

Graphical Abstract
Protein-protein interactions are fundamental to the formation of intricate interaction networks and the assembly of multisubunit protein complexes that represent the functional workhorses of the cell. Thus, a detailed understanding of the structure and dynamics of these multimeric and functional entities is critical towards understanding their biological functions. The vast majority of structural information to date has been contributed by X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, cumulatively totaling over 98% of all Protein Data Bank (PDB) entries (89% and 9%, respectively). However, these traditional techniques are limited by their sample preparation requirements, preventing their application towards large and dynamic protein complexes. Recent progress in cryo-electron microscopy (cryo-EM) detector technology and digital image processing algorithms now permit near-atomic resolution density maps for protein complex structure elucidation, but cryo-EM density maps still only represent approximately 1% of all PDB entries. The growing demand for complementary structural elucidation tools has ushered in the development of alternative methods for protein complex characterization.
Mass spectrometry (MS)-based structural techniques have hit their stride in the last decade, spurred on by the inability of traditional biophysical structure methods to resolve the structures and dynamics of conformationally and compositionally heterogeneous protein complexes. Methodologies such as covalent labeling/footprinting1,2, hydrogen-deuterium exchange3, cross-linking mass spectrometry (XL-MS, sometimes abbreviated CX-MS, CL-MS)4–7, ion-mobility MS8, and native MS9 now constitute valuable assets of the structural biologist’s toolbox, permitting low-resolution characterization of protein complexes that have evaded characterization by traditional means. While individually incapable of providing a complete structure, the advantages of these approaches are their versatility, sensitivity and throughput, which allow them to provide useful information to complement conventional structural analyses. In combination with structural tools such as X-ray crystallography and electron microscopy, hybrid techniques permit the architectural elucidation of multimeric protein complexes that have remained recalcitrant to traditional methodologies alone. These MS-based strategies and their contributions to structural biology have been extensively reviewed in the past several years10–28. Of these tools, XL-MS is unique among MS-based techniques due to its capability to simultaneously capture protein-protein interactions (PPI) from their native environment and uncover their physical interaction contacts, thus permitting the determination of both identity and connectivity of protein-protein interactions in cells29,30
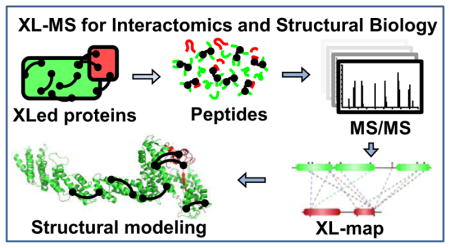
The general XL-MS workflow is illustrated in Fig. 1. Briefly, proteins are first reacted with bifunctional cross-linking reagents that physically tether spatially proximal amino acid residues through the formation of covalent bonds. The cross-linked proteins are then enzymatically digested and the resulting peptide mixtures are separated and analyzed via liquid chromatography–tandem mass spectrometric (LC-MS/MS) analysis. Subsequent database searching of MS data identifies cross-linked peptides and their linkage sites. Given that each cross-linking reagent carries a defined length, the resulting cross-links can be utilized as distance constraints for various applications, ranging from structure validation and integrative modeling31–36 to de novo structure prediction35,37,38. In recent years, significant technological advancements in XL-MS studies have dramatically propelled the field forward, enabling a wide range of applications in vitro and in vivo, not only at the level of protein complexes but also at the proteome scale. Several recent reviews have focused on specific aspects of XL-MS including reagent design39 and applications12,19,27,28,40–42. This review offers an overview of recent developments in XL-MS studies especially during the last three years. Specifically, we describe improvements in cross-linking reagents, sample preparation, XL-MS workflows, and bioinformatics tools to not only address the inherent challenges of XL-MS but also expand the range of cross-linking studies. In addition, representative in vitro and in vivo applications are described to illustrate the effectiveness and potential of XL-MS in defining protein interaction landscapes and architectures of large protein assemblies. Moreover, the current status of comparative XL-MS is outlined to exemplify its role in exploring protein interactions and structural dynamics. Finally, future perspectives on next generation XL-MS strategies are deliberated. Given the limited space in this review, we apologize for not being able to include all literature published in recent years.
Figure 1.

The General XL-MS analysis workflow.
1. Strategies to Overcome Inherent Challenges in XL-MS Studies
Although XL-MS analysis dates back several decades43, this technique has finally blossomed into an accessible and powerful structural tool for mapping protein-protein interactions in recent years. Its effectiveness has long been impeded by three primary obstacles: 1) complex MS/MS fragmentation of cross-linked peptides; 2) low abundance of cross-linked peptides in complex peptide mixtures; 3) heterogeneity of cross-linked products. While the first hurdle makes accurate identification of cross-linked peptides and unambiguous assignment of cross-linked sites difficult, the latter ones hinder effective MS detection of cross-linked peptides. Over the years, enormous efforts have been taken to overcome these challenges, which have led to various technological advancements in XL-MS analyses as highlighted below. These innovations have collectively facilitated the realization of the potential of XL-MS in today’s structural biology.
1.1 Bioinformatics tools for cross-linked peptide analysis
1.1.1 Identification of cross-linked peptides
MS/MS fragmentation of conventional cross-linked peptides is typically convoluted and unpredictable, requiring specialized algorithms or software to accurately sequence the identities of both individual peptides within a cross-link. From a bioinformatics standpoint, this also presents a scaling issue as most database searching platforms function by comparing experimental MS/MS spectra against a computed library of theoretical spectra. For cross-linked peptides, the search space increases exponentially instead of linearly, as all possible peptide combination pairs (n2) need to be considered when building a library44,45. This issue rapidly escalates the computational power and time required for cross-linked peptide identification as the total number of proteins within a sample increases.
To address these problems, various software packages have been designed to enable database searching for identification of cross-linked peptides as summarized in Table 1. One common strategy is to use specialized algorithms to reduce search space, lightening the computational load. For example, xQuest/xProphet was designed to enable large-scale database searches by utilizing isotope-based candidate peptide pre-filtering to minimize the number of permutations that need to be considered46,47. StavroX compares precursor ion masses against a pre-calculated list of theoretical cross-links, using mass to correlate potential matches48. Algorithms such as fast-sorting45 also rely on mass filtering, but utilize array sorting to reduce the number of necessary iterations on the fly, resulting in faster searches. In comparison, software such as ProteinProspector49,50 and pLink44 treat cross-linked peptides as single peptides bearing large, unknown modifications, identifying individual peptide hits then recombining those originating from common spectra into cross-linked hits.
Table 1.
List of Publically Available Software for Cross-link Identification, Quantitation, Visualization and/or Modeling
In recent years, various software suites have been developed to improve cross-link identification. Search engines such as SIM-XL51 and Kojak52 build on strategies to minimize the search space through heuristic approaches. SIM-XL can eliminate possibilities by only considering cross-link combinations that contain at least one peptide identified with a dead-end modification and by only searching spectra that exhibit tell-tale ions exclusively derived from cross-linked peptides (such as those that result from lysine side chain rearrangement following cross-linking)53. Kojak applies a two-pass algorithm that first searches for single candidate peptides with modifications equal to the difference between precursor ion and peptide masses, then pairs top scoring peptides from each spectra to search for cross-linked peptides based on precursor mass52. In comparison, other tools have been designed to search all combinations of peptide pairs. Xilmass generates a library of spectra for all potential cross-links and their fragment ions, which limits its use to single complexes due to the time necessary to run more complex searches54. XLSearch employs a probabilistic scoring method and machine learning to improve cross-link identification accuracy55. Finally, ECL/ECL2 employs a novel algorithm that exhaustively searches all peptide combinations with linear time and space complexity, permitting unfiltered analysis of cross-linked peptides using large-scale databases in several hours56,57. As shown, each software utilizes unique combinations of algorithms and rubrics to provide best-scoring cross-link identifications (Table 1).
1.1.2 Automated visualization of cross-linked residues
Recent bioinformatics tools have also been developed to enable automated visualization of cross-linking data through generation of two-dimensional cross-linked residue networks or mapping of cross-links onto three-dimensional structures. xVis58, xiNET59, ProXL60, and CLMSVault61 are examples of recently developed software that permit visualization of cross-linking data as networks of connected residues. In addition, CLMSVault61 facilitates mapping of cross-links in the context of three-dimensional structures, similar to ProXL60 and XlinkDB 2.062, which also function as public repositories for cross-linking data. While most mapping tools utilize Euclidean distances to determine cross-link length, Xwalk and Jwalk have been developed to determine solvent-accessible surface distances (SASD) for cross-links, the length of the shortest path between two amino acids with respect to protein volume63,64. Tools such as XLMap can be used to score and evaluate protein models based on cross-link satisfaction65. Collectively, these automated tools (Table 1) permit visualization of cross-linking data within primary sequences and existing structures, thus facilitating data interpretation in structural elucidation.
1.2 Development of MS-cleavable cross-linking reagents
Along with software development, major efforts to facilitate the identification and characterization of cross-linked peptides have culminated in the development of new cross-linking reagents, namely cleavable cross-linkers39. These reagents carry labile bonds that can be cleaved by different means based on their unique chemical properties, such as photo- 66, chemical- 5,67, and MS-induced cleavages68–80. Given that the cleavable bonds are localized within the spacer regions of the linkers, the two cross-linked peptide constituents can be physically separated either before or during MS analysis. While chemical-induced and photo-labile cross-linking reagents permit cross-link separation prior to MS analysis66,81–83, MS-cleavable reagents fragment within the mass spectrometer, enabling efficient correlation between separated cross-link peptide constituents and their respective parent ions. This unique feature of MS-cleavable reagents enables the production of characteristic cross-link fragments in MS2, thereby simplifying subsequent MS analysis and data processing for facile and accurate identification of cross-linked peptides. These combined benefits make MS-cleavable cross-linking reagents the most attractive type of cleavable reagents for XL-MS studies.
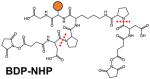
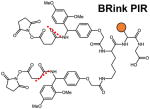
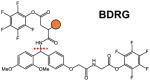
Several classes of cleavable bonds have been incorporated into current MS-cleavable cross-linkers as summarized in Table 2. Among them, CID-cleavable bonds are most popular as they are present in the majority of existing MS-cleavable reagents. As shown, the three commonly used CID-cleavable bonds are: 1) C-S bonds adjacent to sulfoxide74,79,84,85; sulfonium-ion70, or cyanuric ring75; 2) D-P (Asp-Pro) bonds 86–88; 3) C-N bonds associated with Rink68,76,89, urea 73,90,91, or quaternary diamine 92 structures. Depending on the strength of the cleavable bonds, these reagents can be implemented for MSn and/or MS2 based workflows to facilitate the identification of cross-linked peptides as detailed in Table 2.
Table 2.
Representative MS-cleavable Cross-linkers
| Cleavable Bond | Dissociation Type | Reagent | Workflow # | Refs |
|---|---|---|---|---|
| C-S (Sulfoxide) | CID |
|
MSn (CID2, CID3) MS2 (CID2, ETD2) MS2 + MSn |
[74, 95] [101] [102] |
|
|
MSn (CID2, CID3) | [84,96] | ||
|
|
MSn (CID2, CID3) | [85] | ||
|
MSn (CID2, CID3) | [79, 93] | ||
| C-S (Sulfonium) | CID |
|
MSn (CID2, CID3) | [70] |
| C-S (Cyanuric) | CID |
|
MS2 (CID2) | [75] |
| D-P (Asp-Pro) | CID |
|
MS2 (IS1, CID2) MSn (CID2, CID3) |
[69, 86] |
|
MSn (CID2, CID3) | [89, 103] | ||
| CID/ETD |
|
MSn (CID2, CID3) ETD2 validation |
[88] | |
| C-N (Rink) | CID |
|
MSn (CID2, CID3) | [68, 87] |
|
MSn (CID2, CID3) | [76] | ||
| C-N (Urea) | CID |
|
MS2 (CID) | [73] |
|
MS2 (HCD) | [90] | ||
|
|
MS2 (HCD) | [91] | ||
| C-N (Quaternary diamine) | CID |
|
MSn (CID2, CID3) | [92] |
| N=N | CID (FRIPS) |
|
MSn (HCD2, CID3) | [112] |
| N-N (Hydrazone) | ETD |
|
MSn (ETD2, CID3) | [71] |
| C-N | ETD |
|
MS2 (ETD2) | [72] |
1.2.1 MSn-based XL-MS analysis workflow
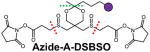
The ideal cross-linkers for MSn analysis should possess MS-cleavable bonds that are significantly more labile than peptide bonds, ensuring selective and preferential fragmentation of the linker with minimal peptide backbone cleavage at the MS2 stage. Such fragmentation should also occur independently of peptide charge and sequence. To this end, we have designed a suite of sulfoxide-containing, MS-cleavable cross-linkers (i.e. DSSO74, DMDSSO84, Azide/Alkyne-A-DSBSO79,93 and DHSO85), and demonstrated that the C-S bonds adjacent to the sulfoxide are robust MS-labile bonds exhibiting the desired features for MSn analysis (Table 2). It is noted that the CID cleavability of the C-S bonds in a sulfonium ion-containing and CBDPS cross-linkers has also been illustrated70,75. While our sulfoxide-containing cross-linkers74,79,84,85,93 each have distinct chemical features that were designed for specific applications, they are all homobifunctional cross-linkers with two symmetric CID-cleavable C-S bonds. As a result, the identification for all sulfoxide-containing MS-cleavable cross-linked peptides is performed using the same MSn-based analytical platform as illustrated in Fig. 2A.
Figure 2. MS analysis workflows for MS-cleavable (A–C) and non-cleavable cross-linked peptides (D).

(A) MSn analysis of a DSSO cross-linked peptide (α-β). (B) MS2-based analysis of a DSSO cross-linked peptide with sequential CID and ETD fragmentation. (C) MS2 analysis workflow of a DSBU cross-linked peptide with HCD. (D). MS2 analysis of a DSS cross-linked peptide with HCD.
Using a DSSO inter-linked peptide α-β as an example (Fig. 2A), low-energy CID during MS2 induces the cleavage of either of the two symmetric C–S bonds adjacent to the sulfoxide functional group, resulting in the physical separation of the cross-link and yielding unique peptide fragment pairs (i.e., αA/βS or αS/βA) with a defined mass relationship74. The resulting α and β peptide fragments are modified with complementary cross-linker remnant moieties, i.e., alkene (A) or sulfenic acid (S). However, the sulfenic acid moiety often undergoes dehydration to become a more stable unsaturated thiol moiety (T). These characteristic and predictable MS2 fragment ion pairs (i.e., αA/βT and αT/βA) are then subjected to MS3 analysis for simplified and unambiguous identification of cross-linked peptides by conventional database searching tools. This workflow enables database searching at the proteome scale with the same level of speed, confidence and accuracy offered in routine proteomics studies. The false discovery rate (FDR) of cross-links is further decreased by the stringency associated with incorporation of three lines of evidence (i.e. MS1 mass mapping, MS2 cross-link fragmentation, MS3 peptide sequencing). In addition to conventional top N acquisition during MS3 analysis, targeted acquisition can be effectively implemented by utilizing the mass difference of alkene- and thiol-modified fragment peptides (e.g. for DSSO cross-linked peptides: Δm = (αT-αA) = (βT-βA) = 31.97 Da)74,94. Both types of acquisition methods provide comparable and complementary results during data-dependent analyses. This MSn-based workflow has been successfully applied to map protein-protein interactions and elucidate architectures of protein complexes in vitro and in vivo74,79,95–100.
While effective, MSn analysis typically suffers from lower sensitivity and requires longer duty cycles compared to MS2-based acquisitions. However, the sensitivity and scan rate of MSn analysis have been significantly improved through the development of new generations of Orbitrap mass spectrometers. Clearly, advanced MS instrumentation contributes to move XL-MS studies forward by enhancing MSn-based identification of low abundant cross-linked peptides from increasingly complex biological samples79,98,99. With the availability of new fragmentation techniques in new instrumentation, the Heck group has recently demonstrated an MS2-based workflow utilizing sequential CID-ETD101,102 to sequence DSSO cross-linked peptides (Fig. 2B). As limited backbone fragmentation of DSSO cross-linked peptides occurs during CID in MS2, ETD is carried out to augment peptide fragmentation and assist in sequence identification74. The XlinkX software was specifically developed for this purpose, utilizing all forms of fragment information in MS2 for database searching (Table 1)101,102. Similar to the MSn workflow, the presence of predictable and dominant DSSO cross-link fragments is essential for accurate identification of cross-linked peptides from MS2 data. While DSSO-based MSn 95,98,99 and MS2 workflows101 have each demonstrated their effectiveness in identifying cross-linked peptides from various complex samples, the integration of MSn and MS2 strategies has been shown to provide more comprehensive and complementary information for systems-level studies102.
