

Abstract
Cancer initiation and progression are the result of genetic and/or epigenetic alterations. Acetylation-mediated histone/non-histone protein modification plays an important role in the epigenetic regulation of gene expression. Histone modification is controlled by the balance between histone acetyltransferase and (HAT) and histone deacetylase (HDAC) enzymes. Imbalance between the activities of these two enzymes is associated with various forms of cancer. Histone deacetylase inhibitors (HDACi) regulate the activity of HDACs and are being used in cancer treatment either alone or in combination with other chemotherapeutic drugs/radiotherapy. The Food and Drug Administration (FDA) has already approved four compounds, namely vorinostat, romidepsin, belinostat, and panobinostat, as HDACi for the treatment of cancer. Several other HDACi of natural and synthetic origin are under clinical trial for the evaluation of efficiency and side-effects. Natural compounds of plant, fungus, and actinomycetes origin, such as phenolics, polyketides, tetrapeptide, terpenoids, alkaloids, and hydoxamic acid, have been reported to show potential HDAC-inhibitory activity. Several HDACi of natural and dietary origin are butein, protocatechuic aldehyde, kaempferol (grapes, green tea, tomatoes, potatoes, and onions), resveratrol (grapes, red wine, blueberries and peanuts), sinapinic acid (wine and vinegar), diallyl disulfide (garlic), and zerumbone (ginger). HDACi exhibit their antitumor effect by the activation of cell cycle arrest, induction of apoptosis and autophagy, angiogenesis inhibition, increased reactive oxygen species generation causing oxidative stress, and mitotic cell death in cancer cells. This review summarizes the HDACs classification, their aberrant expression in cancerous tissue, structures, sources, and the anticancer mechanisms of HDACi, as well as HDACi that are either FDA-approved or under clinical trials.
Keywords: cancer, histone deacetylases, histone deacetylase inhibitors, vorinostat, natural HDACi, apoptosis
1. Introduction
Cancer is the second leading cause of death worldwide and caused 8.8 million deaths in 2015. Globally, 1 out of 6 deaths is because of cancer. Low and middle-income countries are the hotspot of cancer deaths, accounting for approximately 70% of deaths. Lung cancer is the most common cause of cancer death worldwide. According to the National Center for Health Statistics about 1.73 million new cancer cases and 0.6 million cancer deaths are projected to occur in the United States in 2018 [1]. Cancer results from altered cell physiology leading to self-sufficient growth potential, loss of cell cycle control, extended angiogenesis, delay in replicative senescence, dysregulated apoptosis, invasion, and metastasis [2,3]. Progression of the disease is not only governed by genomic and genetic changes, such as translocation, amplification, deletion and point mutation, it also involves epigenetic changes; i.e., alteration in the pattern of gene expression without changing underlying DNA sequence. Methylation of DNA, histone protein modifications and non-coding RNA-mediated gene silencing are the major epigenetic changes, reversible in nature [4].
Chromatin is a compact and highly ordered structure comprised of DNA and histone protein. Nucleosome, the basic unit of chromatin, is made up of 147 bp of DNA superhelix wrapped around histone core protein containing two copies each of H2A, H2B, H3 and H4. H1 is the linker histone. The core plays an important role in establishing interactions between the nucleosomes and within the nucleosome particle itself. N-terminals (histone tails) of core histones are flexible and unstructured while the rest of histone proteins are basically globular and highly ordered. Depending on the epigenetic changes in histone tail, chromatin undergoes various conformational changes responsible for upregulation or downregulation of respective genes [5,6]. The common posttranslational modifications occurring in histones are acetylation, methylation, phosphorylation, sumoylation, ubiquitinylation, and ADP ribosylation.
Acetylation of a lysine residue of histone was discovered by Vincent Allfrey and colleagues in 1964 and based on the finding it has been proposed that acetylation of ε-amino group of lysine residues could play a role in gene expression [7,8]. Acetylation and deacetylation of N-terminal ε-amino group of lysine residues are regulated by two enzymes, namely histone acetyltransferase (HAT) and histone deacetylase (HDAC) (Figure 1). Acetylation neutralizes the positive charge and decreases the affinity between the histone and DNA helix responsible for relaxation of conformation and greater accessibility to transcription machinery [4,9]. Therefore, acetylation is generally associated with gene activation, however, deacetylation catalyzed by HDAC induces chromatin condensation and downregulation of gene expression. N-terminal acetylation of lysine residue also occurs in non-histone proteins, such as cytoplasmic proteins, transcription factors responsible for alteration in gene expression and other cellular processes [10].
Figure 1.

Histone acetylation at the N-terminus lysine by histone acetyltransferases (HATs) and deacetylation by histone deacetylases (HDACs).
Imbalance in the activities of enzymes HATs and HDACs is responsible for the development and progression of wide variety of cancers [2]. Histone deacetylase inhibitors (HDACi) increase the level of acetylated lysine residues of core histone which in turn restarts the expression of silenced regulatory genes in the cancerous cell and therefore, HDACi are now emerging as anticancer agents [11].
Epidemiological studies have suggested that vegetables, fruits, whole grains, microorganism-derived bioactive components, and fatty acids provide protection against some forms of cancer and other diseases without detectable side effects [12,13,14]. Many dietary compounds that have been identified as having HDAC-inhibitory activities implicated in therapeutic potential in the context of a whole food [11,15]. In the present review an effort has been made to highlight the role of HDACs in the tumor initiation and progression and their inhibitors from natural (dietary and non-dietary) as well as synthetic sources in the management of cancer either alone or in combination with other chemotherapeutic drugs/radiotherapy.
2. Classification of HDACs
In humans, 18 HDACs have been identified so far and are divided into two families and four classes based on their sequence homology to Saccharomyces cerevisiae HDACs (Figure 2) [16]. One of the family group members are zinc-dependent, they require Zn++ as a cofactor for their deacetylase activity and include HDAC 1 to HDAC 11. HDACs 1, 2, 3 and 8 are grouped into class 1 having a sequence similarity with yeast reduced potassium dependency-3 (Rpd3) and class II HDACs are subdivided into class IIA and Class IIB that include HDACs 4, 5, 6, 7, 9 and 10 which are reported to have sequence homology with yeast histone deacetylase-1 (hda-1) while HDAC 11 of class IV share sequence similarity with both classes of yeast deacetylase Rpd3 and hda-1.
Figure 2.

Classification of HDAC family.
Another group of the family requires nicotinamide adenine dinucleotide (NAD+) as a cofactor for deacetylase activity classified as class III, has sequence similarity to yeast deacetylase silent information regulator-2 (Sir2) and includes seven members from sirtuins (SIRTs) 1 to 7. Sirtuins are known to regulate several cellular processes; e.g., survival, aging, stress response, and various metabolic processes. The members of class I and IV are located in the nucleus while class IIA is mainly located into the cytoplasm and class IIB is found shuttling between the nucleus and cytoplasm. Cellular localization of class III HDACs are nucleus, cytoplasm, and mitochondria [11,17]. Nomenclature of class I, II and IV HDACs are based on their chronological order of discovery; for example, both HDAC 1 and 2 were discovered in 1996 while HDAC 2 was discovered a few months after HDAC 1 [18,19]. Later on, HDAC 3 was discovered in the subsequent years [20]. While HDACs 4, 5, and 6 were first reported in 1999, the HDAC 7 was discovered in early 2000 and so on [21,22]. Table 1 summarizes the HDACs classification, number of amino acids, cellular and chromosomal locations, biological functions, relevant histone/non-histone target proteins, and their expression pattern [6,23,24].
Table 1.
Histone deacetylase (HDAC) enzymes classification, number of amino acids, localization, function, protein targets and expression pattern.
| Class | HDACs | Number of Amino Acid | Cellular Location | Chromosomal Location | Biological Function | Histone/Non-Histone Protein Target | Pattern of Expression of Gene |
|---|---|---|---|---|---|---|---|
| I | HDAC 1 | 483 | Nucleus | 1p35.2-p35.1 | Proliferation and survival of cells | Histones, pRb, SHP, BRCA1, MECP2, ATM, MEF2, MyoD, p53, NF-κB, AR, DNMT1 | Ubiquitous expression |
| HDAC 2 | 488 | 6q21 | Proliferation of cell and insulin resistance | Histones, BRCA1, NF-κB, MECP, GATA 2, pRb | |||
| HDAC 3 | 428 | 5q31.3 | Proliferation and survival of cells | Histones, HDAC (4, 5, 7, 9), GATA 1, NF-κB, pRb | |||
| HDAC 8 | 377 | Xq13.1 | Proliferation of cell | HSP70 | |||
| IIA | HDAC 4 | 1084 | Nucleus/Cytoplasm | 2q37.3 | Regulation of cytoskeleton dynamics and cell mobility | Histones, HDAC 3, 14-3-3, CaM, MEF 2 | Tissue restricted expression |
| HDAC 5 | 1122 | 17q21.31 | Helps in endothelial cell function, gluconeogenesis, cardiac myocyte growth and function | ||||
| HDAC 7 | 912 | 12q13.11 | Helps in endothelial cell function and glyconeogenesis. | ||||
| HDAC 9 | 1069 | 7p21.1 | Helps in thymocyte differentiation, homologous recombination, cardiac cell function | ||||
| IIB | HDAC 6 | 1215 | Cytoplasm | Xp11.23 | Regulation of cytoskeleton dynamics and cell mobility | HDAC 11, SHP, HSP 90, α tubulin | Tissue restricted expression |
| HDAC 10 | 669 | 2q13.33 | Regulation of autophagy, homologous recombination. | LcoR, PP1 | |||
| III | SIRT 1 | 747 | Nucleus/Cytoplasm | 10q21.3 | Autoimmunity, aging, redox balance, and cell survival | Histones, NF-κB, p53, p300 | Variable expression |
| SIRT 2 | 389 | Nucleus | 19q13.2 | Survival, migration, and invasion of cell | Histone H4, PPAR-ϒ, p53, p300, α-tubulin, FOXO | ||
| SIRT 3 | 399 | Mitochondria | 11p15.5 | Regulate ATP production and metabolism, cell signaling, apoptosis, urea cycle | Complex I of ETC, PGC-1α, p53, Ku70, Acetyl-CoA Synthetase, FOXO | ||
| SIRT 4 | 314 | 12q24.31 | Energy metabolism, Urea cycle, cell signaling | Glutamate dehydrogenase | |||
| SIRT 5 | 310 | 6p23 | Regulate ATP production and metabolism, cell signaling, apoptosis, urea cycle | Carbamoyl phosphate synthetase I, Cytochrome c | |||
| SIRT 6 | 355 | Nucleus | 19p13.3 | Regulate metabolism | Histone H3, TNF-α | ||
| SIRT 7 | 400 | 17q25.3 | Apoptosis | p53, RNA polymerase I | |||
| IV | HDAC 11 | 347 | Nucleus | 3p25.1 | DNA replication, Immunomodulation | HDAC 6 | Ubiquitous in nature |
AR, androgen receptor; ATM, ataxia-telangiectasia-mutated; BRCA, breast cancer; CaM, calmodulin; CoA, co-enzyme A; DNMT, DNA methyltransferase; FOXO, forkhead box O; GATA, GATA binding protein; HIF, hypoxia-inducible factor; HSP, heat shock protein; LcoR, ligand-dependent receptor co-repressor; MECP, methyl-CpG-binding domain protein; MEF, myocyte enhancer factor; NF-κB, nuclear factor-kappa B; PGC, peroxisome proliferator-activated receptor gamma coactivator; PP1, protein phosphatase; PPAR, peroxisome proliferator-activated receptor; pRb, retinoblastoma protein; SHP, Src homology region 2-domain-containing phosphatase; SIRT, sirtuin; TNF, tumor necrosis factor.
