EtCYP112.
To further investigate formation of the lactol form of GA24, the C-7 and C-19 carboxylate groups were methylated in GA12, GA15, and GA24. This was expected to block lactone formation in GA15 and GA9. However, C-19 methylation seems to be unstable in GA15, and is quickly lost by conversion to the δ-lactone upon purification in organic solvents. The instability of the C-19 methyl ester in GA15 is also reflected in assays with the enzymatic system. In particular, MeGA12 was efficiently converted into GA9 and, under an 18O2 atmosphere, all GA9 product contains one 18O label. MeGA15 (closed δ-lactone) also is transformed into GA9, but does not incorporate any 18O label, while MeGA24 is not acted upon by EtCYP112 (Figures 4 and S3, and Table S1).
Figure 4.

In vitro enzyme activity of EtCYP112 with MeGA12 and MeGA15 under an 18O2 atmosphere. (A) GC-MS chromatograms of enzyme assays with purified EtCYP112, spinach Fd and EtFdR under limiting NADPH concentrations with MeGA12 or MeGA15 (closed lactone) as the substrate under an 18O2 atmosphere. (B) MS-spectra of GA9 showing the molecular ion at m/z 330 or 332 and two larger fragments corresponding to the loss of the methylated acid at C-7.
It has been suggested that GA24 can be converted into GA9 in a single catalytic cycle via the atypical use of a ferric-peroxo activated group,[2a] and similar carbon-carbon bond cleavage reactions catalyzed by CYP1A2 and CYP17A1 have been suggested to utilize superoxo or peroxo states of the heme-iron.[9] In CYPs these early stages of the catalytic cycle can be bypassed by using H2O2 or tert-butyl hydroperoxide (tBuOOH) as the reducing agent instead of molecular oxygen and electrons from NADPH (Scheme S2).[10] However, while the overall activity of EtCYP112 with H2O2 is significantly lower, limiting conversion of GA12 to GA15 and GA24, it is able drive the conversion of GA15 to GA24 and GA9, as well as GA24 to GA9. tBuOOH also can drive conversion of GA24 to GA9 (Figure S4).
The incorporation of 18O into a significant proportion of the GA9 γ-lactone ring indicates that one of the oxygens incorporated by EtCYP112 is transferred to the C-19 carboxylate. This must stem from the initial hydroxylation of GA12 to the open form of GA15 that then undergoes dehydration to the δ-lactone ring form.[7b] Indeed, the essentially complete labeling of the GA15 produced from GA12 demonstrates that this reaction invariably involves hydroxylation. The ability of EtCYP112 to catalyze hydroxylation of the δ-lactone form of GA15 to the lactol form of GA24 is demonstrated by both the complete 18O labeling of the GA24 produced from the closed (lactone) form of GA15, and the significant double-labeling of the GA24 produced from GA12. EtCYP112 can also hydroxylate the open form of GA15 to the geminal-diol form of GA24, as demonstrated by the partial labeling of the resulting GA24.