Another representative group of MSn-based MS-cleavable reagents are the PIR (protein interaction reporter) cross-linkers developed by the Bruce lab, which utilize dual Rink68 or D-P bonds89 for CID-induced cleavage. In addition to characteristic cross-link fragmentation, a reporter ion is formed and detected in MS2. To ensure the selection of only cross-linked peptide fragments for subsequent MS3 sequencing, ReACT (Real-time Analysis for Cross-linked peptide Technology) was developed based on unique features of PIR cross-linkers to allow on-the-fly decision making77,103. Coupled with biotin-based enrichment, PIR reagents have been successfully utilized for various large-scale cross-linking applications in bacteria77,89,104–106, murine107, and human systems40,77,108. Collectively, MSn-based workflows using CID-cleavable reagents are effective and robust for mapping PPIs from various types of complex samples.
1.2.2 MS2-based XL-MS workflow
For MS2-based analysis, the prerequisites for MS-cleavable bonds would be less stringent than those designed for MSn analysis. However, characteristic fragmentation of cross-links is still required, as the diagnostic ions generated from the cleavage of the cross-link ensures more accurate identification of cross-linked peptides at the MS2 level compared to identification of non-cleavable cross-linked peptides (Fig. 2C and D). Compared to MSn, MS2-based workflows are more sensitive and have higher scanning rate, thus enhancing the likelihood of sequencing cross-linked peptides. However, some inherent issues associated with MS2-based cross-link analysis remain; fragment ions are present for two peptide sequences, and unequal fragmentation may yield insufficient information to accurately identify both peptides simultaneously. Therefore, database searching of MS2 data—even for MS-cleavable cross-linkers—would be speed-limiting and more prone to higher FDR compared to MSn-based analysis. To eliminate false positives, cautions should be taken and stringent filtering needs to be implemented109. Nevertheless, MS2-based analysis is attractive due to its sensitivity and speed, as well as instrument flexibility and accessibility.
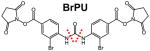
As listed in Table 2, several MS-cleavable cross-linkers are better suited for MS2 analysis as their MS-cleavable bonds have comparable strengths to peptide amide bonds. Therefore, their cleavage requires collision energy similar to that needed for the fragmentation of peptide backbones. One such notable cross-linker is the urea-based MS-cleavable homobifunctional NHS ester, i.e. DSBU (a.k.a. BuUrBu) developed by the Sinz group, which contains cleavable C-N bonds adjacent to a central urea functional group73. Characteristic fragmentation of DSBU (Fig. 2C) aids database searching of MS2 spectra through MeroX software (Table 1)110,111. To further facilitate accurate assignment of cross-linked products via MS2 analyses, the DSBU derivative bromine phenylurea (BrPU) was developed by implementing two bromine atoms in the linker region. This incorporation permits the detection of the unique bromine isotope pattern and mass defect for all cross-linker-modified fragments, improving automated analysis and assignment of cross-linked peptides by MeroX90.
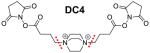
Very recently, the Sinz group has discovered that CDI (1,1′-carbonyldiimidazole), a commercially available compound, carries two symmetric urea-type MS-cleavable bonds with an ultra-short spacer length (~2.6 Å) (Table 2), and thus can function as a “zero length” MS-cleavable cross-linker91. It has been shown that CDI targets amine and hydroxyl groups with similar reactivity near physiological pH, expanding the coverage of protein interactions. In addition, CDI cross-linked peptides fragment similarly to other urea-containing MS-cleavable cross-linkers during CID in MS2, yielding characteristic product ions to facilitate cross-link identification91. Moreover, the heterogeneity of cross-linked products is reduced due to the absence of “dead-end” (type 0) modified peptides.
1.2.3 Other MS-cleavable cross-linkers
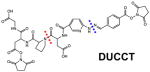
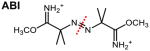
In addition to normal CID-cleavable bonds described above, FRIPS (free radical initiated peptide sequencing)-based cross-linkers have been recently explored 78,80,112. In comparison to TEMPO- 80 and azo-linkers 78, ABI (azobisimidoester)-linker (Table 2) appears to be the most promising as it can induce the FRIPS process during HCD in positive ion mode112. Although interesting, their performance appears to be less favorable than normal CID-cleavable cross-linkers for automated identification of cross-linked peptides due to complex fragmentation90. While most MS-cleavable cross-linkers are CID-cleavable, ETD-cleavable bonds have also been utilized for cross-linking studies (Table 2)71,72. Based on existing cleavable bonds, the dual-cleavable cross-linker DUCCT containing both CID-cleavable and ETD-cleavable sites has been developed88. Integration of two differential cleavable bonds allows complementary cross-link fragmentation obtained by separate CID and ETD analyses in MS2 to assist cross-link identification. Although effective ETD analysis generally requires highly-charged precursor ions, cross-linked peptides are suited for ETD analysis as they carry at least 4+ charges due to the presence of two tryptic peptides. However, in comparison to CID, ETD analysis has had significantly lower sensitivity in the past. Implementation of high-capacity ETD in the Orbitrap Fusion Lumos MS has shown greatly improved sensitivity113, which will undoubtedly facilitate the use of ETD in future XL-MS studies.
1.3 Enrichment of cross-linked peptides to enhance their detection
Another challenge of cross-linking studies is the low abundance of cross-linked peptides compared to non-cross-linked peptides in complex peptide mixtures. This is due to the fact that cross-linking reactions are not homogenous events and often result in heterogeneous cross-linked species. Cross-linking efficiency is dependent on multiple factors including protein concentration, cross-linking chemistry, residue proximity and surface accessibility. Moreover, variations in cross-linking events further complicate the mixture, resulting in dead-end modified (type 0), intra-linked (type 1), and inter-linked (type 2) peptides114. Inter-linked peptides—referred to as cross-linked peptides here—are the most structurally informative species but usually also the least abundant. Ultimately, cross-linked peptide heterogeneity increases exponentially with protein complexity, making the enrichment of cross-linked from linear species a necessity—especially for proteome-wide studies—to enhance the detection of low-abundance cross-linked peptides during data-dependent acquisitions.
1.3.1 Chromatographic separation of cross-linked peptides
Differences in the physicochemical properties of cross-linked versus linear peptides have been exploited for peptide-level separation using chromatographic techniques. Because cross-linked peptides typically have higher charges and are larger than linear peptides (including dead-end modified and intra-linked peptides), strong-cation exchange (SCX)46 and peptide size exclusion chromatography (SEC)115,116 are commonly used to enrich cross-linked peptides. While both methods are effective, SCX separation is more flexible and can be carried out using SCX tips such as StageTips117. However, peptide SEC provides better resolution than SCX in distinguishing cross-linked peptides from linear peptides115. To further enhance SCX separation of cross-linked peptides, charge-based fractional diagonal chromatography (XL-ChaFRADIC) has been recently reported118, which integrates two-dimensional SCX separation. The first dimension isolates highly-charged species from a Lys-C digest, which are then further digested by trypsin and subjected to second-dimensional SCX. Another recent study utilized a combination of protein-level gel filtration coupled with diagonal peptide SCX to facilitate cross-link identifications from proteome-wide mixtures119. While such a combination strategy indeed reduces the complexity of the final peptide mixture, it is noted that multidimensional chromatography can also result in sample loss, particularly for already low abundance cross-linked peptides. Therefore, careful consideration is required when selecting separation methods in order to maximize sample recovery and cross-link identifications.
1.3.2 Affinity purification of cross-linked peptides
Another strategy to improve the sensitivity of XL-MS analysis is to enrich cross-linked peptides by affinity purification. This can be achieved through the design of enrichable cross-linkers that incorporate an affinity handle. Among various affinity tags that have been widely employed in protein purifications, biotin tag appears to be the most popular for peptide purification due to its high affinity to streptavidin (Kd~10−15). Over the years, various types of biotin-tagged cross-linkers have been developed, including non-cleavable and MS-cleavable cross-linking reagents68,75,76,120–124. Although it is convenient to directly use biotin-tagged cross-linking reagents, it is advantageous to design affinity-based cross-linkers without the inclusion of the bulky biotin tag to maintain their small size, accessibility, and membrane permeability. Alternatively, the biotin tag can be incorporated at a later stage by utilizing azide- or alkyne-tagged cross-linkers with ‘click chemistry’-based conjugation79,93,125–129. Once enriched, biotinylated cross-linked peptides can be directly eluted from affinity resins. However, removal of the biotin tag prior to MS analysis is preferred as the presence of biotin can complicate LC-MS analysis of cross-linked peptides124. This can be accomplished through the incorporation of various chemical cleavage sites in the linkers, such as acid-cleavable (e.g. Azide/Alkyne-A-DSBSO)79,93, and azobenzene-based cleavable sites (e.g. Leiker)124.
Apart from incorporating affinity handles in cross-linkers, a recent study has demonstrated the feasibility of an alternative strategy based on the fusion of target proteins with a modified His-tag, i.e. CH (Cysteine–Histidine) tag, composed of a single cysteine followed by a ‘DP’ (aspartic acid-proline) moiety and a histidine tag130. The cysteine residue functions as an anchor site for heterobifunctional cross-linkers with one end targeting cysteines, the His-tag for affinity purification, and the DP bond to assist in MS analysis. This strategy aims to only capture CH tag-containing cross-linked peptides, thus enriching only targeted protein interaction regions for analysis. This work suggests that affinity tags other than biotin tag may be worth exploring for affinity purification of cross-linked peptides in future studies.
2. Developing New Cross-linking Chemistries
To date, the most commonly used cross-linkers are homobifunctional amine (lysine)-targeting reagents consisting of N-hydroxysuccinimidyl (NHS) ester functional groups (e.g. disuccinimidyl suberate (DSS). Targeting lysine residues is preferable for several reasons: their relatively high overall prevalence (~6% of all residues), their distribution across solvent-accessible protein surfaces, and the specificity of primary amine-targeting chemistries. However, targeting lysine residues for cross-linking is less amenable for the capture of hydrophobic surface interactions, which can be buried and lack charged residues. Similarly, proteins with few or no lysine residues can be difficult to characterize using amine-targeting reagents. Over the years, various cross-linking reagents and workflows have been developed, each featuring unique chemical structures and combinations of functional groups to diversify the range in which XL-MS can be applied to study protein structures.
2.1 Acidic residue-specific cross-linkers
Aspartic (Asp) and glutamic (Glu) acid residues constitute approximately 12% of all amino acid residues and commonly occupy surface-exposed regions of proteins, critically important for protein-protein interactions. Therefore, they represent high-potential cross-link targets for mapping protein interaction contacts. Early work by Novak and Kruppa described the feasibility of acidic residue-targeted cross-linking at ~ pH 5.5 using EDC-activated Asp and Glu with dihydrazide cross-linking reagents131. This workflow was later improved by Leitner et al. utilizing DMTMM as a coupling reagent instead of EDC, allowing Asp and Glu cross-linking to occur at neutral pH—better suited for elucidating protein structures under physiological conditions132. Our lab expanded on this cross-linking chemistry by developing a sulfoxide-containing MS-cleavable, acidic residue-specific dihydrazide (i.e. DHSO), which utilizes the same MSn workflow developed for the identification of lysine-reactive DSSO cross-linked peptides (Fig. 2A)85. Due to the higher degree of variability and complexity when comparing acidic residue- to lysine-targeting cross-linking, MS-cleavability of DHSO drastically simplifies identification of Asp/Glu cross-linked peptides, thus providing complementary interaction contacts to facilitate the elucidation of architectures of protein complexes.
2.2 Non-specific, photo-activated cross-linkers
Diazarine-incorporated amino acid analogs are unique from residue-specific cross-linkers in their ability to non-specifically cross-link proximal residues18,133,134. This functionality has an advantage over site-specific cross-linking reagents, as non-specific photo-inducible chemistry permits cross-linking of residues in hydrophobic regions, extending the application of cross-linking to the structural determination of solvent-inaccessible interaction regions e.g. membrane-spanning protein complexes135. Photo-reactive functional groups (i.e. diazarine, phenyl azide, benzophenone) have also been explored in conjunction with NHS esters to create short, heterobifunctional cross-linking reagents consisting of lysine-targeting and non-specific ends136,137. However, while non-specific chemistry is attractive for increasing surface coverage, it often results in highly complex cross-linked products, complicating database searching. In addition, each interaction can be described by multiple species of cross-linked peptides, effectively diluting their individual abundances. Together, these issues pose problems for sensitive MS analysis and accurate identification of cross-linked peptides, limiting their current application to studies of single proteins.
3. XL-MS Strategies for Structural Analysis of Protein Complexes
XL-MS technology is a powerful and effective structural tool because it offers several layers of information through the identification of cross-linked peptides4,6,7,23,38,79,138,139. Within a given protein complex, cross-linking analysis identifies proximal residues between subunits of protein complexes, providing clues on spatial orientation and protein connectivity. For instance, cross-linking has been successfully coupled with phylogenetic sequence alignment to identify potential evolutionarily-conserved, functionally important residues. Subsequent mutation studies of candidate sites confirmed loss-of-function mutants, permitting localization of protein interaction interfaces in Schizosaccharomyces pombe telomere complexes97,100. Similarly, cross-linking data was used to direct truncation/deletion mutagenesis for identification of protein binding domains involved in exocytosis and microtubule complexes140. Identified physical contacts between protein complexes can also help define edges of protein interaction networks, generating experimentally-derived interaction topologies that normally require multiple rounds of reciprocal co-immunoprecipitation in traditional mass spectrometry approaches38. In addition, cross-links contain defined spatial constraints that can be utilized to confirm existing high-resolution structures and/or assist computational modeling. Apart from inter-protein interactions, details about intra-protein interactions are also obtained for each protein complex constituent, which can further contribute to the architectural elucidation of multisubunit assemblies. Given these unique capabilities, XL-MS studies have been employed for structural characterization studies of various protein systems19,28,41,42, dating back to a seminal publication in 2000 where Young et al. demonstrated the potential of XL-MS by coupling cross-link data with computational modeling to predict three-dimensional protein folding of a model protein FGF-237. In the same year, Rappsilber and coworkers employed cross-linking, gel electrophoresis and protein identification to determine the spatial organization of subunits within a protein complex, although cross-linked peptides were not identified141. Despite these early works paving the way for XL-MS studies, successful examples of protein complex structural analysis were not reported until nearly a decade later. In 2010, Chen et al. mapped the interaction between RNA polymerase II and the TFIIF complex via the identification of 352 inter-subunit cross-links, permitting the localization of the Pol II-TFIIF interaction interface32. Since then, XL-MS applications have evolved significantly—particularly in recent years—due to dramatic advancements in bioinformatics tools and cross-linking reagents, as described above. In addition, various XL-MS strategies have also been developed to facilitate structural analysis of protein complexes.
3.1 Combinatory cross-linking strategies to obtain comprehensive information
One of the long-term goals in XL-MS studies is to generate data sufficient for de novo structural modeling of proteins and protein complexes. However, there are two major challenges to de novo computational prediction: sampling of structures and discrimination of inaccurate structures142. Evaluation of the impact of XL-MS data on de novo structural prediction has suggested that a sufficient number of distance restraints generated from XL-MS data is beneficial not only for decreasing the size of the sampling space, but also for increasing discriminating power of the scoring function, thus improving the identification of accurate models142.
