

Abstract
Objective
Serotoninergic neurotransmission may modulate beta-amyloid peptide (Aβ) metabolism through up-regulation of alpha-secretase. Early Parkinson disease (PD) shows variable serotoninergic denervation, which may impact Aβ deposition.
Methods
We conducted three analyses to explore associations between serotoninergic neurotransmission and cerebral Aβ burden in PD. The first was a cross-sectional imaging study of PD subjects (n=23) using the serotoninergic transporter positron emission tomography (PET) ligand [11C]DASB and amyloid PET Pittsburgh compound B ([11C]PiB). The second was a baseline study of Parkinson’s Progression Markers Initiative (PPMI) subjects exploring the influence of serotoninergic medications on cerebrospinal fluid (CSF) Aβ-42 levels (n=389), controlling for age, sex, Geriatric Depression scale (GDS), disease duration, and education. Third, we fit an interval-censored proportional hazard model with longitudinal PPMI data (n=367) to test whether serotoninergic medication use associates with reduced risk of PD-cognitive-decline, defined as time to reach a Montreal Cognitive Assessment score ≤20, adjusting for baseline caudate dopamine transporter (DaT)[123I]Ioflupane single photon emission computed tomography and cerebrospinal fluid Aβ-42 levels.
Results
Serotoninergic DASB distribution volume ratio (DVR) inversely associated with PiB DVR in the cerebral cortex (Pearson’s-r=−0.478,p=0.021) but not the striatum (r=−0.264,p=0.224). In the baseline PPMI analysis, serotoninergic medication use for ≥6 months associated with a lower level of CSF Aβ-42 (t=−2.20,p=0.029). In the longitudinal PPMI model, baseline serotoninergic medication use associated with a reduced risk of cognitive decline (t=−2.03,p=0.043) after controlling for covariates.
Interpretation
Cortical Aβ burden in PD associates inversely with serotoninergic innervation. Serotoninergic medications may alter Aβ metabolism and reduce the risk of PD cognitive decline.
Keywords: Parkinson disease, SSRIs, amyloid, cognition, PPMI
Introduction
Progressive cerebral beta-amyloid peptide (Aβ) plaque burden is linked to clinically meaningful outcomes–including cognitive decline1, 2 and gait impairments3, 4–in Parkinson disease (PD) and synucleionpathy-associated dementias. Aβ plaque deposition, assessed by amyloid positron emission tomography (PET), is associated with prevalent cognitive impairment in parkinsonian conditions.5 Reduced cerebrospinal fluid (CSF) Aβ-42 oligomers and factors influencing their metabolism are also consistently reported as risk factors for subsequent cognitive decline in PD without dementia.6–8 Developing strategies to alter the natural history of Aβ burden in PD may represent an untapped disease-modifying approach and is an emerging research priority.
Variable loss of serotoninergic nerve terminals in the forebrain, striatum, and brainstem is described in Parkinson disease.9, 10 Significant loss of serotoninergic terminals is an early feature in a substantial fraction of PD subjects.7,8 Preclinical experiments in Alzheimer disease (AD) mouse models by Cirrito et al. showed that chronic exposure to either serotonin itself or the selective-serotonin reuptake inhibitor (SSRI) citalopram reduced extracellular cerebral Aβ peptide concentrations and plaque burden in short- and long-term in vivo studies.11, 12 Serotonin and SSRIs are suggested to stimulate a serotonin G-protein-coupled receptor-linked intracellular cascade that favors the cleavage of Amyloid Precursor Protein (APP) by alpha-secretase, leading to reductions in toxic Aβ peptide amyloid generation and reduced plaque formation.11 Serotonin medications may also promote efflux of toxic Aβ peptide species from the brain into the blood.13 Other preclinical research supports the concept that serotoninergic neurotransmission can favorably modulate APP processing.14–17 If the same protective association between serotonin receptor activation and reduced amyloid burden exists in humans, a disease state such as PD with reductions in serotoninergic tone offers a potential model for testing this hypothesis. In a small cohort, we previously reported that regional density of cortical and striatal serotoninergic terminals in PD, measured by in vivo positron emission tomography (PET), showed inverse correlations with Aβ plaque burden.18
We present three analyses exploring associations between serotoninergic neurotransmission and Aβ burden in PD, using different experimental designs to address 3 related hypotheses. First, using an identical multimodal imaging approach to our earlier study in a new cohort, we sought to replicate our findings that decreased cortical serotoninergic innervation is inversely associated with the severity of cortical Aβ plaque burden in PD. Second, using baseline data from the multicenter Parkinson Progression Markers Initiative (PPMI), we explored associations between serotonin medication use and CSF Aβ-42 levels. Third, we conducted a longitudinal analysis of PPMI data to test whether baseline serotoninergic medication use associates with a protective effect on cognitive decline in PD, independent of existing heterogeneity in caudate dopamine transporter binding or baseline cerebral amyloid burden.
Methods
Study 1: Cross-sectional prospective PET imaging study
In a previous PD cohort (n=13), we reported inverse associations between cortical and striatal serotoninergic terminal and amyloid PET findings.18 To determine whether these findings were due to cohort specific effects, we conducted a separate cross-sectional multimodal PET imaging study of 23 PD subjects. Inclusion criteria included age ≥45, a diagnosis of PD as determined by the UK Parkinson’s Disease Society Brain Bank Clinical diagnostic criteria,19 no evidence of atypical parkinsonism or neuroleptic-associated PD, no contraindications to undergo magnetic resonance imaging (MRI) such as indwelling metal hardware in the body, and no exposure to serotoninergic medications, anticholinergic medications, or cholinesterase inhibitors in the last 2 months. This study was reviewed by the University of Michigan IRBMED and all subjects signed informed consent prior to study enrollment.