3. Cellular Targets of Histone/Non-Histone Protein Acetylation
The acetylation or deacetylation status of histone proteins and transcription factors modulate the gene expression pattern. Hyperacetylation of lysine residues of histone proteins promotes the relaxed state of chromatin and activates gene expression [25]. Besides this, acetylation of transcription factors affects their cellular localization. For example, signal transducer and activator of transcription 1 (STAT 1) and nuclear factor-κB (NF-κB) are internalized into the nucleus from cytosol after the acetylation of specific lysine residues where they activate transcription of respective genes. The activity of other transcription factors, such as p53 and FOXO, is also positively regulated by the acetylation process. Acetylation process also affects the stability of proteins, i.e., acetylation of p53, p73, and mothers against decapentaplegic homolog 7 (SMAD 7) prevents their ubiquitinylation and degradation. Interestingly, cell mobility is also affected by the acetylation pattern of α-tubulin and cortactin. It has been reported that HDAC 6 and SIRT2 cause deacetylation of α-tubulin, which promotes microtubule depolymerization and therefore increases microtubule dynamics and cell mobility. Acetylation also affects the activity of retinoblastoma protein (pRB) by blocking its cyclin E-cdk2 dependent phosphorylation so acetylation-dependent hypophosphorylation causes cell cycle arrest [26,27].
4. HDAC Mutations in Cancer
Mutations of HDACs have also been observed. HDAC2 mutation in human epithelial cancer resulted in microsatellite instability. Interestingly, a truncating mutation of HDAC2 in human cancers confers resistance to HDACi. These findings suggest that the HDAC2 mutational status of patients should be assessed before therapies using HDACi [28]. HDAC3 are associated with DNA damage control response. Inactivation of HDAC3 causes genomic instability. HDAC4 acts s transcriptional repressor and its mutations have been identified at significant frequency in breast and colorectal cancers. SIRT 2 acts as a tumor suppressor and mutation in its catalytic domain eliminates its enzymatic activity, which compromises the mitotic checkpoint, contributing to genomic instability and tumorigenesis. HDAC9 and 10 are reported to be involved in homologous recombination, and depletion in HDAC 9 and 10 resulted in inhibition of homologous recombination [11].
5. HDACs and Cancer: Expression Pattern and Function
Altered acetylation level and mutation/or aberrant expression of various HDACs have been observed frequently in numerous human diseases including cancer, hence making them an important drug target [2]. Fraga et al. (2005) reported that change in genome-wide patterns of acetylation may lead to the initiation and progression of cancer by demonstrating that cancer cells undergo a loss of acetylation at lysine 16 of H4 [29]. HDACs have various histone and non-histone protein targets that not only regulate the chromatin activity, but also control apoptosis, cell cycle progression and differentiation. Association of HDACs with regulatory processes reflects their involvement in cancer phenotypes [30].
5.1. Class I HDACs
5.1.1. HDAC 1
HDAC 1 overexpression has been reported in Hodgkin’s lymphoma (HL), gastric, ovarian, and prostate cancers [11,23]. Choi and co-workers (2001) have shown the overexpression of HDAC 1 in 60% cases compared with normal tissue [31]. This study was further validated by a recent study including 293 gastric cancer samples showing upregulation in expression of HDACs 1, 2, and 3 [32,33]. Elevated level of expression of HDAC 1, 2, and 3 was reported in pancreatic cancer involving 192 samples and this overexpression was responsible for dedifferentiation and enhanced proliferation of pancreatic cancer cell [34]. Overexpression of HDAC 1, 2, and 3 is associated with mortality rate in colorectal cancer. Expression of HDAC 2 has emerged as an independent prognostic marker in colorectal cancer [35]. Furthermore, overexpression of HDAC 1 is reported in hepatocellular carcinoma [36], lung cancer [37] and breast cancer [38]. Direct correlation between HDAC 1, 3 expression and estrogen and progesterone receptor expression have been reported by Krusche et al. [39].
HDAC 1 induces cell proliferation and inhibition of differentiation and apoptosis [23,38]. HDAC 1 and 3 knockdowns (KD) resulted in the inhibition of cell proliferation in Hela cells [40]. HDAC 1 KD has been shown to result in cell cycle arrest either at the G1 phase or at the G2/M transition phase, causing loss of mitotic cells, inhibition of cell growth, and an increase in the number of apoptotic cells in osteosarcoma and breast cancer cells. However, HDAC 2 KD showed no such effects in these cells [41]. HDAC1 might also be involved in multidrug resistance as they showed overexpression in a pattern in chemotherapeutically resistant neuroblastoma cells [42].
5.1.2. HDAC 2
Overexpression of HDAC2 reported in uterine, cervical, gastric, cutaneous T cell lymphoma (CTCL), HL, prostate and colorectal cancers [38]. Elevated expression of HDAC 2 along with HDAC 1 and 3 are associated with advance stage of disease and prognosis of gastric, colorectal, and prostate cancers [33,34,35].
Knockdown of HDAC2 in cervical cancer causes increased apoptosis and the differentiated phenotype of cells associated with increased p21Cip1/WAF1 expression that was independent of p53 [43]. In breast carcinoma cells, HDAC 2 KD results into the increased DNA binding activity of tumor suppressor protein p53, which causes the inhibition of cell proliferation and induction of cellular senescence [44]. HDAC 2 KD also causes decreased viability, growth arrest, and increased apoptosis in colorectal and breast cancer cells [45].
5.1.3. HDAC 3
HDAC 3 overexpression has been reported in HL, ovarian and lung cancers, colon cancer, and chronic lymphocyte leukemia (CLL) [11,38,46]. An upregulated expression of HDAC1 along with HDAC3 was paradoxically related to disease-free survival in invasive breast cancer patients [24]. HDAC3 elevated expression together with HDAC 1 and 2 significantly causes poor prognosis in gastric, colon, and prostate cancers [33,34,35]. Decreased expression of HDAC 3 has been observed in liver cancer [45].
HDAC 3 KD in acute promyelocytic leukemia (APL) cells causes restoration of expression of a retinoic acid-dependent gene whose transcription repression was caused by promyelocytic leukemia retinoic acid receptor alpha (PML-RARα) [47]. While HDAC3 KD in colon cancer causes a decrease in the viability of cell by increasing the rate of apoptosis [45].
5.1.4. HDAC 8
Expression of HDAC 8 has been reported to increase in childhood neuroblastoma. HDAC KD shows reduction in proliferation of lung, colon, and cervical cancer. In childhood neuroblastoma cell HDAC 8 KD causes cell cycle arrest, reduction in cell proliferation [48,49]. HDAC 8 has also been reported in controlling telomerase activity [50].
5.2. Class IIA HDACs
5.2.1. HDAC4
A breast cancer sample showed overexpressed genotype of HDAC 4 [51]. However, lung and colon cancer analysis showed downregulated expression of HDAC 4 [11].
In APL, HDAC 4 was found to repress the expression of differentiation-associated genes by interacting with a leukemic fusion protein, PLZF-RARa [52]. HDAC 4 also regulates the activity of hypoxia inducing factor-1α (HIF-1α). HDAC4 has also been shown to help prostate cancer cells overcome hypoxic conditions by stabilizing HIF-1α. The binding of HDAC4 to HIF-1α generates a complex that regulates glycolysis and the cytotoxic stress of cell adaptation to hypoxic conditions [53]. HDAC4 KD in colon and glioblastoma cells causes an increased apoptosis rate and reduced growth rate [54].
5.2.2. HDAC 5
HDAC 5 overexpression has been reported in medulloblastoma [55] and colon cancer [51] while interestingly, the lung cancer sample showed a downregulated genotypic expression of HDAC5 [11]. HDAC 5 traverses from the nucleus to the cytosol upon interacting with transcription factor GATA-1 during differentiation of mouse erythroleukemia cells [56]. Knock down of HDAC 5 has been shown to cause reduced growth and viability of medulloblastoma cells [55].
5.2.3. HDAC 7
They are highly expressed in ALP, CLL, and colon cancer [46,51] and downregulated expression is reported in lung cancer samples [45,57]. HDAC 7 silencing in endothelial cells altered their morphology, their migration, and their capacity to form capillary tube-like structures in vitro but did not affect cell adhesion, proliferation, or apoptosis, suggesting that HDAC7 may represent a rational target for anti-angiogenesis in cancer [58].
5.2.4. HDAC 9
Elevated expression of HDAC 9 was reported in ALL and medulloblastoma [59,60] and in cervical cancer [31]. Silencing HDAC 9 results into inhibition of homologous recombination, sensitivity towards DNA damage and decreased viability and growth of medulloblastoma [45].
5.3. Class IIB HDACs
5.3.1. HDAC 6
A significantly higher expression of HDAC 6 was reported in oral squamous cell carcinoma, hepatocellular carcinoma, acute myeloid leukemia, CLL, breast cancer, CTCL and ovarian cancer, whereas its expression increased in the advanced stages of cancer compared to the early stages of cancer [5,61].
HDAC 6 overexpression causes increased migration of the fibroblast cell while the inhibition of HDAC 6 results in decreased fibroblast cell migration [62]. HDAC 6 targeted inhibition causes HSP 90 protein acetylation and disruption of its chaperone activity resulting in decreased viability of K562 leukemic cell [63]. HDAC4/6 has the potential to help prostate cancer cells overcome hypoxic conditions by stabilizing HIF-1 α [53]. HDAC 6 is also involved in metastasis, epithelial to mesenchymal transition in lung cancer cells by compromising TGF-β SMAD 3 pathway [64].
5.3.2. HDAC 10
Osada and colleague [57] described the potential role of HDAC 10 in cancer initiation and progression and reported the downregulated expression of HDAC 10 in non-small lung carcinoma cells and this is related with poor prognosis in lung cancer patients. Its role in gastric cancer and CLL is also reported [65].
HDAC 10 are shown to regulate the production of reactive oxygen species in gastric cancer cells [66]. HDAC 10 KD causes reduced vascular endothelial growth factor of receptor 1 and 2 (VEGF 1 and 2) and increased sensitivity towards DNA damage in cancer cells [67].
5.4. Class III HDACs
Sirtuins
Growing evidence supports the relation between cancer and sirtuins. As other HDACs they are also both tumor suppressors as well as pro-oncogenic in nature. SIRT 1 expression was shown to be upregulated in acute myeloid leukemia (AML), non-melanoma skin cancer and prostate cancer [68,69,70] while downregulated in colorectal cancer [51]. SIRT 2 expression decreased in glioma and gastric cancer [71]. However, a mutation in the catalytic domain of SIRT 2 has also been reported [72]. Expression pattern of SIRT 3 has been found to increase or decrease in different kinds of breast cancer tissues [73]. SIRT 6 expression was reported to decrease in liver cancer while increased expression pattern in CLL [46]. SIRT 7 shows an upregulated expression pattern in breast cancer [73].
SIRT 2 functions as a tumor suppressor protein and the mutation in its catalytic domain results in compromised cell cycle checkpoints leading to genomic instability and tumorigenesis [74]. In many cancerous cells, increased expression of SIRT 1 induces the p-glycoprotein expression responsible for chemotherapeutically resistant cancer cells whereas siRNA-mediated SIRT 1 KD causes a reversal of drug-resistant phenotype [75]. SIRT 3 functions both as a tumor suppressor and a tumor promoter protein. Higher expression of SIRT 3 was responsible for preventing bladder cancer cells from p53-mediated cell growth arrest [76]. SIRT 7 was shown to stabilize cancer cell phenotypes. SIRT 7 KD results in the tumorigenic potential of human cancer xenograft in mice [77].