The most interesting transformation is however the conversion of GA24 into GA9, requiring both carbon-carbon bond scission and γ-lactone ring formation. While MacMillan has proposed several reaction mechanisms,[2a] these were focused on the plant dioxygenases. Nevertheless, CYP monooxygenases also use an iron co-factor to bind molecular oxygen, and both of these oxygenases generally utilize activated ferryl-oxo complexes (Compound I) for catalysis.[10–11] Thus, the proposed mechanisms are adaptable to CYPs (Schemes S3 and S4). In addition, CYPs are known to catalyze reactions via ferryl-hydroxo (Compound II), ferric-superoxo, ferric-peroxo,[9] or ferric-hydroperoxo species (Compound 0) as well (Scheme S2).[12] However, the ability of EtCYP112 to use H2O2 or tBuOOH to drive this transformation precludes the use of either the superoxo or peroxo complexes, or Compound 0, respectively. Thus, EtCYP112 uses Compound I in the transformation of GA24 to GA9, which is consistent with the implied use of Compound I for the hydroxylation reactions shown here for the two preceding transformations, as well as the mechanism proposed for the similar reaction catalyzed by CYP19A1.[13] However, release of C-20 as CO2 rules out transformation of GA24 to GA9 via a single oxidation cycle, even with consideration of alternative reactivity with Compound II, despite the inability of the C-20 carboxylate GA25 to serve as a substrate. It has been suggested that the plant GA20ox also uses two oxidation cycles, but proceeds via a covalent intermediate.[4b] However, there is no evidence for such a mechanism with EtCYP112. On the other hand, the intermediacy of a C-20 geminal-diol cyclic anhydride equivalent may explain why the open C-20 carboxylate GA25 is not a substrate. Consistent with a requirement for a cyclic anhydride intermediate is the inability of EtCYP112 to use MeGA24 as a substrate, as it cannot form such a heterocycle.
The results presented here indicate that, similar to CYP170A1 in albaflavenone biosynthesis,[14] EtCYP112 utilizes two diverging routes in transformation of GA12 to GA9. This is demonstrated by the partial labeling of the GA9 produced from GA12, and contrast with the complete labeling observed with MeGA12, which is biased towards formation of the closed (δ-lactone) form of GA15. Accordingly, rather than choose, EtCYP112 partitions the travel of its reactants between two mechanistic roads that diverge at GA15 (Scheme 2). Initial hydroxylation to the open form of GA15 can be followed by dehydration to the δ-lactone form, with subsequent hydroxylation of either producing the gem-diol or lactol forms of GA24, respectively (Scheme S4). These distinct intermediates can then undergo separate conversion to GA9 via two sequential oxidation reactions. This presumably involves formation of the C-20 gem-diol cyclic anhydride equivalent in both cases, reflecting either direct hydroxylation of δ-lactol, or cyclization of gem-diol GA24 mediated by the ability of Compound II to abstract an additional hydrogen. The resulting anhydride gem-diol then undergoes coupled carbon-carbon bond scission and γ-lactone ring formation, with release of CO2, again relying on abstraction of another hydrogen by Compound II. Altogether, despite their independent evolution, plants, fungi and bacteria catalyze the same series of reactions in the coupled demethylation and γ-lactone ring formation transformation step of gibberellin biosynthesis.[3–4] The C-20 carboxylate equivalent GA25 does not serve as a productive intermediate for any of the relevant oxygenases, and the C-20 gem-diol cyclic anhydride equivalent indicated here for the bacterial CYP112 has also been suggested for plants.[2a] Thus, despite their independent evolutionary origins, these oxygenases seem to have converged on similar mechanistic routes, perhaps reflecting the chemical constraints of this complex transformation as well as biochemical constraints of the utilized iron-dependent oxygenases.
Scheme 2.