Regional serotonin terminal density was assessed with the serotonin transporter PET ligand [11C]3-amino-4-(2-dimethylaminomethyl-phenylsulfaryl)-benzonitrile (DASB) and cerebral amyloid plaque deposition was measured with [11C]Pittsburgh compound B (PiB) PET as described previously.18 For 22 of the 23 subjects all MRI, DASB, and PiB PET scans took place within 1 week of each other. In one subject, the DASB and PiB scans were separated by 41 days. All PET studies were performed with bolus and infusion dynamic imaging protocols. Cortical and subcortical segmentation was performed with FreeSurfer version 5 in the fully automated mode. All image frames were spatially coregistered within participants with a rigid-body transformation. Motion-corrected PET frames were spatially coregistered to the T1-weighted MRI using standard coregistration procedures in NeuroStat (https://neurostat.neuro.utah.edu/). Time activity curves for each VOI were generated from the spatially aligned PET frames. DASB and PiB distribution volume ratios (DVRs) were estimated by the Logan plot graphical analysis method20 with the time-activity curves as the input function and with the inferior posterior cerebellum as reference tissue for both PiB and DASB. Pearson correlation coefficients were used to assess the relationship of PiB and DASB DVRs in the total cortical and striatal regions of interest. Using subject age, disease duration, years of education, and cortical DASB DVR as covariates, we also conducted a multivariable linear regression model with cortical PiB DVR as the outcome variable and a covariate significance level for retention in the final model of 0.1 using a backwards-selection process. Interaction terms were tested in the covariates retained in the final model and were included at a p-value threshold of 0.1.
Study 2a: Cross-sectional PPMI Baseline Analysis
The Parkinson’s Progression Markers Initiative (PPMI) is a multicenter longitudinal observational study of PD (http://www.ppmi-info.org/). Curated PPMI datasets containing Aβ-42 measurements, primary diagnoses, and dopamine transporter (DaT) [123I]Ioflupane single photon emission computed tomography (SPECT) results were downloaded on 8/22/17. Datasets containing demographics and clinical non-motor testing were downloaded between 10/10/2016 and 1/13/2017. PPMI subjects underwent screening evaluations (month 0), followed by baseline evaluations (typically at month 1), visit 1 evaluations (month 4), and subsequent evaluations on a scheduled basis—initially at 3 month intervals and then at 6 month intervals after visit 4 (month 13)–through a possible visit 12 (month 61). All subjects signed informed consent upon enrollment into the PPMI. This project using publically available coded PPMI data was granted not-regulated status by the University of Michigan IRBMED. Analyses were conducted in STATA 15 (College Station, TX) and SAS 9.4 (Cary, NC).
Eligible subjects for our baseline analyses were those who were coded as having a primary study diagnosis of “idiopathic PD” at the time of screening visit, a screening visit DaT scan evaluation, who had CSF Aβ-42 tested at the baseline visit, documentation of serotonin medication use at the time, and who did not accrue an alternative primary study diagnosis at a subsequent study visit. Figure 1 provides an overview of our PPMI study cohorts. We categorized serotoninergic medications to include selective serotonin reuptake inhibitors (SSRIs)21, serotonin-norepinephrine reuptake inhibitors (SNRIs)22, tricyclic antidepressants (TCAs)17, bupropion23, lithium24, and St. John’s Wort25 given that each of these compounds has been shown to influence serotoninergic neurotransmission. In this cross-sectional baseline study aimed at understanding the influence of serotonin medications on CSF Aβ-42, we categorized subjects as those taking serotonin medications for ≥6 months (n=45) vs. others (n=344). Serotonin-medication-use was treated as a categorical variable (yes/no) and was not adjusted for dosing or weight. Age was defined as the difference between year of birth and year of study enrollment and disease duration was defined at the number of years between when symptoms were first noticed by the subject and year of study enrollment. CSF Aβ-42 levels26 were measured at the baseline visit.
Figure 1.

Diagram of subject selection from Parkinson’s Progression Markers Initiative (PPMI).
Baseline CSF Aβ-42 was treated as the outcome variable of interest in a multivariable linear regression using baseline cross-sectional PPMI data. Covariates included serotonin medication use, age, sex, disease duration, self-reported years of education, and baseline Geriatric Depression Scale (GDS) score on the 15-item GDS. We aimed to control for the degree of depressive symptoms using the GDS since late-life depression is known to associate both with dementia risk and with CSF Aβ-42 burden.27, 28 We controlled for years of education to account for the possibility that subjects receiving prescription serotonin medications might have unmeasured differences in socio-economic status relative to those not on serotonin medications.
Study 2b: Longitudinal PPMI Proportional Hazard Analysis
We conducted a survival analysis in the PPMI cohort to explore the effects of serotoninergic medications on cognitive decline in PD. PD subjects who had 2 or more Montreal Cognitive Assessment (MoCA) scores recorded over the duration of the study, who underwent DaT SPECT at screening, and who had a screening MoCA score of >20 at study entry were eligible for inclusion (Figure 1).
