

SUMMARY
Intracellular nicotinamide phosphoribosyltransferase (iNAMPT) is the rate-limiting enzyme of the mammalian NAD+ biosynthesis salvage pathway. Using inducible and conditional knockout (cKO) mice, we show that Nampt gene deletion in adult projection neurons leads to a progressive loss of body weight, hypothermia, motor neuron (MN) degeneration, motor function deficits, paralysis, and death. Nampt deletion causes mitochondrial dysfunction, muscle fiber type conversion, and atrophy, as well as defective synaptic function at neuromuscular junctions (NMJs). When treated with nicotinamide mononucleotide (NMN), Nampt cKO mice exhibit reduced motor function deficits and prolonged lifespan. iNAMPT protein levels are significantly reduced in the spinal cord of amyotrophic lateral sclerosis (ALS) patients, indicating the involvement of NAMPT in ALS pathology. Our findings reveal that neuronal NAMPT plays an essential role in mitochondrial bioenergetics, motor function, and survival. Our study suggests that the NAMPT-mediated NAD+ biosynthesis pathway is a potential therapeutic target for degenerative MN diseases.
In Brief
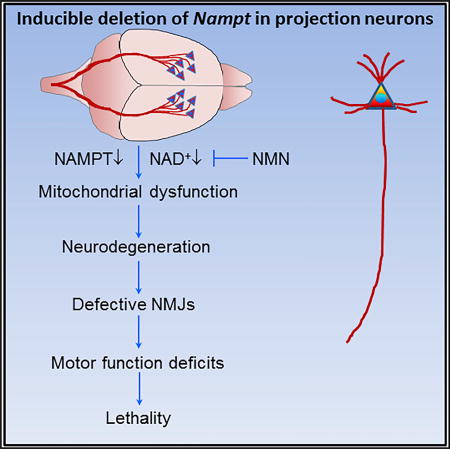
Wang et al. find that projection neuron NAMPT is essential for mitochondrial bioenergetics, motor function, and survival of adult mice and that iNAMPT is reduced in ALS patients. NMN improves health and extends the lifespan of Nampt knockout mice. Their findings suggest therapeutic avenues for motor neuron degenerative diseases.
INTRODUCTION
Nicotinamide phosphoribosyltransferase (NAMPT), originally known as pre-B cell colony-enhancing factor (PBEF), exists in intracellular NAMPT (iNAMPT) and extracellular NAMPT (eNAMPT) forms. iNAMPT is the rate-limiting enzyme of the mammalian NAD+ salvage pathway. In this pathway, iNAMPT combines nicotinamide (NAM) with 5-phosphoribosyl pyrophosphate (PRPP) to form nicotinamide mononucleotide (NMN), which is subsequently adenylated by nicotinamide nucleotide adenylyltransferase1-3 (NMNAT1-3) to produce NAD+. NAD+ is a key cellular factor for intermediary metabolism. In addition to facilitating redox reactions, NAD+ is also a co-substrate of several important NAD+-consuming enzymes, including sirtuins (SIRTs), poly(ADP-ribose) polymerases (PARPs), and CD38 (Cantó et al., 2015; Verdin, 2015). SIRTs, PARPs, and CD38 generate NAM, which, in turn, serves as a precursor for NAD+ biosynthesis. While it has been well established that iNAMPT is a rate-limiting NAD+ biosynthetic enzyme, the functions of eNAMPT are still debated and unclear, especially in the CNS.
Increasing evidence indicates that non-CNS NAMPT dysregulation occurs in various pathological conditions (Garten et al., 2015; Imai and Yoshino, 2013; Shackelford et al., 2013; Verdin, 2015), but its physiological and pathological roles in the CNS are less well understood. In our previous studies, we demonstrated that NAMPT is mainly expressed in neurons in the mouse brain; heterozygous deletion of global Nampt in mice exacerbates focal ischemic stroke-induced neuronal death and brain damage (Zhang et al., 2010). Using in vitro ischemic models, we also demonstrated that NAMPT’s neuronal protective effect is dependent on its NAD+ synthetic activity (Bi et al., 2012; Wang et al., 2016). Other studies have shown that transgenic overexpression of NAMPT in mouse neurons can reduce infarct volume, protect against white matter injury, and increase neuroregeneration after ischemic stroke (Jing et al., 2014; Zhao et al., 2015). Treatment of NAD+ precursors also effectively protects neurons after ischemic injury (Bi et al., 2012; Wang et al., 2008a, 2014). Recently, a NAMPT enzymatic activity enhancer, P7C3, was reported to prevent neuronal degeneration in motor neuron (MN)-related diseases, including amyotrophic lateral sclerosis (ALS) and Parkinson’s disease (PD) models (De Jesús-Cortés et al., 2012; Tesla et al., 2012), indicating that NAMPT may play a role in neurodegenerative diseases. However, whether and how neuronal NAMPT affects motor function and neurodegeneration under physiological conditions remain understudied.
In the mammalian CNS, projection neurons in the cortex extend axons to distant intracortical, subcortical, and subcerebral targets and are responsible for controlling sensory input, motor output, and cognitive functions (Custo Greig et al., 2013). To define the potential role of NAMPT in the function of projection neurons, we generated projection-neuron-specific and inducible Nampt conditional knockout (cKO) mice. Our findings show that projection neuron NAMPT plays an essential role in mitochondrial metabolic bioenergetics, motor function, and survival and identify the NAMPT-mediated NAD+ salvage pathway as a potential therapeutic target for neuromuscular and MN degenerative diseases.
RESULTS
Characterization of Projection-Neuron-Specific Nampt cKO Mice
To study the effect of NAMPT on motor function, we used CreERT2-LoxP recombination technology to delete Nampt in the projection neurons of adult mice. By crossing Thy1-YFP-CreERT2 mice (Feng et al., 2000; Young et al., 2008) with floxed Nampt (Namptf/f) mice (Rongvaux et al., 2008), we obtained Thy1-YFP-CreERT2:Nampt+/f and Thy1-YFP-CreERT2:Namptf/f bitransgenic mice (Figures S1A–S1C). These mice developed normally and were indistinguishable from their littermate controls, Nampt+/f and Namptf/f mice. Nampt gene deletion was achieved by tamoxifen (TAM) oral gavage (Figure 1A). We refer to these TAM-treated mice as Thy1-YFP-Nampt+/− and Thy1-YFP-Nampt−/− cKO mice. The Thy1 promoter mediates recombination primarily located in cortical layer 5 (L5) and in the hippocampal CA1 region (Figure S1D). Double immunostaining of NAMPT with NeuN, Iba1, and S100β confirmed that YFP expression was restricted to neurons (Young et al., 2008) (Figure S1E).
Figure 1. Characterization of Thy1-YFP-Nampt−/− cKO Mice.

(A) Timeline for experimental design.
(B and C) Photograph (B) and average brain weights (C) (N = 4).
(D–F) Cortical NAD+ (D) and NADH (E) levels and NAD+/NADH ratio (F) (N = 6). **p < 0.01, t test.
(G) Western blots for NAMPT using mouse and rabbit anti-NAMPT antibodies. Upper and lower arrows indicate eNAMPT and iNAMPT, respectively. NAMPT(m) and NAMPT (r) were NAMPT detected by mouse and rabbit antibodies, respectively.
(H–K) iNAMPT(m) and eNAMPT(m) levels in cortex (H and J) and hippocampus (I and K) and eNAMPT(m)/iNAMPT(m) ratios.
(L and M) iNAMPT(r) levels in cortex (L) and hippocampus (M).
(N and O) iNAMPT(m) (N) and eNAMPT(m) (O) levels in the motor cortex.
In (H)–(O), N = 4 mice. *p < 0.05; **p < 0.01, t test. Data are expressed as means ± SEM.
(P–R) Immunofluorescent images showing NAMPT expression in the motor cortex using a rabbit antibody; (P) Namptf/f, (Q) Thy1-YFP-Nampt−/−, and (R) Thy1-YFP-CreERT2. (a–c) Epi-fluorescent images (top row) and confocal maximal projection images (second and third rows). (a’–c’) Confocal single-frame images. White asterisks indicate YFP+ but NAMPT− cells. Error bars represent ± SEM. Notice that most of the YFP+ cells in the motor cortex were NAMPT− in Thy1-YFP-Nampt−/− mice.
See also Figure S1.
