

A novel role for S100B, a recognized brain distress marker, as a chaperone-like suppressor of Aβ42 aggregation and toxicity.
Abstract
Amyloid-β (Aβ) aggregation and neuroinflammation are consistent features in Alzheimer’s disease (AD) and strong candidates for the initiation of neurodegeneration. S100B is one of the most abundant proinflammatory proteins that is chronically up-regulated in AD and is found associated with senile plaques. This recognized biomarker for brain distress may, thus, play roles in amyloid aggregation which remain to be determined. We report a novel role for the neuronal S100B protein as suppressor of Aβ42 aggregation and toxicity. We determined the structural details of the interaction between monomeric Aβ42 and S100B, which is favored by calcium binding to S100B, possibly involving conformational switching of disordered Aβ42 into an α-helical conformer, which locks aggregation. From nuclear magnetic resonance experiments, we show that this dynamic interaction occurs at a promiscuous peptide-binding region within the interfacial cleft of the S100B homodimer. This physical interaction is coupled to a functional role in the inhibition of Aβ42 aggregation and toxicity and is tuned by calcium binding to S100B. S100B delays the onset of Aβ42 aggregation by interacting with Aβ42 monomers inhibiting primary nucleation, and the calcium-bound state substantially affects secondary nucleation by inhibiting fibril surface–catalyzed reactions through S100B binding to growing Aβ42 oligomers and fibrils. S100B protects cells from Aβ42-mediated toxicity, rescuing cell viability and decreasing apoptosis induced by Aβ42 in cell cultures. Together, our findings suggest that molecular targeting of S100B could be translated into development of novel approaches to ameliorate AD neurodegeneration.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia in the aging population, resulting in a progressive decline of cognitive functions. Pathological hallmarks include senile plaques, neurofibrillary tangles, and severe astrogliosis in the cerebral cortex and hippocampus. The extracellular senile plaques are formed by amyloid-β (Aβ) peptide deposits, notably composed of the 1–42 variant (Aβ42), as a result of a pathological cascade of conversion of monomeric Aβ into oligomers and fibrils. The amyloid cascade is a highly complex phenomenon which develops very gradually over decades prior to clinical manifestations (1). Therefore, Aβ aggregation must involve age-related biochemical changes within the extracellular milieu.
An increased neuroinflammatory response is a further characteristic feature of AD, and its role in disease is not yet well understood. Microglia and reactive astrocytes are the main cellular components found around and within the senile plaques (2). Exogenous Aβ induces massive production of cytokines and chemokines by astrocytes, resulting in autocrine activation (3) and persistent secretion of proinflammatory cytokines, up-regulation of amyloid precursor protein (APP) mRNA, and enhanced Aβ generation. The cross-talk between Aβ, astrocytes, and cytokines creates a vicious cycle in AD immunopathology (3). The proinflammatory protein S100B is one of these cytokines [or damage associated molecular patterns (DAMPs)] that is highly up-regulated in AD (4, 5) and is released by astrocytes in the close vicinity of Aβ deposits (6). The molecular details of the relationship between aggregation and inflammatory cascades in AD, however, remain to be fully elucidated.
S100B is expressed primarily by astrocytes and is one of the most abundant soluble proteins in the brain (0.5%) (7). S100B is a small dimeric metal-binding protein whose structure and functional regulatory interactions with other proteins are modulated by calcium binding through two EF-hand motifs and by zinc binding to ligands at the protein dimer interface (8). We and others have established that these structural changes in S100 proteins expose regions with high potential to trigger aggregation reactions and to influence the aggregation of other proteins (9–11). S100B functions are linked to the dynamic and broad range of physiological concentrations in which this protein is found. At low concentrations (nanomolar), S100B acts as a prosurvival and differentiation factor on neurons, promoting neurite extension and protecting neurons during development against cellular stress. However, as a response to aging processes or tissue damage, S100B reaches high extracellular concentrations (micromolar) and acts as a proinflammatory cytokine, for example, via signaling through the RAGE receptor, resulting in a positive feedback cycle via nuclear factor κB activation (12).