We chose progression to MoCA scores of 20 or less as our endpoint given that this transition value (21->20) has been previously validated against full neuropsychological testing in PD as an optimal diagnostic cutoff point (as opposed to a “screening” cutoff value) for meeting neuropsychological criteria of impairment in at least 2 cognitive domains suggestive of dementia.29 We aimed to define our MoCA endpoint as manifestation of a clear stage-progression milestone, and tried to differentiate this endpoint from a transient drop in scoring that might improve at the next visit. Subsequently, we defined our endpoint as having been met if a subject had 2 or more consecutive visits with MoCA scores ≤20 or if their final recorded MoCA score was ≤20. We qualified time-to-event as the month correlating with the planned visit according to the PPMI schedule of activities: screening visit (month 0), visit 4 (month 13), visit 6 (month 25), visit 8 (month 37), visit 10 (month 49), through visit 12 (month 61). To account for the possibility of missed study visits and the likelihood of that not all study visits took place at the identical time interval, we conducted a survival analysis using interval censoring with a Weibull proportional hazard distribution. Subjects whose MoCA scores progressed from >20 at screening visit (month 0) to ≤20 by visit 4 (month 13) were identified as having progressed to the MoCA endpoint in an interval between 1 and 13 months in order to categorized as interval-censored rather than left censored. All other subjects who did not progress to a MoCA score of 20 or less were considered right censored at the time of their final recorded MoCA score. Of note, an inclusion criteria for enrollment in the PPMI was that subjects were “not expected to require PD medication within at least 6 months from baseline” (http://www.ppmi-info.org/study-design/).30 Subsequently, none of our longitudinal PPMI subjects were on dopaminergic PD medications at study enrollment.
We first tested the unadjusted bivariate associations between several different potential explanatory variables including the use of serotoninergic medications at the time of baseline evaluation (Supplementary Table 1) in separate Cox-proportional hazard analyses. Next, we tested associations in a multivariable model after adjusting for several baseline confounders. The nigrostriatal dopaminergic system is known to play a role in PD cognitive impairment, specifically dopamine terminal loss in the caudate nucleus.31, 32 We estimated this using mean bilateral caudate nucleus DaT SPECT measurements from the PPMI.33 We sought to control for heterogeneity in baseline amyloid status by using baseline CSF Aβ-42 levels as a covariate as well. Covariates were tested to explore the assumption of proportional hazards. To test whether a serotonin-medication effect on MoCA decline might vary across drug classes of differing specificity for serotonin-receptor modulation, we conducted two separate sensitivity analyses where the serotonin medication use categorical variable was restricted to either only those subjects taking SSRIs or those subjects taking either SSRIs or TCAs.
Results
Demographic characteristics for the Study 1 cohort are presented in Table 1. In the multi-modal PET imaging study, cortical serotoninergic terminal density correlated inversely with cortical amyloid burden (Pearson’s r=−0.478, p=0.021; Figure 2). Striatal serotoninergic terminal density did not significantly correlate with striatal amyloid deposition (Pearson’s r=−0.264, p=0.224). In our multivariable linear regression model of cortical PiB DVR, disease duration and cortical DASB DVR were retained at a covariate threshold of p<0.1. The final model (F = 5.11, p =0.0161) showed an R2 of 0.338 with a significant association seen for cortical DASB DVR (t=8.13, p =0.0099) and a non-significant associative trend for duration of disease (t=3.32, p −0.0836). There were no significant interactions between cortical DASB DVR and disease duration.
Table 1.
Subjects Characteristics
| Study 1: PD PET imaging cohort | Study 2a: PPMI Cross-sectional Baseline cohort | Study 2b: PPMI Longitudinal cohort | |
|---|---|---|---|
| Mean (SD) [min-max] or Count (%) | |||
| N | 23 | 389 | 367 |
| Baseline Age in years | 66.2 (6.7) [48-79] |
61.8 (9.6) [34-85] |
61.7 (9.7) [34-85] |
| Male Sex | 17 (74%) | 253 (65%) | 238 (65%) |
| Disease Duration in years | 4.9 (4.1) [1-12] |
1.9 (2.0) [0-21] |
1.9 (2.0) [0-21] |
| Hoehn and Yahr score | HY1=6 HY2 =15 HY3 =2 |
HY1=191 HY2=198 |
HY1=180 HY2=187 |
| Years of education | 16.9 (3.1) [12-25] |
15.6 (3.0) [5-26] |
15.5 (2.9) [5-26] |
| Montreal Cognitive Assessment (MoCA) Score at Screening | 25.6 (2.4) [20-29] |
27.2 (2.3) [17-30] |
27.2 (2.1) [21-30] |
| Final MoCA Score at the time of event/censoring | NA | NA | 26.4 (3.3) [15-31] |
| Mean absolute decline in MoCA score between screening and final score at the time of event/censoring | NA | NA | 0.8 (3.0) [−6 - +13] |
| Geriatric Depression score at baseline | 7.2 (4.5)* [1-15] |
5.1 (1.6) [0-10] |
5.1 (1.6) [0-10] |
| Cerebrospinal Fluid Aβ-42 (pg/ml) levels at baseline | — | 371.5 (101.5) [129.2-796.5] |
373.2 (102.2) [129.2-796.5] |
| Mean Cortical Amyloid PiB PET Distribution Volume Ratio | 1.06 (0.021) [1.04-1.11] |
— | — |
This score represents the 30-item Geriatric Depression Scale score, whereas the PPMI cohorts used the 15-item Geriatric Depression Scale.
NA = Not Applicable
Figure 2.