The average brain size of the Thy1-YFP-Nampt−/− cKO mice was comparable to that of the control mice 3 weeks post-TAM administration (PTA) (Figures 1B and 1C). Compared to levels in the Namptf/f mice, iNAMPT levels in the whole cortex and hippocampus were reduced by more than 50% in the cKO mice (Figures 1G–1I, 1L, and 1M), while eNAMPT(m) levels were similar (Figures 1J and 1K). The ratio of eNAMPT(m)/iNAMPT(m) was, however, much higher in the cKO mice (Figures 1J and 1K, right panels), suggesting enhanced release of eNAMPT in the cKO mice. A similar iNAMPT reduction was observed in the motor cortex (Figures 1N and 1O). Spinal cord iNAMPT levels trended lower in the cKO mice, but this change was not statistically significant (Figures S1F and S1G). Notably, iNAMPT levels in non-CNS organs remained unchanged in the cKO mice (Figures S1F and S1H–S1L), confirming neuronal deletion of Nampt in the cKO mice. Consistently, the cKO mice exhibited a dramatic decline in NAMPT immunoreactivity in YFP+ neurons in the motor cortex (Figures 1P–1R); almost all of the NAMPT–cells were YFP+ neurons in the cKO mice (Figure 1Q). Similarly, there was a significant decrease of iNAMPT levels in Thy1-YFP-Namp+/− cKO mice (Figures S2A–S2C). NAD+ measurements show that Thy1-YFP-Nampt−/− cKO mice exhibited dramatic reductions in both NAD+ and NADH levels in the cortex 3 weeks PTA, while the NAD+/NADH ratio remained the same (Figures 1D–1F). Thus, Nampt was successfully deleted in projection neurons, which led to reduced NAD+ levels.
Loss of Projection Neuron NAMPT Causes Motor Abnormalities, Muscle Atrophy, and Death
Namptf/f control mice grew normally; however, both female and male Thy1-YFP-Nampt−/− cKO mice exhibited a progressive decline in body weight around day 10 PTA (Figure 2A). cKO mice exhibited paralysis at about day 14 PTA and developed severe motor deficits around day 18 PTA. They walked or ran with a substantially reduced pelvic elevation, gait impairment, and dragging of hindlimbs and became moribund (Movie S1). At the end-stage (days 21–25 PTA), some mice developed cramps and spasticity (Movie S2), a typical symptom observed in ALS patients (Hobson and McDermott, 2016). These cKO mice had a median survival of 22 days PTA (Figure 2B). We further characterized motor abnormalities and muscle atrophy. The cKO mice displayed frequent limb clasping starting day 14 PTA, which was accompanied by piloerection (e.g., poor grooming, rough coat), hunched stance, and notable kyphosis (Figures 2C and 2D). Mice with various CNS lesions may display a limb-clasping posture; hindlimb clasping and kyphosis are common manifestations of a number of well-known neurodegenerative diseases (Lalonde and Strazielle, 2011). Thus, these abnormal reflexes suggest that cKO mice develop MN degeneration in the neocortex and/or spinal cord. At end-stage, a dramatic muscle wasting in the trunk and limbs was observed (Figure 2E), and there was a remarkable wasting of gastrocnemius and soleus muscles (Figures 2F and 2G). Significant reductions in heart, lung, liver, and kidney mass were also observed (Figures S1M–S1P), suggesting that neuronal Nampt mediates tissue nonautonomous effects.
Figure 2. Thy1-YFP-Nampt−/− cKO Mice Exhibit General Abnormalities and Motor Function Deficits and Have a Short Lifespan.

(A) Body weight curves.
(B) Survival curves.
(C) Worsening limb clasping reflexes of the cKO mice.
(D) Piloerection and hunched stance; paralysis of hindquarter accompanied by the dull and ungroomed coat of the cKO mice.
(E) Trunk and limb muscle atrophy in the cKO mice.
Photos in (C–E) were taken at day 21 PTA.
(F and G) Photos (F) and average weights (G) of gastrocnemius and soleus muscles (N = 6). Mice were sacrificed at 23–25 days PTA.
(H) Time courses of abdominal temperature.
(I) Lack of interscapular BAT in the cKO mice.
(J) Time courses of general condition scores.
(K–M) Time courses for pole (K), cylinder (L), and hanging wire (M) tests.
(N and O) Grip strength tests for forelimbs (N) and four limbs (O) before and 3 weeks PTA.
(P) Time courses for accelerating rotarod test.
(Q) The open-field size. Center area: 12.5 cm × 12.5 cm; corner area: 10.7 cm × 16.7 cm × 4 cm.
(R) Movement trajectories of 20-min recording.
(S–X) Time courses of 6 parameters in an open-field test including total travel distance (S), total immobile time (T), the ratio of corner travel distance to total travel distance (U), the time to explore the corners (V), the ratio of center distance to total distance (W), and the time to explore the center (X).
*p < 0.05; **p < 0.01, t test. Error bars represent ± SEM.
See also Figure S2.
Thy1-YFP-Nampt−/− cKO mice also displayed a progressive decline in their resting abdominal temperature, starting around day 10 PTA. The temperature of the cKO mice decreased to 27.7°C at the end-stage, while the control mice maintained a constant temperature of ~31.1°C (Figure 2H). Brown adipose tissue (BAT) is the major site for thermogenesis and functions to produce heat to maintain body temperature in rodents (Bartelt and Heeren, 2014). In examining interscapular BAT, we found that BAT was undetectable in the cKO mice but abundant in the control mice (Figure 2I, arrowheads).
In contrast to Thy1-YFP-Nampt−/− cKO mice, Thy1-YFP-Nampt+/− cKO mice were phenotypically normal and survived like control mice (Figures S2D and S2E). This suggests that NAD+ was not reduced to levels that compromised basic metabolic function. Thus, our results indicate that NAMPT is essential for survival.
Thy1-YFP-Nampt−/− cKO Mice Exhibit Progressive Motor Function Deficits
To further evaluate disease progression, we assessed general motor performance using a 5-point (1–5) behavioral scoring system (Knippenberg et al., 2010). Symptom onset for the Thy1-YFP-Nampt−/− cKO mice commenced around day 7 PTA; around days 13–14 PTA, most exhibited signs of hindlimb paralysis and an uneven pace, followed by a dramatic progression of aggregated symptoms (Figure 2J). The complex motor function phenotype was further assessed using a battery of motor behavioral tests, including (1) pole, (2) cylinder, (3) hanging wire, (4) grip strength, and (5) accelerating rotarod tests to assess simple motor function, voluntary forelimb use, grasping ability, forelimb and four-limb strengths, motor coordination, and balance. The cKO mice exhibited significant motor function deficits, which progressed over time (Figures 2K–2P).
Next, we performed open-field testing to assess general motor activity, exploration, anxiety-like behavior (i.e., thigmotaxis), and motor impairment (Brooks and Dunnett, 2009) (Figures 2Q and 2R). Shorter total travel distance and longer immobile time were first detected at day 5 PTA in Thy1-YFP-Nampt−/− cKO mice (Figures 2S and 2T). On thigmotaxis analysis, there was a significant increase in time to explore the corners for the cKO mice, as well as the ratio of the corner to the total travel distance (Figures 2U and 2V). No difference in the ratios of the center to the total travel distance and the time for visiting the center was detected between the two genotypes (Figures 2W and 2X). The general condition scores of Thy1-YFP-Nampt+/− cKO mice, however, were not different from those of the control mice (Figure S2F). Taken together, homozygous deletion of Nampt in the projection neurons leads to motor dysfunction and enhancement of anxiety-like behaviors.