5.5. Class IV HDACs
HDAC 11
The involvement of HDAC 11 in cancer is not well understood. However aberrant expression of HDAC 11 were reported in HL and Philadelphia-negative chronic myeloproliferative neoplasms (CMPN) [78].
siRNA-mediated HDAC 11, KD results in an increased apoptosis rate in HL, colon, prostate, breast, and ovarian cell lines [45,79]. Selective inhibition of HDAC 11 with histone deacetylase inhibitor (HDACi) reduces the chance of splenomegaly and other metabolic disorders reported in CMPN affected people [78].
6. Histone Deacetylase Inhibitors as an Anticancer Agent
The aberrant or altered expression of HDACs or their functions is frequently observed in a variety of cancer types and is the reason for targeting HDACs in cancer therapy. The availability of HDACi not only has accelerated our understanding of HDAC functions and its mechanism of actions but also presented a promising new class of compounds for cancer treatment. HDACi belong to a large and diverse family of both natural and synthetic compounds and can be categorized into four groups; i.e., hydroximic acid, benzamides, cyclic peptides, and aliphatic fatty acids [80]. Several natural or synthetic compounds are isolated and characterized for their potential as histone deacetylase inhibitors. The first significant compound identified as HDACi was n-butyrate, responsible for the accumulation of hyperacetylated histone inside the nucleus [81]. Subsequently, trichostatin A (TSA) and trapoxin A were found to be reversible and irreversible inhibitors of HDACs, respectively [11,82]. Schreiber et al. (1996) were first to discover and clone the human HDAC by using trapoxin A [80]. While developing these compounds as anticancer agents, parameters like their specificity, efficiency, pharmacokinetic and toxicological properties were analyzed [83]. Some HDACi, such as TSA, lycorine and zerumbone, are pan-HDAC inhibitors since they act on all isoform of zinc-dependent HDAC classes, while others are class specific.
HDCAs can reversibly modify the acetylation pattern of histone/non-histone protein resulted in abnormal gene expression without changing the DNA sequence. HDACi can overcome this problem and can resume the expression of tumor the suppressor genes responsible for apoptosis, cell cycle arrest, and the inhibition of angiogenesis and metastasis. Ungerstedt and co-workers [84] reported that cancerous cells are more sensitive than normal cells towards HDACi-induced apoptosis.
6.1. Natural HDACi
Altogether natural compounds provide pleiotropic and potent inhibitors of all hallmarks of cancer. Many of the HDACi discovered to date are of natural origin; for example in 1976, Tsuji et al. [85] isolated a naturally occurring HDACi, TSA from Streptomyces hygroscopicus. FK322, a cyclic peptide isolated from Chromobacterium violaceum selectively inhibits the activity of HDAC 1 and 2. TSA causes differentiation of cell and arrests the cell cycle of both normal and cancerous cells, resulting in the accumulation of acetylated histones [86]. Depudecin and trapoxin A and B are also the examples of naturally occurring HDACi extracted from a fungus. Marine organisms are also the source of natural HDACi, such as largazole and azumamides, and they are reported to be active even at nanomolar concentrations [11]. Other well-characterized naturally occurring HDACi, such as butein, kaempferol, protocatechuic aldehyde, sinapinic acid, resveratrol and zerumbone, are isolated from plant, fruits or vegetables (Table 2). Molecular modelling studies revealed the HDACi like activity of other dietary compounds; i.e., vitamin E, α-lipoic acid, and biotin [48]. For the first time, the clinical validation of natural HDACi was done by Riggs and colleague [81] in 1977. They analyzed the effect of butyrate on histone modificationin HeLa and Friend erythroleukemia cell lines [80]. Later in 1980, McKnight et al. [87] reported the effect of propionate on histone deacetylation in chick oviduct and showed it to have lesser activity than butyrate. Both these compounds were active at millimolar concentrations and synthesized by colonic bacteria. Valproic acid, a longer chain aliphatic fatty acid, also reported to have significant HDACi activity. Valproic acid inhibits HDACs activity by binding to its active site [87]. Detailed features of other natural histone deacetylase inhibitors and their sources are shown in Table 2.
Table 2.
Examples of the natural compound with histone deacetylase inhibitory activity.
| S.N | Class of Compounds | Name of the Compound | HDAC Target | Source (Species/Family) | Structure | Reference |
|---|---|---|---|---|---|---|
| 1. | Phenolics | Aceroside VIII | HDAC6 | Betula platyphylla |
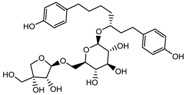
|
[87] |
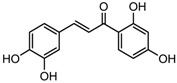
| Homobutein | Class I, II and IV | Butea frondosa |
|
[88] | ||
| Isoliquiritigenin | Class I, II and IV | Glycyrhiza glabra |
|
[89] | ||
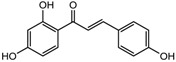
| Butein | Class I, II and IV | Toxicodendron vernicifluum |
|
[88] | ||
| Kaempferol | Class I, II and IV | Aloe vera |
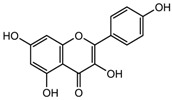
|
[89] | ||
| Marein | Class I, II and IV | Coreopsis maritima |

|
[88] | ||
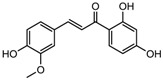
| Protocatechuic aldehyde | HDAC2 | Hordeum vulgare |
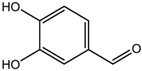
|
[23] | ||
| Psammaplin A | Class I | Poecillastra spp. and Jaspis spp. |
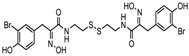
|
[23] | ||
| Sinapinic acid | Pan-HDAC | Hydnophytum formicarum Jack |
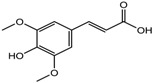
|
[90] | ||
| Resveratrol | Class I, II and IV | Vitis vinifera |
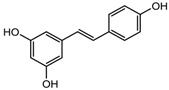
|
[91] | ||
| 2. | Polyketides | Depudecin | HDAC 1 | Alternaria brassicicola |
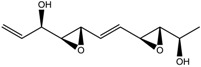
|
[92] |
| 3. | Tetrapeptide | Apicidin | Class I HDAC | Fusarium spp. |
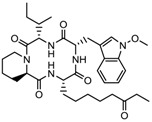
|
[93] |
| Azumamide E | Class I | Mycale izuensis |

|
[94] | ||
| Chlamydocin | HDAC 1, 6 | Diheterospora chlamydosporia |
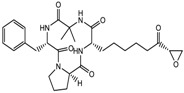
|
[95] | ||
| Trapoxin A | Class I | Helicoma ambiens RF-1023 |
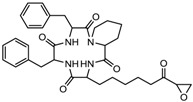
|
[96] | ||
| 4. | Terpenoids | Zerumbone | Pan HDAC | Zingiber zerumbet |
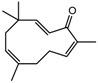
|
[23] |
| β-Thujaplicin | HDAC 2 | Cupressaceae spp. |
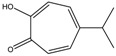
|
[23] | ||
| 6-methoxy-2E,9E-humuladien-8-one | Pan HDAC | Zingiber zerumbet |
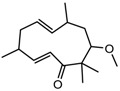
|
[23] | ||
| 5. | Alkaloid | Lycorine | Pan HDAC | Amaryllidaceae |
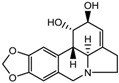
|
[97] |
| 6. | Fatty acid | 9-Hydroxystearic acid | Class I | Lipid peroxidation product |
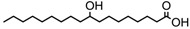
|
[98] |
| 7. | Organosulphur compounds | Diallyl disulfide | Acetylation Level increased | Allium sativum |
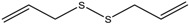
|
[99,100] |
| (S)-allylmercaptocysteine | Allium sativum |
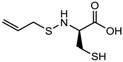
|
[101] | |||
| 8. | Hydroxamic acid | Trichostatin A | Class I and II | Streptomyces hygroscopicus |
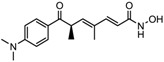
|
[2,11] |
| 9. | Desipeptides | FK228 | HDAC 1, 2 | Chromobacterium violaceum |
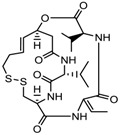
|
[11] |
| Largazole | Class I | Symploca spp. |
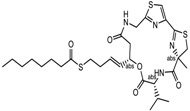
|
[99] |
6.2. FDA-Approved and Under Clinical Trial HDACi
Aberrant expression of HDACs gene is reported in various cancer types. So, the scientists across the globe are searching for a therapeutic alternative, which not only can inhibit the increased activity of HDACs, but also reverse the malignant phenotype. The histone deacetylase inhibitors chiefly cause the gene to be in a hyperacetylated state, which in turn restarts the gene expression, and are also involved in chromatin stability, mitosis, and the DNA repair mechanism [23]. However, it has been reported that both normal and cancerous cells show accumulation of acetylated histone after treatment with HDACi, but healthy cells appear to be much less susceptible to apoptosis and growth inhibition than a cancerous cell. Four synthetic compounds, viz., vorinostat, romidepsin, belinostat, and panobinostat have been approved as HDACi for cancer treatment to date by the United States Food and Drug Administration (FDA). In addition, many other HDACi are under clinical trials in patients suffering from various types of cancer [101,102]. FDA-approved and other HDACi under clinical trials are presented in Table 3.
Table 3.
FDA approved and under clinical trials histone deacytylase inhibitors (HDACi).
| S.N | Chemical Class | Name of the Compounds | HDAC Target | Cancer Specificity | Trial Stage | Structure of the Compound | Reference |
|---|---|---|---|---|---|---|---|
| 1. | Hydroxamic acid | SAHA (Vorinostat) | Class I, II and IV | CTCL | FDA approved (2006) |
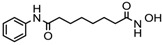
|
[107] |
| Belinostat | Class I, II and IV | PTCL | FDA approved (2014) |
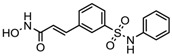
|
[108] | ||
| Panobinostat | Class I, II and IV | MM | FDA approved in 2015 |
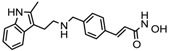
|
[109] | ||
| Resminostat | Class I and II | Colorectal, HCC, HL | Phase II trial |

|
[6] | ||
| Givinostat | Class I and II | CLL, HL, MM | Phase II trial |
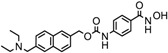
|
[110] | ||
| Pracinostat | Classes I, II and IV | AML | Phase II trial |
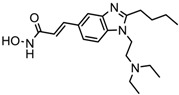
|
[111] | ||
| Abexinostat | Class I and II | CLL, HL, Non-HL, Solid tumors | Phase I trial |
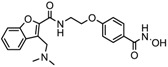
|
[45] | ||
| Quisinostat | Class I and II | Solid tumor, CTCL | Phase I and II trial |
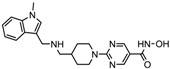
|
[45] | ||
| MPT0E028 | HDAC 1, 2 and 6 | Solid tumor, B-cell lymphoma | Phase I trial |
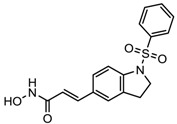
|
[112] | ||
| CHR 3996 | Class I | Solid tumors | Phase I trial |

|
[113] | ||
| CUDC 101 | Class I and II | Solid tumor | Phase I trial |

|
[6] | ||
| CUDC 907 | Class I and II | MM; lymphoma; solid tumor | Phase I trial |
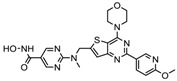
|
[6] | ||
| 2. | Benzamides | Entinostat | Class I | Solid tumors | Phase I and II trial |
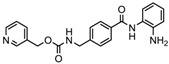
|
[113] |
| Chidamide | HDAC 1, 2,3 and 10 | Breast cancer | Phase II and III trial |
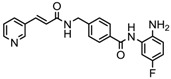
|
[114] | ||
| Ricolinostat | HDAC 6 | MM, Lymphoma | Phase I and II trial |
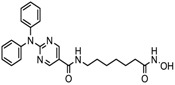
|
[115] | ||
| Tacedinaline | Class I | Lung and pancreatic cancer, MM | Phase II and III trial |

|
[6] | ||
| Mocetinostat | Class I and IV | Solid malignancies | Phase I and II trial |
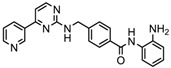
|
[116] | ||
| 3. | Cyclic peptides | Romidepsin | Class I | CTCL, PTCL | FDA approved in 2009 |
|
[6,117] |
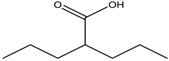
| 4. | Fatty acids | Valproic acid | Class I and II | Solid and hematological tumors | Phase I and II trial |
|
[118] |
| AR-42 | Class I and IIb | AML | Phase I trial |
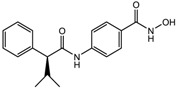
|
[45] | ||
| Phenyl butyrate | Class I and II | Solid and hematological tumors | Phase I and II trial |
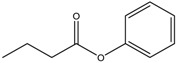
|
[118] | ||
| Pivanex | Class I and II | NSCLC, Myeloma, CLL | Phase II trial |
|
[113] |
CTCL, cutaneous T cell lymphoma treatment; PTCL, peripheral T cell lymphoma; MM, multiple myeloma; HCC, hepatocellular Carcinoma; HL, Hodgkin lymphoma; CLL, chronic lymphocyte leukemia; AML, acute myeloid leukemia; NSCLC, non-small cell lung cancer.