To enhance the yield of structural data acquirable through XL-MS analyses, various cross-linking reagents targeting different residues can be employed to obtain more comprehensive information. For instance, the Chait group showed that combination of lysine-reactive reagents such as DSS (amine-to-amine) with the zero-length cross-linker EDC (amine-to-carboxyl) resulted in complementary structural information to one another, as evidenced in their characterization of the Nup84 complex36. Similar combinations have also been adopted by other labs17,143,144. In addition, the integration of NHS esters and acidic residue-targeting dihydrazides provides complementary maps of protein interactions as well85,132. Other combinations such as lysine-reactive reagents with photo-induced unnatural amino acids have also been successful, due to their orthogonal abilities to target solvent-accessible and solvent-inaccessible regions, respectively145,146. Novel heterobifunctional cross-linkers combining lysine-targeting NHS chemistry with non-specific photo-activatable diazarenes (sulfo-SDA, sulfo-SBP) have served to further increase the amount of information obtainable by XL-MS, yielding ‘high-density’ datasets with greater potential for de novo modeling137,147. Collectively, integration of different cross-linking chemistries has demonstrated its effectiveness for expanding the coverage of interaction maps.
In addition to residue-targeting chemistry, cross-linker length can also impact the number of obtainable cross-linked peptides and their informative interaction content6,132,142. While shorter cross-linking reagents often result in fewer cross-links, their translation to spatial information represents ‘tighter’ distance restraints for structural modeling. On the other hand, longer cross-linkers typically yield higher numbers of cross-links that may not be as structurally informative, making them better suited for interaction capture studies142. Since XL-MS enables the capture of all types of interactions (i.e stable, weak, dynamic/transient) in a single experiment, the resulting cross-link data generally represents the average of multiple structural ensembles148. Therefore, it is conceptually conceivable that integration of cross-linking reagents with various lengths can increase the depth of structural information by capturing protein interactions within different conformations of protein complexes for more accurate structural prediction142. Although different lengths of linkers have been shown to provide complementary results132,148,149, it remains unclear how much additional valuable information can be obtained to improve computational modeling. Commonly used cross-linkers that carry optimal cross-linking lengths (10~15Å)6,142 have proven their effectiveness in elucidating architectures of protein complexes at the systems level38,101,102, whereas significantly longer PIR cross-linkers have been equally successful for mapping protein interactions40,105–108,139,150,151. Clearly, further exploration on the impact of cross-linker lengths on structural characterization is necessary to help optimize XL-MS experiments.
3.2 Sample preparation for in vitro XL-MS analysis of protein complexes
3.2.1 In vitro on-bead cross-linking strategies
Most in vitro XL-MS studies to date have relied on recombinant protein complexes as a source of highly pure and homogenous materials for in-solution XL-MS experiments (Fig. 3A). Developments in sample preparation have extended the application of XL-MS to more heterogeneous studies, such as affinity-purified protein complexes from native cells. A landmark study by Herzog et al. demonstrated the feasibility of performing on-bead cross-linking and digestion of His-tagged protein complexes affinity purified from human cells (Fig. 3B), which was shown to enhance both reaction efficiency and analysis sensitivity38. As a result, the optimized XL-MS protocol enabled a systematic analysis of human phosphatase 2A (PP2A) complexes and the generation of a comprehensive interaction network topology of PP2A complexes containing 176 inter-protein and 570 intra-protein cross-links. In addition, the cross-link data provided distance restraints to model the interactions between PP2A regulatory proteins using ROSETTA.
Figure 3. In vitro cross-linking strategies for protein complexes.

(A) 1- and 2-step in-solution in vitro cross-linking protocols for protein complexes. (B) 1- and 2-step on-bead in vitro cross-linking protocols for affinity purified protein complexes. 2-step protocols in (A) and (B) involve in vitro mild FA cross-linking prior to the second cross-linking step. (C) 2-step cross-linking protocol by coupling in vivo mild FA prefixing with on-bead in vitro cross-linking of affinity purified protein complexes.
In addition to the His tag, biotin tag can be used for purifications under both native and fully denaturing conditions. Over the years, we have developed several new HB (i.e. Histidine-Biotin)-tag based affinity strategies to effectively isolate protein complexes from human cells for XL-MS studies79,99,152,153. Similarly, we have also employed on-bead cross-linking and digestion of HB-tagged proteasome complexes, which was proven robust and effective with single-step Streptavidin purification98,99. The extremely tight binding affinity between biotin and Streptavidin permits purifications with higher specificity and less background compared to His tag-based purification in mammalian cells. It is worth noting that Streptavidin beads do not appear to interfere with XL-MS analysis. Altogether, our results have further demonstrated the benefits of on-bead processing of protein complexes for cross-linking studies.
In general, antibody-based purification is not preferred for XL-MS experiments due to the sensitivity of antibodies to buffer conditions and their interference in peptide analysis. To circumvent these problems, the Chait group recently created lysine-less GFP nanobodies to facilitate purification of GFP-tagged protein complexes and on-bead cross-linking with lysine-reactive cross-linkers prior to subsequent SDS-PAGE and in-gel digestion138. This workflow has been successfully applied to define architectures of several protein complexes e.g. exosomes138. Recently, it has been shown that a small commercially-available GFP nanobody (12 kDa) can also be used for similar XL-MS analysis154. However, extensive washing and rigorous data filtering are needed to minimize spurious identifications due to nanobody interference. Collectively, on-bead cross-linking of affinity purified protein complexes (Fig. 3B) appears to be more attractive than in-solution cross-linking due to the flexibility of sample handling to ensure optimal cross-linking of protein complexes at low concentrations with minimal sample loss.
3.2.2 Two-step cross-linking protocols
Although most XL-MS studies of proteins and protein complexes employ effective single-step reactions with a selected cross-linker, not all transient interactions may be sufficiently captured and characterized. To improve detection of weak interactions and generate sufficient cross-linked products for MS analysis, a two-step cross-linking protocol has been successfully applied to dissect the dynamics of Mediator-RNA Polymerase II Pre-Initiation Complexes (Fig. 3A)155. This strategy employed a substoichiometric formaldehyde (FA) fixation step as the first step to freeze configurational dynamics prior to SBAT (i.e. 1-hydroxy-7-azabenzotriazole analog of DSS) cross-linking155. This fixation step did not seem to interfere with subsequent cross-linking and identification of cross-linked peptides, and the results indicate that such stabilization greatly assisted structural characterization of Mediator-polymerase complexes by XL-MS. Furthermore, it has also been shown that SBAT has faster lysine-reactive chemistry than DSS155,156. Together, this revised cross-linking procedure allowed the architectural elucidation of the entire complex and individual subcomplexes, along with their structural dynamics155.
In addition to in vitro fixation, FA cross-linking has been widely used in vivo for stabilizing protein-protein interactions in intact cells prior to cell lysis and protein purification under both native98,99,157,158 and denaturing conditions159–162. Recently, we have developed two affinity purification strategies, namely XAP (in vivo cross-linking (X) assisted Affinity Purification)98 and XBAP (in vivo cross-linking (X) assisted Bimolecular tandem Affinity Purification)153 to study dynamic interactors of protein complexes and subcomplexes, respectively. These approaches employ mild in vivo FA (<0.1%) cross-linking, which enables better preservation of dynamic interactions for affinity purification under native conditions. In addition, we have demonstrated that HB-tagged proteasome complexes purified with the XAP method can be directly subjected to in vitro on-bead DSSO cross-linking and LC-MSn analysis, thus allowing us to dissect interaction-mediated regulation of proteasomes98. This two-step protocol combines mild in vivo FA fixing prior to in vitro DSSO cross-linking (Fig. 3C) and is beneficial to study affinity-purified complexes due to its ability to preserve weak, transient and/or dynamic interactions. Apart from FA, glutaraldehyde cross-linking has also been used for stabilizing protein interactions after cryomilling of flash-frozen cells prior to affinity purification163. Potentially, such pretreatment can also be integrated with two-step cross-linking procedures when processing cells at low temperature. Clearly, two-step sequential cross-linking procedures present certain benefits for in vitro XL-MS studies, especially for preserving dynamic/weak interactions. However, systematic comparisons to better understand the impact of prefixing would undoubtedly contribute to optimization of future experimental protocols.
3.2.3 In vivo cross-linking of protein complexes
In recent years, in vitro XL-MS studies of protein complexes have produced enormous amounts of molecular details for understanding their architectures, functions and regulation11,12,19,27,28,36,40–42,155,164,165. However, it is more biologically relevant to perform in vivo cross-linking experiments as they can provide a more authentic map of protein-protein interactions (PPIs) occurring in native cellular environments30. This is because PPIs in cells are highly dynamic, and can be modulated by various cellular cues including posttranslational modifications. While stable interactions can survive various purification conditions, transient and weakly interacting proteins are often lost during the process. In addition, native cell lysis can lead to PPI reorganization and yield non-physiological interactions. Therefore, to obtain a true snapshot of protein interaction networks in living cells, it is essential to stabilize/freeze these protein interactions prior to cell lysis, which can be achieved through in vivo cross-linking, a unique capability that other structural tools do not possess. Until now, only a limited number of in vivo XL-MS studies have been reported, mainly due to the challenges discussed above. While the Bruce lab has focused on in vivo cross-linking at the proteome level using their PIR cross-linkers 40,77,89,104–106,108,139,151, our lab has mostly focused on in vivo XL-MS analysis of protein complexes 79,99,159,162,166,167. Recently, we have developed a membrane permeable, sulfoxide-containing MS-cleavable and enrichable NHS ester cross-linker Azide-A-DSBSO (Table 2)79. This multifunctional cross-linker carries several unique features (i.e. small size, proper spacer length (~14 Å), bio-orthogonal affinity handle, robust MS-cleavable bonds, and an acid cleavage site), making it an attractive reagent for defining PPIs in cells. For mapping the interaction network topology of protein complexes in mammalian cells, in vivo Azide-A-DSBSO cross-linking of intact cells is first carried out, followed by HB tag-based tandem affinity purification159 to extract in vivo cross-linked protein complexes under fully denaturing conditions (i.e. 8M urea), eliminating non-specific background. Cross-links are then biotinylated through click chemistry-based conjugation before protein digestion, followed by subsequent peptide affinity enrichment. Cross-linked peptides are eluted through the acid cleavage site while the biotin tag-containing portion remains bound to the resin. The addition and removal of biotin tag eliminates its intervention during cross-linking and MS analysis, respectively. This strategy has been successfully applied to the study of proteasome complexes and has identified in vivo inter-subunit and intra-subunit proteasome interactions for the first time79. In addition, Azide-A-DSBSO is capable of dissecting in vivo PPIs at the proteome scale79. Recently, we have also explored the feasibility of using DSSO for in vivo cross-linking in mammalian cells99 coupled with HB-tagged purification of cross-linked proteasome complexes. Through these studies we have been able to identify extensive interactions within proteasome subunits by utilizing various cell lines and advanced Orbitrap instrumentation. Nonetheless, Azide-A-DSBSO is better suited for in vivo cross-linking studies due to its enrichment capability, which is critical for proteome-wide studies.
4. Elucidation of Protein Complex Architectures
While restraints provided by XL-MS studies have played a pivotal role in probing the topologies of various protein complexes19,28,41,42, cross-linking data alone is typically insufficient for structural derivatization of protein complexes. This is still largely due to the limited structural information that can be yielded by cross-linking due to residue specificity of reagents and respective residue availability in proteins, despite recent advancements in cross-linking methodologies. Furthermore, cross-linking data typically represents a dataset describing the “average” state of proteins, which can include multiple conformations. Software such as XL-MOD have been designed for the analysis of conflicting, high-confidence cross-links168,169, whereas DynaXL is another example of software developed to interpret conflicting cross-linking data by modeling excited-state structures that may result from distance-violating cross-links170. However, without other sources of information, translation of sparse cross-linking data to definitive structures and models remains difficult. Combining restraints derived from XL-MS experiments with complementary data from other structural techniques has been shown to be most effective for examining various aspects of protein structure.
4.1 Coupling XL-MS with other MS-based structural tools
Combinatory bottom-up analysis of cross-linked proteins and top-down analysis via native/IM-MS has been shown to be an effective means of determining the three-dimensional structures of multiprotein assemblies171–175. While native mass spectrometry approaches are capable of determining overall complex topology and subunit stoichiometry, they are usually unable to determine subunit connectivity and map interaction interfaces. On the other hand, XL-MS techniques are well-suited for determining topological ordering of protein complexes, identifying protein interacting regions, and preserving dynamic interactions. Clearly, the two MS-based approaches can provide unique and complementary structural information that cannot be easily assessed by either alone. Thus, the combination of these two strategies continues to be attractive for macromolecular complex structure determination144,146,176,177, though slightly limited in application due to the difficulty in extracting endogenous protein assemblies in high enough concentration and purity for native MS analysis.
XL-MS has also been utilized alongside surface-probing methodologies such as radiolytic footprinting178,179, covalent labeling176,180,181, HDX (hydrogen-deuterium exchange)182–184 and limited proteolysis185,186 to study the structures of proteins, as well as their dynamics and conformational changes. The basis of these techniques lies in their ability to identify the solvent-accessible surfaces of proteins, which can be interpreted for protein-folding studies and identifying solvent-inaccessible contact interfaces. Footprinting and labeling methods modify solvent-exposed amino acid residue side chains by oxidation or chemical modifications that can be identified by standard proteomics, whereas HDX exploits the in-solution exchange of protons for deuterium. Limited or partial proteolysis methodologies are likely the least sensitive, relying on short incubations with enzymatic proteases to determine exposed loop and flexible regions. Consequently, these techniques can also be used to compare and contrast the structural conformations of multiple (e.g. wild-type, ligand-bound, PTM-modified, PPI) protein states based on their differential surface dynamics. Various tools have been developed to facilitate integration of spatial restraints from XL-MS with other MS-based structural methods (see Table 1), for instance with covalent labeling to probe solvent-accessible surfaces for protein modeling64,180.
4.2 Integrative modeling of macromolecular machines
As a result of a recent surge in instrumentation and image interpreting software advancement, cryo-EM has emerged as an alternative method for the structural elucidation of macromolecular assemblies at (near) atomic-resolution. Compared to x-ray crystallography and NMR, the sample preparation process for cryo-EM is significantly less cumbersome. However, analysis of conformationally or compositionally heterogeneous complexes still poses a challenge, as there are often areas of unassigned electron densities due to limited resolution in EM maps. As a result, accurate localization of individual subunits within a protein complex can be difficult without other forms of structural information. In these scenarios, XL-MS data can provide orthogonal information to low-resolution EM density maps to guide the special organization of individual subunits187,188.
Ultimately, most protein complexes remain recalcitrant to any single structural method, giving rise to an era dominated by combinatory workflows utilizing computational software to consolidate various forms of complementary structural information (Fig. 4). From high-resolution partial structures contributed by X-ray crystallography/NMR and low-resolution density maps from cryo-EM/SAXS studies to structural information from XL-MS and other structural MS strategies, ‘hybrid’ platforms such as IMP (Integrative Modeling Platform)189,190 have been designed to integrate diverse forms of biophysical and proteomic data for structural determination. Data from various structural methodologies are translated into a set of spatial restraints that cumulatively describe a scoring function. Starting from initial random configurations, the space of conformations is explored iteratively to minimize the scoring function, yielding an optimized ensemble that maximizes satisfaction of the original data. As summarized in Table 1, Xlink Analyzer191, ROSETTA192 and HADDOCK193 have also been successfully employed for similar studies, paving the way for a new archetype in structural biology in the form of ‘integrative structural modeling’. XL-MS has been instrumental in the integrative modeling of a number of multi-protein assemblies in recent years. Notable examples include the SAGA transcriptional co-activator complex194–196, Elongator197,198, GPCRs145,199, ribosomal complexes35,164,188, and mitochondrial complex I200. Three other macromolecular assemblies that have been extensively studied using integrative approaches include the 26S proteasome, nuclear pore complex (NPC), and RNA polymerase complexes, as reviewed recently23. However, these complexes have been continually studied in recent years, further revealing more intricate details on their structure and function that are worth mentioning. In addition, the strategies used for studying these complexes can be applied to other protein complexes.
Figure 4. General scheme for integrative structural modeling.