Thy1-YFP-Nampt−/− cKO Mice Exhibit Neurodegeneration and Global Reactive Gliosis
Since Thy1-YFP-Nampt+/− cKO mice did not differ in lethality and general condition from Nampt+/f or Namptf/f mice, we used Thy1-YFP-Nampt−/− cKO mice for further mechanistic studies. To understand the underlying cause of motor deficits after Nampt deletion, we evaluated neurodegeneration of the end-stage Thy1-YFP-Nampt−/− cKO mice. Nissl staining revealed a large number of degenerated MNs in the motor cortex (Figures 3A and 3B). These MNs exhibited a triangular shape, dark color, and shrunken Nissl body (soma), with a condensation of karyoplasm. Neurons in the sensory cortex and hippocampus were less susceptible to Nampt deletion (Figures S3A and S3B). Reduction and loss of NeuN immunoreactivity, which is used to reveal neuronal degeneration, were observed in a small area of the cKO mouse motor and sensory cortices (Figures 3C and S3C). Most of YFP+ cells are NeuN− (Figures 3C and S3C, asterisks) in the motor cortex of the cKO mice. However, there was no detectable change of NeuN immunoreactivity in the hippocampus (Figure S3D). Overall, NeuN fluorescence was significantly reduced in the motor and sensory cortices of the cKO mice where YFP was highly expressed (Figure 3D). Next, we examined microtubule-associated protein 2 (MAP2), which is a neuron-specific cytoskeletal protein known to stabilize dendritic morphology. Control mice exhibited long, string-like MAP2+ dendrites in the cortex and hippocampus. In contrast, faint, curly, and fragmented MAP2+ dendrites were observed in YFP+ neurons in the cKO mice (Figures 3E, S3E, and S3F), indicating the loss of MAP2 reactivity and abnormal dendritic structure. Similarly, significant decreases of MAP2 immunoreactivity in the motor and sensory cortices of the cKO mice were observed (Figure 3F). Taken together, Nampt deletion in the projection neurons induces neurodegeneration in the brain, especially in the motor cortex.
Figure 3. Deletion of Nampt in Thy1-YFP-Nampt−/− cKO Mice Induces Neurodegeneration in the Motor Cortex.

(A–F) Shown in (A) and (B): Nissl staining images of brain sections, including the motor cortices. (C and E) Confocal images of NeuN and MAP2 staining; (a) epi-fluorescent (top) and confocal maximal projection images (bottom); (a’) confocal single frame images. (D and F) Normalized mean NeuN and MAP2 fluorescence signals within a standardized region of interest (187 × 280 µm2 in L4/5 and hippocampus CA1 region; N = 3–5 mice). *p < 0.05; **p < 0.01, t test. Error bars represent ± SEM.
See also Figure S3.
Lower MNs in the spinal cord innervate muscle fibers. However, we found no significant difference in the number of choline acetyl transferase (ChAT)+ MNs or NeuN+ large sensory-supplied α-MNs from the ventral horn of lumbar 3–5 (L3–5) spinal cords (Friese et al., 2009) of Namptf/f and Thy1-YFP-Nampt−/− cKO mice (Figures S4A–S4D).
Reactive gliosis is associated with almost all CNS diseases and brain injury (Choudhury and Ding, 2016). Upregulation of GFAP and Iba1 expressions (both immunofluorescence intensities and the densities of GFAP+ astrocyte and Iba1+ microglia) as well as altered astrocyte and microglia morphology were detected in various regions in the CNS of Thy1-YFP-Nampt−/−cKO mice (Figures S4E, S4F, S5A–S5F, and S5K–S5N). Consistently, western blot analysis demonstrated that GFAP expression levels were significantly elevated in the cKO mice (Figures S5G and S5H). However, the levels of glutamate type 1 transporter (GLT1) protein, typically expressed in mature astrocytes, remained equivalent (Figures S5I and S5J). Taken together, even though NAMPT loss only occurs in neurons, its effect on reactive gliosis is global, indicating a non-cell-autonomous effect.
NAMPT Is Crucial for Mitochondrial Homeostasis and Metabolic Function
To investigate underlying mechanisms through which Nampt deletion in projection neurons leads to neuronal degeneration and lethality, we conducted a series of experiments using different approaches. Western blot analysis showed that Thy1-YFP-Nampt−/− cKO mice exhibited a progressive decrease of iNAMPT, but similar eNAMPT, levels, as compared with the control mice (Figures 4A–4C). Notably, a large reduction of iNAMPT was evident 1 week PTA, at a time when the cKO mice were still phenotypically normal. This suggests that iNAMPT reduction is the initial cause of disease symptoms.
Figure 4. Neuronal Deletion of Nampt Causes Mitochondrial Dysfunction.

(A) Western blots for NAMPT at different PTA times.
(B and C) The time-dependent changes of iNAMPT(m) and eNAMPT(m) levels in cortex (B) and hippocampus (C) (N = 4).
(D–H) Western blots for the indicated proteins from cortical whole-cell lysates (D–F) and cortical mitochondrial fractions (G and H).
(I) Protein levels from mitochondrial fractions (N = 4).
(J and K) Cortical PG (J) and CL (K) lipid contents (N = 3).
(L–N) Vmax activities of complexes I (L), complex IV (M), and CS (N) (N = 3).
(O and P) Western blot analysis (O) of NAMPT 48 hr after scrambled and Nampt-targeting siRNA transfection in SH-SY5Y cells (n = 3 independent experiments).
(Q–S) OCR traces (Q), analyses of OCR at different states (R), and coupling efficiency of SH-SY5Y cells (S) (n = 6 and 7 replicates for scrambled and Nampt-targeting siRNA-treated cells). Oligomycin is an ATP synthase inhibitor, FCCP is a mitochondrial uncoupler, and rotenone and antimycin A are complex I and complex III inhibitors to completely shut down mitochondrial respiration.
(T) The cortical mtDNA content relative to nucDNA based on real-time PCR analysis (N = 5).
*p < 0.05; **p < 0.01; ***p < 0.005, t test.
See also Figure S6.
Next, we examined the effect of neuronal Nampt deletion on pre- and post-synaptic protein expression, including synaptophysin (SYP), PSD95, and the 2B sub-unit of NMDA receptor (NMDAR2B) in whole-cell cortical lysates. Nampt deletion did not affect the expression levels of these proteins (Figure 4D). Similar results were obtained for LC3 I and II, caspase 9, and PARP1 product PAR (Figures 4D and 4E), suggesting that Nampt deletion did not cause autophagy, apoptosis, or DNA damage. Moreover, Nampt deletion did not affect expression levels of proteins related to mitochondrial fusion (mitofusin-2), fission (Drp1 and p-Drp1), biogenesis (Sirt1, PGC1α, and NRF-1), protein acetylation (Sirt1, Sirt3, and acetylated lysine), or mitochondrial Nmnat3 (Figure 4F).
Because whole-cell lysates dilute mitochondrial proteins and thereby affect the sensitivity of western blot analyses, we isolated mitochondrial fractions from cortical tissues. Similar to whole-cell lysates, the Thy1-YFP-Nampt−/− cKO mice exhibited an approximate 50% reduction of iNAMPT in their mitochondrial fractions (Figures 4G and 4I). Importantly, we observed that the cKO mice exhibited a significant decrease of mitofusin-2, a significant increase of p-Drp1, and increased acetylation of mitochondrial proteins (Figures 4G and 4I). These data indicate that deletion of Nampt causes mitochondrial fragmentation and alters posttranslational protein modifications. Despite this, protein levels of Sirt3, Nmnat3, Caspase 9, Sirt1, PGC1α, NRF-1, and TOM20 (which approximates mitochondrial content) in the mitochondrial fractions were not changed in the cKO mice (Figure 4H).
We further interrogated the effect of Nampt deletion on mitochondrial function by evaluating the metabolism of cardiolipin (CL) and phosphatidylglycerol (PG) lipids using shotgun lipidomics analysis (Yang et al., 2009; Yang and Han, 2011). CL and PG lipids are synthesized and localized in mitochondria (PG is the precursor for CL) and are crucial for mitochondrial respiration and bioenergetics (Han, 2016; Paradies et al., 2014). We found that many CL and PG family species were reduced in the cKO mice (Figures 4J, 4K, and S6).
Next, we measured the activities of complexes I and IV, electron transport chain (ETC) enzymes that contain mtDNA-encoded subunits. Nampt deletion led to significant reductions of their activities (Figures 4L and 4M). Activity of citrate synthase (CS), a matrix-soluble enzyme whose levels tend to reflect mitochondrial mass, trended lower in the cKO mice (Figure 4N). Reduced ETC enzymatic activities could indicate a concomitant decrease in mitochondrial respiration. We, therefore, determined the effect of NAMPT knockdown on the respiration of transformed neuronal SH-SY5Y cells. Western blot analysis confirmed a significant NAMPT knockdown by Nampt-targeting small interfering RNA (siRNA) (Figures 4O and 4P). We then measured the oxygen consumption rate (OCR) by conducting an extracellular flux analysis (Wilkins et al., 2016). OCR time courses were generated by injecting ETC inhibitors to probe mitochondrial oxygen consumption under different states (Figure 4Q). Indeed, NAMPT knockdown significantly decreased maximal respiration, spare respiratory capacity, basal respiration, and ATP production-related respiration (Figure 4R). The coupling efficiency (the ratio of ATP production OCR to basal respiration OCR) was not affected (Figure 4S). Overall, NAMPT appears essential for normal mitochondrial metabolic function. In addition to NAD+ depletion and fragmentation resulting from Nampt deletion or knockdown, a decrease in mitochondrial mass could also cause mitochondrial dysfunction that manifests as reduced ETC activities and OCR. However, our results excluded this possibility, because the relative contents of mtDNA to nuclear DNA (nucDNA) were similar in Namptf/f and Thy1-YFP-Nampt−/− cKO mice (Figure 4T). This result is consistent with the CS activity assay and TOM20 western blot analysis.