Proposed bifurcation and reconversion of pathways from GA12 to GA9 catalyzed by EtCYP112.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (GM109773) and NSF (CHE-1609917) to R.J.P., along with a postdoctoral fellowship to R.N. from the Deutsche Forschungsgemeinschaft (DFG) NA 1261/1-2. We wish to thank ISU Chemical Instrumentation Facility staff member Steve Veysey for assistance with the Waters GCT GC-MS analyses, and Prof. Peter Hedden (Rothamsted Research) for GA12 and GA25 standards.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Hedden P, Sponsel V. J Plant Growth Regul. 2015;34:740–760. doi: 10.1007/s00344-015-9546-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nett RS, Montanares M, Marcassa A, Lu X, Nagel R, Charles TC, Hedden P, Rojas MC, Peters RJ. Nat Chem Biol. 2017;13:69–74. doi: 10.1038/nchembio.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nagel R, Turrini PC, Nett RS, Leach JE, Verdier V, Van Sluys MA, Peters RJ. New Phytol. 2017;214:1260–1266. doi: 10.1111/nph.14441. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Nagel R, Peters RJ. Mol Plant-Microbe Interact. 2017;30:343–349. doi: 10.1094/MPMI-01-17-0001-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacMillan J. Nat Prod Rep. 1997;14:221–243. [Google Scholar]; b) Tudzynski B, Rojas MC, Gaskin P, Hedden P. J Biol Chem. 2002;277:21246–21253. doi: 10.1074/jbc.M201651200. [DOI] [PubMed] [Google Scholar]
- 3.Dockerill B, Hanson JR. Phytochemistry. 1978;17:701–704. [Google Scholar]
- 4.a) Kamiya Y, Takahashi N, Graebe JE. Planta. 1986;169:524–528. doi: 10.1007/BF00392102. [DOI] [PubMed] [Google Scholar]; b) Ward JL, Gaskin P, Brown RGS, Jackson GS, Hedden P, Phillips AL, Willis CL, Beale MH. J Chem Soc, Perkin Trans. 2002;1:232–241. [Google Scholar]; c) Ward JL, Jackson GJ, Beale MH, Gaskin P, Hedden P, Mander LN, Phillips AL, Seto H, Talon M, Willis CL, Wilson TM, Zeevaart JAD. Chem Commun. 1997:13–14. [Google Scholar]
- 5.a) Gibbons BH, Edsall JT. J Biol Chem. 1963;238:3502–3507. [PubMed] [Google Scholar]; b) Mills GA, Urey HC. J Am Chem Soc. 1940;62:1019–1026. [Google Scholar]; c) Usdowski E, Hoefs J. Chem Geol: Isotope Geosci. 1990;80:109–118. [Google Scholar]
- 6.Gaskin P, MacMillan J. GC-MS of the gibberellins and related compounds : methodology and a library of spectra. Univ. of Bristol (Cantocks Enterprises Ltd); Bristol: 1991. [Google Scholar]
- 7.a) Harrison DM, MacMillan J. J Chem Soc C: Organic. 1971:631–636. [Google Scholar]; b) Harrison DM, MacMillan J, Galt RHB. Tetrahedron Lett. 1968;9:3137–3139. [Google Scholar]
- 8.Nagel R, Peters RJ. Org Biomol Chem. 2017;15:7566–7571. doi: 10.1039/c7ob01819c. [DOI] [PubMed] [Google Scholar]
- 9.a) Varfaj F, Zulkifli SNA, Park H-G, Challinor VL, De Voss JJ, Ortiz de Montellano PR. Drug Metabol Dispos. 2014;42:828–838. doi: 10.1124/dmd.114.056903. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gregory MC, Denisov IG, Grinkova YV, Khatri Y, Sligar SG. J Am Chem Soc. 2013;135:16245–16247. doi: 10.1021/ja4086403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Ortiz de Montellano PR. Cytochrome P450 - Structure, Mechanism, and Biochemistry. 4. Springer International Publishing; 2015. [Google Scholar]; b) Hiroya K, Murakami Y, Shimizu T, Hatano M, de Montellano PRO. Arch Biochem Biophys. 1994;310:397–401. doi: 10.1006/abbi.1994.1184. [DOI] [PubMed] [Google Scholar]
- 11.Martinez S, Hausinger RP. J Biol Chem. 2015 doi: 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krest CM, Onderko EL, Yosca TH, Calixto JC, Karp RF, Livada J, Rittle J, Green MT. J Biol Chem. 2013;288:17074–17081. doi: 10.1074/jbc.R113.473108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshimoto FK, Guengerich FP. J Am Chem Soc. 2014;136:15016–15025. doi: 10.1021/ja508185d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao B, Lin X, Lei L, Lamb DC, Kelly SL, Waterman MR, Cane DE. J Biol Chem. 2008;283:8183–8189. doi: 10.1074/jbc.M710421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.