Vorinostat, a hydroximic acid-based drug, is also known as suberanilohydroxamic acid (SAHA) and is marketed with different names. Zolinza was the first FDA-approved vorinostat drug for cutaneous T cell lymphoma treatment (CTCL). It inhibits the HDACs except class III sirtuins and was developed by Merck & Co. (Kenilworth, NJ, USA). In phase II clinical trial study (NCT00958074) on 74 CTCL patients with a daily oral dose of vorinostat (400 mg), an objective response rate (ORR) of 30% was observed [11]. Vorinostat is also reported to be useful in other cancer types, such as brain metastasis, refractory colorectal, advanced solid tumors, melanoma, pancreatic, lung cancer and multiple myeloma [6]. A combinatorial therapy of vorinostat with temozolomide and radiotherapy is under clinical trial for the treatment of early stage of glioblastoma multiformae (NCT00731731).
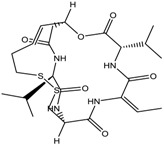
Romidepsin, a cyclic tetrapeptide, was the second FDA-approved drug for treatment of CTCL in 2009 and in 2011, for treatment of peripheral T cell lymphoma (PTCL) with the response rate of 34% and 25%, respectively. However, romidepsin treatment was associated with side effects, such as nausea, vomiting, cardiac toxicity, and myelotoxicity [11,37]. Romidepsin has been evaluated for treatment of T-cell lymphoma either as a single agent (NCT00426764) or in combination with other drugs (NCT03141203) in 30 clinical trials. Romidepsin intake produced fatigue, nausea, vomiting, diarrhea, constipation, phlebitis, headache, and dyspnea as side effects [6]. Romidepsin either singly or in combination with paclitaxel exhibited elimination of both primary tumors and a metastatic lesion at multiple sites formed by the SUM149 IBC cell line in the Mary-X preclinical model [103].
In 2014, belinostat was the third drug approved by the FDA for PTCL treatment. It is a hydroxamate that inhibits the activity of class I and II HDACs. Clinical trial study (NCT00865969) on 120 PTCL patients showed an ORR of 26% [6]. Belinostat is now being investigated in more than 15 clinical trials for the treatment of CTCL (NCT00274651), multiple myeloma (NCT00431340), Burkitt lymphoma (NCT00303953), and solid tumors as in fallopian tube cancer (NCT00301756) [23].
Panobinostat was approved by the FDA in 2015 for the treatment of multiple myelomas [37]. It is a hydroxamate derivative causing the inhibition of class I, II, and IV HDACs. An ORR of 27% was reported for panobinostat. Diarrhoea and cardio-toxicity are side effects associated with Panobinostat. It has been used for treating other cancer types, such as CTCL (NCT00490776), AML (NCT01613976), Hodgkin’s lymphoma (NCT00742027), MDS (NCT00594230), thyroid carcinoma (NCT01013597), and colorectal and prostate cancers (NCT00663832) in more than 50 clinical trials [23].
In contrast to the above mentioned clinical trials and studies reporting efficacy of HDACi in the treatment of various lymphomas, leukemia and myeloma, solid tumors have shown limited response against HDACi. The study conducted by Paller and colleagues [104] reported that HDACi VPA and vorinostat in combination with AMG 900, a pan-aurora kinase inhibitor, significantly enhanced cellular senescence, polyploidy, and apoptosis in prostate cancer cell lines (DU-145, LNCaP and PC3) as compared with a single agent treatment. Combination therapy with Janus kinase (JAK) inhibitor INCB018424 has been shown to improve the clinical efficacy of vorinostat in triple-negative breast cancer patients [105]. Similarly, combination therapies targeting HDACs and IκB kinase have shown potential against ovarian cancer [106].
7. Mechanisms of Action of HDACi
HDACi induces cell cycle arrest, differentiation, and apoptosis as well as inhibits angiogenesis [119]. The mechanism of the anticancer effect of HDACi depends upon the cancer type, individual, stage of cancer, dose, and some other factors [120]. The antiproliferative mechanisms of HDACi action are described below.
7.1. Cell Cycle Arrest
Various mechanisms are involved in HDACi-mediated cell cycle arrest. One of the most important mechanisms is the increased expression of the cyclin-dependent kinase (CDK) inhibitor gene CDKN1A (p21, WAF1/CIP1). An interesting fact is that the HDACi-mediated overexpression of p21 is independent of p53 [121,122]. The concentration-dependent cell cycle inhibitory effects of HDACi have been observed. At lower concentrations HDACi predominately induces G1 arrest while at higher concentrations induces both G1 and G2/M arrest [123]. p21 is mainly associated with G1 and G2/M arrest, inhibits the activity of CDKs (i.e., CDK 4/6), which regulates the progression of the G1 stage of cell cycle, CDK 2 responsible for G1/S transition and cdc2/CDK 1 causes G2/M transition. The p21 mutation abolishes the HDACi induced G1 arrest [124]. However, HDACi-mediated G1 arrest is also observed in a cell without p21. Hitomi et al. [125] reported that TSA causes G1 arrest in human colon p21 mutant cell by induction of p15 (INK4b), which subsequently causes inhibition of cyclin D-dependent kinases resulting in the absence of CDK 2. Protein p53 interacts with the p21 promoter by competing with HDAC1 and alters the expression of the p21 gene. HDAC1 is the transcriptional repressor of p21, which gets detached from the Sp1 (promoter-specific RNA polymerase II transcription factor) after HDACi treatment, resulting in increased p21 expression. Moreover, HDACi also causes an increase in the half-life of protein p53 thereby improving its interaction with p21 and increased levels of p21 inside the cell mediates cell cycle arrest and apoptosis [126]. HDACi treatment also compromises the CDK activity, which may account for the dephosphorylation of the retinoblastoma protein (Rb), which blocks the elongation factor, E2F function in the transcription of genes for G1 progression and G1/S transition [118].
7.2. Induction of Apoptosis in Transformed Cell
HDACi induces the rate of apoptosis in a transformed cell by regulating both pro-apoptotic and anti-apoptotic genes (Figure 3) and this involves the activation of both extrinsic and intrinsic apoptotic pathways. HDACi induced extrinsic pathway initiation involves the binding of death receptor such as Fas (Apo-1 or CD95), tumor necrosis factor (TNF) receptor-1 (TNFR-1), TNF-related apoptosis-inducing ligand (TRAIL) receptors (DR-4 and DR-5), DR-3 (Apo3) and DR-6 to their ligand FasL, TNF, TRAIL, and TL1A resulting into activation of caspase 8 and 10. In vitro and in vivo studies suggest that HDACi upregulates the expression of both death receptor and their ligand in transformed cells but no such effect is observed in normal cells [127]. Upregulated expression of Fas and FasL have been reported in the treatment of human neuroblastoma cells with m-carboxycinnamic acid bihydroxamide, nude mice xenograft of osteocarcinoma with FK228, acute promyelocytic leukemia model of a rat with valproic acid, and so on [128,129]. FK228 also induces the expression of TNF α in HL 60 and K562 cells. cFLIP, an inhibitor of the activity of the death receptor has been reported to be downregulated after HDACi treatment in cancer cells [127]. HDACi-mediated apoptosis also involves the activation of the intrinsic apoptotic pathway, which causes the release of inter mitochondrial membrane protein, such as cytochrome c, Smac, apoptosis-inducing factor (AIF), and the subsequent activation of caspase. HDACi activate the intrinsic apoptotic pathways by regulating the transcription of pro-apoptotic genes; i.e., Bid (BH3 interacting domain death agonist protein), Bad (Bcl-2 associated agonist of cell death protein), and Bim [130]. The mechanism of HDACi-mediated intrinsic pathway activation is not well understood, but it is suggested that HDACi causes leaking of mitochondrial intermembrane proteins, cytochrome c, AIF and Smac. The release of cytochrome c from mitochondria in turn causes activation of caspase-9 [131,132]. The intrinsic apoptotic pathway is regulated by changing the HDACi-mediated expression of factors. It can be concluded that HDACi upregulate the expression of proapoptotic genes and pathways (BAX, Apaf1 and BAK) and downregulate the expression of the antiapoptotic Bcl-2 family proteins, Bcl-2, Bcl-XL and Mcl-1 [133].
Figure 3.

Multiple anti-tumor pathways such as, cell cycle arrest, induction of autophagy and apoptosis, DNA damage repair, ROS generation, angiogenesis inhibitor and mitotic cell death are activated by the action of HDACi in cancer cells. Arrows (↑ and ↓) indicates the increase and decrease, respectively, in the obtained variables. HIF, Hypoxia inducing factor; VEGF, Vascular endothelial growth factor; HR, homologous recombination; NHEJ, Non-homologous end joining; Trx, Thioredoxin; TBP, Thioredoxin binding protein.
7.3. Autophagic Cell Death
The role of HDACi in autophagy is not well understood. Recent findings suggest that autophagy serves as cell death mechanism, therefore, autophagy inhibitors or knockdown of autophagy-related gene decreases the anticancer effect of HDACi. When HeLa cell line having an Apaf-1 knockout or Bcl-XL overexpression was cultured with vorinostat or butyrate, it undergoes autophagic cell death by forming autophagosome inside cytoplasm [134]. Treatment of a colon carcinoma cell HCT116 with vorinostat resulted in the inhibition of cell growth as well as senescence type phenotype [135]. SAHA causes cell death in endometrial stromal sarcoma cell via autophagy [136]. In p53 mutant cancer cells, SAHA induces autophagy and cell death. HDACi-mediated autophagy involves several signaling pathways, one such pathway is the mechanistic target of the rapamycin (mTOR) pathway that is the main suppressor of autophagy via phosphorylation and the inactivation of the Unc51 like autophagy activating kinase 1 (ULK1) complex. SAHA inhibits the activity of mTOR and resumes the activity of ULK1, upstream component of autophagy pathway [137]. SAHA up-regulates the expression of autophagy-related protein by stimulating NF-κβ activity [138]. SAHA also induces autophagy via ROS production in leukemic and hepatocellular carcinoma cells. Romidepsin induces autophagy in HeLa cells [139]. SAHA inhibited the growth of glioblastoma cells xenograft in nude mice by inducing autophagy via downregulation of AKT-mTOR signaling [140]. In some cancer cells showing resistance towards apoptosis, HDACi can induce cell death via the induction of autophagy by bypassing the apoptosis. Hence, HDACi induced autophagy stimulation could be a promising anticancer strategy.