Loss of NAMPT Induces Widespread Abnormalities of NMJs
The neuromuscular junction (NMJ) is a chemical synapse that regulates motor function by transmitting signals from MNs to skeletal muscles. To provide further mechanistic insight into why and how Nampt deletion in projection neurons causes muscle atrophy, motor dysfunction, and paralysis, we examined the integrity of skeletal NMJs. Using high-resolution confocal imaging, we analyzed NMJs in the semitendinosus muscles isolated from Thy1-YFP-Nampt−/− cKO mice and age-matched Namptf/f and Thy1-YFP-CreERT2 control mice. In control mice, α-bungarotoxin (α-BTX) labeling revealed that aggregated acetylcholine receptors (AChRs) cluster in postsynaptic sites, which assumed a pretzel-like shape. Each cluster was precisely innervated by a single presynaptic axon (Figure 5A). However, only 35% of the NMJs from the cKO mice remained intact (183 intact NMJs in a total of 523 from 3 mice), and the other 65% presented one or more abnormal features (described later).
Figure 5. Morphological Alterations and Abnormalities of Individual NMJs in Thy1-YFP-Nampt−/− Mice.

(A) Maximal projection confocal images of Alexa-Fluor-594-conjugated α-BTX (α-BTX-Alexa 594) (red) labeled NMJs in the semitendinosus muscles.
(B) Average values of stained area, perimeter, breadth and length of individual NMJs.
(C and D) Confocal images of normal (C) and fragmented (D) NMJs from Thy1-YFP-Nampt−/− cKO mice.
(E) The average numbers of islands and frequency distributions of island number.
Data in (B) and (E) were obtained from N = 3 mice for each genotype, and ns = 104, 112, and 103 NMJs from Thy1-YFP-Nampt−/−, Namptf/f, and Thy1-YFP-CreERT2 mice, respectively.
(F) Faintly labeled AChRs with decreased intensity of α-BTX staining (arrowhead).
(G) Statistical analyses of faint AChRs (N = 3 mice; n = 523 and 698 NMJs for Thy1-YFP-Nampt−/− and Namptf/f mice). *p < 0.05; and **p < 0.01 versus Namptf/f and Thy1-YFP-CreERT2 control mice, one-way ANOVA test.
(H–M) Maximal projection confocal images of innervated (H), non-innervated (I), multiple innervated (arrowheads) (J), fully denervated (K), swelling axon (L), and sprouting (M) NMJs. NMJs from Thy1-YFP-Nampt−/− cKO mice were labeled with α-BTX-Alexa 594 (red) and anti-neurofilament (green; H and I) or YFP (green; J–M).
(N–R) Analyses of innervation (N), multi-innervation (O), denervation (P), swelling axon (Q), and sprouting (R). In (N), N = 3 mice, and n = 218 and 157 NMJs for Thy1-YFP-Nampt−/− and Namptf/f mice. In (O), (Q), and (R), N = 3 mice, and n = 506 and 311 NMJs for Thy1-YFP-Nampt−/− and Thy1-YFP-CreERT2 mice. In (P), N = 3 mice, and n = 506 and 144 NMJs for Thy1-YFP-Nampt−/− and Thy1-YFP-CreERT2 mice.
*p < 0.05; **p < 0.01, t test. The semitendinosus muscles in Thy1-YFP-Nampt−/− cKO mice were isolated 3 weeks PTA. Error bars represent ± SEM.
See also Figure S7.
We initially studied morphological alterations by evaluating individual NMJ area, perimeter, breadth, and length. Significant decreases in the values of these parameters were revealed in Thy1-YFP-Nampt−/− cKO mice (Figures 5B and S7A). Next, we analyzed NMJs at the postsynaptic level. The normal postsynaptic endplate was pretzel shaped as a whole and brightly stained with α-BTX, and the AChR clusters were exactly innervated by a single YFP-labeled axon (Figure 5C). However, a large portion of AChR clusters in the cKO mice exhibited irregular, fragmented, or separated AChR clusters (Figure 5D). The average island numbers were significantly increased in the cKO mice (Figure 5E, left), and more NMJs in the cKO mice had higher fragments (Figure 5E, right). On the other hand, there were more postsynaptic sites with faint AChRs in the cKO mice than in Namptf/f and Thy1-YFP-CreERT2 control mice (Figures 5F and 5G), but no significant difference in postsynaptic loss was detected between the cKO mice and Thy1-YFP-CreERT2 mice (Figures S7B and S7C). We also observed dystrophic NMJs in the cKO mice, albeit with a very small percentage (3 out of 523 NMJs from 3 mice) (Figure S7D). This type of NMJ was not observed in either type of the control mice.
At the presynaptic level, the overwhelming majority of NMJs in Namptf/f mice (94.6% ± 3.1%) were aligned with presynaptic terminals, while only 68.3% ± 2.3% NMJs in Thy1-YFP-Nampt−/− cKO mice remained innervated (Figures 5H, 5I, and 5N). Meanwhile, a striking increase in the number of multiple innervated NMJs was observed in the cKO mice (Figures 5J and 5O). Completely and partially denervated NMJs together accounted for 23.4% ± 1.1% of total NMJs in the cKO mice, while only 10.9% ± 0.5% of partially denervated NMJs, but no completely denervated NMJs, were detected in Thy1-YFP-CreERT2 mice (Figures 5K, 5P, and S7E). Additionally, both partial denervation and complete denervation were often associated with non-innervation. Ectasis or swollen axons occurred in 12.4% ± 2.8% of presynaptic axons in the cKO mice but only in 0.3% ± 0.3% presynaptic axons in Thy1-YFP-CreERT2 mice (Figures 5L and 5Q). Finally, the percentage of sprouting NMJs was significantly increased in the cKO mice (Figures 5M and 5R).
Overall, the aforementioned analyses demonstrate that loss of NAMPT in the projection neurons results in various NMJ abnormalities, which disrupt synaptic connectivity and likely cause defective synaptic transmission.
Synaptic Transmission at the NMJ Is Defective in Thy1-YFP-Nampt−/− cKO Mice
Skeletal muscle exhibits plasticity, which includes muscle fiber type conversion (Lin et al., 2002). Semitendinosus muscle contains a mixture of slow- and fast-twitch or type I and II fibers (Lin et al., 2002; Rafuse et al., 2000). Since Thy1-YFP-Nampt−/− cKO mice exhibit motor function deficits and muscle wasting, we analyzed fiber type conversion and cross-section area (CSA). The cKO mice exhibited an increased percentage of myosin+ slow-twitch fibers and reduced CSAs of both myosin+ and myosin– fibers (Figures 6A–6C), consistent with the overall presence of muscle atrophy and motor dysfunction.
Figure 6. Defective Synaptic Transmission in NMJ in Thy1-YFP-Nampt−/− cKO Mice.

(A) Fluorescent images of cross-section semitendinosus muscles stained with slow skeletal myosin heavy chain. Arrows indicate myosin+ slow-twitch muscle fibers.
(B) Percentage of slow-twitch muscle fibers (N = 3 mice, and 9 muscle slices were from each genotype). **p < 0.01, t test.
(C) Distributions of CSA of myosin+ and myosin− fibers. Inserts indicate the average CSA values (N = 3 mice; 60–113 myosin+ fibers and 125–199 myosin− fibers of three muscles from each genotype).
(D and E) Confocal images of endplates labeled with α-BTX-Alexa 488 and FM1-43; (D) Namptf/f ; (E) Thy1-YFP-Nampt−/−.
(F and G) Normalized FM1-43 (F) and α-BTX-Alexa 488 (G) intensities of NMJs. n = 30 endplates for each genotype of mice.