Integrative structural determination of the protein complexes proceeds through four main stages: (1) data is first gathered from various structural elucidation methodologies; (2) subunits are represented from high-resolution structures while other forms of low-resolution structural data are translated into spatial restraints; (3) configurational sampling produces an ensemble of structures that satisfies translated restraints; and (4) the ensemble structure is analyzed and validated. Adapted from Wang, et al 98 (This research was originally published in J. Biol. Chem. Wang, X., Chemmama, I.E., Yu, C., Huszagh, A., Xu, Y., Viner, R., Block, S.A., Cimermancic, P., Rychnovsky, S.D., Ye, Y., Sali, A., Huang, L. The Proteasome-Interacting Ecm29 Protein Disassembles the 26S Proteasome in Response to Oxidative Stress. J Biol. Chem. 2017, 292(39):16310–16320. © the American Society for Biochemistry and Molecular Biology).
4.2.1 26S Proteasome
The 26S proteasome is the macromolecular machine responsible for controlled degradation of ubiquitinated substrates, and is composed of 33 subunits that form two subcomplexes: the 20S catalytic core particle (CP) and 19S regulatory particle (RP)201. While the 20S CP is responsible for proteolytic activities, the 19S RP assists protein degradation through various functions including substrate recognition, protein deubiquitination, unfolding, and 20S gating. In comparison to the 20S CP, the 19S RP is much more dynamic and flexible—and can be further divided into the base and lid subcomplexes. Because of its compositional and conformational heterogeneity and dynamic nature, structural analysis of the 26S proteasome holocomplex has been challenging for decades. With the advancement of cryo-EM and hybrid structural tools, high-resolution structures of 26S proteasomes have been determined in recent years33,202–205. Through this process, XL-MS analysis has contributed significantly to the elucidation of proteasome architectures and to uncovering structural details underlying proteasome function and regulation23,33,95,98,99,206–209. In an early study, XL-MS and EM were used to define the structural topology of the AAA-ATPase module in the S.pombe 19S RP, as well as its spatial relationship to the α-ring of the 20S core particle206. To characterize the architecture of the yeast 26S proteasome, Lasker et al. employed integrative structural modeling by utilizing structural information from XL-MS, EM, X-ray crystallography and literatures33. This combinatory approach enabled the determination of the topological ordering and shape of the 19S lid as well as its interaction with the 20S CP and AAA-ATPase33. These results corroborated very well with the architecture of the complete 19S lid from yeast obtained using only EM and a heterologous expression system203. Although the later study did not attempt to identify any cross-links, site-specific cross-linking was performed to validate interactions observed in EM maps203. Coincidentally, we have also delineated the topological ordering of the yeast 19S base and lid subcomplexes based on DSSO cross-link data and probabilistic modeling95, obtaining the same spatial organization as reported203. In addition, our cross-linking data complemented previous reports by providing physical contacts between subunit Rpn12, Rpn13 and Rpn15 within the 26S proteasome, further confirming their interactions and aiding their localization in final models. This was important as the small size of Rpn15 had prevented its assignment in EM maps33,203,206. In addition to structural elucidation, an integrated approach combining biochemical, EM and XL-MS analyses has been successfully utilized to study 20S biogenesis by investigating the subunit arrangement and chaperone localization in proteasomal precursors207. The results have shown that the yeast Pba1-Pba2 heterodimer is recycled during 20S maturation due to substantial conformational changes within the alpha and beta rings of the 20S CP. Interestingly, the fate of the yeast Pba1-Pba2 heterodimer is different from its human counterpart, PAC1-PAC2, which is degraded after the completion of 20S assembly210. A similar integrated strategy has also been employed to dissect 19S RP biogenesis209, which implemented quantitative XL-MS for comparative analysis of the structures of the 19S lid intermediate and full lid. The results have revealed an extensive remodeling of the 19S lid precursor mediated by Rpn12, thus facilitating RP assembly. Protein-protein interactions are critically important not only for proteasome assembly and structure, but also for proteasome regulation. Ubp6 (Usp14 in mammals) is a proteasome-bound deubiquitinase important in modulating proteasomal degradation201. Structural analysis of the Ubp6-26S proteasome complex by EM and XL-MS has suggested that Ubp6’s active site may contribute to the conformational landscape of the 26S proteasome208.
In comparison to the yeast proteasomes, reports on the human 26S proteasome have been sparse. Only very recently, two high-resolution Cryo-EM structures (3.9 and 3.5 Å) of the human 26S proteasome were reported204,205. While the overall architecture of the 26S holocomplex is highly conserved from yeast to human, there are differences in conformational states, subunit assignments and structural dynamics. To further understand dynamic structures and regulation of the human 26S proteasome, we have employed DSSO-based in vivo and in vitro XL-MS workflows to comprehensively examine PPIs within the 26S proteasome99. As a result, we have obtained the largest cross-link dataset for proteasome complexes, with 447 unique lysine-to-lysine linkages delineating 67 inter-protein and 26 intra-protein interactions. In combination with EM maps and computational modeling, we have resolved architectures of the human 26S proteasome having global RMSDs ≤ 1.3 with reported high-resolution structures204,205, indicating minimal structural differences. Importantly, structural dynamics of subunits Rpn1, Rpn6 and Rpt6 been inferred based on less-resolved structures and multiple forms of Rpn1 and Rpn6 detected in EM maps, and a large number of violating cross-links (>35 Å) for Rpn1 and Rpt699. These results suggest conformational heterogeneity of the human 26S proteasome. Apart from the 26S holocomplex, additional cross-links were identified to demonstrate direct physical interactions between proteasome subunits and 15 proteasome-interacting proteins, including 9 known and 6 new interactors99. Among them, UBLCP1, is the only known proteasome phosphatase211, whose dynamic interaction with Rpn1 has been confirmed by XL-MS and integrative modeling with a proposed model inferring the action modes of UBLCP1 on regulating proteasomes. In addition, validation of the selected PPIs using reciprocal purification and XL-MS suggests that cross-links can serve as direct evidence for pair-wise protein interactions without further biochemical—confirmation as often required for conventional AP-MS experiments. Moreover, we have demonstrated that the combination of in vivo and in vitro XL-MS analyses has provided more comprehensive interaction data for structural analysis of proteasome complexes. While in vitro studies yield abundant cross-links for stable interactions, in vivo experiments are advantageous for characterizing dynamic and transient interactions.
Oxidative stress is known to trigger eukaryotic 26S proteasome disassembly, releasing more free 20S CP for the removal of oxidized proteins99,212. Proteasome-interacting protein Ecm29 has been recognized as the key regulator that modulates 26S proteasome disassembly. To determine how Ecm29 regulates the 26S proteasome, we set out to use XL-MS to identify their interaction contacts. Because the Ecm29-proteasome interaction is weak and sensitive to cellular/experimental conditions, we coupled XAP with DSSO-based in vitro XL-MS (i.e. two-step cross-linking (see Fig. 3B and Section 3.2.2)) to obtain sufficient Ecm29-proteasome complexes for structural analysis. In total, respective residue-residue interactions between Ecm29 and five 19S base subunits, Rpt1, Rpt4, Rpt5, Rpn1, and Rpn10 have been identified. Using integrative structural modeling through IMP, the architecture of the Ecm29-proteasome complex was determined, along with a proposed model for Ecm29-dependent dissociation of the 26S proteasome in response to oxidative stress98.
4.2.2 Nuclear pore complex
The nuclear pore complex is a massive 50 MDa nucleocytoplasmic transport assembly comprising roughly 30 distinct nucleoporins, for a total of at least 456 individual proteins. The model of Saccharomyces cerevisiae NPC was the first developed by an integrative modeling approach—delineating the NPC architecture as a highly conserved, symmetrical organization of distinct, multisubunit modules31. In the following years, several integrative studies utilizing XL-MS were employed to characterize those individual modules. A study by the Beck lab utilizing electron tomography, single-particle electron microscopy, and cross-linking mass spectrometry determined the structure of the human Nup107 subcomplex, a major scaffolding motif of the NPC spanning two reticulated rings via 32 copies34. Using a combination of X-ray crystallography, EM, and multi-linker cross-linking, the Chait lab proposed an integrative model for the budding yeast Nup84 subcomplex, a hetero-heptameric assembly that comprises the outer-ring of the NPC via 16 stable copies36. A publication on the Nup84 complex from Chaetomium thermophilum using cryo-EM and XL-MS revealed a dimeric module more similar to the orthologous Nup107-Nup160 complex in higher eukaryotes213, while further research into the budding yeast NPC by the Sali and Rout groups revealed a multi-state model for Nup133 using a combination of X-ray crystallographic data, SAXS, and negative-stain EM validated by cross-linking restraints214. Recently, the structure of the Nup82-Nup84 complex was solved using multi-linker cross-linking, EM, X-ray crystallography, and SAXS215. Integration of cryo-electron tomographic data, homology modeling, and fitting of high-resolution structures and cross-linking restraints were used to determine a comprehensive architecture for the human NPC216. The convergence between such elucidated structures and the biological function of the nuclear pore complex were very recently reviewed, shedding light on the molecular mechanisms underlying nucleocytoplasmic transport217.
4.2.3 RNA polymerase complexes
RNA polymerase, along with general transcription factors (GTFs) that comprise the preinitiation complex (PIC) and the multi-protein Mediator complex, constitute the basic eukaryotic transcriptional apparatus. In addition to Pol II-TFIIF32, XL-MS studies have been employed for the structural elucidation of individual RNA polymerases Pol I218,219, II76, III220, along with PIC-bound Pol II221,222 and the Pol II capping complex223. Additionally, the structures of the Mediator middle module224 and RNA Pol II-bound head module225 have also been targeted by XL-MS studies. In recent years, integrative studies have been extended to study the composite architectures of RNA polymerase in complex with GTFs226,227 and Mediator complex165,228, culminating in the structural determination of the complete Mediator-PIC155. This structure reported a previously undetermined protein kinase complex involving TFIIK and the Mediator-activator interaction region of the Med-PIC structure, revealing the molecular mechanism underlying transcriptional regulation by Mediator. Other interesting studies within the last year also reported structural insights on the initiation of yeast RNA polymerase I229 and the molecular dynamics of viral polymerases that permit adoption of multiple functional configurations230.
5. Proteome-wide XL-MS studies
While the structural elucidation of isolated assemblies are without a doubt critical for their functional understanding, the interplay between these protein complexes and their regulators required for homeostatic maintenance represent an avenue of research that has only begun to be breached. Thus, there is a pressing need for systematic, proteome-wide characterization of PPIs to fully understand the roles and functions of proteins and protein complexes. Various studies have shown the potential of cross-linking methodologies to not only provide structural details of endogenous interactions, but to offer a glimpse towards comprehensive interaction profiling of whole proteomes.
5.1 In vitro proteome-wide studies
The simplification of MS analysis for identification of cross-linked peptides enabled by MS-cleavable reagents has been particularly effective in increasing the yield of XL-MS data from highly heterogeneous mixtures such as cell lysates. Early in vitro studies at the proteome-level using non-cleavable cross-linkers yielded limited information even when coupled with offline separation techniques. Several attempts at E. coli lysate cross-linking were conducted, with the first two yielding less than a hundred cross-links each, and a third identifying 394 cross-links44,231,232. The study by Yang et al. contributing the most data from E. coli lysate cross-linking also reported 39 cross-links from the more complex C. elegans lysates. In comparison, MS-cleavable cross-linkers have been successfully utilized for large-scale studies of various organisms. For example, BAMG has been effectively employed to profiling HeLa cell nuclear extracts when coupled with diagonal SCX separation of cross-linked peptides233, identifying 247 cross-links at an estimated FDR of 0.4%. The Heck group also demonstrated the feasibility of using DSSO and SCX fractionation for proteome-wide analysis of HeLa and Escherichia coli cell lysates101,102, identifying 3,301 and 1,158 unique cross-links, respectively at 1% FDR. These studies suggest that MS-labile cross-linking reagents coupled with off-line peptide enrichment are more effective for in vitro XL-MS studies at the proteome-level.
Recently, the Leiker reagents developed by Tan et al. have been employed for cross-linking studies on lysates from E. coli and Caenorhabditis elegans124. Although Leiker cross-linkers are not MS-labile, they are biotinylated reagents that permit selective enrichment of cross-linked peptides. In addition, chemical removal of the biotin tag results in the formation of a functional group that generates unique reporter ions from Leiker cross-linked peptides during MS analysis, thus aiding in the confidence of their identification. As a result, 3,130 and 893 lysine linkages were identified at 5% FDR from E. coli and C. elegans, respectively124, indicating that cross-link enrichment through affinity purification is beneficial for large-scale XL-MS analysis.
5.2 In vivo proteome-wide studies
In comparison to in vitro studies, in vivo XL-MS experiments can better preserve native protein-protein interactions that are dynamic and highly dependent on their molecular and cellular environment. The Bruce lab and their collaborators have been instrumental in the application of XL-MS strategies for in-culture studies and have successfully employed their MS-cleavable, enrichable PIR cross-linkers for proteome-wide studies of various organisms including Shewanella oneidensis89, Escherichia coli104, Pseudomonas aeruginosa105, and Acinetobacter baumannii106 and human cells40,77. Recent analyses of murine mitochondria107 have resulted in the identification of 2,427 cross-linked peptide pairs from 327 mitochondrial proteins with FDR at 1.9%. In addition to PIR linkers, Azide-A-DSBSO79 has been applied to map PPIs from human cells through click chemistry-based conjugation and biotin-based enrichment, while a recent study employed BAMG for in vivo analysis of Bacillus subtilis119 using a combination of protein- and peptide-level separation. Collectively, these studies have demonstrated the effectiveness of combining cross-link enrichment and MS-cleavability for in vivo studies and have established a solid foundation for future endeavor towards full characterization of PPI network topologies in living systems.
6. Quantitative XL-MS strategies and their applications
Most cross-linking studies have been geared towards the elucidation of static protein structures. However, protein complexes are naturally dynamic, existing in a myriad of conformational and compositional states—each with their own unique properties, characteristics and functions. To dissect interaction and structural dynamics of protein complexes, quantitative XL-MS (QXL-MS) is needed to enable comparative analysis of various protein complex conformational states obtained at different physiological conditions. Changes in relative abundances of individual cross-links will allow us to assess condition-dependent structural changes of proteins and protein complexes.
6.1 Cross-linking reagent labeling
Similar to quantitative proteomics, stable isotopes (i.e. 2H, 13C, 15N, 18O) can be incorporated into cross-linked peptides for comparative analysis. The most common strategy to label cross-linked peptides for quantitation involves the use of isotope-coded cross-linkers (e.g. d0/d4-BS3 or d0/d12-DSS). Prior to their applications for QXL-MS, isotope-coded cross-linkers have long been used for facilitating the identification of non-cleavable cross-linked peptides based on their unique isotopic peptide patterns234. Their application for QXL-MS was first explored by the Rappsilber235 and Robinson groups using d0/d4-BS3 236,237. The general workflow comprises separate cross-linking of the samples to be compared, in which one reacts with the light-labeled reagent, and the other with the heavy-labeled reagent (Fig. 5A). Following equivalent mixing, the proteins are digested and submitted for MS analysis and database searching. The identified cross-linked peptides can be quantified at MS1 level by measuring the relative abundances of light- and heavy-labeled cross-linked peptide pairs. By comparing the cross-link profiles of untreated and dephosphorylated forms, Schmidt et al. were able to distinguish the effects of phosphorylation on spinach chloroplast ATPase topology, providing new insights on phosphorylation-mediated regulation mechanisms236,237. While early QXL-MS studies required manual quantitation and were thus limited in their application, significant process has been made in recent years to accommodate the analysis of QXL-MS datasets. Software such as xTract238 have been specifically developed for automatic quantitation of cross-linked peptides, while existing tools such as MaxQuant239, PinpointTM 240, pQuant124, and mMass241 have been adapted for analysis of QXL-MS data (Table 1). Automation of QXL-MS analysis has resulted in the ability to quickly analyze larger datasets, expanding the range of quantitative studies from single proteins240,241 to larger protein complexes such as TRiC238 and the 19S proteasome lid209. Recently, Tan et al. demonstrated the feasibility of extracting QXL-MS data from complex mixtures using d0/d6-Leiker cross-linkers and pQuant124.
Figure 5. Workflows for proteome-level in vitro and in vivo QXL-MS experiments using MS-cleavable cross-linkers and MSn-based acquisition.