7.4. Inhibition of Angiogenesis
HDACi mediated inhibition of angiogenesis can interfere with the metastasis. HDAC inhibitors downregulate proangiogenic genes, such as the vascular endothelial growth factor gene (VEGF) and endothelial nitric oxide synthase gene [141]. The inhibition of the activity of hypoxia-inducible factor (HIF) by HDACi can block the angiogenesis. Hypoxia is a commonly occurring condition in tumor cells responsible for the overexpression of class I HDACs, except HDAC 8, which in turn causes activation of HIF 1α and promotes angiogenesis [142]. Several compounds viz., TSA, vorinostat, FK228, butyrate and LAQ824, have been reported to inhibit the angiogenesis process and thereby decrease the expression of proangiogenic (HIF 1α and VEGF) factors. Several mechanisms are responsible for the HDACi-mediated degradation of HIF 1α. These include the degradation of HIF 1α by acetylation at Lys532 leading to ubiquitination, class II HDACs associate with HIF 1α and cause siRNA-mediated degradation of HIF 1α [143,144]. HDACi also degrades HIF 1α through compromising its transactivation potential, and also reduces the sensitivity of cancerous cells towards the angiogenic signal generated by VEGF [145,146]. Vorinostat and TSA downregulate the expression of VEGF receptors. Valproic acid impedes angiogenesis by increasing the production of thrombospondin-1 and activin A, the antiangiogenic proteins [147]. Use of combination therapies with VEGF inhibitors finds support from the above reports.
7.5. ROS Generation
Oxidative stress has been implicated in cell death [148,149,150,151]. Experimental facts support the treatment of transformed cells with HDACi, such as TSA, vorinostat, butyrate or MS275, leading to the accumulation of reactive oxygen species (ROS) inside cells that subsequently induce cell death [152]. Ruelfi and co-workers [153] in 2001 reported that the quenching of ROS by N-acetylcysteine, a free radical scavenger decreases HDACi induced apoptosis, suggesting the role of HDACi in cell death. Thioredoxin (Trx), a hydrogen ion donor, is a ROS scavenger [154]. It is required in several redox reactions and is responsible for the activity of many protein, such as ribonucleotide reductase, which play an important role in the synthesis of DNA and the transcription factor. HDACi inhibits the activity of Trx by inducing the expression of Trx binding protein 2 (TBP2), which binds to it and inhibits its activity in cancer cells but not in normal cells [155]. Trx binds and inhibits the activity of ASK1. ASK1, an apoptosis signal regulating kinase, increases the rate of apoptosis by the activation of the SET1-JNK and MKK3/MKK6-p38 signaling pathways, and by elevating the expression of pro-apoptotic protein Bim. Therefore, higher expression of TBP2 by HDACi prevents the activity of Trx resulting in the increased expression of ASK1 and ultimately leading to apoptosis [156].
7.6. Mitotic Cell Death
HDACi treatment causes an abnormal acetylation pattern of histone protein in heterochromatin and centromere region. Newly synthesized chromatin contains acetylated histones and TSA treatment in transformed cells causes newly synthesized chromatin to remain acetylated, leading to the structural and functional disorder of centromere and pericentric heterochromatin [157], and this structural disturbance interferes with the phosphorylation of histone as well as disrupts the activity of mitotic spindle checkpoint proteins; i.e., BubR1, hBUB1, CENP-F, and CENP-E [123]. And this causes cell division arrest at the prometaphase stage followed by abnormal mitosis, such as the disaggregation and chromosomal loss leading to cell death either through apoptosis or mitotic cell death [2,124,158].
8. Potential Limitation and Side-Effects of HDACi
There are potential limitations to selective HDACi therapy. The effects of class I HDACi on DNA damage and repair pathways suggest that prolonged exposure to these agents could lead to unacceptable toxicities and secondary malignancies [159]. Class I HDACs may also play an oncogenic role depending on the context. Sontoro et al. [160] reported that in mouse tumor model, a single putative barrier to full transformation is surprisingly provided by HDAC 1 and HDAC2. Knock-down of HDAC 1 resulted in the both blockade of cellular differentiation and the increased genomic instability mediated by PML-RAR in hematopoietic progenitors. Either or both biological deregulations could be sufficient to cooperate with the tumor-promoting activities of oncoproteins, such as PML-RAR and Myc, and would provide a functional explanation for the observed increase in frequency of transformation to leukemia in HDAC1-deficient cells. Epigenetic changes are important for reprogramming somatic cells into pluripotent stem cells. Therefore, several inhibitors of epigenetic-modifying enzymes, including HDACi, are able to reprogram somatic cells into the pluripotent stem cells by modifying a chromatin structure and making it more permissive to transcription factors [119].
9. Conclusions
The present review summarizes the biological activities of HDAC. Further, emphasis has been given on the structural and functional features of the natural and synthetic inhibitors of HDAC as well as on their antitumor potential. HDACs are multisubstrate (histone and non-histone) enzyme involved in many biological processes; i.e., cell proliferation, differentiation, apoptosis, and senescence. According to scientific reports, histone deacetylase inhibitors have shown their efficacy as inhibitors of cancer initiation and progression. Generally, normal cells are resistant to HDACi, which selectively modulates gene expression in cancerous cells. Pan-HDAC inhibitors, such as vorinostat, belinostat, and panobinostat stimulate antitumor pathways, suggesting their therapeutic potential. FDA-approved HDACi are being used for the treatment of several types of cancers. Many HDACi are under clinical trial stages. HDACi show their anticancer action involving various mechanisms; i.e., cell cycle arrest, induction of apoptosis and autophagy in transformed cells, inhibition of angiogenesis, ROS as mediators of cell death, and mitotic cell death. Several natural HDACi are present in our diet: kaempferol (grapes, green tea, tomatoes, potatoes, and onions), resveratrol (grapes, red wine, blueberries and peanuts), sinapinic acid (wine and vinegar), diallyl disulfide (garlic) and zerumbone (ginger). Hence a better understanding of dietary HDACi regarding their target specificity and toxicity may underscore potential health benefits through nutritional intervention. Combination therapy might be another important direction to enhance the therapeutic efficacy of HDACi. Elucidation and validation of the detailed mechanistic aspects of HDACi action will provide a bright future for the use of HDACi as one of the important tools in the fight against cancer.
Acknowledgments
A.K.S. acknowledges the CSIR New Delhi for providing financial support in the form of Junior Research Fellowship. A.K.S. and A.K.P. also acknowledge the UGC-SAP and DST-FIST facilities of the Department of Biochemistry, University of Allahabad, Allahabad, India.
Author Contributions
A.K.S. and A.K.P. have equally contributed to the structure, preparation, and finalization of manuscript. A.K.S. conducted the literature search and prepared the first draft of the manuscript. A.K.P. and A.B. thoroughly reviewed the manuscript and made desired corrections in the final draft. All the authors have read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Kumar S., Ahmad M.K., Waseem M., Pandey A.K. Drug Targets for Cancer Treatment: An Overview. Med. Chem. 2015;5:115–123. doi: 10.4172/2161-0444.1000252. [DOI] [Google Scholar]
- 3.Sharma U.K., Sharma A.K., Pandey A.K. Medicinal attributes of major phenylpropanoids present in cinnamon. BMC Complement. Altern. Med. 2016;16:156. doi: 10.1186/s12906-016-1147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar S., Pandey A.K. Oxidative stress-related microRNAs as diagnostic markers: A newer Insight in diagnostics. In: Maurya P., Chandra P., editors. Oxidative Stress: Diagnostic Methods and Applications in Medical Science. Springer; Singapore: 2017. pp. 113–125. [Google Scholar]
- 5.Zahonero B.B., Parra M. Histone deacetylases and cancer. Mol. Oncol. 2012;6:579–589. doi: 10.1016/j.molonc.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mottamal M., Zheng S., Huang T.L., Wang G. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules. 2016;20:3898–3941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allfrey V.G., Faulkner R., Mirsky A.E. Acetylation and methylation of histones and their possible role in the regulation of Rna synthesis. Proc. Natl. Acad. Sci. USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gershey E.L., Vidali G., Allfrey V.G. Chemical studies of histone acetylation. The occurrence of epsilon-N-acetyllysine in the f2a1 histone. J. Biol. Chem. 1968;243:5018–5022. [PubMed] [Google Scholar]
- 9.Haberland M., Montgomery R.L., Olson E.N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng L., Seto E. Deacetylation of Nonhistone Proteins by HDACs and the Implications in Cancer. In: Yao T.P., Seto E., editors. Histone Deacetylases: The Biology and Clinical Implication. Volume 206. Springer; Berlin/Heidelberg, Germany: 2011. pp. 39–56. Handbook of Experimental Pharmacology. [DOI] [PubMed] [Google Scholar]
- 11.Basset S.A., Barnett M.P. The Role of Dietary Histone Deacetylases (HDACs) Inhibitors in Health and Disease. Nutrients. 2014;6:4273–4301. doi: 10.3390/nu6104273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller P.E., Snyder D.C. Phytochemicals and cancer risk: A review of the epidemiological evidence. Nutr. Clin. Pract. 2012;27:599–612. doi: 10.1177/0884533612456043. [DOI] [PubMed] [Google Scholar]
- 13.Hou I.C., Amarnani S., Chong M.T., Bishayee A. Green tea and the risk of genetic cancer: Epidemiological evidence. World J. Gastroenterol. 2013;19:3713–3722. doi: 10.3748/wjg.v19.i24.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ivey K.L., Jensen M.K., Hodgson J.M., Eliassen A.H., Cassidy A., Rimm E.B. Association of flavonoid-rich foods and flavonoids with risk of all-cause mortality. Br. J. Nutr. 2017;117:1470–1477. doi: 10.1017/S0007114517001325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar S., Pandey A.K. Chemistry and biological activities of flavonoids: an overview. Sci. World J. 2013;2013:162750. doi: 10.1155/2013/162750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim H.J., Bae S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011;3:166–179. [PMC free article] [PubMed] [Google Scholar]
- 17.Lawson M., Uciechowska U., Schemies J., Rumpf T., Jung M., Sippl W. Inhibitors to understand molecular mechanisms of NAD+-dependent deacetylases (sirtuins) Biochim. Biophys. Acta. 2010;1799:726–739. doi: 10.1016/j.bbagrm.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Taunton J., Hassig C.A., Schreiber S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 19.Yang W.M., Inouye C., Zeng Y., Bearss D., Seto E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA. 1996;93:12845–12850. doi: 10.1073/pnas.93.23.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang W.M., Yao Y.L., Sun J.M., Davie J.R., Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 1997;272:28001–28007. doi: 10.1074/jbc.272.44.28001. [DOI] [PubMed] [Google Scholar]