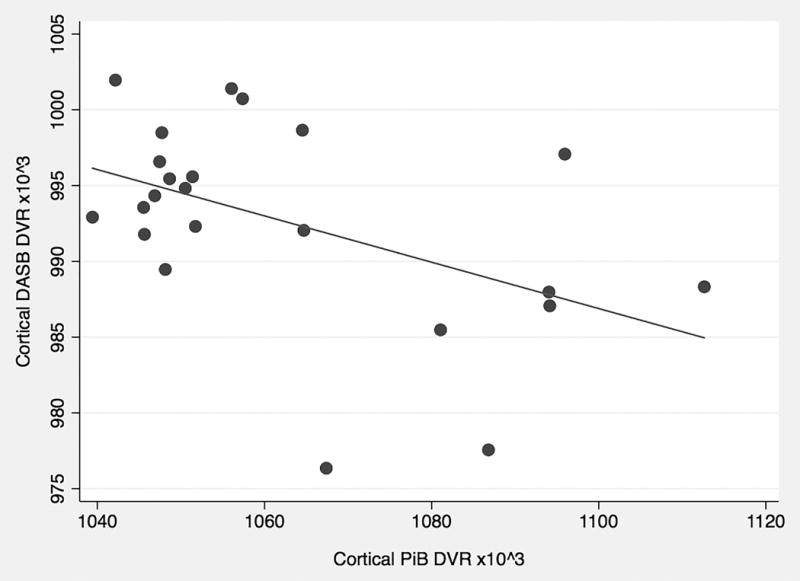
Cortical PiB (amyloid) vs DASB (serotonin) PET DVR (n=23).
In Study 2a, subjects on serotonin medications for at least 6 months at baseline (n=45) did not differ significantly in CSF Aβ-42 levels from subjects not receiving serotonin medications in unadjusted analyses (mean +/− SE: 348.5 pg/ml +/− 15.0 vs. 374.5 pg/ml +/− 5.5; t=1.62, p=0.11). After adjusting for confounders, the multivariable linear regression analyses (n=389; Table 2) showed that male sex and use of serotonin medications for at least 6 months both associated with lower levels of CSF Aβ-42 at baseline. Age, disease duration, years of education, and GDS score, did not show significant associations.
Table 2.
Baseline PPMI cross-sectional multivariable linear regression analysis (n=389)
| Dependent Variable: Baseline CSF Aβ-42 (pg/ml); Overall Model F=2.35, p=0.0305 | |||||
|---|---|---|---|---|---|
| Covariates | Beta | Standard Error | t-score | p-value | 95% Confidence Interval for Beta values |
| Age in years | −0.810 | 0.533 | −1.52 | 0.129 | −1.859, 0.238 |
| Male Sex | −28.971 | 11.002 | −2.63 | 0.009 | −50.603, −7.338 |
| Disease duration in years | 1.241 | 2.533 | 0.49 | 0.625 | −3.740, 6.221 |
| Years of Education | 0.067 | 1.718 | 0.04 | 0.969 | −3.311, 3.446 |
| Geriatric Depression scale (GDS-15) score | −4.262 | 3.142 | −1.36 | 0.176 | −10.440, 1.915 |
| (+) Serotonin medication status for ≥6 months | −35.874 | 16.341 | −2.20 | 0.029 | −68.004, −3.745 |
367 subjects met inclusion criteria for the survival analysis (Study 2b) predicting time to a decline in MoCA score of ≤20. Of these 367 subjects, 70 (19.1%) were using serotoninergic medications at the time of their baseline visit. 28 of the total 367 subjects progressed to develop a MoCA score of 20 as assessed at a subsequent study visit. In a bivariate Cox-proportional hazard analysis, taking a serotoninergic medication at baseline did not show a significant association with decline to a MoCA of ≤20 (Supplementary Table 1). Table 3 shows the results of a multivariable interval-censored model controlling for the confounder effects of screening caudate nucleus DaT SPECT binding ratio and baseline CSF Aβ-42 levels. In this model, serotoninergic medication use at baseline showed a protective association with reduced likelihood of MoCA decline. Sensitivity analyses of this interval-censored multivariable model using different definitions of serotonin-medications revealed comparable but non-significant trends between both SSRI (n=49 out of 367; Hazard ratio = 0.185, SE = 0.189 [95% CI: 0.025, 1.372], Z=−1.65, p = 0.099) use and/or the combination of either SSRI or TCA (n=54 out of 367; Hazard ratio = 0.170, SE = 0.173 [95% CI: 0.023, 1.257], Z = −1.74, p = 0.083) use with the risk for MoCA decline.
Table 3.
Study 2b Multivariable Proportional Hazard model of time to reach Montreal Cognitive Assessment Score of ≤20.
| Overall Model Likelihood Ratio Chi square = 22.70, p<0.0001, 3 degrees of freedom | |||||
|---|---|---|---|---|---|
| Covariates | Hazard Ratio | Standard Error | Z-score | p-value | 95% Confidence Interval for Hazard Ratio |
| Mean bilateral caudate DaT SPECT binding ratio at screening | 0.456 | 0.183 | −1.96 | 0.050 | 0.208,1.000 |
| Serotoninergic medication status at baseline | 0.223 | 0.165 | −2.03 | 0.043 | 0.052, 0.952 |
| Mean baseline Aβ-42 (pg/ml) | 0.993 | 0.002 | −3.25 | 0.001 | 0.989, 0.997 |
Discussion
We present findings from converging lines of evidence supporting the hypothesized relationship between serotoninergic neurotransmission and cerebral amyloid burden in PD. Study 1 confirms our previous imaging findings of a relationship between reduced serotonin terminals in the cortex and the increased severity of cerebral amyloid plaque burden in PD. Study 2a shows an association between serotoninergic medication exposure for ≥6 months and lower CSF Aβ-42 levels. Study 2b suggests that serotoninergic medication exposure at baseline is associated with a reduced risk for progression to a MoCA score of ≤20. Collectively, these findings support the concept that serotoninergic medications favorably associate and/or alter cerebral Aβ peptide activity in PD.