(H) The percentage of NMJ with FM1-43 colocalized with α-BTX-Alexa 488. n = 14 and 16 muscle slices, and 101 and 100 NMJs from the control and the cKO mice.
(I) eEPPs recorded with stimulations of indicated frequencies.
(J) Number of eEPP within a 1-s train as a function of stimulation frequency. n = 16 and 22 stimulations for the control and cKO mice.
(K and L) Paired pulse facilitation (PPF). Representative eEPPs after a paired pulse stimulation with a pulse interval (tin) of 10 ms (K) and paired pulse ratio (PPR), i.e., the ratio of the second eEPP amplitude to the first eEPP amplitude, as a function of tin (L). n = 6 and 5 stimulations for the control and cKO mice.
(M) Representative mEPPs.
(N and O) The mEPP amplitude (N) and frequency (O). The values were averaged of 1,618 and 916 mEPPs from n = 12 and 15 recordings of the control and cKO mice.
Data in (I–O) were collected from N = 6 control and N = 3 cKO mice. *p < 0.05; **p < 0.01, t test. Error bars represent ± SEM.
Synaptic transmission is the most vulnerable step in neuronal signaling. To further characterize muscle function in the cKO mice, we studied synaptic transmission in semitendinosus muscle at pre- and post-synaptic levels. We loaded the cycling pool of presynaptic vesicles with FM1-43 and labeled postsynaptic AChRs with α-BTX simultaneously (Jevsek et al., 2006). In Namptf/f mice, FM1-43 revealed presynaptic vesicles with bright fluorescence confined to the endplate (Figure 6D), while in Thy1-YFP-Nampt−/− cKO mice, FM1-43 fluorescence was smeared and not confined to many endplates (Figure 6E). Further analysis showed that both FM1-43 and α-BTX fluorescent signals at the endplate were significantly reduced in the cKO mice (Figures 6F and 6G). Moreover, the percentage of NMJs with FM1-43 confined to the endplate based on α-BTX staining, which we defined as colocalization, was also significantly reduced in the cKO mice (Figure 6H). These data suggest the presence of a small or impaired vesicle pool in the presynaptic terminals of the cKO mice.
Next, we determined the effect of Nampt deletion on synaptic transmission at NMJs by recording evoked endplate potentials (eEPPs), spontaneous miniature EPPs (mEPPs), and paired pulse facilitation (PPF). In Namptf/f mice, electrical stimulations of muscle nerve induced normal and reliable EPP responses, even at high stimulation frequencies (up to 200 Hz) (Figures 6I and 6J). In the cKO mice, synaptic transmission was sustained only at low frequencies (<20 Hz), and EPP failure dramatically increased with high stimulation frequency (Figures 6I and 6J). PPF is a form of short-term synaptic plasticity. The control mice exhibited a significant increase in their paired pulse ratio (PPR) at different pulse intervals, while significant decreases of PPR were observed in the cKO mice (Figures 6K and 6L). This suggests that Nampt deletion caused synaptic depression at the NMJ. The cKO mice also exhibited a similar amplitude but higher mEPP frequency than the control mice (Figures 6M–6O), suggesting a higher turnover or an impaired or a disrupted vesicle pool and higher Ca2+ concentration and sensitivity at the NMJ presynaptic terminals of cKO mouse. Together, these data demonstrate defective synaptic transmission at the cKO mouse NMJs, consistent with the analysis of NMJ abnormalities. Overall, muscle fiber type conversion, NMJ abnormalities, and defective synaptic transmission together might contribute to muscle atrophy and motor function deficits in cKO mice.
NAMPT Is Involved in ALS Disease
Thy1-YFP-Nampt−/− cKO mice recapitulate key features of ALS, including MN loss, muscle weakness, muscle atrophy, and NMJ abnormalities. It was also previously reported that the NAMPT enzymatic activity enhancer P7C3 blocks MN death and ameliorates motor behavioral deficits in a mouse ALS model (Tesla et al., 2012). These findings collectively suggest that NAMPT is relevant to the pathogenesis of rodent ALS models. However, direct evidence of NAMPT pathology in human ALS is lacking. Therefore, we measured iNAMPT and eNAMPT levels in lumbar spinal cords from ALS patients and age-matched controls. A western blot analysis found that iNAMPT levels were significantly reduced and that eNAMPT levels were significantly increased in all human ALS samples (Figure 7A). On average, iNAMPT was reduced by 18.2%, while eNAMPT was increased by 39.9% (Figures 7B and 7C), and the ratio of eNAMPT/iNAMPT was increased by 71.2% (Figure 7D).
Figure 7. Altered iNAMPT and eNAMPT Levels in ALS Human Samples and the Effect of NMN Administration on Survival and Motor Dysfunction of Thy1-YFP-Nampt−/− cKO Mice.

(A–D) Western blots (A) and analysis of iNAMPT (B) and eNAMPT levels (C) and eNAMPT/iNAMPT ratio (D) in lumbar spinal cords of 10 ALS patients and 10 age-matched controls. Each well was loaded with 20 µg protein. **p < 0.01, t test.
(E–J) NMN administration prolongs survival and relieves motor deficits of Thy1-YFP-Nampt−/− cKO mice. Time courses of normalized body weight (E), general condition scores (F), survival rate (G), hanging wire test (H), average time spent on an accelerating rotarod (I), and pole test (J).
(K and L) Grip strength tests of forelimbs (K) and four limbs (L) before and 3 weeks PTA.
In (E)–(L), one group of Thy1-YFP-Nampt−/− cKO mice (N = 12) was daily injected with NMN (400 mg/kg) starting at day 11 PTA, and the other groups of Thy1-YFP-Nampt−/− cKO mice (N = 9) and Namptf/f mice (N = 23) were injected with saline. Red asterisks and blue asterisks indicate the comparisons of NMN and saline-injected Thy1-YFP-Nampt−/− mice with Namptf/f mice, respectively. # indicates the comparison of NMN and saline-injected Thy1-YFP-Nampt−/−cKO mice. *p < 0.05; **p < 0.01, versus control mice, t test. #p < 0.05; ##p < 0.01, versus saline-injected Thy1-YFP-Nampt−/− cKO mice, t test. Error bars represent ± SEM. (M and N) Effect of NMN on NAD+ levels in the cortex (M) and liver (N) (N = 4). Adult control mice were administrated NMN and sacrificed 1 hr later for NAD+ assay.
**p < 0.01, ANOVA test.
NMN Alleviates Disease Severity, Rescues Motor Behavioral Deficits, and Extends the Lifespan of Thy1-YFP-Nampt−/− cKO Mice
Since NMN is the immediate product of the NAMPT, we investigated whether NMN could delay or alleviate disease progression in Thy1-YFP-Nampt−/− cKO mice and rescue motor behavior deficits. We injected 400 mg/kg NMN in the cKO mice starting on day 11 PTA, at which point a significant iNAMPT reduction was present (see Figures 4A–4C) but the cKO mice were still phenotypically normal. This intervention significantly attenuated body weight loss as well as general condition score decline in the cKO mice (Figures 7E and 7F). Importantly, NMN treatment prolonged survival; 83% of the NMN-treated cKO mice were still alive at the point when all of the saline-treated cKO mice had died (Figure 7G). NMN treatment also improved performance in hanging wire, rotarod, and pole tests (Figures 7H–7J) and benefited grip forces (Figures 7K and 7L). Using an NAD+ assay, we also found that both cortical and liver NAD+ levels significantly increased after an injection of 400 mg/kg NMN (Figures 7M and 7N). In this assay, liver was used as a positive control, since there is no blood-brain barrier (BBB) in this organ (Yoshino et al., 2011).
DISCUSSION
Here, we demonstrate that NAMPT is essential to projection neuron function and viability. We found that eliminating Nampt in adult mice led to body weight loss, MN degeneration, hypothermia, motor dysfunction and paralysis, reduced general motor activity, and anxiety-like behaviors. Progressive motor dysfunction and muscle atrophy were associated with concurrent MN degeneration in the motor cortex and global reactive gliosis. Notably, iNAMPT reduction in cKO mice preceded disease symptoms and death. Mechanistically, neuronal Nampt deletion causes mitochondrial dysfunction and loss of mitochondrial homeostasis, NMJ abnormalities, and defective NMJ transmission. Remarkably, NMN, an NAD+ precursor, improved health span, restored motor function, and extended lifespan, which suggests that the phenotype that emerges following Nampt deletion in projection neurons is mediated by NAD+ depletion and consequent degenerative changes in the motor cortex and neuromuscular system. The pertinence of this to ALS is emphasized by our findings that ALS patients show decreased iNAMPT and increased eNAMPT, which links NAMPT and the NAMPT-mediated NAD+ salvage pathway to ALS pathologies.