- 21.Grozinger C.M., Hassig C.A., Schreiber S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kao H.Y., Downes M., Ordentlich P., Evans R.M. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 2000;14:55–66. [PMC free article] [PubMed] [Google Scholar]
- 23.Losson H., Schnekenburger M., Dicato M., Diederich M. Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signalling Pathway Modulators and Clinical Applications in Cancer. Molecules. 2016;21:1608. doi: 10.3390/molecules21111608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lane A.A., Chabner B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 25.Minucci S., Pelicci P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 26.Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X.F., Yao T.P. HDAC6 is a microtubule associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 27.Seidel C., Schnekenburger M., Dicato M., Diederich M. Histone deacetylase modulators provided by Mother Nature. Genes Nutr. 2012;7:357. doi: 10.1007/s12263-012-0283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ropero S., Fraga M.F., Ballestar E., Hamelin R., Yamamoto H., Boix-Chornet M., Caballero R., Alaminos M., Setien F., Paz M.F., et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 2006;38:566–569. doi: 10.1038/ng1773. [DOI] [PubMed] [Google Scholar]
- 29.Fraga M.F., Ballestar E., Villar-Garea A., Boix-Chornet M., Espada J., Schotta G., Bonaldi T., Haydon C., Ropero S., Petrie K., et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 30.Mai A., Massa S., Rotili D., Cerbara I., Valente S., Pezzi R., Simeoni S., Ragno R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005;25:261–309. doi: 10.1002/med.20024. [DOI] [PubMed] [Google Scholar]
- 31.Choi J.H., Kwon H.J., Yoon B.I., Kim J.H., Han S.U., Joo H.J., Kim D.Y. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn. J. Cancer Res. 2001;92:1300–1304. doi: 10.1111/j.1349-7006.2001.tb02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Witt O., Deubzer H.E., Milde T., Oehme I. HDAC family: What are the cancer relevant targets? Cancer Letters. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 33.Weichert W., Roske V.A., Gekeler T., Beckers M.P., Ebert M., Pross M., Dietel C., Denkert C., Rocken C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008;9:139–148. doi: 10.1016/S1470-2045(08)70004-4. [DOI] [PubMed] [Google Scholar]
- 34.Weichert W., Roske A., Gekeler V., Beckers T., Stephan C., Jung K., Fritzsche F.R., Niesporek S., Denkert C., Dietel M., Kristiansen G. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer. 2008;98:604–610. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weichert W., Roske A., Niesporek S., Noske A., Buckendahl A.C., Dietel M., Gekeler V., Boehm M., Beckers T., Denkert C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008;14:1669–1677. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- 36.Rikimaru T., Taketomi A., Yamashita Y., Shirabe K., Hamatsu T., Shimada M., Maehara Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology. 2007;72:69–74. doi: 10.1159/000111106. [DOI] [PubMed] [Google Scholar]
- 37.Sasaki H., Moriyama S., Nakashima Y., Kobayashi Y., Kiriyama M., Fukai I., Yamakawa Y., Fujii Y. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer. 2004;46:171–178. doi: 10.1016/j.lungcan.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 38.Yoon S., Eom G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016;52:1–11. doi: 10.4068/cmj.2016.52.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krusche C.A., Wulfing P., Kersting C., Vloet A., Bocker W., Kiesel L., Beier H.M., Alfer J. Histone deacetylase-1 and -3 protein expression in human breast cancer: A tissue microarray analysis. Breast Cancer Res. Treat. 2005;90:15–23. doi: 10.1007/s10549-004-1668-2. [DOI] [PubMed] [Google Scholar]
- 40.Glaser K.B., Li J., Staver M.J., Wei R.Q., Albert D.H., Davidsen S.K. Role of class I and class II histone deacetylases in carcinoma cells using siRNA. Biochem. Biophys. Res. Commun. 2003;310:529–536. doi: 10.1016/j.bbrc.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 41.Senese S., Zaragoza K., Minardi S., Muradore I., Ronzoni A.S., Passafaro L., Bernard G.F., Draetta M., Alcalay C., Seiser S., et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol. Cell. Biol. 2007;27:4784–4795. doi: 10.1128/MCB.00494-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keshelava N., Davicioni E., Wan Z., Ji L., Sposto R., Triche T.J., Reynolds C.P. Histone deacetylase 1 gene expression and sensitization of multidrug-resistant neuroblastoma cell lines to cytotoxic agents by depsipeptide. J. Natl. Cancer Inst. 2007;99:1107–1119. doi: 10.1093/jnci/djm044. [DOI] [PubMed] [Google Scholar]
- 43.Huang B.H., Laban M., Leung C.H., Lee L., Lee C.K., Salto-Tellez M., Raju G.C., Hooi S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005;12:395–404. doi: 10.1038/sj.cdd.4401567. [DOI] [PubMed] [Google Scholar]
- 44.Harms K.L., Chen X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007;67:3145–3152. doi: 10.1158/0008-5472.CAN-06-4397. [DOI] [PubMed] [Google Scholar]
- 45.West A.C., Johnstone R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J.C., Kafeel M.I., Avezbakiyev B., Chen C., Sun Y., Rathnasabapathy C., Kalavar M., He Z., Burton J., Lichter S. Histone deacetylase in chronic lymphocytic leukemia. Oncology. 2011;81:325–329. doi: 10.1159/000334577. [DOI] [PubMed] [Google Scholar]
- 47.Atsumi A., Tomita A., Kiyoi H., Naoe T. Histone deacetylase 3 (HDAC3) is recruited to target promoters by PML-RARa as a component of the N-CoR co-repressor complex to repress transcription in vivo. Biochem. Biophys. Res. Commun. 2006;345:1471–1480. doi: 10.1016/j.bbrc.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 48.Vannini A., Volpari C., Filocamo G., Casavola E.C., Brunetti M., Renzoni D., Chakravarty P., Paolini C., De Francesco R., Gallinari P., et al. Crystal structure of a eukaryotic zinc dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oehme I., Deubzer H.E., Wegener D., Pickert D., Linke J.P., Hero B., Kopp-Schneider A., Westermann F., Ulrich S.M., Von Deimling A., Fischer M., Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009;15:91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- 50.Lee H., Sengupta N., Villagra A., Rezai-Zadeh N., Seto E. Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (hEST1B) protein from ubiquitin-mediated degradation. Mol. Cell. Biol. 2006;26:5259–5269. doi: 10.1128/MCB.01971-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ozdag H., Teschendorff A.E., Ahmed A.A., Hyland S.J., Blenkiron C., Bobrow L., Veerakumarasivam A., Burtt G., Subkhankulova T., Arends M.J., et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genom. 2006;7:90. doi: 10.1186/1471-2164-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chauchereau A., Mathieu M., de Saintignon J., Ferreira R., Pritchard L.L., Mishal Z., Dejean A., Harel-Bellan A. HDAC4 mediates transcriptional repression by the acute promyelocytic leukaemiaassociated protein PLZF. Oncogene. 2004;23:8777–8784. doi: 10.1038/sj.onc.1208128. [DOI] [PubMed] [Google Scholar]
- 53.Geng H., Harvey C.T., Pittsenbarger J., Liu Q., Beer T.M., Xue C., Qian D.Z. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J. Biol. Chem. 2011;286:38095–38102. doi: 10.1074/jbc.M111.257055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilson A.J., Byun D.S., Nasser S., Murray L.B., Ayyanar K., Arango D., Figueroa M., Melnick A., Kao G.D., Augenlicht L.H., et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol. Biol. Cell. 2008;19:4062–4075. doi: 10.1091/mbc.e08-02-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milde T., Oehme I., Korshunov A., Kopp-Schneider A., Remke M., Northcott P., Deubzer H.E., Lodrini M., Taylor M.D., von Deimling A., et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010;16:3240–3252. doi: 10.1158/1078-0432.CCR-10-0395. [DOI] [PubMed] [Google Scholar]
- 56.Watamoto K., Towatari M., Ozawa Y., Miyata Y., Okamoto M., Abe A., Naoe T., Saito H. Altered interaction of HDAC5 with GATA-1 during MEL cell differentiation. Oncogene. 2003;22:9176–9184. doi: 10.1038/sj.onc.1206902. [DOI] [PubMed] [Google Scholar]
- 57.Osada H., Tatematsu Y., Saito H., Yatabe Y., Mitsudomi T., Takahashi T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int. J. Cancer. 2004;112:26–32. doi: 10.1002/ijc.20395. [DOI] [PubMed] [Google Scholar]
- 58.Mottet D., Bellahcene A., Pirotte S., Waltregny D., Deroanne C., Lamour V., Lidereau R., Castronovo V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ. Res. 2007;101:1237–1246. doi: 10.1161/CIRCRESAHA.107.149377. [DOI] [PubMed] [Google Scholar]
- 59.Kotian S., Liyanarachchi S., Zelent A., Parvin J.D. Histone deacetylases 9 and 10 are required for homologous recombination. J. Biol. Chem. 2011;286:7722–7726. doi: 10.1074/jbc.C110.194233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moreno D.A., Scrideli C.A., Cortez M.A., de Paula Queiroz R., Valera E.T., da Silva Silveira V., Yunes J.A., Brandalise S.R., Tone L.G. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010;150:665–673. doi: 10.1111/j.1365-2141.2010.08301.x. [DOI] [PubMed] [Google Scholar]
- 61.Seidel C., Schnekenburger M., Dicato M., Diederich M. Histone deacetylase 6 in health and disease. Epigenomics. 2015;7:103–118. doi: 10.2217/epi.14.69. [DOI] [PubMed] [Google Scholar]
- 62.Sakuma T., Uzawa K., Onda T., Shiiba M., Yokoe H., Shibahara T., Tanzawa H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int. J. Oncol. 2006;29:117–124. doi: 10.3892/ijo.29.1.117. [DOI] [PubMed] [Google Scholar]
- 63.Haggarty S.J., Koeller K.M., Wong J.C., Grozinger C.M., Schreiber S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bali P., Pranpat M., Bradner J., Balasis M., Fiskus W., Guo F., Rocha K., Kumaraswamy S., Boyapalle S., Atadja P., et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 65.Shan B., Yao T.P., Nguyen H.T., Zhuo Y., Levy D.R., Klingsberg R.C., Palmer M.L., Holder K.N., Lasky J.A. Requirement of HDAC6 for TGF-β 1-induced epithelial-mesenchymal transition. J. Biol. Chem. 2008;283:21065–21073. doi: 10.1074/jbc.M802786200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee J.H., Jeong E.G., Choi M.C., Kim S.H., Park J.H., Song S.H., Park J., Bang Y.J., Kim T.Y. Inhibition of histone deacetylase 10 induces thioredoxin-interacting protein and causes accumulation of reactive oxygen species in SNU-620 human gastric cancer cells. Mol. Cell. 2010;30:107–112. doi: 10.1007/s10059-010-0094-z. [DOI] [PubMed] [Google Scholar]
- 67.Johnson E.K., Wilgus A.T. Vascular endothelial growth factor and angiogenesis in the regulation of cutaneous wound repair. Adv. Wound Care (New Rochelle), 2014;3:647–661. doi: 10.1089/wound.2013.0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park J.H., Kim S.H., Choi M.C., Lee J., Oh D.Y., Im S.A., Bang Y.J., Kim T.Y. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem. Biophys. Res. Commun. 2008;368:318–322. doi: 10.1016/j.bbrc.2008.01.056. [DOI] [PubMed] [Google Scholar]