Pair-wise QXL-MS can be carried out using (A) cross-linking labeling with isotope-coded reagents and (B) SILAC-based metabolic labeling. Cross-linked peptides are identified in MS3, while corresponding parent ions are quantified in MS1 based on their spectral abundance. (C) QMIX-based multiplexed QXL-MS strategy, in which each peptide digest is labeled with a distinct isobaric labeling reagent and then mixed equivalently prior to LC-MSn analysis. Cross-linked peptides are both identified and quantified in MS3, based on sequencing ions and quantitative reporter ions released during HCD fragmentation. Note: Reporter ions in MS3 can be significantly enhanced using synchronous precursor selection (SPS) acquisition to increase quantitation sensitivity and accuracy.
Although the majority of cross-link labeling-based QXL-MS studies have employed commercially available isotope-coded non-cleavable cross-linkers (i.e. d0/d4-BS3 or d0/d12-DSS), we have attempted to integrate quantitative ability with MS-cleavability to facilitate simplified, accurate cross-link identification and quantitation simultaneously. Therefore, we developed d0- and d10-labeled DMDSSO, a pair of isotope-coded, sulfoxide-containing MS-cleavable cross-linkers (Table 2)84. Similar to DSSO, d0/d10-DMDSSO cross-linked peptides are sequenced in MS3, but are quantified at the MS1 level (Fig. 5A). Using Skyline and later our in-house developed software xl-Tools, we characterized the neddylation-dependent conformational changes associated with SCF ubiquitin ligase activation and proposed mechanistic models for deactivation of SCFs by bacterial effector proteins through Nedd8 deamidation96. In addition, a set of isotope-labeled PIR cross-linking reagents (i,e. d0/d8-BDP-NHP) have been recently employed for quantitative analysis of in vivo cross-linked E. coli cells40, which resulted in the quantification of 941 out of 1213 identified peptide pairs, further demonstrating the feasibility of QXL-MS for large scale identification and quantitation.
6.2 Metabolic labeling
In addition to reagent labeling, incorporation of stable isotopes into cross-linked peptides can be accomplished through SILAC labeling242. Coincidentally, lysine is not only one of the most commonly used amino acids for SILAC labeling, but also the most popular targeted residue for cross-linking. Similar to isotope-coded reagent-based approaches, SILAC quantitation is also accomplished by comparing the relative abundances of light- and heavy-labeled cross-linked peptide pairs at MS1 level (Fig. 5B). In comparison, SILAC-based methods not only provide global labeling with minimal variance, but also eliminate the potential effects of isotope incorporation on cross-linking reactions and peptide separation. Isotope-coded cross-linkers are most often labeled with deuterium, which alters their chromatographic profiles compared to their non-labeled counterparts, complicating automated quantitation. In contrast, 13C/12C and 15N/14N labeled amino acids are typically used for SILAC labeling, allowing both labeled and unlabeled cross-links to co-elute chromatographically. Unfortunately, metabolic labeling also produces samples much more complex than cross-link labeling, due to global isotope incorporation (including non-cross-linked peptides). While it is possible to only label lysines to quantify cross-linked peptides, SILAC labeling with both lysine and arginine is required for cross-linkers targeting other residues such as aspartic/glutamic acids to ensure quantitation of all tryptic cross-linked peptides. Although SILAC-based methods are limited to cell culture systems in general, the idea of using labeled cell cultures as internal references to compare non-labeled samples in proteomic studies243 can potentially be borrowed and integrated with QXL-MS for extended applications. While SILAC-based methods have been widely used in quantitative proteomics for more than a decade242,244, they have only been integrated with XL-MS analysis recently40,108,139,150. SILAC-based QXL-MS approaches have been applied to quantify in vivo interactions and structural dynamics of Hsp90 and proteomes in response to drug treatments108,150. These applications have established a solid foundation for future QXL-MS experiments using metabolic labeling.
6.3 Isobaric labeling for multiplexed quantitation
Although both cross-link and SILAC labeling-based QXL-MS strategies have enabled successful quantitative studies, these workflows are typically restricted to binary (pair-wise) comparisons due to insufficient differentiating isotope labels, increased spectral complexity, and decreased detectability of low abundance cross-linked peptides. Inspired by the enormous success of multiplexed quantitation in proteomic studies using isobaric labeling reagents (e.g. iTRAQ245 and TMT reagents246,247), we have recently attempted to extend multiplexed capability to QXL-MS analysis94. Isobaric labeling-based quantitative methods permit the parallel analysis of up to 11 experiments with commercially available reagents (i.e. TMT 11-plex), significantly increasing throughput and quantitation accuracy without increasing sample complexity. In contrast to isotope-labeling based methods, quantitation is achieved through the detection of unique reporter ions resulting from the fragmentation of isobaric labels during HCD at the MS2 level. Because all peptides are modified by the same isobaric labels, peptide interference can occur during MS2-level quantitation, resulting in underestimation of quantitative changes among compared samples248. This distortion can be effectively eliminated by conducting quantitation in MS3 248, where the intensities of reporter ions can be significantly enhanced by synchronous precursor selection (SPS) acquisition to increase quantitation sensitivity249. In order to establish a robust workflow enabling comparative analysis of multiple cross-linked samples simultaneously, we have developed a multiplexed QXL-MS strategy, namely QMIX (Quantitation of Multiplexed, Isobaric-labeled cross (X)-linked peptides) by integrating MS-cleavable cross-linkers with isobaric labeling reagents (Fig. 5C)94. In a proof-of-concept study, we combined DSSO-based XL-MS with TMT reagents and showed that the identification and quantitation of isobaric-labeled, cross-linked peptides can be conducted in MS3 simultaneously94. The established QMIX strategy will facilitate high-throughput comparative analysis to determine interaction and structural dynamics of protein complexes and proteomes. It is noted that QMIX is a general strategy compatible with all cross-linking reagents regardless of their residue-targeting chemistries or chemical functionalities. Although isobaric labeling in theory can be applied to conventional non-cleavable cross-linkers, coupling this multiplexing strategy with MS-cleavable cross-linking reagents is optimal due to compatibility of MSn-based acquisitions and resulting simplification of cross-linked peptide identification94.
7. Future Perspectives and Conclusions
Five key steps are normally involved in XL-MS studies for interactomics and structural biology: 1) choice of cross-linkers; 2) sample preparation; 3) MS analysis; 4) data processing; 5) interaction mapping and structural characterization. The advancement of XL-MS analysis has relied on improvements and innovations in each aspect, which has made today’s XL-MS the method of choice for defining PPI landscapes at the systems level and elucidating architectures of large protein assemblies. With the advent of robust cross-linking reagents, ultra-high performance mass spectrometers, and powerful bioinformatics tools, XL-MS analysis has become much more accessible and doable to the research community than before. While the XL-MS field is entering the spotlight of proteomics research, new developments in all of the five key steps are still needed to further mature the field, and thus to make XL-MS experiments possible for routine analysis with sensitivity, accuracy, reliability, depth, speed and throughput. While conventional non-cleavable cross-linkers will continue to play a part in XL-MS studies with the aid of new database searching tools, we anticipate MS-cleavable cross-linkers will further thrive to advance simplified and robust XL-MS workflows for large-scale PPI profiling in vitro and in vivo. The commercial availability of MS-cleavable cross-linkers such as DSSO, DSBU and CDI will surely facilitate various applications of MS-cleavable cross-linkers in the near future. In addition, exploration in new cross-linking chemistries will continue to expand the XL-MS toolbox with additional capabilities to complement existing reagents. This is important as faster kinetics in cross-linking reactions will enable better capture of transient/dynamic interactions in cells and new residue-targeting chemistries will allow a broader coverage of interaction interfaces. While recent assessment of non-specific cross-linkers with single proteins has shown promises in obtaining a wide range of distance restraints137, the feasibility and potential of these reagents in XL-MS analysis of complex systems need to be carefully examined. Although peptide purification strategies based on enrichable cross-linkers and separation techniques have been developed, analytical improvements in sensitivity, specificity and selectivity are still required to further enhance MS detectability and identification of low abundance cross-linked peptides in complex mixtures. While biotin tag has been exclusively used for cross-linked peptide purification, a recent report130 suggests that other affinity tags (e.g. CH tag) may also be useful for specific XL-MS applications. This implies that strategies in protein affinity purification could be adopted for peptide enrichment. The accessibility of multiple peptide fragmentation techniques (e.g, CID, HCD, ETD, EThCD, UVPD) provides more flexibility in MS analysis, and has made it possible to re-examine existing cross-linkers (e.g. DSSO) and thus establish new complementary workflows (Figs. 2A and 2B) for obtaining more comprehensive cross-link data. Combination of different fragmentation techniques would not only maximize the potential of known cross-linkers, but also provide guidance on future designs of new cross-linking reagents.
Apart from static structural analysis, QXL-MS represents the next generation of XL-MS to enable the characterization of dynamic proteomes under different physiological conditions in different organisms. It has been demonstrated that methods in quantitative proteomics can be directly transferred to QXL-MS analysis, thus greatly facilitating its development and applications. In addition to pair-wise quantitation, multiplexed QXL-MS platforms have been recently established based on isobaric labeling94. Moreover, the feasibility of PRM (parallel reaction monitoring)-based targeted quantification has also been illustrated for XL-MS studies40,139. Thus, we envision that the combination of experimental developments with powerful multiplexed relative and targeted quantitation will allow quantitative assessments of PPI dynamics at the protein complex and the proteome-wide scale with speed and accuracy. However, challenges do exist regarding to automated data processing to allow fast and accurate identification and quantitation simultaneously with multi-layer data acquired at different MS stages. In addition, how to correctly interpret relative abundance changes in cross-links to infer protein interaction and conformational changes needs carefully inspected, as residue cross-linkability is dependent on many factors including their proximity, chemical environments and solvent accessibility. Nonetheless, this process can be facilitated with structural modeling tools to better dissect the nature of quantitative changes. On the other hand, the measured quantitative changes of cross-links can serve as another type of constraints to refine structural models derived with distance restraints. Clearly, continuous evolvement of XL-MS software is highly anticipated to expedite cross-link identification, quantification, and validation as well as visualization, data interpretation and modeling.
While major efforts are taken at in vitro studies, in vivo applications are the ultimate goal for XL-MS technologies. Abundant evidence has shown that aberrant protein-protein interactions can lead to human disease and cancer, and protein interaction interfaces describe a new class of attractive targets for drug development250–253. Therefore, the ability to obtain authentic PPI network maps, and define their temporal and spatial dynamics from living systems will not only uncover molecular details underlying the function and regulation of macromolecular machines, but also provide a unique means to assist the development of interaction-dependent therapeutics. With continuous breakthrough in the field, we envision XL-MS will make more significant contributions to structural biology, but more importantly, to advanced biomedical research.
Acknowledgments
This work was supported by National Institutes of Health grants R01GM074830 to L.H., R01GM106003 to L.H. and S. R.
ABBREVIATIONS
- 19S RP
19S regulatory particle
- 20S CP
20S core particle
- CID
Collision-induced dissociation
- CDI
1,1′-carbonyldiimidazole
- Cryo-EM
Cryogenic electron microscopy
- DHSO
Dihydrazide sulfoxide
- DSSO
Disuccinimidyl sulfoxide
- DSBSO
Disuccinimidyl bissulfoxide
- DSS
Disuccinimidyl suberate
- EDC
(1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride)
- ETD
Electron-transfer dissociation
- FA
Formaldehyde
- FDR
False discovery rate
- HB
Histidine-biotin (tag)
- HCD
Higher-energy collisional dissociation
- HDX
Hydrogen-deuterium exchange
- LC-MS
Liquid chromatography-mass spectrometry
- MS
Mass spectrometry
- MS/MS or MS2
Tandem mass spectrometry
- MS3
3rd stage mass spectrometry
- MSn
Multi-stage mass spectrometry
- NHS
N-hydroxysuccinimidyl
- NPC
Nuclear pore complex
- NMR
Nuclear magnetic resonance
- PIR
Protein-interaction reporter
- PPI
Protein-protein interaction
- QMIX
Quantitation of multiplexed, isobaric-labeled cross (x)-linked peptides
- QXL-MS
Quantitative cross (x)-linking mass spectrometry
- SCX
Strong-cation exchange
- SEC
Size exclusion chromatography
- SILAC
Stable isotope-labeling with amino acids in cell culture
- SPS
Synchronous precursor selection
- TMT
Tandem mass tag
- XAP
in vivo cross-linking (x) assisted affinity purification
- XBAP
in vivo cross-linking (x) assisted bimolecular tandem affinity purification
- XL-MS
Cross (x)-linking mass spectrometry
Biographies
Clinton Yu is a research scientist in the Department of Physiology & Biophysics, School of Medicine, University of California, Irvine (UCI). He received his B.S. with a double major in biology and Chemistry in 2011, and Ph.D. in biological mass spectrometry/proteomics in 2017 from UCI with Professor Lan Huang. During his graduate studies, he has developed multiple new cross-linking mass spectrometry methodologies for mapping protein-protein interactions and quantifying structural dynamics of protein complexes. His work has contributed to the understanding of the structure and regulation of several protein complexes (e.g. proteasomes cullin-Ring E3 ligases) in the ubiquitin-proteasome pathway and other biological systems. His current research continues to focus on the development and application of XL-MS strategies for the study of protein interactomes and structural biology.
Lan Huang is Professor of Physiology & Biophysics in School of Medicine, University of California, Irvine. Her research focuses on developing novel, integrated mass spectrometry-based proteomic strategies to characterize dynamic proteomes of macromolecular protein complexes and understand their functions, particularly those in the ubiquitin-proteasome system. Through the years, the Huang lab has developed various novel methodologies to capture, purify and quantify protein-protein interactions of protein complexes in living cells. In addition, her lab has developed a new class of cross-linking reagents, i.e. sulfoxide-containing MS-cleavable cross-linkers (e.g. DSSO), and thus established a robust cross-linking mass spectrometry (XL-MS) platform enabling fast and accurate identification of cross-linked peptides. These XL-MS strategies have been successfully employed to determine protein interaction interfaces and derive structural topologies of protein complexes in vitro and in vivo. Moreover, her lab has developed several quantitative cross-linking mass spectrometry strategies for comparative analysis of conformational changes in protein complexes. The strategies developed by her group have proven highly effective as general proteomic tools for studying protein complexes.