Recent studies demonstrate conditional as opposed to inducible Nampt deletion results in functional changes across a spectrum of cell and tissue types (Frederick et al., 2016; Lin et al., 2016; Rongvaux et al., 2008; Stein et al., 2014; Stromsdorfer et al., 2016). Results from the present study underscore the importance of projection neuron NAMPT in energy metabolism and survival. It is interesting to note that neuron degeneration is prevalent in the motor cortex after Nampt deletion. This may reflect the fact that the motor cortex exhibits high energy demand, shows a substantial degree of connectivity, and projects over long distances (McCloskey et al., 2001; Tomasi and Volkow, 2012; Tomasi et al., 2013).
Although progressive motor deficits and cortical MN degeneration were observed in Thy1-YFP-Nampt−/− cKO mice, we did not detect significant lumbar spinal cord MN loss at the end-stage of disease. On the other hand, enhanced reactive gliosis was observed in cKO mouse spinal cords. It is possible to reconcile these findings, as reactive gliosis can be triggered in response to mild injuries that do not cause neuronal death (Choudhury and Ding, 2016; Cunha-Santos et al., 2016; Li et al., 2014), and reactive astrogliosis could become evident before disease onset and NM death in an ALS model (Vargas and Johnson, 2010). Enhanced reactive gliosis also suggests that both cell-autonomous and non-cell-autonomous mechanisms may contribute to the demise of Thy1-YFP-Nampt−/− cKO mice.
Multiple lines of evidence from this study indicate that Nampt deletion perturbs mitochondrial homeostasis and impairs mitochondrial function. Our data show that Nampt deletion disrupts the balance between fission/fusion and enhances mitochondrial fragmentation, which could lead to mitochondrial structural damage and subsequent neuronal degeneration (Song et al., 2011; Wang et al., 2008b). NAD+ is a substrate for various enzymes involved in survival. SIRT-mediated deacetylation consumes NAD+ (Houtkooper et al., 2012). Emerging evidence shows that increasing Sirt1 and Sirt3 deacetylation activities play a neuroprotective role in MN degenerative diseases, including ALS (Fu et al., 2012; Jiang et al., 2011; Watanabe et al., 2014), and Sirt3 deacetylation activity is essential for activating mitochondrial enzymes that, ultimately, promote neuron survival (Fu et al., 2012; Lin et al., 2016). Compared with control mice, Thy1-YFP-Nampt−/− cKO mice exhibited increased acetylation of mitochondrial proteins but similar Sirt1 and Sirt3 levels. The mitochondrial protein hyperacetylation that follows Nampt deletion may reflect an NAD+-mediated reduction in Sirt3 activity. We further found that many species of mitochondrial CL and PG lipid families were reduced in the cortex of the cKO mice, suggesting that NAMPT regulates lipid synthesis. cKO mouse mitochondria also exhibited significantly reduced complex I and complex IV activities, as well as reduced oxygen consumption. The ability of NMN to improve health span, benefit motor performance, and extend the Thy1-YFP-Nampt−/− cKO mouse lifespan argues that motor function deficits in this models arise, at least partially, as a consequence of NAD+ depletion. Taken together, these results provide compelling evidence that NAD+-related perturbations in mitochondrial homeostasis and bioenergetics cause MN degeneration, motor function deficits, and, eventually, death.
Thy1-YFP-Nampt−/− cKO mice also showed a progressive reduction in abdominal temperature. This could reflect multifactorial causes, including low locomotor activity, mitochondrial dysfunction, and reduced thermogenic potential (Cannon and Nedergaard, 2011; Drougard et al., 2016; Weydt et al., 2006). Indeed, in an open-field test, the cKO mice were less active than the control mice. Reduced ETC enzyme activities and respiration could potentially reduce ATP, which, in turn, contributes to lower body temperature (Rango et al., 2014). The loss of interscapular BAT in end-stage cKO mice could also represent another explanation for why Nampt deletion disrupted thermogenic homeostasis. BAT loss could be time dependent and also parallel Nampt deletion-induced body weight reductions. As mitochondrial uncoupling protein 1 (UCP1) in brown fat cells contributes to heat generation and body core temperature maintenance (Bartelt and Heeren, 2014), future studies that examine the effects of Nampt deletion on UCP1 should be considered.
Both MN integrity and the synaptic connectivity between the MN and muscle are required for proper motor unit function (Seijffers et al., 2014). NMJ defects preclude proper synaptic transmission, which leads to denervation and impaired muscle contraction and motor function (Sanes and Lichtman, 1999; Valdez et al., 2010). We found that, compared to control mice, semitendinosus NMJs in the Thy1-YFP-Nampt−/− cKO mice showed abnormal pre- and post-synaptic features and defective NMJ synaptic transmission. cKO mouse semitendinosus muscles had more slow-twitch muscle fibers and smaller slow- and fast-twitch fiber CSAs than control mice, indicating that projection neuron Nampt deletion affects muscle plasticity. NAMPT, therefore, appears to play a role in maintaining synaptic connectivity, distal axonal integrity, NMJ synaptic transmission, and muscle fiber type identity in a non-cell-autonomous manner.
Thy1-YFP-Nampt−/− cKO mice recapitulate key features of ALS, including MN loss, muscle atrophy, and NMJ abnormalities (Robberecht and Philips, 2013; Valdez et al., 2012). Consistent with the present study, a recent study reported that P7C3, which enhances NAMPT activity, preserves MNs in a mouse ALS model (Tesla et al., 2012). Relative to control mice, our cKO mice exhibited a large reduction of iNAMPT but comparable levels of eNAMPT. This is, to some extent, consistent with our finding that ALS patient spinal cords have reduced iNAMPT, but higher eNAMPT, levels than those of age-matched controls. Although the physiological and pathological roles of eNAMPT in the CNS are controversial, a large body of evidence suggests that eNAMPT acts as a cytokine (Jacques et al., 2012) as well as a proinflammatory cytokine in non-CNS (Liu et al., 2009). This raises the possibility that eNAMPT might stimulate neuroinflammation, a characteristic pathological feature in ALS. Our data from human ALS subjects potentially link NAMPT to ALS and suggest that targeting the NAMPT-mediated NAD+ salvage pathway may represent a reasonable therapeutic approach.
In summary, our present study establishes that NAMPT is essential for projection neuron function and survival and links NAMPT to ALS pathologies. Our findings suggest that the NAMPT-NAD+ pathway is a potential therapeutic target for MN degenerative diseases, including ALS. Enhancing the NAD+ salvage pathway might also benefit other neurodegenerative diseases, including PD and Alzheimer’s disease (AD), as P7C3 also attenuates neuronal loss in a PD mouse model, and NAD+ precursors (NAM and NMN) mitigate cognitive decline in AD mouse models (De Jesús-Cortés et al., 2012; Green et al., 2008).
EXPERIMENTAL PROCEDURES
Animals
Mice were housed in the animal facility at the University of Missouri. All experimental procedures were performed according to the NIH Guide for the Care and Use of Laboratory Animals and approved by the University of Missouri Animal Care Quality Assurance Committee. Adult male and female mice were used in the present study. Projection neuron-specific and inducible Nampt heterozygous and homozygous cKO mice, i.e., Thy1-YFP-Nampt+/− and Thy1-YFP-Nampt−/− cKO mice, were generated by crossing Thy1-CreERT2-YFP mice (Jackson Laboratory) (Young et al., 2008) with Namptf/f mice (Rongvaux et al., 2008). Positive mice at 8 weeks old were administered TAM by oral gavage to delete Nampt.
General Conditional Score and Analysis of End-Stage
To assess the general condition of knockout mice, we set up a 5-point (1–5) behavioral scoring system adapted from an ALS model (Knippenberg et al., 2010). The time of disease end-stage was defined when the paralyzed mice were unable to right themselves within 20 s after being placed on their side or found in lateral recumbency (Boillée et al., 2006). Mice were euthanized at end-stage.