- 69.Bradbury C.A., Khanim F.L., Hayden R., Bunce C.M., White D.A., Drayson M.T., Craddock C., Turner B.M. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19:1751–1759. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 70.Hida Y., Kubo Y., Murao K., Arase S. Strong expression of a longevity-related protein, SIRT1, in Bowen’s disease. Arch. Dermatol. Res. 2007;299:103–106. doi: 10.1007/s00403-006-0725-6. [DOI] [PubMed] [Google Scholar]
- 71.Huffman D.M., Grizzle W.E., Bamman M.M., Kim J.S., Eltoum I.A., Elgavish A., Nagy T.R. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612–6618. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 72.Hiratsuka M., Inoue T., Toda T., Kimura N., Shirayoshi Y., Kamitani H., Watanabe T., Ohama E., Tahimic C.G., Kurimasa A., et al. Proteomics-based identification of differentially expressed genes in human gliomas: Down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 2003;309:558–566. doi: 10.1016/j.bbrc.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 73.Lennerz V., Fatho M., Gentilini C., Frye R.A., Lifke A., Ferel D., Wolfel C., Huber C., Wolfel T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA. 2005;102:16013–16018. doi: 10.1073/pnas.0500090102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ashraf N., Zino S., Macintyre A., Kingsmore D., Payne A.P., George W.D., Shiels P.G. Altered sirtuin expression is associated with node-positive breast cancer. Br. J. Cancer. 2006;95:1056–1061. doi: 10.1038/sj.bjc.6603384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dryden S.C., Nahhas F.A., Nowak J.E., Goustin A.S., Tainsky M.A. Role for human SIRT2 NAD-dependent class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev. 2003;14:45–54. [Google Scholar]
- 76.Chu F., Chou P.M., Zheng X., Mirkin B.L., Rebbaa A. Control of multidrug resistance gene mdr1 and cancer resistance to chemotherapy by the longevity gene sirt1. Cancer Res. 2005;65:10183–10187. doi: 10.1158/0008-5472.CAN-05-2002. [DOI] [PubMed] [Google Scholar]
- 77.Yang H., Yang T., Baur J.A., Perez E., Matsui T., Carmona J.J., Lamming D.W., Souza-Pinto N.C., Bohr V.A., Rosenzweig A., et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bell E.L., Emerling B.M., Ricoult S.J., Guarente L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011;30:2986–2996. doi: 10.1038/onc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Skov V., Larsen T.S., Thomassen M., Riley C.H., Jensen M.K., Bjerrum O.W., Kruse T.A., Hasselbalch H.C. Increased gene expression of histone deacetylases in patients with Philadelphia-negative chronic myeloproliferative neoplasms. Leuk. Lymphoma. 2012;53:123–129. doi: 10.3109/10428194.2011.597905. [DOI] [PubMed] [Google Scholar]
- 80.Gao L., Cueto M.A., Asselbergs F., Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002;277:25748–25755. doi: 10.1074/jbc.M111871200. [DOI] [PubMed] [Google Scholar]
- 81.Riggs M.G., Whittaker R.G., Neumann J.R., Ingram V.M. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268:462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 82.Kijima M., Yoshida M., Sugita K., Horinouchi S., Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993;268:22429–22435. [PubMed] [Google Scholar]
- 83.Li Y., Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016;6:10. doi: 10.1101/cshperspect.a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ungerstedt J.S., Sowa Y., Xu W.S., Shao Y., Dokmanovic M., Perez G., Ngo L., Holmgren A., Jiang X., Marks P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA. 2005;102:673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tsuji N., Kobayashi M., Nagashima K., Wakisaka Y., Koizumi K. A new antifungal antibiotic, trichostatin. J. Antibiot. 1976;29:1–6. doi: 10.7164/antibiotics.29.1. [DOI] [PubMed] [Google Scholar]
- 86.Yoshida M., Kijima M., Akita M., Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 87.Mcknight G.S., Hanger L., Palmiter R.D. Butyrate and related inhibitors of histone deacetylation block the induction of egg white genes by steroid harmones. Cell. 1980;22:469–477. doi: 10.1016/0092-8674(80)90357-8. [DOI] [PubMed] [Google Scholar]
- 88.Orlikova B., Schnekenburger M., Zloh M., Golais F., Diederich M., Tasdemir D. Natural chalcones as dual inhibitors of HDACs and NF-κB. Oncol. Rep. 2012;28:797–805. doi: 10.3892/or.2012.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Berger A., Venturelli S., Kallnischkies M., Bocker A., Busch C., Weiland T., Noor S., Leischner C., Weiss T.S., Lauer U.M., et al. Kaempferol, a new nutrition-derived pan-inhibitor of human histone deacetylases. J. Nutr. Biochem. 2013;24:977–985. doi: 10.1016/j.jnutbio.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 90.Senawong T., Misuna S., Khaopha S., Nuchadomrong S., Sawatsitang P., Phaosiri C., Surapaitoon A., Sripa B. Histone deacetylase (HDAC) inhibitory and antiproliferative activities of phenolic-rich extracts derived from the rhizome of Hydnophytum formicarum Jack.: Sinapinic acid acts as HDAC inhibitor. BMC Complement. Altern. Med. 2013;13:232. doi: 10.1186/1472-6882-13-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Venturelli S., Berger A., Bocker A., Busch C., Weiland T., Noor S., Leischner C., Schleicher S., Mayer M., Weiss T.S., et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLoS ONE. 2013;8:73097. doi: 10.1371/annotation/5b9a8614-1009-40ca-b90b-db817fe445c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ryu H.W., Lee D.H., Shin D.H., Kim S.H., Kwon S.H. Aeroside VIII is a natural selective HDAC6 inhibitor that synergistically enhances the anticancer activity of HDAC inhibitor in HT29 cells. Planta Med. 2015;81:222–227. doi: 10.1055/s-0034-1396149. [DOI] [PubMed] [Google Scholar]
- 93.Jones P., Altamura S., Chakravarty P.K., Cecchetti O., de Francesco R., Gallinari P., Ingenito R., Meinke P.T., Petrocchi A., Rowley M., et al. A series of novel, potent, and selective histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2006;16:5948–5952. doi: 10.1016/j.bmcl.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 94.Maulucci N., Chini M.G., Micco S.D., Izzo I., Cafaro E., Russo A., Gallinari P., Paolini C., Nardi M.C., Casapullo A., et al. Molecular Insights into Azumamide E Histone Deacetylases Inhibitory Activity. J. Am. Chem. Soc. 2007;129:3007–3012. doi: 10.1021/ja0686256. [DOI] [PubMed] [Google Scholar]
- 95.De Schepper S., Bruwiere H., Verhulst T., Steller U., Andries L., Wouters W., Janicot M., Arts J., vanHeusden J. Inhibition of histone deacetylases by chlamydocin induces apoptosis and proteasome-mediated degradation of survivin. J. Pharmacol. Exp. Ther. 2003;304:881–888. doi: 10.1124/jpet.102.042903. [DOI] [PubMed] [Google Scholar]
- 96.Itazaki H., Nagashima K., Sugita K., Yoshida H., Kawamura Y., Yasuda Y., Matsumoto K., Ishii K., Uotani N., Nakai H., et al. Isolation and structural elucidation of new cyclotetrapeptides, trapoxins A and B, having detransformation activities as antitumor agents. J. Antibiot. 1990;43:1524–1532. doi: 10.7164/antibiotics.43.1524. [DOI] [PubMed] [Google Scholar]
- 97.Li L., Dai H.J., Ye M., Wang S.L., Xiao X.J., Zheng J., Chen H.Y., Luo Y.H., Liu J. Lycorine induces cell-cycle arrest in the G0/G1 phase in K562 cells via HDAC inhibition. Cancer Cell Int. 2012;12:49. doi: 10.1186/1475-2867-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Parolin C., Calonghi N., Presta E., Boga C., Caruana P., Naldi M., Andrisano V., Masotti L., Sartor G. Mechanism and stereoselectivity of HDAC I inhibition by (R)-9-hydroxystearic acid in colon cancer. Biochim. Biophys. Acta. 2012;1821:1334–1340. doi: 10.1016/j.bbalip.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 99.Ghosh A.K., Kulkarni S. Enantioselective total synthesis of (+)-largazole, a potent inhibitor of histone deacetylase. Org. Lett. 2008;10:3907–3909. doi: 10.1021/ol8014623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Druesne N., Pagniez A., Mayeur C., Thomas M., Cherbuy C., Duee P.H., Martel P., Chaumontet C. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis. 2004;25:1227–1236. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- 101.Lea M.A., Rasheed M., Randolph V.M., Khan F., Shareef A., desBordes C. Induction of histone acetylation and inhibition of growth of mouse erythroleukemia cells by S-allylmercaptocysteine. Nutr. Cancer. 2002;43:90–102. doi: 10.1207/S15327914NC431_11. [DOI] [PubMed] [Google Scholar]
- 102.Ververis K., Hiong A., Karagiannis T.C., Licciardi P.V. Histone deacetylase inhibitors (HDACIs): Multitargeted anticancer agents. Biologics. 2013;7:47–60. doi: 10.2147/BTT.S29965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Robertson F.M., Chu K., Boley K.M., Ye Z., Liu H., Wright M.C., Moraes R., Zhang X., Green T.L., Barsky S.H., et al. The class I HDAC inhibitor romidepsin targets inflammatory breast cancer tumor emboli and synergizes with paclitaxel to inhibit metastasis. J. Exp. Ther. Oncol. 2013;10:219–233. [PubMed] [Google Scholar]
- 104.Paller C.J., Wissing M.D., Mendonca J., Sharma A., Kim E., Kim H.S., Kortenhorst M.S.Q., Gerber S., Rosen M., Shaikh F., et al. Combining the pan-aurora kinase inhibitor AMG 900 with histone deacetylase inhibitors enhances antitumor activity in prostate cancer. Cancer Med. 2014;3:1322–1335. doi: 10.1002/cam4.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ferrarelli L.K. HDAC inhibitors in solid tumors and blood cancers. Sci. Signal. 2016;9:ec216. doi: 10.1126/scisignal.aaj2316. [DOI] [Google Scholar]
- 106.Vancurova I., Uddin M.M., Zou Y., Vancura A. Combination Therapies Targeting HDAC and IKK in Solid Tumors. Trends Pharmacol. Sci. 2018;39:295–306. doi: 10.1016/j.tips.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marks P.A., Breslow R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 108.Giaccone G., Rajan A., Berman A., Kelly R.J., Szabo E., Lopez-Chavez A., Trepel J., Lee M.J., Cao L., Espinoza-Delgado I., et al. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J. Clin. Oncol. 2011;29:2052–2059. doi: 10.1200/JCO.2010.32.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Duvic M., Dummer R., Becker J.C., Poulalhon N., Ortiz Romero P., Grazia Bernengo M., Lebbé C., Assaf C., Squier M., Williams D., et al. Panobinostat activity in both bexarotene- exposed and -naive patients with refractory cutaneous T-cell lymphoma: Results of a phase II trial. Eur. J. Cancer. 2013;49:386–394. doi: 10.1016/j.ejca.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 110.Rambaldi A., Dummer R., Becker J.C., Poulalhon N., Ortiz R.P., Grazia B.M., Lebbe C., Assaf C., Squier M., Williams D., et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br. J. Haematol. 2010;150:446–455. doi: 10.1111/j.1365-2141.2010.08266.x. [DOI] [PubMed] [Google Scholar]
- 111.Razak A.R., Hotte S.J., Siu L.L., Chen E.X., Hirte H.W., Powers J., Walsh W., Stayner L.A., Laughlin A., Novotny-Diermayr V., et al. Phase I clinical, pharmacokinetic and pharmacodynamic study of SB939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br. J. Cancer. 2011;104:756–762. doi: 10.1038/bjc.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pili R., Salumbides B., Zhao M., Altiok S., Qian D., Zwiebel J., Carducci M.A., Rudek M.A. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br. J. Cancer. 2012;106:77–84. doi: 10.1038/bjc.2011.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Banerji U., van Doorn L., Papadatos-Pastos D., Kristeleit R., Debnam P., Tall M., Stewart A., Raynaud F., Garrett M.D., Toal M., et al. A phase I pharmacokinetic and pharmacodynamics study of CHR-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin. Cancer Res. 2012;18:2687–2694. doi: 10.1158/1078-0432.CCR-11-3165. [DOI] [PubMed] [Google Scholar]