Post-mortem studies of PD show reductions in serotonin terminal markers in numerous cortical and subcortical regions.34, 35 Consistent with in vivo PET imaging results, analysis of post-mortem specimens by Buddhala et al. showed that serotoninergic terminal loss in PD exhibits considerable variation with many PD subjects exhibiting regional serotonin terminal markers levels comparable to those found in control subjects and other showing marked loss of serotonin terminal markers.34 PET studies suggest that regional serotonin terminal loss is an early event in PD.10, 36 This early and heterogeneous loss of serotoninergic terminals may suggest a possible causal role for serotoninergic modulation of Aβ peptide generation in PD, the latter of which is a later-stage disease feature that also shows substantial heterogeneity in cortical plaque burden.5 In contrast to our initial findings, we did not find a significant correlation between striatal DASB and PiB DVRs in the current cohort. This may reflect stage-specific associations not seen in our cohort given that striatal amyloid burden has been described more commonly in PD with advanced cognitive impairment as opposed to early PD.37, 38 Within the range of values seen in our Study 1 cohort however, disease duration did not appear to influence the association between cortical DASB and PiB DVRs. Understanding temporal associations between early regional serotonin terminal loss and progressive amyloid plaque deposition leading to local neuronal dysfunction may be a fruitful goal for future PD natural history studies.
Our Study 2a analysis results indicating lower CSF Aβ-42 levels in PD subjects taking serotoninergic medications are consistent with recent preclinical and clinical data suggesting that increasing serotoninergic neurotransmission reduces Aβ peptide generation. Cirrito et al demonstrated that chronic oral administration of citalopram relative to placebo in an AD model mice diminished extracellular Aβ peptide levels and led to a 62% reduction in relative cortical amyloid plaque burden after 4 months.11 In a retrospective analysis of 177 healthy controls, the duration of serotonin medication exposure correlated with reduced cortical amyloid PET binding.11 A separate prospective randomized trial of young healthy adults showed that those receiving an acute 60mg dose of citalopram rather than placebo experienced a 38% reduction over about 2 days in CSF Aβ peptide concentrations and the kinetics of this reduction was consistent with reduced Aβ peptide generation.12 Although higher Aβ-42 CSF levels are typically associated with better cognitive outcomes in PD, interpretation of Aβ-42 CSF level data is not straightforward.
CSF Aβ-42 CSF levels likely reflect complex interactions between Aβ-42 peptide generation, clearance, and possibly sequestration in amyloid plaques. Some evidence suggests that reduced Abeta-42 levels would be consistent with reduced risk of dementia. In vitro data indicates that the protective APP A673T mutation is associated with reduced Aβ-42 generation.39 Recent evidence from APP A673T mutation carriers show reduced serum levels of Aβ-42, which is expected to correlate with CSF Aβ-42 levels.40 Similarly, increased Aβ-42 generation is predicted to increase dementia risk. Statistical modelling of CSF Aβ-42 data from autosomal dominant Alzheimer disease (ADAD) mutant allele carriers followed in the Dominantly Inherited Alzheimer Network (DIAN) study suggests early elevation of CSF Aβ-42 levels followed by decline around the estimated onset of manifest disease.41 In this model, declining CSF Aβ-42 levels in ADAD subjects is secondary to neurodegeneration causing diminished Aβ-42 peptide generation and/or Aβ-42 peptide sequestration in amyloid plaques. The implication is that the association between lower CSF Aβ-42 levels and dementia risk is a consequence of more advanced pathology.
Our finding (Study 2b) that baseline serotoninergic medication exposure reduced risk of progression to MoCA <20 is also consistent with a beneficial modulatory effect of serotoninergic agents on Aβ-42 peptide generation. Similar results were reported recently in a retrospective analysis of conversion from Mild Cognitive Impairment to dementia in the Alzheimer Disease Neuroimaging (ADNI) cohort.42 Antidepressant medications were also suggested to reduce dementia incidence in analyses from Kessing et al. in datasets drawn from large Danish registries of prescription drugs.43, 44 Our sensitivity analyses did not show significant associations between SSRIs and SSRIs/TCAs with time to MoCA decline. Given the relatively similar hazard ratios but increased hazard ratio standard errors for the serotonin-medication-use variable in these subgroup analyses, these findings may be a manifestation of an expected reduction in statistical power with fewer subjects subsequently identified as having the exposure of interest.45 Alternatively, they may reflect either the possibility that non-serotonin neuronal systems are preferentially involved in altering the risk or PD cognitive decline or the possibility that only a low-degree of serotonin-receptor modulation may be needed to yield a protective cognitive effect.
Serotoninergic medications are used early and often in PD to treat symptoms of depression, anxiety, sleep disorders, or other non-motor features. In the PPMI cohort, about 20% of subjects were on serotoninergic medications at baseline. A community-based PD registry study in Sweden estimated the rate to be 22% in home-dwelling individuals and 50% in those residing in an institution.46 To date, PD trials that have studied the efficacy of SSRIs, SNRIs, or TCAs, typically employed relatively short study assessment periods (weeks to months) focused on symptomatic modification of self-reported affective symptoms and have not measured cerebral amyloid burden.47, 48 One post-hoc analysis of a 4-month randomized PD-antidepressant trial showed no clear benefit to paroxetine or nortriptyline on cognitive performance relative to placebo but was limited to a small sample size and short follow-up duration.49 The possible beneficial effect of certain TCAs on amyloid-linked cognitive decline may also be confounded by their anticholinergic properties. A systematic review by Moraros et al. investigated the association between antidepressant drugs and dementia risk and found a higher unadjusted dementia risk in subjects initiated on antidepressants before age 65.50 These findings are likely to be influenced by the severity and variable causes of comorbid depression. They do, however, raise the possibility that there may exist a critical time window for serotonin-neurodegeneration-induced acceleration of cerebral amyloid burden. This may be relevant for the design of future interventional studies. Our Study 2b clinical findings would benefit from validation not only in other PD cohorts but also in prospective longitudinal studies of aging that could control for drug class and dosing.