Footnotes
Competing financial interests: The authors declare no competing financial interests
References
- 1.Mendoza VL, Vachet RW. Mass Spectrom Rev. 2009;28:785–815. doi: 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kiselar JG, Chance MR. J Mass Spectrom. 2010;45:1373–1382. doi: 10.1002/jms.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konermann L, Pan J, Liu YH. Chemical Society reviews. 2011;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- 4.Sinz A. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- 5.Petrotchenko E, Borchers C. BMC bioinformatics. 2010;11:64. doi: 10.1186/1471-2105-11-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leitner A, Walzthoeni T, Kahraman A, Herzog F, Rinner O, Beck M, Aebersold R. Mol Cell Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rappsilber J. Journal of structural biology. 2011;173:530–540. doi: 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJ. Chemical Society reviews. 2010;39:1633–1655. doi: 10.1039/b914002f. [DOI] [PubMed] [Google Scholar]
- 9.Lorenzen K, van Duijn E. Current protocols in protein science. 2010;(Unit17):12. doi: 10.1002/0471140864.ps1712s62. Chapter 17. [DOI] [PubMed] [Google Scholar]
- 10.Rajabi K, Ashcroft AE, Radford SE. Methods. 2015;89:13–21. doi: 10.1016/j.ymeth.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Politis A, Borysik AJ. Proteomics. 2015;15:2792–2803. doi: 10.1002/pmic.201400507. [DOI] [PubMed] [Google Scholar]
- 12.Liu F, Heck AJ. Current opinion in structural biology. 2015;35:100–108. doi: 10.1016/j.sbi.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Sinz A, Arlt C, Chorev D, Sharon M. Protein Sci. 2015;24:1193–1209. doi: 10.1002/pro.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dailing A, Luchini A, Liotta L. Expert Rev Proteomics. 2015;12:457–467. doi: 10.1586/14789450.2015.1079487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holding AN. Methods. 2015;89:54–63. doi: 10.1016/j.ymeth.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 16.Srinivasa S, Ding X, Kast J. Methods. 2015;89:91–98. doi: 10.1016/j.ymeth.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 17.Rivera-Santiago RF, Sriswasdi S, Harper SL, Speicher DW. Methods. 2015;89:99–111. doi: 10.1016/j.ymeth.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piotrowski C, Ihling CH, Sinz A. Methods. 2015;89:121–127. doi: 10.1016/j.ymeth.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 19.Hurt E, Beck M. Curr Opin Cell Biol. 2015;34:31–38. doi: 10.1016/j.ceb.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Faini M, Stengel F, Aebersold R. J Am Soc Mass Spectrom. 2016;27:966–974. doi: 10.1007/s13361-016-1382-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chait BT, Cadene M, Olinares PD, Rout MP, Shi Y. J Am Soc Mass Spectrom. 2016;27:952–965. doi: 10.1007/s13361-016-1385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen F, Gulbakan B, Weidmann S, Fagerer SR, Ibanez AJ, Zenobi R. Mass Spectrom Rev. 2016;35:48–70. doi: 10.1002/mas.21462. [DOI] [PubMed] [Google Scholar]
- 23.Leitner A, Faini M, Stengel F, Aebersold R. Trends Biochem Sci. 2016;41:20–32. doi: 10.1016/j.tibs.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 24.Lossl P, van de Waterbeemd M, Heck AJ. Embo j. 2016;35:2634–2657. doi: 10.15252/embj.201694818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran BQ, Goodlett DR, Goo YA. Biochim Biophys Acta. 2016;1864:123–129. doi: 10.1016/j.bbapap.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 26.Maes E, Dyer JM, McKerchar HJ, Deb-Choudhury S, Clerens S. Chemistry (Weinheim an der Bergstrasse, Germany) 2017:1–13. [Google Scholar]
- 27.Barysz HM, Malmstroem J. Molecular & cellular proteomics. 2017 doi: 10.1074/mcp.R116.061663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lipstein N, Goth M, Piotrowski C, Pagel K, Sinz A, Jahn O. Anal Chem. 2017;14:223–242. doi: 10.1080/14789450.2017.1275966. [DOI] [PubMed] [Google Scholar]
- 29.Sinz A. Anal Bioanal Chem. 2010;397:3433–3440. doi: 10.1007/s00216-009-3405-5. [DOI] [PubMed] [Google Scholar]
- 30.Bruce JE. Proteomics. 2012;12:1565–1575. doi: 10.1002/pmic.201100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Rout MP, Sali A. Nature. 2007;450:683–694. doi: 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- 32.Chen ZA, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowski-Wills JC, Nilges M, Cramer P, Rappsilber J. Embo J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W. Proc Natl Acad Sci U S A. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Senko MW, Remes PM, Canterbury JD, Mathur R, Song Q, Eliuk SM, Mullen C, Earley L, Hardman M, Blethrow JD, Bui H, Specht A, Lange O, Denisov E, Makarov A, Horning S, Zabrouskov V. Anal Chem. 2013;85:11710–11714. doi: 10.1021/ac403115c. [DOI] [PubMed] [Google Scholar]
- 35.Erzberger JP, Stengel F, Pellarin R, Zhang S, Schaefer T, Aylett CH, Cimermancic P, Boehringer D, Sali A, Aebersold R, Ban N. Cell. 2014;158:1123–1135. doi: 10.1016/j.cell.2014.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi Y, Fernandez-Martinez J, Tjioe E, Pellarin R, Kim SJ, Williams R, Schneidman D, Sali A, Rout MP, Chait BT. Mol Cell Proteomics. 2014;13:2927–2943. doi: 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. Proc Natl Acad Sci U S A. 2000;97:5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herzog F, Kahraman A, Boehringer D, Mak R, Bracher A, Walzthoeni T, Leitner A, Beck M, Hartl FU, Ban N, Malmstrom L, Aebersold R. Science (New York, NY) 2012;337:1348–1352. doi: 10.1126/science.1221483. [DOI] [PubMed] [Google Scholar]
- 39.Sinz A. Anal Bioanal Chem. 2017;409:33–44. doi: 10.1007/s00216-016-9941-x. [DOI] [PubMed] [Google Scholar]
- 40.Zhong X, Navare AT, Chavez JD, Eng JK, Schweppe DK, Bruce JE. J Proteome Res. 2017;16:720–727. doi: 10.1021/acs.jproteome.6b00752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weisz DA, Gross ML, Pakrasi HB. Mol Cell Proteomics. 2016;7:617. [Google Scholar]
- 42.Kar UK, Simonian M, Whitelegge JP. Expert Rev Proteomics. 2017;14:715–723. doi: 10.1080/14789450.2017.1359545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davies GE, Stark GR. Proc Natl Acad Sci U S A. 1970;66:651–656. doi: 10.1073/pnas.66.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang B, Wu YJ, Zhu M, Fan SB, Lin J, Zhang K, Li S, Chi H, Li YX, Chen HF, Luo SK, Ding YH, Wang LH, Hao Z, Xiu LY, Chen S, Ye K, He SM, Dong MQ. Nat Methods. 2012;9:904–906. doi: 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]
- 45.Petrotchenko EV, Borchers CH. Proteomics. 2014;14:1987–1989. doi: 10.1002/pmic.201300486. [DOI] [PubMed] [Google Scholar]
- 46.Rinner O, Seebacher J, Walzthoeni T, Mueller LN, Beck M, Schmidt A, Mueller M, Aebersold R. Nat Methods. 2008;5:315–318. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walzthoeni T, Claassen M, Leitner A, Herzog F, Bohn S, Forster F, Beck M, Aebersold R. Nat Methods. 2012;9:901–903. doi: 10.1038/nmeth.2103. [DOI] [PubMed] [Google Scholar]
- 48.Gotze M, Pettelkau J, Schaks S, Bosse K, Ihling CH, Krauth F, Fritzsche R, Kuhn U, Sinz A. J Am Soc Mass Spectrom. 2012;23:76–87. doi: 10.1007/s13361-011-0261-2. [DOI] [PubMed] [Google Scholar]
- 49.Chu F, Baker PR, Burlingame AL, Chalkley RJ. Mol Cell Proteomics. 2009 doi: 10.1074/mcp.M800555-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trnka MJ, Baker PR, Robinson PJ, Burlingame AL, Chalkley RJ. Mol Cell Proteomics. 2014;13:420–434. doi: 10.1074/mcp.M113.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lima DB, de Lima TB, Balbuena TS, Neves-Ferreira AGC, Barbosa VC, Gozzo FC, Carvalho PC. Journal of proteomics. 2015;129:51–55. doi: 10.1016/j.jprot.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 52.Hoopmann MR, Zelter A, Johnson RS, Riffle M, MacCoss MJ, Davis TN, Moritz RL. J Proteome Res. 2015;14:2190–2198. doi: 10.1021/pr501321h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iglesias AH, Santos LF, Gozzo FC. Anal Chem. 2010;82:909–916. doi: 10.1021/ac902051q. [DOI] [PubMed] [Google Scholar]
- 54.Yilmaz S, Drepper F, Hulstaert N, Cernic M, Gevaert K, Economou A, Warscheid B, Martens L, Vandermarliere E. Anal Chem. 2016;88:9949–9957. doi: 10.1021/acs.analchem.6b01585. [DOI] [PubMed] [Google Scholar]
- 55.Ji C, Li S, Reilly JP, Radivojac P, Tang H. J Proteome Res. 2016;15:1830–1841. doi: 10.1021/acs.jproteome.6b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu F, Li N, Yu W. BMC Bioinformatics. 2016;17:217. doi: 10.1186/s12859-016-1073-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu F, Li N, Yu W. J Proteome Res. 2017;16:3942–3952. doi: 10.1021/acs.jproteome.7b00338. [DOI] [PubMed] [Google Scholar]
- 58.Grimm M, Zimniak T, Kahraman A, Herzog F. Nucleic Acids Res. 2015;43:W362–369. doi: 10.1093/nar/gkv463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Combe CW, Fischer L, Rappsilber J. Mol Cell Proteomics. 2015;14:1137–1147. doi: 10.1074/mcp.O114.042259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riffle M, Jaschob D, Zelter A, Davis TN. Mol Cell Proteomics. 2016;15:2863–2870. doi: 10.1021/acs.jproteome.6b00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Courcelles M, Coulombe-Huntington J, Cossette E, Gingras AC, Thibault P, Tyers M. J Proteome Res. 2017;16:2645–2652. doi: 10.1021/acs.jproteome.7b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schweppe DK, Zheng C, Chavez JD, Navare AT, Wu X, Eng JK, Bruce JE. Bioinformatics. 2016;32:2716–2718. doi: 10.1093/bioinformatics/btw232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kahraman A, Malmstrom L, Aebersold R. Bioinformatics. 2011;27:2163–2164. doi: 10.1093/bioinformatics/btr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matthew Allen Bullock J, Schwab J, Thalassinos K, Topf M. Mol Cell Proteomics. 2016;15:2491–2500. doi: 10.1074/mcp.M116.058560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schweppe DK, Chavez JD, Bruce JE. Bioinformatics. 2016;32:306–308. doi: 10.1093/bioinformatics/btv519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang L, Tang X, Weisbrod C, Munske G, Eng J, von Haller P, Kaiser N, Bruce J. Anal Chem. 2010:663–682. doi: 10.1021/ac902615g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bennett KL, Kussmann M, Björk P, Godzwon M, Mikkelsen M, Sørensen P, Roepstorff P. Protein Sci. 2000;9:1503–1518. doi: 10.1110/ps.9.8.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang X, Munske GR, Siems WF, Bruce JE. Anal Chem. 2005;77:311–318. doi: 10.1021/ac0488762. [DOI] [PubMed] [Google Scholar]
- 69.Soderblom EJ, Bobay BG, Cavanagh J, Goshe MB. Rapid Commun Mass Spectrom. 2007;21:3395–3408. doi: 10.1002/rcm.3213. [DOI] [PubMed] [Google Scholar]
- 70.Lu Y, Tanasova M, Borhan B, Reid GE. Anal Chem. 2008;80:9279–9287. doi: 10.1021/ac801625e. [DOI] [PubMed] [Google Scholar]
- 71.Gardner MW, Brodbelt JS. Anal Chem. 2010;82:5751–5759. doi: 10.1021/ac100788a. [DOI] [PubMed] [Google Scholar]
- 72.Trnka MJ, Burlingame AL. Mol Cell Proteomics. 2010;9:2306–2317. doi: 10.1074/mcp.M110.003764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Muller MQ, Dreiocker F, Ihling CH, Schafer M, Sinz A. Anal Chem. 2010;82:6958–6968. doi: 10.1021/ac101241t. [DOI] [PubMed] [Google Scholar]
- 74.Kao A, Chiu CL, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, Huang L. Mol Cell Proteomics. 2011;10:M110.002212. doi: 10.1074/mcp.M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petrotchenko EV, Serpa JJ, Borchers CH. Mol Cell Proteomics. 2011;10:M110.001420. doi: 10.1074/mcp.M110.001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luo J, Fishburn J, Hahn S, Ranish J. Mol Cell Proteomics. 2012;11:M111 008318. doi: 10.1074/mcp.M111.008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chavez JD, Weisbrod CR, Zheng C, Eng JK, Bruce JE. Mol Cell Proteomics. 2013;12:1451–1467. doi: 10.1074/mcp.M112.024497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Falvo FFL, Schafer M. International Journal of Mass Spectrometry. 2013;354–355:26–32. [Google Scholar]
- 79.Kaake RM, Wang X, Burke A, Yu C, Kandur W, Yang Y, Novtisky EJ, Second T, Duan J, Kao A, Guan S, Vellucci D, Rychnovsky SD, Huang L. Mol Cell Proteomics. 2014 doi: 10.1074/mcp.M114.042630. pii: mcp.M114.042630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ihling C, Falvo F, Kratochvil I, Sinz A, Schafer M. J Mass Spectrom. 2015;50:396–406. doi: 10.1002/jms.3543. [DOI] [PubMed] [Google Scholar]
- 81.Gardner MW, Vasicek LA, Shabbir S, Anslyn EV, Brodbelt JS. Anal Chem. 2008;80:4807–4819. doi: 10.1021/ac800625x. [DOI] [PubMed] [Google Scholar]
- 82.Gardner MW, Brodbelt JS. Anal Chem. 2009;81:4864–4872. doi: 10.1021/ac9005233. [DOI] [PubMed] [Google Scholar]
- 83.Petrotchenko EV, Xiao K, Cable J, Chen Y, Dokholyan NV, Borchers CH. Mol Cell Proteomics. 2009;8:273–286. doi: 10.1074/mcp.M800265-MCP200. [DOI] [PubMed] [Google Scholar]
- 84.Yu C, Kandur W, Kao A, Rychnovsky S, Huang L. Anal Chem. 2014;86:2099–2106. doi: 10.1021/ac403636b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gutierrez CB, Yu C, Novitsky EJ, Huszagh AS, Rychnovsky SD, Huang L. Anal Chem. 2016;88:8315–8322. doi: 10.1021/acs.analchem.6b02240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soderblom EJ, Goshe MB. Anal Chem. 2006;78:8059–8068. doi: 10.1021/ac0613840. [DOI] [PubMed] [Google Scholar]
- 87.Zhang H, Tang X, Munske GR, Zakharova N, Yang L, Zheng C, Wolff MA, Tolic N, Anderson GA, Shi L, Marshall MJ, Fredrickson JK, Bruce JE. J Proteome Res. 2008;7:1712–1720. doi: 10.1021/pr7007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chakrabarty JK, Naik AG, Fessler MB, Munske GR, Chowdhury SM. Anal Chem. 2016;88:10215–10222. doi: 10.1021/acs.analchem.6b02886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang H, Tang X, Munske GR, Tolic N, Anderson GA, Bruce JE. Mol Cell Proteomics. 2009;8:409–420. doi: 10.1074/mcp.M800232-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hage C, Falvo F, Schafer M, Sinz A. J Am Soc Mass Spectrom. 2017 doi: 10.1007/s13361-13017-11712-13361. [DOI] [PubMed] [Google Scholar]
- 91.Hage C, Iacobucci C, Rehkamp A, Arlt C, Sinz A. Angew Chem Int Ed Engl. 2017 doi: 10.1002/anie.201708273. [DOI] [PubMed] [Google Scholar]