- 114.Dong M., Ning Z.Q., Xing P.Y., Xu J.L., Cao H.X., Dou G.F., Meng Z.Y., Shi Y.K., Lu X.P., Feng F.Y. Phase I study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemother. Pharmacol. 2012;69:1413–1422. doi: 10.1007/s00280-012-1847-5. [DOI] [PubMed] [Google Scholar]
- 115.Eckschlager T., Plch J., Stiborova M., Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017;18:1414. doi: 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Younes A., Oki Y., Bociek R.G., Kuruvilla J., Fanale M., Neelapu S., Copeland A., Buglio D., Galal A., Besterman J., et al. Mocetinostat for relapsed classical Hodgkin’s lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2011;12:1222–1228. doi: 10.1016/S1470-2045(11)70265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Coiffier B., Pro B., Prince H.M., Foss F., Sokol L., Greenwood M., Caballero D., Borchmann P., Morschhauser F., Wilhelm M., et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012;30:631–636. doi: 10.1200/JCO.2011.37.4223. [DOI] [PubMed] [Google Scholar]
- 118.Bolden J.E., Peart M.J., Johnstone R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 119.Bao L., Diao H., Dong N., Xu S., Wang B., Mo Q., Yu H., Wang X., Chen C. Histone deacetylase inhibitor induces cell apoptosis and cycle arrest in lung cancer cell via mitochondrial injury and p53 up-acetylation. Cell Biol. Toxicol. 2016;32:469–482. doi: 10.1007/s10565-016-9347-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kretsovali A., Hadjimichael C., Charmpilas N. Histone deacetylase inhibitors in cell pluripotency, differentiation, and reprogramming. Stem Cells Int. 2012;2012:184154. doi: 10.1155/2012/184154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vrana J.A., Decker R.H., Johnson C.R., Wang Z., Jarvis W.D., Richon V.M., Ehinger M., Fisher P.B., Grant S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene. 1999;18:7016–7025. doi: 10.1038/sj.onc.1203176. [DOI] [PubMed] [Google Scholar]
- 122.Richon V.M., Sandhoff T.W., Rifkind R.A., Marks P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xu W.S., Parmigiani R.B., Marks P.A. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 124.Singh A.K., Kumar R., Pandey A.K. Hepatocellular carcinoma: Causes, mechanism of progression and biomarkers. Curr. Chem. Genom. Transl. Med. 2018 doi: 10.2174/2213988501812010009. Accepted-in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hitomi T., Matsuzaki Y., Yokota T., Takaoka Y., Sakai T. p15 (INK4b) in HDAC inhibitor-induced growth arrest. FEBS Lett. 2003;554:347–350. doi: 10.1016/S0014-5793(03)01186-4. [DOI] [PubMed] [Google Scholar]
- 126.Suzuki T., Yokozaki H., Kuniyasu H., Hayashi K., Naka K., Ono S., Ishikawa T., Tahara E., Yasui W. Effect of trichostatin A on cell growth and expression of cell cycle- and apoptosis-related molecules in humangastric and oral carcinoma cell lines. Int. J. Cancer. 2000;88:992–997. doi: 10.1002/1097-0215(20001215)88:6<992::AID-IJC24>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 127.Nakata S., Yoshida T., Horinaka M., Shiraishi T., Wakada M., Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene. 2004;23:6261–6271. doi: 10.1038/sj.onc.1207830. [DOI] [PubMed] [Google Scholar]
- 128.Glick R.D., Swendeman S.L., Coffey D.C., Rifkind R.A., Marks P.A., Richon V.M., Michael P., La-Quaglia M.P. Hybrid polar histone deacetylase inhibitor induces apoptosis and CD95/ CD95 ligand expression in human neuroblastoma. Cancer Res. 1999;59:4392–4399. [PubMed] [Google Scholar]
- 129.Insinga A., Monestiroli S., Ronzoni S., Gelmetti V., Marchesi F., Viale A., Altucci L., Nervi C., Minucci S., Pelicci P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005;11:71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 130.Zhao Y., Tan J., Zhuang L., Jiang X., Liu E.T., Yu Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA. 2005;102:16090–16095. doi: 10.1073/pnas.0505585102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ruefli A.A., Bernhard D., Tainton K.M., Kofler R., Smyth M.J., Johnstone R.W. Suberoylanilide hydroxamic acid (SAHA) overcomes multidrug resistance and induces cell death in P-glycoprotein-expressing cells. Int. J. Cancer. 2002;99:292–298. doi: 10.1002/ijc.10327. [DOI] [PubMed] [Google Scholar]
- 132.Sharma A.K., Kumar S., Chashoo G., Saxena A.K., Pandey A.K. Cell cycle inhibitory activity of Piper longum against A549 cell line and its protective effect against metal-induced toxicity in rats. Ind. J. Biochem. Biophys. 2014;51:358–364. [PubMed] [Google Scholar]
- 133.Rosato R.R., Maggio S.C., Almenara J.A., Payne S.G., Atadja P., Spiegel S., Dent P., Grant S. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid sphingomyelinase-dependent generation of ceramide. Mol. Pharmacol. 2006;69:216–225. doi: 10.1124/mol.105.017145. [DOI] [PubMed] [Google Scholar]
- 134.Shao Y., Gao Z., Marks P.A., Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA. 2004;101:18030–18035. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang J., Ng S., Wang J., Zhou J., Tan S.H., Yang N., Lin Q., Xia D., Shen H.M. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy. 2015;11:629–642. doi: 10.1080/15548627.2015.1023981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hrzenjak A., Kremser M.L., Strohmeier B., Moinfar F., Zatloukal K., Denk H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008;216:495–504. doi: 10.1002/path.2434. [DOI] [PubMed] [Google Scholar]
- 137.Liu Y.L., Yang P.M., Shun C.T., Wu M.S., Weng J.R., Chen C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy. 2010;6:1057–1065. doi: 10.4161/auto.6.8.13365. [DOI] [PubMed] [Google Scholar]
- 138.Shulak L., Beljanski V., Chiang C., Dutta M., Van Grevenynghe J., Belgnaoui S.M., Nguyen L., Di Lenardo T., Semmes O.J., Lin R., et al. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-κB-dependent autophagy. J. Virol. 2014;88:2927–2940. doi: 10.1128/JVI.03406-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Oh M., Choi I.K., Kwon H.J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 2008;369:1179–1183. doi: 10.1016/j.bbrc.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 140.Chiao M.T., Cheng W.Y., Yang Y.C., Shen C.C., Ko J.L. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy. 2013;9:1509–1526. doi: 10.4161/auto.25664. [DOI] [PubMed] [Google Scholar]
- 141.Zupkovitz G., Tischler J., Posch M., Sadzak I., Ramsauer K., Egger G., Grausenburger R., Schweifer N., Chiocca S., Decker T., et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol. Cell Biol. 2006;26:7913–7928. doi: 10.1128/MCB.01220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liang D., Kong X., Sang N. Effects of histone deacetylase inhibitors on HIF-1. Cell Cycle. 2006;5:2430–2435. doi: 10.4161/cc.5.21.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jeong J.W., Bae M.K., Ahn M.Y., Kim S.H., Sohn T.K., Bae M.H., Yoo M.A., Song E.J., Lee K.J., Kim K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111:709–720. doi: 10.1016/S0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 144.Qian D.Z., Kachhap S.K., Collis S.J., Verheul H.M., Carducci M.A., Atadja P., Pili R. Class II histone deacetylases arenassociated with VHL-independent regulation of hypoxiainducible factor 1{alpha} Cancer Res. 2006;66:8814–8821. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 145.Fath D.M., Kong X., Liang D., Lin Z., Chou A., Jiang Y., Fang J., Caro J., Sang N. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J. Biol. Chem. 2006;281:13612–13619. doi: 10.1074/jbc.M600456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Deroanne C.F., Bonjean K., Servotte S., Devy L., Colige A., Clausse N., Blacher S., Verdin E., Foidart J.M., Nusgens B.V., et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene. 2002;21:427–436. doi: 10.1038/sj.onc.1205108. [DOI] [PubMed] [Google Scholar]
- 147.Cinatl J., Kotchetkov R., Blaheta R., Driever P.H., Vogel J.U., Cinatl J. Induction of differentiation and suppression of malignant phenotype of human neuroblastoma BE(2)-C cells by valproic acid: Enhancement by combination with interferon α. Int. J. Oncol. 2002;20:97–106. doi: 10.3892/ijo.20.1.97. [DOI] [PubMed] [Google Scholar]
- 148.Sharma U.K., Kumar R., Gupta A., Ganguly R., Pandey A.K. Renoprotective effect of cinnamaldehyde in food color induced toxicity. 3 Biotech. 2018;8:212. doi: 10.1007/s13205-018-1241-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sharma U.K., Sharma A.K., Gupta A., Kumar R., Pandey A., Pandey A.K. Pharmacological activities of cinnamaldehyde and eugenol: antioxidant, cytotoxic and anti-leishmanial studies. Cell. Mol. Biol. (Noisy-le-grand) 2017;63:73–78. doi: 10.14715/cmb/2017.63.6.15. [DOI] [PubMed] [Google Scholar]
- 150.Kumar S., Pandey S., Pandey A.K. In vitro antibacterial, antioxidant, cytotoxic activities of Parthenium hysterophorus and characterization of extracts by LC MS analysis. BioMed Res. Int. 2014;2014:495154. doi: 10.1155/2014/495154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sharma A.K., Sharma U.K., Pandey A.K. Protective effect of Bauhinia variegata leaf extracts against oxidative damage, cell proliferation and bacterial growth. Proc. Natl. Acad. Sci. India, Sect. B Biol. Sci. 2017;87:47–51. doi: 10.1007/s40011-015-0578-x. [DOI] [Google Scholar]
- 152.Rosato R.R., Almenara J.A., Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003;63:3637–3645. [PubMed] [Google Scholar]
- 153.Ruefli A.A., Ausserlechner M.J., Bernhard D., Sutton V.R., Tainton K.M., Kofler R., Smyth M.J., Johnstone R.W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroximic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA. 2001;98:10833–10838. doi: 10.1073/pnas.191208598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kumar S., Pandey A.K. Free radicals: Health implications and their mitigation by herbals. Br. J. Med. Med. Res. 2015;7:438–457. doi: 10.9734/BJMMR/2015/16284. [DOI] [Google Scholar]
- 155.Xu W., Ngo L., Perez G., Dokmanovic M., Marks P.A. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc. Natl. Acad. Sci. USA. 2006;103:15540–15545. doi: 10.1073/pnas.0607518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Saitoh M., Nishitoh H., Fujii M., Takeda K., Tobiume K., Sawada Y., Kawabata M., Miyazono K., Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cimini D., Mattiuzzo M., Torosantucci L., Degrassi F. Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol. Biol. Cell. 2003;14:3821–3833. doi: 10.1091/mbc.e03-01-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kumar S., Chashoo G., Saxena A.K., Pandey A.K. Parthenium hysterophorus: A Probable Source of Anticancer, Antioxidant and Anti-HIV Agents. BioMed Res. Int. 2013;2013:810734. doi: 10.1155/2013/810734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Khabele D. The Therapeutic Potential of Class I Selective Histone Deacetylase Inhibitors in Ovarian Cancer. Front. Oncol. 2014;4:111. doi: 10.3389/fonc.2014.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Santoro F., Botrugno O.A., Dal Z.R., Pallavicini I., Matthews G.M., Cluse L., Barozzi I., Senese S., Fornasari L., Moretti S., et al. dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood. 2013:3459–3468. doi: 10.1182/blood-2012-10-461988. [DOI] [PubMed] [Google Scholar]