Behavioral Tests
A battery of motor behavioral tests, including pole, cylinder, hanging wire, grip strength, accelerating rotarod, and open field, were conducted to assess simple motor function, voluntary forelimb use, grasping ability, forelimb and four-limb strength, motor coordination and balance, and general locomotor activity and thigmotaxis (Brooks and Dunnett, 2009; Li et al., 2015).
Characterization of NMJs
Maximum intensity projections of stacking confocal images were created. Four parameters, i.e., the stained area, the perimeter, the length, and the breadth of a single postsynaptic endplate, were evaluated. We also analyzed nine different types of abnormal or irregular morphology of NMJs (Seijffers et al., 2014; Valdez et al., 2010), namely: (1) fragmentation, (2) faint AChRs, (3) postsynaptic loss, (4) denervation, (5) non-innervation, (6) multi-innervation, (7) swollen axon, (8) sprouting, and 9) dystrophy.
Shotgun Lipidomics Analysis of Lipids from Mouse Tissues
Shotgun lipidomics analysis of lipid extracts of mouse cortices was performed as described previously (Yang et al., 2009; Yang and Han, 2011).
FM1-43 Labeling of Synaptic Vesicles and Electrophysiological Recordings of NMJs
Synaptic vesicle labeling with FM1-43 and standard intracellular electrophysiological recordings of EPP and mEPP were described previously (Maeno-Hikichi et al., 2011; Polo-Parada et al., 2005).
Mitochondrial Function Assays
Complexes I and IV and CS Vmax activities of brain cortices were measured using an Infinite M200 plate reader (Ghosh et al., 2007). Mitochondrial OCRs in SH-SY5Y cells were measured using a Seahorse XF24 extracellular flux analyzer (Wilkins et al., 2016).
Statistical Analysis
All data were expressed as means ± SEM. Statistical comparisons were made by Student’s t test for two groups and a one-way ANOVA test followed by Bonferroni test for multiple groups using Excel and OriginPro 8 software. The statistical significance was set at *p < 0.05, **p < 0.01, and ***p < 0.005.
Supplementary Material
Highlights.
Deletion of projection neuron Nampt leads to motor dysfunction and death of mice
Nampt deletion impairs mitochondrial function and NMJ synaptic transmission
ALS patients exhibit reduced iNAMPT and increased eNAMPT protein levels
NMN rescues motor dysfunction and prolongs lifespan of Nampt knockout mice
Acknowledgments
We thank Dr. O. Leo for providing Namptf/f mice. The study was supported by NIH grants R01NS069726 and R01NS094539 and an American Heart Association (AHA) Grant in Aid award (13GRANT17020004) to S.D., NIH grant P30AG035982 (which supports the University of Kansas Alzheimer’s Disease Center), and NIH grant 1R01NS089604 to X.W. (CWRU).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two movies and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.08.022.
AUTHOR CONTRIBUTIONS
X.W. designed and conducted experiments, analyzed data, and helped to write the manuscript draft. Q.Z. designed and conducted experiments and analyzed data. R.B. conducted experiments and analyzed data. N.Z. conducted experiments and analyzed data. Y.W. conducted experiments and analyzed data. L.P.-P. conducted experiments, analyzed data, and edited the manuscript. A.T. conducted experiments. A.A. conducted experiments. H.M.W. conducted experiments, analyzed data, and edited the manuscript. R.H.S. edited the manuscript, designed experiments, and analyzed data. X.W. (Case Western Reserve University; CWRU) conducted experiments. S.D. conceived and supervised the project, designed and conducted experiments, analyzed data, wrote the manuscript, and acquired funding for the project. All authors read and approved the final manuscript.
References
- Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 2014;10:24–36. doi: 10.1038/nrendo.2013.204. [DOI] [PubMed] [Google Scholar]
- Bi J, Li H, Ye SQ, Ding S. Pre-B-cell colony-enhancing factor exerts a neuronal protection through its enzymatic activity and the reduction of mitochondrial dysfunction in in vitro ischemic models. J. Neurochem. 2012;120:334–346. doi: 10.1111/j.1471-4159.2011.07566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Brooks SP, Dunnett SB. Tests to assess motor phenotype in mice: a user’s guide. Nat. Rev. Neurosci. 2009;10:519–529. doi: 10.1038/nrn2652. [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Nonshivering thermogenesis and its adequate measurement in metabolic studies. J. Exp. Biol. 2011;214:242–253. doi: 10.1242/jeb.050989. [DOI] [PubMed] [Google Scholar]
- Cantó C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury GR, Ding S. Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol. Dis. 2016;85:234–244. doi: 10.1016/j.nbd.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha-Santos J, Duarte-Neves J, Carmona V, Guarente L, Pereira de Almeida L, Cavadas C. Caloric restriction blocks neuropathology and motor deficits in Machado-Joseph disease mouse models through SIRT1 pathway. Nat. Commun. 2016;7:11445. doi: 10.1038/ncomms11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custo Greig LF, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 2013;14:755–769. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jesús-Cortés H, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, Britt J, Tesla R, Morlock L, Naidoo J, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proc. Natl. Acad. Sci. USA. 2012;109:17010–17015. doi: 10.1073/pnas.1213956109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drougard A, Fournel A, Marlin A, Meunier E, Abot A, Bautzova T, Duparc T, Louche K, Batut A, Lucas A, et al. Central chronic apelin infusion decreases energy expenditure and thermogenesis in mice. Sci. Rep. 2016;6:31849. doi: 10.1038/srep31849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Frederick DW, Loro E, Liu L, Davila A, Jr, Chellappa K, Silverman IM, Quinn WJ, 3rd, Gosai SJ, Tichy ED, Davis JG, et al. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 2016;24:269–282. doi: 10.1016/j.cmet.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese A, Kaltschmidt JA, Ladle DR, Sigrist M, Jessell TM, Arber S. Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc. Natl. Acad. Sci. USA. 2009;106:13588–13593. doi: 10.1073/pnas.0906809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Jin J, Cichewicz RH, Hageman SA, Ellis TK, Xiang L, Peng Q, Jiang M, Arbez N, Hotaling K, et al. trans-(−)-ε-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington disease. J. Biol. Chem. 2012;287:24460–24472. doi: 10.1074/jbc.M112.382226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015;11:535–546. doi: 10.1038/nrendo.2015.117. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Patel N, Rahn D, McAllister J, Sadeghi S, Horwitz G, Berry D, Wang KX, Swerdlow RH. The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells. Mol. Pharmacol. 2007;71:1695–1702. doi: 10.1124/mol.106.033845. [DOI] [PubMed] [Google Scholar]
- Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, Thompson LM, LaFerla FM. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 2008;28:11500–11510. doi: 10.1523/JNEUROSCI.3203-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X. Lipidomics for studying metabolism. Nat. Rev. Endocrinol. 2016;12:668–679. doi: 10.1038/nrendo.2016.98. [DOI] [PubMed] [Google Scholar]
- Hobson EV, McDermott CJ. Supportive and symptomatic management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2016;12:526–538. doi: 10.1038/nrneurol.2016.111. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 2013;15(Suppl 3):26–33. doi: 10.1111/dom.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques C, Holzenberger M, Mladenovic Z, Salvat C, Pecchi E, Berenbaum F, Gosset M. Proinflammatory actions of visfatin/nicotinamide phosphoribosyltransferase (Nampt) involve regulation of insulin signaling pathway and Nampt enzymatic activity. J. Biol. Chem. 2012;287:15100–15108. doi: 10.1074/jbc.M112.350215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevsek M, Jaworski A, Polo-Parada L, Kim N, Fan J, Landmesser LT, Burden SJ. CD24 is expressed by myofiber synaptic nuclei and regulates synaptic transmission. Proc. Natl. Acad. Sci. USA. 2006;103:6374–6379. doi: 10.1073/pnas.0601468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Wang J, Fu J, Du L, Jeong H, West T, Xiang L, Peng Q, Hou Z, Cai H, et al. Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nat. Med. 2011;18:153–158. doi: 10.1038/nm.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Z, Xing J, Chen X, Stetler RA, Weng Z, Gan Y, Zhang F, Gao Y, Chen J, Leak RK, Cao G. Neuronal NAMPT is released after cerebral ischemia and protects against white matter injury. J. Cereb. Blood Flow Metab. 2014;34:1613–1621. doi: 10.1038/jcbfm.