Limitations of our study include the possibility of differential censoring. Only a small fraction of eligible subjects progressed to a MoCA score of 20 or less, raising the possibility that some subjects with declining cognition might have been lost to follow-up and would thereby not be captured in this dataset. It is possible that such censoring occurred and may influence these data. Subjects enrolled in the PPMI may represent a skewed population given that their parkinsonian symptoms were not severe enough to require the use of dopaminergic medications at the time of enrollment. This cohort factor may limit the generalizability of our findings. Because of power concerns, we were not able to control for a greater number of covariates given the limited number of subjects who did progress to the MoCA endpoint. Some PPMI subjects showed variability in MoCA scoring from one visit to the next. We chose our endpoint specifically for purpose of accurately identifying newly developed probable dementia rather than as a screening cut-off for subjects at risk for dementia. This effort to more precisely define a cognitive state transition point limited the number of PPMI subjects who met our primary endpoint. This trade-off, however, comes with improved precision as it relates to our assessment of baseline risk factors associated with longitudinal cognitive decline. Our previously published cross-sectional DASB-PiB PD PET imaging cohort18 had an even higher mean age (68.38) and lower mean MoCA (24.3) than the 23 subject imaging cohort in Study 1. In the present analyses, subjects enrolled in Study 1 were older, had longer disease duration, and lower MoCA scores than subjects in the PPMI. This may limit how translatable findings from Study 1 are to the cohort in Study 2 and vice versa. On the other hand, these findings may also collectively shed light into different aspects of the natural history of serotoninergic neurodegeneration across different disease stages in PD.
We acknowledge that our findings do not directly test the protective causal mechanism that we hypothesize exists between serotoninergic medications, alpha-secretase activity, and the degree of cerebral amyloid burden in PD. It is possible that subjects in this cohort receiving serotoninergic medications from their personal clinicians may have other unmeasured factors that influence amyloid burden and/or cognition, including severity and subtypes of affective symptoms or factors that associate with greater access to care and prescription medications. Given the wide variety, dosing, and duration of use of serotoninergic medications by subjects, we were not able to explore a dose-response relationship.
Developing treatments for different causes of cerebral amyloid burden is a goal relevant not only to PD but to a majority of patients affected by neurodegenerative diseases. Our PD findings are strengthened by the multi-center nature of the PPMI but would benefit from replication in other representative PD datasets. Serotoninergic medications are well tolerated in PD patients and trials of SSRIs targeted against cerebral amyloid burden merit consideration for future PD clinical trials. Such trials might be usefully enhanced by use of serotonin terminal imaging to select or stratify the subject population.
Supplementary Material
Acknowledgments
We thank all study participants who generously donated their time and effort.
Footnotes
Author contributions
VK, RLA, and NIB designed the study, VK, CS, RLA, and RAK acquired and analyzed the data. VK, NIB, and RLA drafted the manuscript which was reviewed and revised by all coauthors.
Potential Conflicts of Interest
None to report
References
- 1.Aarsland D, Creese B, Politis M, et al. Cognitive decline in Parkinson disease. Nature reviews Neurology. 2017 Apr;13(4):217–31. doi: 10.1038/nrneurol.2017.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petrou M, Bohnen NI, Muller ML, Koeppe RA, Albin RL, Frey KA. Abeta-amyloid deposition in patients with Parkinson disease at risk for development of dementia. Neurology. 2012 Sep 11;79(11):1161–7. doi: 10.1212/WNL.0b013e3182698d4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rochester L, Galna B, Lord S, et al. Decrease in Abeta42 predicts dopa-resistant gait progression in early Parkinson disease. Neurology. 2017 Apr 18;88(16):1501–11. doi: 10.1212/WNL.0000000000003840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller ML, Frey KA, Petrou M, et al. beta-Amyloid and postural instability and gait difficulty in Parkinson’s disease at risk for dementia. Movement disorders: official journal of the Movement Disorder Society. 2013 Mar;28(3):296–301. doi: 10.1002/mds.25213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrou M, Dwamena BA, Foerster BR, et al. Amyloid deposition in Parkinson’s disease and cognitive impairment: a systematic review. Movement disorders: official journal of the Movement Disorder Society. 2015 Jun;30(7):928–35. doi: 10.1002/mds.26191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terrelonge M, Jr, Marder KS, Weintraub D, Alcalay RN. CSF beta-Amyloid 1-42 Predicts Progression to Cognitive Impairment in Newly Diagnosed Parkinson Disease. Journal of molecular neuroscience: MN. 2016 Jan;58(1):88–92. doi: 10.1007/s12031-015-0647-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brockmann K, Lerche S, Dilger SS, et al. SNPs in Abeta clearance proteins: Lower CSF Abeta 1-42 levels and earlier onset of dementia in PD. Neurology. 2017 Nov 08; doi: 10.1212/WNL.0000000000004705. [DOI] [PubMed] [Google Scholar]