- 92.Clifford-Nunn B, Showalter HD, Andrews PC. J Am Soc Mass Spectrom. 2012;23:201–212. doi: 10.1007/s13361-011-0288-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Burke AM, Kandur W, Novitsky EJ, Kaake RM, Yu C, Kao A, Vellucci D, Huang L, Rychnovsky SD. Organic & biomolecular chemistry. 2015;13:5030–5037. doi: 10.1039/c5ob00488h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu C, Huszagh A, Viner R, Novitsky EJ, Rychnovsky SD, Huang L. Anal Chem. 2016;88:10301–10308. doi: 10.1021/acs.analchem.6b03148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kao A, Randall A, Yang Y, Patel VR, Kandur W, Guan S, Rychnovsky SD, Baldi P, Huang L. Mol Cell Proteomics. 2012;11:1566–1577. doi: 10.1074/mcp.M112.018374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu C, Mao H, Novitsky EJ, Tang X, Rychnovsky SD, Zheng N, Huang L. Nature communications. 2015;6:10053. doi: 10.1038/ncomms10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu J, Yu C, Hu X, Kim JK, Bierma JC, Jun HI, Rychnovsky SD, Huang L, Qiao F. Cell reports. 2015;12:2169–2180. doi: 10.1016/j.celrep.2015.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang X, Chemmama IE, Yu C, Huszagh A, Xu Y, Viner R, Block SA, Cimermancic P, Rychnovsky SD, Ye Y, Sali A, Huang L. J Biol Chem. 2017;292:16310–16320. doi: 10.1074/jbc.M117.803619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang X, Cimermancic P, Yu C, Schweitzer A, Chopra N, Engel JL, Greenberg C, Huszagh AS, Beck F, Sakata E, Yang Y, Novitsky EJ, Leitner A, Nanni P, Kahraman A, Guo X, Dixon JE, Rychnovsky SD, Aebersold R, Baumeister W, et al. Mol Cell Proteomics. 2017;16:840–854. doi: 10.1074/mcp.M116.065326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Scott H, Kim JK, Yu C, Huang L, Qiao F, Taylor DJ. J Mol Biol. 2017;429:2863–2872. doi: 10.1016/j.jmb.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu F, Rijkers DT, Post H, Heck AJ. Nat Methods. 2015;12:1179–1184. doi: 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- 102.Liu F, Lossl P, Scheltema R, Viner R, Heck AJR. Nature communications. 2017;8:15473. doi: 10.1038/ncomms15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weisbrod CR, Chavez JD, Eng JK, Yang L, Zheng C, Bruce JE. J Proteome Res. 2013;12:1569–1579. doi: 10.1021/pr3011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zheng C, Yang L, Hoopmann MR, Eng JK, Tang X, Weisbrod CR, Bruce JE. Mol Cell Proteomics. 2011;10:M110 006841. doi: 10.1074/mcp.M110.006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Navare AT, Chavez JD, Zheng C, Weisbrod CR, Eng JK, Siehnel R, Singh PK, Manoil C, Bruce JE. Structure. 2015;23:762–773. doi: 10.1016/j.str.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wu X, Chavez JD, Schweppe DK, Zheng C, Weisbrod CR, Eng JK, Murali A, Lee SA, Ramage E, Gallagher LA, Kulasekara HD, Edrozo ME, Kamischke CN, Brittnacher MJ, Miller SI, Singh PK, Manoil C, Bruce JE. Nature communications. 2016;7:13414. doi: 10.1038/ncomms13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schweppe DK, Chavez JD, Lee CF, Caudal A, Kruse SE, Stuppard R, Marcinek DJ, Shadel GS, Tian R, Bruce JE. Proc Natl Acad Sci U S A. 2017;114:1732–1737. doi: 10.1073/pnas.1617220114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chavez JD, Schweppe DK, Eng JK, Bruce JE. Cell chemical biology. 2016;23:716–726. doi: 10.1016/j.chembiol.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Iacobucci C, Sinz A. Expert Rev Proteomics. 2017;89:7832–7835. [Google Scholar]
- 110.Arlt C, Gotze M, Ihling CH, Hage C, Schafer M, Sinz A. Clinical epigenetics. 2016;88:7930–7937. doi: 10.1021/acs.analchem.5b04853. [DOI] [PubMed] [Google Scholar]
- 111.Gotze M, Pettelkau J, Fritzsche R, Ihling CH, Schafer M, Sinz A. J Am Soc Mass Spectrom. 2015;26:83–97. doi: 10.1007/s13361-014-1001-1. [DOI] [PubMed] [Google Scholar]
- 112.Iacobucci C, Hage C, Schafer M, Sinz A. J Am Soc Mass Spectrom. 2017 doi: 10.1007/s13361-13017-11744-13366. [DOI] [PubMed] [Google Scholar]
- 113.Riley NM, Westphall MS, Coon JJ. J Am Soc Mass Spectrom. 2017 doi: 10.1007/s13361-017-1808-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schilling B, Row RH, Gibson BW, Guo X, Young MM. J Am Soc Mass Spectrom. 2003;14:834–850. doi: 10.1016/S1044-0305(03)00327-1. [DOI] [PubMed] [Google Scholar]
- 115.Leitner A, Reischl R, Walzthoeni T, Herzog F, Bohn S, Forster F, Aebersold R. Mol Cell Proteomics. 2012;11:M111 014126. doi: 10.1074/mcp.M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leitner A, Walzthoeni T, Aebersold R. Nat Protoc. 2014;9:120–137. doi: 10.1038/nprot.2013.168. [DOI] [PubMed] [Google Scholar]
- 117.Schmidt R, Sinz A. Anal Bioanal Chem. 2017;409:2393–2400. doi: 10.1007/s00216-017-0185-1. [DOI] [PubMed] [Google Scholar]
- 118.Tinnefeld V, Venne AS, Sickmann A, Zahedi RP. J Proteome Res. 2017;16:459–469. doi: 10.1021/acs.jproteome.6b00587. [DOI] [PubMed] [Google Scholar]
- 119.de Jong L, de Koning EA, Roseboom W, Buncherd H, Wanner MJ, Dapic I, Jansen PJ, van Maarseveen JH, Corthals GL, Lewis PJ, Hamoen LW, de Koster CG. J Proteome Res. 2017;16:2457–2471. doi: 10.1021/acs.jproteome.7b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Trester-Zedlitz M, Kamada K, Burley SK, Fenyo D, Chait BT, Muir TW. J Am Chem Soc. 2003;125:2416–2425. doi: 10.1021/ja026917a. [DOI] [PubMed] [Google Scholar]
- 121.Fujii K, Nakano T, Hike H, Usui F, Bando Y, Tojo H, Nishimura T. J Chromatogr A. 2004;1057:107–113. doi: 10.1016/j.chroma.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 122.Chu F, Mahrus S, Craik CS, Burlingame AL. J Am Chem Soc. 2006;128:10362–10363. doi: 10.1021/ja0614159. [DOI] [PubMed] [Google Scholar]
- 123.Tang X, Bruce JE. Mol Biosyst. 2010;6:939–947. doi: 10.1039/b920876c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tan D, Li Q, Zhang MJ, Liu C, Ma C, Zhang P, Ding YH, Fan SB, Tao L, Yang B, Li X, Ma S, Liu J, Feng B, Liu X, Wang HW, He SM, Gao N, Ye K, Dong MQ, et al. eLife. 2016:5. doi: 10.7554/eLife.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kasper PT, Back JW, Vitale M, Hartog AF, Roseboom W, de Koning LJ, van Maarseveen JH, Muijsers AO, de Koster CG, de Jong L. Chembiochem. 2007;8:1281–1292. doi: 10.1002/cbic.200700150. [DOI] [PubMed] [Google Scholar]
- 126.Chowdhury SM, Du X, Tolić N, Wu S, Moore RJ, Mayer MU, Smith RD, Adkins JN. Anal Chem. 2009;81:5524–5532. doi: 10.1021/ac900853k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nessen MA, Kramer G, Back J, Baskin JM, Smeenk LE, de Koning LJ, van Maarseveen JH, de Jong L, Bertozzi CR, Hiemstra H, de Koster CG. J Proteome Res. 2009;8:3702–3711. doi: 10.1021/pr900257z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vellucci D, Kao A, Kaake RM, Rychnovsky SD, Huang L. J Am Soc Mass Spectrom. 2010;21:1432–1445. doi: 10.1016/j.jasms.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Buncherd H, Nessen MA, Nouse N, Stelder SK, Roseboom W, Dekker HL, Arents JC, Smeenk LE, Wanner MJ, van Maarseveen JH, Yang X, Lewis PJ, de Koning LJ, de Koster CG, de Jong L. Journal of proteomics. 2012;75:2205–2215. doi: 10.1016/j.jprot.2012.01.025. [DOI] [PubMed] [Google Scholar]
- 130.Trahan C, Oeffinger M. Nucleic Acids Res. 2016;44:1354–1369. doi: 10.1093/nar/gkv1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Novak P, Kruppa GH. Eur J Mass Spectrom (Chichester, Eng) 2008;14:355–365. doi: 10.1255/ejms.963. [DOI] [PubMed] [Google Scholar]
- 132.Leitner A, Joachimiak LA, Unverdorben P, Walzthoeni T, Frydman J, Forster F, Aebersold R. Proc Natl Acad Sci U S A. 2014;111:9455–9460. doi: 10.1073/pnas.1320298111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Suchanek M, Radzikowska A, Thiele C. Nat Methods. 2005;2:261–267. doi: 10.1038/nmeth752. [DOI] [PubMed] [Google Scholar]
- 134.Schwarz R, Tanzler D, Ihling CH, Muller MQ, Kolbel K, Sinz A. J Med Chem. 2013;56:4252–4263. doi: 10.1021/jm400446b. [DOI] [PubMed] [Google Scholar]
- 135.Jecmen T, Ptackova R, Cerna V, Dracinska H, Hodek P, Stiborova M, Hudecek J, Sulc M. Methods. 2015;89:128–137. doi: 10.1016/j.ymeth.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 136.Brodie NI, Makepeace KA, Petrotchenko EV, Borchers CH. Journal of proteomics. 2015;118:12–20. doi: 10.1016/j.jprot.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 137.Belsom A, Mudd G, Giese S. 2017;89:5319–5324. doi: 10.1021/acs.analchem.6b04938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shi Y, Pellarin R, Fridy PC, Fernandez-Martinez J, Thompson MK, Li Y, Wang QJ, Sali A, Rout MP, Chait BT. Nat Methods. 2015;12:1135–1138. doi: 10.1038/nmeth.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chavez JD, Eng JK, Schweppe DK, Cilia M, Rivera K, Zhong X, Wu X, Allen T, Khurgel M, Kumar A, Lampropoulos A, Larsson M, Maity S, Morozov Y, Pathmasiri W, Perez-Neut M, Pineyro-Ruiz C, Polina E, Post S, Rider M, et al. PLoS One. 2016;11:e0167547. doi: 10.1371/journal.pone.0167547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Liu Q, Remmelzwaal S, Heck AJR, Akhmanova A, Liu F. Scientific reports. 2017;7:13453. doi: 10.1038/s41598-017-13663-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rappsilber J, Siniossoglou S, Hurt EC, Mann M. Anal Chem. 2000;72:267–275. doi: 10.1021/ac991081o. [DOI] [PubMed] [Google Scholar]
- 142.Hofmann T, Fischer AW, Meiler J, Kalkhof S. Methods. 2015;89:79–90. doi: 10.1016/j.ymeth.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rampler E, Stranzl T, Orban-Nemeth Z, Hollenstein DM, Hudecz O, Schloegelhofer P, Mechtler K. J Proteome Res. 2015;14:5048–5062. doi: 10.1021/acs.jproteome.5b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Arlt C, Flegler V, Ihling CH, Schafer M, Thondorf I, Sinz A. Angew Chem Int Ed Engl. 2017;56:275–279. doi: 10.1002/anie.201609826. [DOI] [PubMed] [Google Scholar]
- 145.Plimpton RL, Cuellar J, Lai CW, Aoba T, Makaju A, Franklin S, Mathis AD, Prince JT, Carrascosa JL, Valpuesta JM, Willardson BM. Proc Natl Acad Sci U S A. 2015;112:2413–2418. doi: 10.1073/pnas.1419595112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Glas M, van den Berg van Saparoea HB, McLaughlin SH, Roseboom W, Liu F, Koningstein GM, Fish A, den Blaauwen T, Heck AJ, de Jong L, Bitter W, de Esch IJ, Luirink J. The Journal of biological chemistry. 2015;290:21498–21509. doi: 10.1074/jbc.M115.654756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Belsom A, Schneider M, Fischer L, Brock O, Rappsilber J. Mol Cell Proteomics. 2016;15:1105–1116. doi: 10.1074/mcp.M115.048504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ding YH, Fan SB, Li S, Feng BY, Gao N, Ye K, He SM, Dong MQ. Anal Chem. 2016;88:4461–4469. doi: 10.1021/acs.analchem.6b00281. [DOI] [PubMed] [Google Scholar]
- 149.Zeng-Elmore X, Gao XZ, Pellarin R, Schneidman-Duhovny D, Zhang XJ, Kozacka KA, Tang Y, Sali A, Chalkley RJ, Cote RH, Chu F. J Mol Biol. 2014;426:3713–3728. doi: 10.1016/j.jmb.2014.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chavez JD, Schweppe DK, Eng JK, Zheng C, Taipale A, Zhang Y, Takara K, Bruce JE. Nature communications. 2015;6:7928. doi: 10.1038/ncomms8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wu X, Held K, Zheng C, Staudinger BJ, Chavez JD, Weisbrod CR, Eng JK, Singh PK, Manoil C, Bruce JE. Mol Cell Proteomics. 2015;14:2126–2137. doi: 10.1074/mcp.M115.050161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang X, Chen CF, Baker PR, Chen PL, Kaiser P, Huang L. Biochemistry. 2007;46:3553–3565. doi: 10.1021/bi061994u. [DOI] [PubMed] [Google Scholar]
- 153.Yu C, Yang Y, Wang X, Guan S, Fang L, Liu F, Walters KJ, Kaiser P, Huang L. Mol Cell Proteomics. 2016;15:2279–2292. doi: 10.1074/mcp.M116.058271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Makowski MM, Willems E, Jansen PW, Vermeulen M. Nature communications. 2016;15:854–865. doi: 10.1074/mcp.M115.053082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Robinson PJ, Trnka MJ, Bushnell DA, Davis RE, Mattei PJ, Burlingame AL, Kornberg RD. Protein Sci. 2016;166:1411–1422. e1416. doi: 10.1016/j.cell.2016.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bich C, Maedler S, Chiesa K, DeGiacomo F, Bogliotti N, Zenobi R. Anal Chem. 2010;82:172–179. doi: 10.1021/ac901651r. [DOI] [PubMed] [Google Scholar]
- 157.Vasilescu J, Guo X, Kast J. Proteomics. 2004;4:3845–3854. doi: 10.1002/pmic.200400856. [DOI] [PubMed] [Google Scholar]
- 158.Schmitt-Ulms G, Hansen K, Liu J, Cowdrey C, Yang J, DeArmond SJ, Cohen FE, Prusiner SB, Baldwin MA. Nat Biotechnol. 2004;22:724–731. doi: 10.1038/nbt969. [DOI] [PubMed] [Google Scholar]
- 159.Guerrero C, Tagwerker C, Kaiser P, Huang L. Mol Cell Proteomics. 2006;5:366–378. doi: 10.1074/mcp.M500303-MCP200. [DOI] [PubMed] [Google Scholar]
- 160.Tagwerker C, Flick K, Cui M, Guerrero C, Dou Y, Auer B, Baldi P, Huang L, Kaiser P. Mol Cell Proteomics. 2006;5:737–748. doi: 10.1074/mcp.M500368-MCP200. [DOI] [PubMed] [Google Scholar]
- 161.Kaake RM, Wang X, Huang L. Mol Cell Proteomics. 2010 doi: 10.1074/mcp.R110.000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fang L, Kaake RM, Patel VR, Yang Y, Baldi P, Huang L. Mol Cell Proteomics. 2012;11:138–147. doi: 10.1074/mcp.M111.016352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Subbotin RI, Chait BT. Mol Cell Proteomics. 2014;13:2824–2835. doi: 10.1074/mcp.M114.041095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D, Ban N. Science (New York, NY) 2015;348:303–308. doi: 10.1126/science.aaa3872. [DOI] [PubMed] [Google Scholar]
- 165.Plaschka C, Lariviere L, Wenzeck L, Seizl M, Hemann M, Tegunov D, Petrotchenko EV, Borchers CH, Baumeister W, Herzog F, Villa E, Cramer P. Nature. 2015;518:376–380. doi: 10.1038/nature14229. [DOI] [PubMed] [Google Scholar]
- 166.Guerrero C, Milenkovic T, Przulj N, Kaiser P, Huang L. Proc Natl Acad Sci U S A. 2008;105:13333–13338. doi: 10.1073/pnas.0801870105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kaake RM, Milenkovic T, Przulj N, Kaiser P, Huang L. J Proteome Res. 2010;9:2016–2029. doi: 10.1021/pr1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gong Z, Ding YH, Dong X, Liu N, Zhang EE, Dong MQ, Tang C. Biophysics reports. 2015;1:127–138. doi: 10.1007/s41048-015-0015-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ferber M, Kosinski J, Ori A, Rashid UJ, Moreno-Morcillo M, Simon B, Bouvier G, Batista PR, Müller CW, Beck M, Nilges M. Nat Methods. 2016;13:515–520. doi: 10.1038/nmeth.3838. [DOI] [PubMed] [Google Scholar]