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knippenberg S, Thau N, Dengler R, Petri S. Significance of behavioural tests in a transgenic mouse model of amyotrophic lateral sclerosis (ALS) Behav. Brain Res. 2010;213:82–87. doi: 10.1016/j.bbr.2010.04.042. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Strazielle C. Brain regions and genes affecting limb-clasping responses. Brain Res. Brain Res. Rev. 2011;67:252–259. doi: 10.1016/j.brainresrev.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Li H, Zhang N, Lin HY, Yu Y, Cai QY, Ma L, Ding S. Histological, cellular and behavioral assessments of stroke outcomes after photothrombosis-induced ischemia in adult mice. BMC Neurosci. 2014;15:58. doi: 10.1186/1471-2202-15-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Xie Y, Zhang N, Yu Y, Zhang Q, Ding S. Disruption of IP3R2-mediated Ca2+ signaling pathway in astrocytes ameliorates neuronal death and brain damage while reducing behavioral deficits after focal ischemic stroke. Cell Calcium. 2015;58:565–576. doi: 10.1016/j.ceca.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, et al. Transcriptional co-activator PGC-1 α drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Lin JB, Kubota S, Ban N, Yoshida M, Santeford A, Sene A, Nakamura R, Zapata N, Kubota M, Tsubota K, et al. NAMPT-mediated NAD(+) biosynthesis is essential for vision In mice. Cell Rep. 2016;17:69–85. doi: 10.1016/j.celrep.2016.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Li H, Cepeda J, Zhang LQ, Cui X, Garcia JGN, Ye SQ. Critical role of PBEF expression in pulmonary cell inflammation and permeability. Cell Biol. Int. 2009;33:19–30. doi: 10.1016/j.cellbi.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeno-Hikichi Y, Polo-Parada L, Kastanenka KV, Landmesser LT. Frequency-dependent modes of synaptic vesicle endocytosis and exocytosis at adult mouse neuromuscular junctions. J. Neurosci. 2011;31:1093–1105. doi: 10.1523/JNEUROSCI.2800-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloskey DP, Adamo DS, Anderson BJ. Exercise increases metabolic capacity in the motor cortex and striatum, but not in the hippocampus. Brain Res. 2001;891:168–175. doi: 10.1016/s0006-8993(00)03200-5. [DOI] [PubMed] [Google Scholar]
- Paradies G, Paradies V, De Benedictis V, Ruggiero FM, Petrosillo G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta. 2014;1837:408–417. doi: 10.1016/j.bbabio.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Polo-Parada L, Plattner F, Bose C, Landmesser LT. NCAM 180 acting via a conserved C-terminal domain and MLCK is essential for effective transmission with repetitive stimulation. Neuron. 2005;46:917–931. doi: 10.1016/j.neuron.2005.05.018. [DOI] [PubMed] [Google Scholar]
- Rafuse VF, Polo-Parada L, Landmesser LT. Structural and functional alterations of neuromuscular junctions in NCAM-deficient mice. J. Neurosci. 2000;20:6529–6539. doi: 10.1523/JNEUROSCI.20-17-06529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rango M, Arighi A, Bonifati C, Del Bo R, Comi G, Bresolin N. The brain is hypothermic in patients with mitochondrial diseases. J. Cereb. Blood Flow Metab. 2014;34:915–920. doi: 10.1038/jcbfm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Rongvaux A, Galli M, Denanglaire S, Van Gool F, Dréze PL, Szpirer C, Bureau F, Andris F, Leo O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J. Immunol. 2008;181:4685–4695. doi: 10.4049/jimmunol.181.7.4685. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Seijffers R, Zhang J, Matthews JC, Chen A, Tamrazian E, Babaniyi O, Selig M, Hynynen M, Woolf CJ, Brown RH., Jr ATF3 expression improves motor function in the ALS mouse model by promoting motor neuron survival and retaining muscle innervation. Proc. Natl. Acad. Sci. USA. 2014;111:1622–1627. doi: 10.1073/pnas.1314826111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford RE, Mayhall K, Maxwell NM, Kandil E, Coppola D. Nicotinamide phosphoribosyltransferase in malignancy: a review. Genes Cancer. 2013;4:447–456. doi: 10.1177/1947601913507576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA, Hayden MR, et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011;17:377–382. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Wozniak DF, Dearborn JT, Kubota S, Apte RS, Izumi Y, Zorumski CF, Imai S. Expression of Nampt in hippocampal and cortical excitatory neurons is critical for cognitive function. J. Neurosci. 2014;34:5800–5815. doi: 10.1523/JNEUROSCI.4730-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromsdorfer KL, Yamaguchi S, Yoon MJ, Moseley AC, Franczyk MP, Kelly SC, Qi N, Imai S, Yoshino J. NAMPT-mediated NAD(+) biosynthesis in adipocytes regulates adipose tissue function and multi-organ insulin sensitivity in mice. Cell Rep. 2016;16:1851–1860. doi: 10.1016/j.celrep.2016.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, McDaniel L, Knobbe W, Burket A, Tran S, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA. 2012;109:17016–17021. doi: 10.1073/pnas.1213960109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi D, Volkow ND. Aging and functional brain networks. Mol. Psychiatry. 2012;17:471–558. doi: 10.1038/mp.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi D, Wang GJ, Volkow ND. Energetic cost of brain functional connectivity. Proc. Natl. Acad. Sci. USA. 2013;110:13642–13647. doi: 10.1073/pnas.1303346110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Kang H, Clemenson GD, Jr, Gage FH, Lichtman JW, Sanes JR. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. USA. 2010;107:14863–14868. doi: 10.1073/pnas.1002220107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR. Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS ONE. 2012;7:e34640. doi: 10.1371/journal.pone.0034640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Johnson JA. Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurotherapeutics. 2010;7:471–481. doi: 10.1016/j.nurt.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Wang S, Xing Z, Vosler PS, Yin H, Li W, Zhang F, Signore AP, Stetler RA, Gao Y, Chen J. Cellular NAD replenishment confers marked neuroprotection against ischemic cell death: role of enhanced DNA repair. Stroke. 2008a;39:2587–2595. doi: 10.1161/STROKEAHA.107.509158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA. 2008b;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li H, Ding S. The effects of NAD+ on apoptotic neuronal death and mitochondrial biogenesis and function after glutamate excitotoxicity. Int. J. Mol. Sci. 2014;15:20449–20468. doi: 10.3390/ijms151120449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li H, Ding S. Pre-B-cell colony-enhancing factor protects against apoptotic neuronal death and mitochondrial damage in ischemia. Sci. Rep. 2016;6:32416. doi: 10.1038/srep32416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Ageta-Ishihara N, Nagatsu S, Takao K, Komine O, Endo F, Miyakawa T, Misawa H, Takahashi R, Kinoshita M, Yamanaka K. SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol. Brain. 2014;7:62. doi: 10.1186/s13041-014-0062-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1α in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Wilkins HM, Koppel S, Carl SM, Ramanujan S, Weidling I, Michaelis ML, Michaelis EK, Swerdlow RH. Oxaloacetate enhances neuronal cell bioenergetic fluxes and infrastructure. J. Neurochem. 2016;137:76–87. doi: 10.1111/jnc.13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Han X. Accurate quantification of lipid species by electrospray ionization mass spectrometry—meets a key challenge in lipidomics. Metabolites. 2011;1:21–40. doi: 10.3390/metabo1010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Cheng H, Gross RW, Han X. Automated lipid identification and quantification by multidimensional mass spectrometry-based shotgun lipidomics. Anal. Chem. 2009;81:4356–4368. doi: 10.1021/ac900241u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P, Qiu L, Wang D, Zhao S, Gross J, Feng G. Single-neuron labeling with inducible Cre-mediated knockout in transgenic mice. Nat. Neurosci. 2008;11:721–728. doi: 10.1038/nn.2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xie Y, Wang T, Bi J, Li H, Zhang LQ, Ye SQ, Ding S. Neuronal protective role of PBEF in a mouse model of cerebral ischemia. J. Cereb. Blood Flow Metab. 2010;30:1962–1971. doi: 10.1038/jcbfm.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Guan YF, Zhou XM, Li GQ, Li ZY, Zhou CC, Wang P, Miao CY. Regenerative neurogenesis after ischemic stroke promoted by nicotinamide phosphoribosyltransferase-nicotinamide adenine dinucleotide cascade. Stroke. 2015;46:1966–1974. doi: 10.1161/STROKEAHA.115.009216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.