- 8.Johar I, Mollenhauer B, Aarsland D. Cerebrospinal Fluid Biomarkers of Cognitive Decline in Parkinson’s Disease. International review of neurobiology. 2017;132:275–94. doi: 10.1016/bs.irn.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Albin RL, Koeppe RA, Bohnen NI, Wernette K, Kilbourn MA, Frey KA. Spared caudal brainstem SERT binding in early Parkinson’s disease. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008 Mar;28(3):441–4. doi: 10.1038/sj.jcbfm.9600599. [DOI] [PubMed] [Google Scholar]
- 10.Pagano G, Niccolini F, Fusar-Poli P, Politis M. Serotonin transporter in Parkinson’s disease: A meta-analysis of positron emission tomography studies. Annals of neurology. 2017 Feb;81(2):171–80. doi: 10.1002/ana.24859. [DOI] [PubMed] [Google Scholar]
- 11.Cirrito JR, Disabato BM, Restivo JL, et al. Serotonin signaling is associated with lower amyloid-beta levels and plaques in transgenic mice and humans. Proceedings of the National Academy of Sciences of the United States of America. 2011 Sep 06;108(36):14968–73. doi: 10.1073/pnas.1107411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheline YI, West T, Yarasheski K, et al. An antidepressant decreases CSF Abeta production in healthy individuals and in transgenic AD mice. Science translational medicine. 2014 May 14;6(236):236re4. doi: 10.1126/scitranslmed.3008169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brenn A, Grube M, Jedlitschky G, et al. St. John’s Wort reduces beta-amyloid accumulation in a double transgenic Alzheimer’s disease mouse model-role of P-glycoprotein. Brain Pathol. 2014 Jan;24(1):18–24. doi: 10.1111/bpa.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nitsch RM, Deng M, Growdon JH, Wurtman RJ. Serotonin 5-HT2a and 5-HT2c receptors stimulate amyloid precursor protein ectodomain secretion. The Journal of biological chemistry. 1996 Feb 23;271(8):4188–94. doi: 10.1074/jbc.271.8.4188. [DOI] [PubMed] [Google Scholar]
- 15.Postina R. Activation of alpha-secretase cleavage. Journal of neurochemistry. 2012 Jan;120(Suppl 1):46–54. doi: 10.1111/j.1471-4159.2011.07459.x. [DOI] [PubMed] [Google Scholar]
- 16.Lezoualc’h F. 5-HT4 receptor and Alzheimer’s disease: the amyloid connection. Experimental neurology. 2007 Jun;205(2):325–9. doi: 10.1016/j.expneurol.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Wang Q, Hu T, et al. A tricyclic antidepressant, amoxapine, reduces amyloid-beta generation through multiple serotonin receptor 6-mediated targets. Scientific reports. 2017 Jul 10;7(1):4983. doi: 10.1038/s41598-017-04144-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kotagal V, Bohnen NI, Muller ML, Koeppe RA, Frey KA, Albin RL. Cerebral amyloid deposition and serotoninergic innervation in Parkinson disease. Archives of neurology. 2012 Dec;69(12):1628–31. doi: 10.1001/archneurol.2012.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. Journal of neurology, neurosurgery, and psychiatry. 1992 Mar;55(3):181–4. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1996 Sep;16(5):834–40. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Dempsey CM, Mackenzie SM, Gargus A, Blanco G, Sze JY. Serotonin (5HT), fluoxetine, imipramine and dopamine target distinct 5HT receptor signaling to modulate Caenorhabditis elegans egg-laying behavior. Genetics. 2005 Mar;169(3):1425–36. doi: 10.1534/genetics.104.032540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong DT. Duloxetine (LY 248686): an inhibitor of serotonin and noradrenaline uptake and an antidepressant drug candidate. Expert opinion on investigational drugs. 1998 Oct;7(10):1691–9. doi: 10.1517/13543784.7.10.1691. [DOI] [PubMed] [Google Scholar]
- 23.Pandhare A, Pappu AS, Wilms H, Blanton MP, Jansen M. The antidepressant bupropion is a negative allosteric modulator of serotonin type 3A receptors. Neuropharmacology. 2017 Feb;113(Pt A):89–99. doi: 10.1016/j.neuropharm.2016.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Januel D, Massot O, Poirier MF, Olie JP, Fillion G. Interaction of lithium with 5-HT(1B) receptors in depressed unipolar patients treated with clomipramine and lithium versus clomipramine and placebo: preliminary results. Psychiatry research. 2002 Aug 30;111(2–3):117–24. doi: 10.1016/s0165-1781(02)00136-1. [DOI] [PubMed] [Google Scholar]
- 25.Butterweck V, Nahrstedt A, Evans J, et al. In vitro receptor screening of pure constituents of St. John’s wort reveals novel interactions with a number of GPCRs. Psychopharmacology. 2002 Jul;162(2):193–202. doi: 10.1007/s00213-002-1073-7. [DOI] [PubMed] [Google Scholar]
- 26.Kang JH, Irwin DJ, Chen-Plotkin AS, et al. Association of cerebrospinal fluid beta-amyloid 1-42, T-tau, P-tau181, and alpha-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA neurology. 2013 Oct;70(10):1277–87. doi: 10.1001/jamaneurol.2013.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Defrancesco M, Marksteiner J, Kemmler G, Fleischhacker WW, Blasko I, Deisenhammer EA. Severity of Depression Impacts Imminent Conversion from Mild Cognitive Impairment to Alzheimer’s Disease. Journal of Alzheimer’s disease: JAD. 2017;59(4):1439–48. doi: 10.3233/JAD-161135. [DOI] [PubMed] [Google Scholar]