- 170.Ding YH, Gong Z, Dong X, Liu K, Liu Z, Liu C, He SM, Dong MQ, Tang C. J Biol Chem. 2017;292:1187–1196. doi: 10.1074/jbc.M116.761841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, Robinson CV, Latz E, Gay NJ. The Journal of biological chemistry. 2009;284:25404–25411. doi: 10.1074/jbc.M109.022392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Politis A, Stengel F, Hall Z, Hernandez H, Leitner A, Walzthoeni T, Robinson CV, Aebersold R. Nat Methods. 2014;11:403–406. doi: 10.1038/nmeth.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Konijnenberg A, Butterer A, Sobott F. Biochim Biophys Acta. 2013;1834:1239–1256. doi: 10.1016/j.bbapap.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 174.Snijder J, van de Waterbeemd M, Damoc E, Denisov E, Grinfeld D, Bennett A, Agbandje-McKenna M, Makarov A, Heck AJ. J Am Chem Soc. 2014;136:7295–7299. doi: 10.1021/ja502616y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liko I, Allison TM, Hopper JT, Robinson CV. Current opinion in structural biology. 2016;40:136–144. doi: 10.1016/j.sbi.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 176.Song Y, Nelp MT, Bandarian V, Wysocki VH. ACS central science. 2015;1:477–487. doi: 10.1021/acscentsci.5b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lossl P, Brunner AM, Liu F, Leney AC, Yamashita M, Scheltema RA, Heck AJ. ACS central science. 2016;2:445–455. doi: 10.1021/acscentsci.6b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Baud A, Gonnet F, Salard I, Le Mignon M, Giuliani A, Mercere P, Sclavi B, Daniel R. Nat Methods. 2016;473:1805–1819. doi: 10.1042/BCJ20160225. [DOI] [PubMed] [Google Scholar]
- 179.Mummadisetti MP, Frankel LK, Bellamy HD, Sallans L, Goettert JS, Brylinski M, Bricker TM. Biochemistry. 2016;55:3204–3213. doi: 10.1021/acs.biochem.6b00365. [DOI] [PubMed] [Google Scholar]
- 180.Schmidt C, Macpherson JA, Lau AM, Tan KW, Fraternali F, Politis A. Anal Chem. 2017;89:1459–1468. doi: 10.1021/acs.analchem.6b02875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.de Koster CG, Woehl JL, Ramyar KX, Katz BB, Walker JK, Geisbrecht BV. J Proteome Res. 2017;26:1595–1608. doi: 10.1002/pro.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Liu WH, Roemer SC, Zhou Y, Shen ZJ, Dennehey BK, Balsbaugh JL, Liddle JC, Nemkov T, Ahn NG, Hansen KC, Tyler JK, Churchill ME. eLife. 2016;5:e18023. doi: 10.7554/eLife.18023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mysling S, Kristensen KK, Larsson M, Beigneux AP, Gardsvoll H, Fong LG, Bensadouen A, Jorgensen TJ, Young SG, Ploug M. eLife. 2016;5:e12095. doi: 10.7554/eLife.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zanphorlin LM, Lima TB, Wong MJ, Balbuena TS, Minetti CA, Remeta DP, Young JC, Barbosa LR, Gozzo FC, Ramos CH. The Journal of biological chemistry. 2016;291:18620–18631. doi: 10.1074/jbc.M115.710137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Birolo L, Sacchi S, Smaldone G, Molla G, Leo G, Caldinelli L, Pirone L, Eliometri P, Di Gaetano S, Orefice I, Pedone E, Pucci P, Pollegioni L. Febs j. 2016;283:3353–3370. doi: 10.1111/febs.13809. [DOI] [PubMed] [Google Scholar]
- 186.Pence N, Tokmina-Lukaszewska M, Yang ZY, Ledbetter RN, Seefeldt LC, Bothner B, Peters JW. The Journal of biological chemistry. 2017;292:15661–15669. doi: 10.1074/jbc.M117.801548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rouillon C, Zhou M, Zhang J, Politis A, Beilsten-Edmands V, Cannone G, Graham S, Robinson CV, Spagnolo L, White MF. Mol Cell. 2013;52:124–134. doi: 10.1016/j.molcel.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, Ban N. Nature. 2014;505:515–519. doi: 10.1038/nature12890. [DOI] [PubMed] [Google Scholar]
- 189.Forster F, Lasker K, Nickell S, Sali A, Baumeister W. Mol Cell Proteomics. 2010;9:1666–1677. doi: 10.1074/mcp.R000002-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Russel D, Lasker K, Webb B, Velazquez-Muriel J, Tjioe E, Schneidman-Duhovny D, Peterson B, Sali A. PLoS Biol. 2012;10:e1001244. doi: 10.1371/journal.pbio.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kosinski J, von Appen A, Ori A, Karius K, Muller CW, Beck M. Journal of structural biology. 2015;189:177–183. doi: 10.1016/j.jsb.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kahraman A, Herzog F, Leitner A, Rosenberger G, Aebersold R, Malmstrom L. PLoS One. 2013;8:e73411. doi: 10.1371/journal.pone.0073411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Karaca E, Bonvin AM. Methods. 2013;59:372–381. doi: 10.1016/j.ymeth.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 194.Han Y, Luo J, Ranish J, Hahn S. EMBO J. 2014;33:2534–2546. doi: 10.15252/embj.201488638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nguyen-Huynh NT, Sharov G, Potel C, Fichter P, Trowitzsch S, Berger I, Lamour V, Schultz P, Potier N, Leize-Wagner E. Protein Sci. 2015;24:1232–1246. doi: 10.1002/pro.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Setiaputra D, Ross JD, Lu S, Cheng DT, Dong MQ, Yip CK. The Journal of biological chemistry. 2015;290:10057–10070. doi: 10.1074/jbc.M114.624684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Setiaputra DT, Cheng DT, Lu S, Hansen JM, Dalwadi U, Lam CH, To JL, Dong MQ, Yip CK. EMBO Rep. 2017;18:280–291. doi: 10.15252/embr.201642548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Dauden MI, Kosinski J, Kolaj-Robin O, Desfosses A, Ori A, Faux C, Hoffmann NA, Onuma OF, Breunig KD, Beck M, Sachse C, Seraphin B, Glatt S, Muller CW. EMBO Rep. 2017;18:264–279. doi: 10.15252/embr.201643353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Komolov KE, Du Y, Duc NM, Betz RM, Rodrigues J, Leib RD, Patra D, Skiniotis G, Adams CM, Dror RO, Chung KY, Kobilka BK, Benovic JL. Anal Chem. 2017;169:407–421. e416. doi: 10.1016/j.cell.2017.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fiedorczuk K, Letts JA, Degliesposti G, Kaszuba K, Skehel M, Sazanov LA. Nature. 2016;538:406–410. doi: 10.1038/nature19794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Finley D. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Beck F, Unverdorben P, Bohn S, Schweitzer A, Pfeifer G, Sakata E, Nickell S, Plitzko JM, Villa E, Baumeister W, Forster F. Proc Natl Acad Sci U S A. 2012;109:14870–14875. doi: 10.1073/pnas.1213333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. Nature. 2012;482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Huang X, Luan B, Wu J, Shi Y. Nat Struct Mol Biol. 2016;23:778–785. doi: 10.1038/nsmb.3273. [DOI] [PubMed] [Google Scholar]
- 205.Schweitzer A, Aufderheide A, Rudack T, Beck F, Pfeifer G, Plitzko JM, Sakata E, Schulten K, Forster F, Baumeister W. Proc Natl Acad Sci U S A. 2016;113:7816–7821. doi: 10.1073/pnas.1608050113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bohn S, Beck F, Sakata E, Walzthoeni T, Beck M, Aebersold R, Förster F, Baumeister W, Nickell S. Proc Natl Acad Sci U S A. 2010;107:20992–20997. doi: 10.1073/pnas.1015530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kock M, Nunes MM, Hemann M, Kube S, Dohmen RJ, Herzog F, Ramos PC, Wendler P. Nature communications. 2015;6:6123. doi: 10.1038/ncomms7123. [DOI] [PubMed] [Google Scholar]
- 208.Aufderheide A, Beck F, Stengel F, Hartwig M, Schweitzer A, Pfeifer G, Goldberg AL, Sakata E, Baumeister W, Forster F. Proc Natl Acad Sci U S A. 2015;112:8626–8631. doi: 10.1073/pnas.1510449112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tomko RJ, Jr, Taylor DW, Chen ZA, Wang HW, Rappsilber J, Hochstrasser M. Cell. 2015;163:432–444. doi: 10.1016/j.cell.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hirano Y, Hendil KB, Yashiroda H, Iemura S, Nagane R, Hioki Y, Natsume T, Tanaka K, Murata S. Nature. 2005;437:1381–1385. doi: 10.1038/nature04106. [DOI] [PubMed] [Google Scholar]
- 211.Guo X, Engel JL, Xiao J, Tagliabracci VS, Wang X, Huang L, Dixon JE. Proc Natl Acad Sci U S A. 2011;108:18649–18654. doi: 10.1073/pnas.1113170108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wang X, Yen J, Kaiser P, Huang L. Sci Signal. 2010;3:ra88. doi: 10.1126/scisignal.2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Thierbach K, von Appen A, Thoms M, Beck M, Flemming D, Hurt E. Structure. 2013;21:1672–1682. doi: 10.1016/j.str.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 214.Kim SJ, Fernandez-Martinez J, Sampathkumar P, Martel A, Matsui T, Tsuruta H, Weiss T, Shi Y, Markina-Inarrairaegui A, Bonanno JB, Sauder JM, Burley SK, Chait BT, Almo SC, Rout MP, Sali A. Mol Cell Proteomics. 2014;13:2911–2926. doi: 10.1074/mcp.M114.040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Fernandez-Martinez J, Kim SJ, Shi Y, Upla P, Pellarin R, Gagnon M, Chemmama IE, Wang J, Nudelman I, Zhang W, Williams R, Rice WJ, Stokes DL, Zenklusen D, Chait BT, Sali A, Rout MP. Cell. 2016;167:1215–1228. doi: 10.1016/j.cell.2016.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.von Appen A, Kosinski J, Sparks L, Ori A, DiGuilio AL, Vollmer B, Mackmull MT, Banterle N, Parca L, Kastritis P, Buczak K, Mosalaganti S, Hagen W, Andres-Pons A, Lemke EA, Bork P, Antonin W, Glavy JS, Bui KH, Beck M. Nature. 2015;526:140–143. doi: 10.1038/nature15381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Beck M, Hurt E. Nat Rev Mol Cell Biol. 2017;18:73–89. doi: 10.1038/nrm.2016.147. [DOI] [PubMed] [Google Scholar]
- 218.Blattner C, Jennebach S, Herzog F, Mayer A, Cheung AC, Witte G, Lorenzen K, Hopfner KP, Heck AJ, Aebersold R, Cramer P. Genes Dev. 2011;25:2093–2105. doi: 10.1101/gad.17363311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Jennebach S, Herzog F, Aebersold R, Cramer P. Nucleic Acids Res. 2012;40:5591–5601. doi: 10.1093/nar/gks220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wu CC, Herzog F, Jennebach S, Lin YC, Pai CY, Aebersold R, Cramer P, Chen HT. Proc Natl Acad Sci U S A. 2012;109:19232–19237. doi: 10.1073/pnas.1211665109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Murakami K, Elmlund H, Kalisman N, Bushnell DA, Adams CM, Azubel M, Elmlund D, Levi-Kalisman Y, Liu X, Gibbons BJ, Levitt M, Kornberg RD. Science (New York, NY) 2013;342:1238724. doi: 10.1126/science.1238724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Muhlbacher W, Sainsbury S, Hemann M, Hantsche M, Neyer S, Herzog F, Cramer P. Nature communications. 2014;5:4310. doi: 10.1038/ncomms5310. [DOI] [PubMed] [Google Scholar]
- 223.Martinez-Rucobo FW, Kohler R, van de Waterbeemd M, Heck AJ, Hemann M, Herzog F, Stark H, Cramer P. Mol Cell. 2015;58:1079–1089. doi: 10.1016/j.molcel.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 224.Lariviere L, Plaschka C, Seizl M, Petrotchenko EV, Wenzeck L, Borchers CH, Cramer P. Nucleic Acids Res. 2013;41:9266–9273. doi: 10.1093/nar/gkt704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Robinson PJ, Bushnell DA, Trnka MJ, Burlingame AL, Kornberg RD. Proc Natl Acad Sci U S A. 2012;109:17931–17935. doi: 10.1073/pnas.1215241109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Murakami K, Tsai KL, Kalisman N, Bushnell DA, Asturias FJ, Kornberg RD. Proc Natl Acad Sci U S A. 2015;112:13543–13548. doi: 10.1073/pnas.1518255112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Luo J, Cimermancic P, Viswanath S, Ebmeier CC, Kim B, Dehecq M, Raman V, Greenberg CH, Pellarin R, Sali A, Taatjes DJ, Hahn S, Ranish J. Mol Cell. 2015;59:794–806. doi: 10.1016/j.molcel.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Robinson PJ, Trnka MJ, Pellarin R, Greenberg CH, Bushnell DA, Davis R, Burlingame AL, Sali A, Kornberg RD. eLife. 2015;4:e08719. doi: 10.7554/eLife.08719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sadian Y, Tafur L, Kosinski J, Jakobi AJ, Wetzel R, Buczak K, Hagen WJ, Beck M, Sachse C, Muller CW. EMBO J. 2017;36:2698–2709. doi: 10.15252/embj.201796958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Thierry E, Guilligay D, Kosinski J, Bock T, Gaudon S, Round A, Pflug A, Hengrung N, El Omari K, Baudin F, Hart DJ, Beck M, Cusack S. Mol Cell. 2016;61:125–137. doi: 10.1016/j.molcel.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Pflieger D, Junger MA, Muller M, Rinner O, Lee H, Gehrig PM, Gstaiger M, Aebersold R. Mol Cell Proteomics. 2008;7:326–346. doi: 10.1074/mcp.M700282-MCP200. [DOI] [PubMed] [Google Scholar]
- 232.Xu H, Hsu PH, Zhang L, Tsai MD, Freitas MA. J Proteome Res. 2010;9:3384–3393. doi: 10.1021/pr100369y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Buncherd H, Roseboom W, de Koning LJ, de Koster CG, de Jong L. Journal of proteomics. 2014;108:65–77. doi: 10.1016/j.jprot.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 234.Sinz A. Angew Chem Int Ed Engl. 2006;46:660–662. doi: 10.1002/anie.200602549. [DOI] [PubMed] [Google Scholar]
- 235.Fischer L, Chen ZA, Rappsilber J. Journal of proteomics. 2013;88:120–128. doi: 10.1016/j.jprot.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Schmidt C, Zhou M, Marriott H, Morgner N, Politis A, Robinson CV. Nature communications. 2013;4:1985. doi: 10.1038/ncomms2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Schmidt C, Robinson CV. Nat Protoc. 2014;9:2224–2236. doi: 10.1038/nprot.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Walzthoeni T, Joachimiak LA, Rosenberger G. 2015;12:1185–1190. doi: 10.1038/nmeth.3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Chen ZA, Fischer L, Cox J, Rappsilber J. Mol Cell Proteomics. 2016;15:2769–2778. doi: 10.1074/mcp.M115.056481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Chen ZA, Pellarin R, Fischer L, Sali A, Nilges M, Barlow PN, Rappsilber J. Mol Cell Proteomics. 2016;15:2730–2743. doi: 10.1074/mcp.M115.056473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kukacka Z, Rosulek M, Strohalm M, Kavan D, Novak P. Methods. 2015;89:112–120. doi: 10.1016/j.ymeth.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 242.Ong SE, Foster LJ, Mann M. Methods. 2003;29:124–130. doi: 10.1016/s1046-2023(02)00303-1. [DOI] [PubMed] [Google Scholar]
- 243.Ishihama Y, Sato T, Tabata T, Miyamoto N, Sagane K, Nagasu T, Oda Y. Nat Biotechnol. 2005;23:617–621. doi: 10.1038/nbt1086. [DOI] [PubMed] [Google Scholar]
- 244.Mann M. Nat Rev Mol Cell Biol. 2006;7:952–958. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- 245.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 246.Dayon L, Hainard A, Licker V, Turck N, Kuhn K, Hochstrasser DF, Burkhard PR, Sanchez JC. Anal Chem. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- 247.McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, Gygi SP. Anal Chem. 2012;84:7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Ting L, Rad R, Gygi SP, Haas W. Nat Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. Anal Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 251.Popowicz GM, Czarna A, Wolf S, Wang K, Wang W, Domling A, Holak TA. Cell Cycle. 2010;9:1104–1111. doi: 10.4161/cc.9.6.10956. [DOI] [PubMed] [Google Scholar]
- 252.Dubrez L, Berthelet J, Glorian V. Onco Targets Ther. 2013;9:1285–1304. doi: 10.2147/OTT.S33375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Arkin MR, Tang Y, Wells JA. Chem Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]