- 28.Nascimento KK, Silva KP, Malloy-Diniz LF, Butters MA, Diniz BS. Plasma and cerebrospinal fluid amyloid-beta levels in late-life depression: A systematic review and meta-analysis. Journal of psychiatric research. 2015 Oct;69:35–41. doi: 10.1016/j.jpsychires.2015.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoops S, Nazem S, Siderowf AD, et al. Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology. 2009 Nov 24;73(21):1738–45. doi: 10.1212/WNL.0b013e3181c34b47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The Parkinson Progression Marker Initiative (PPMI) Progress in neurobiology. 2011 Dec;95(4):629–35. doi: 10.1016/j.pneurobio.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pellecchia MT, Picillo M, Santangelo G, et al. Cognitive performances and DAT imaging in early Parkinson’s disease with mild cognitive impairment: a preliminary study. Acta neurologica Scandinavica. 2015 May;131(5):275–81. doi: 10.1111/ane.12365. [DOI] [PubMed] [Google Scholar]
- 32.Marquie M, Locascio JJ, Rentz DM, et al. Striatal and extrastriatal dopamine transporter levels relate to cognition in Lewy body diseases: an (11)C altropane positron emission tomography study. Alzheimer’s research & therapy. 2014;6(5–8):52. doi: 10.1186/s13195-014-0052-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schrag A, Siddiqui UF, Anastasiou Z, Weintraub D, Schott JM. Clinical variables and biomarkers in prediction of cognitive impairment in patients with newly diagnosed Parkinson’s disease: a cohort study. The Lancet Neurology. 2017 Jan;16(1):66–75. doi: 10.1016/S1474-4422(16)30328-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buddhala C, Loftin SK, Kuley BM, et al. Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Annals of clinical and translational neurology. 2015 Oct;2(10):949–59. doi: 10.1002/acn3.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ogawa T, Matson WR, Beal MF, et al. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology. 1992 Sep;42(9):1702–6. doi: 10.1212/wnl.42.9.1702. [DOI] [PubMed] [Google Scholar]
- 36.Politis M, Wu K, Loane C, et al. Staging of serotonergic dysfunction in Parkinson’s disease: an in vivo 11C-DASB PET study. Neurobiology of disease. 2010 Oct;40(1):216–21. doi: 10.1016/j.nbd.2010.05.028. [DOI] [PubMed] [Google Scholar]
- 37.Kalaitzakis ME, Walls AJ, Pearce RK, Gentleman SM. Striatal Abeta peptide deposition mirrors dementia and differentiates DLB and PDD from other parkinsonian syndromes. Neurobiology of disease. 2011 Feb;41(2):377–84. doi: 10.1016/j.nbd.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 38.Shah N, Frey KA, Muller ML, et al. Striatal and Cortical beta-Amyloidopathy and Cognition in Parkinson’s Disease. Movement disorders: official journal of the Movement Disorder Society. 2016 Jan;31(1):111–7. doi: 10.1002/mds.26369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maloney JA, Bainbridge T, Gustafson A, et al. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. The Journal of biological chemistry. 2014 Nov 7;289(45):30990–1000. doi: 10.1074/jbc.M114.589069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martiskainen H, Herukka SK, Stancakova A, et al. Decreased plasma beta-amyloid in the Alzheimer’s disease APP A673T variant carriers. Annals of neurology. 2017 Jul;82(1):128–32. doi: 10.1002/ana.24969. [DOI] [PubMed] [Google Scholar]
- 41.Fagan AM, Xiong C, Jasielec MS, et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Science translational medicine. 2014 Mar 5;6(226):226ra30. doi: 10.1126/scitranslmed.3007901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartels C, Wagner M, Wolfsgruber S, Ehrenreich H, Schneider A. Impact of SSRI Therapy on Risk of Conversion From Mild Cognitive Impairment to Alzheimer’s Dementia in Individuals With Previous Depression. The American journal of psychiatry. 2017 Nov 28; doi: 10.1176/appi.ajp.2017.17040404. appiajp201717040404. [DOI] [PubMed] [Google Scholar]
- 43.Kessing LV, Forman JL, Andersen PK. Do continued antidepressants protect against dementia in patients with severe depressive disorder? International clinical psychopharmacology. 2011 Nov;26(6):316–22. doi: 10.1097/YIC.0b013e32834ace0f. [DOI] [PubMed] [Google Scholar]
- 44.Kessing LV, Sondergard L, Forman JL, Andersen PK. Antidepressants and dementia. Journal of affective disorders. 2009 Sep;117(1–2):24–9. doi: 10.1016/j.jad.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 45.Hajian-Tilaki K. Sample size estimation in epidemiologic studies. Caspian journal of internal medicine. 2011 Fall;2(4):289–98. [PMC free article] [PubMed] [Google Scholar]
- 46.Haasum Y, Fastbom J, Johnell K. Use of antidepressants in Parkinson’s disease: A Swedish register-based study of over 1.5 million older people. Parkinsonism & related disorders. 2016 Jun;27:85–8. doi: 10.1016/j.parkreldis.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 47.Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology. 2009 Mar 10;72(10):886–92. doi: 10.1212/01.wnl.0000336340.89821.b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richard IH, McDermott MP, Kurlan R, et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology. 2012 Apr 17;78(16):1229–36. doi: 10.1212/WNL.0b013e3182516244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobkin RD, Menza M, Bienfait KL, et al. The impact of antidepressant treatment on cognitive functioning in depressed patients with Parkinson’s disease. The Journal of neuropsychiatry and clinical neurosciences. 2010 Spring;22(2):188–95. doi: 10.1176/appi.neuropsych.22.2.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moraros J, Nwankwo C, Patten SB, Mousseau DD. The association of antidepressant drug usage with cognitive impairment or dementia, including Alzheimer disease: A systematic review and meta-analysis. Depression and anxiety. 2017 Mar;34(3):217–26. doi: 10.1002/da.22584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.