The role of accumulating S100B near amyloid plaques remains unclear. However, there are several studies demonstrating conflicting results regarding the contribution of increased S100B levels in AD models. Exogenous Aβ can stimulate S100B production in primary astrocyte cultures and glioma cells (13). However, in AD transgenic mice, overexpression of S100B occurs months before the formation of Aβ plaques (14). AD transgenic mice overexpressing S100B exhibit elevated Aβ levels as well as an overall increased levels of proinflammatory cytokines (15) and are more susceptible to neuronal dysfunction, although no changes were noted in Aβ plaques (16). However, a reduction of S100B expression in AD transgenic mice leads to reduction of amyloid plaques and gliosis (17), with the exception of plaques localized in the hippocampus (18). Although several studies indicate that S100B may have a noxious role in AD, others suggest that this toxic role is dependent on its concentration and that low levels of S100B protect neuronal cell lines against Aβ neurotoxicity (19). These observations led us to hypothesize that S100B may interact with the Aβ peptide and play an active role in Aβ aggregation. Here, we report that S100B binds Aβ42; we demonstrate a novel regulatory effect of S100B on Aβ42 aggregation, suggesting a new chaperone-like function for the protein S100B.
RESULTS
Monomeric Aβ42 interacts preferentially with calcium-bound S100B
Considering the high extracellular levels of S100B and Aβ42 in AD and the fact that S100B colocalizes with plaques, we postulated that these two proteins physically interact. From isothermal titration calorimetry (ITC) experiments, we observed that titration of monomeric Aβ42 with S100B yielded an exothermic process indicative of an interaction, which could not, however, be reliably quantitatively fitted to a binding model (fig. S1). Because calcium binding to S100B promotes interactions with other proteins and peptides (20), we investigated the interaction of Aβ42 with Ca2+-S100B. In this case, titration of monomeric Aβ42 with Ca2+-S100B resulted in an endothermic isotherm whose fitting to the simplest one binding site model describes the formation of a tight complex [dissociation constant (Kd) ≈ 0.6 μM] (Fig. 1A). The Aβ42/S100B complex is in excellent agreement with those determined for other S100B-binding peptides (21, 22). The interaction between monomeric Aβ42 and S100B was also analyzed using biolayer interferometry (BLI), from which the binding and dissociation rate constants of complex formation were determined, corroborating the formation of a dynamic Aβ42/S100B complex, both in the presence and absence of calcium (fig. S2).
Fig. 1. S100B binds to Aβ42.
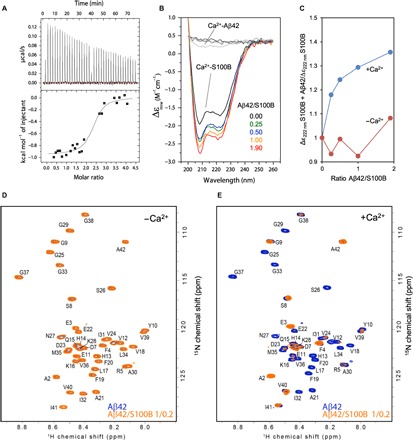
(A) Isothermal titration calorimetry analysis of S100B binding to Aβ42 in the presence of calcium. The upper panel shows the raw heats of binding, and the lower panel shows the integrated data obtained after subtracting the heat of dilution from the buffer. Data fitting was performed with the nonlinear regression analyses of one binding site model obtaining the thermodynamic parameters N = 2.37 ± 0.07, Kd = 0.59 ± 0.23 μM, ΔH = −947.1 ± 29.48 cal mol−1, and ΔS = 25.3 cal mol−1 deg−1. (B) Far UV-CD spectra of 4 μM Ca2+-S100B alone (black) and with a molar ratio Aβ42/S100B of 0.25 (green), 0.5 (blue), 1 (orange), and 1.9 (red) after incubation overnight at 4°C. (C) Ratio of ellipticity at 222 nm of S100B alone and S100B + Aβ42 in the presence (blue) and absence (red) of 1.1 mM CaCl2. (D) Heteronuclear single-quantum coherence (HSQC) spectra of 100 μM 15N-labeled Aβ42 in the absence (blue) and in the presence (orange) of 20 μM S100B. (E) HSQC spectra of 100 μM 15N-Aβ42 in the absence (blue) and in the presence (orange) of 20 μM S100B with 10 mM CaCl2. ppm, parts per million.
To evaluate conformational changes arising upon interaction of Aβ42 with S100B, we carried out titration experiments monitored by far ultraviolet circular dichroism (far UV-CD) spectroscopy. The far UV-CD spectrum of S100B is typical of an α-helical protein, in agreement with its crystallographic structure (8). Addition of up to twofold of freshly prepared monomeric Aβ42 to S100B results in a change in ellipticity at 222 nm, upon overnight incubation at 4°C, which is compatible with a gain in α-helical structure (Fig. 1B). Under the tested conditions, the low concentration of Aβ42 used (from 4.5 to 34 ng/ml) did not contribute to any signal in the far UV-CD spectrum, as shown in control measurements. Identical experiments performed in the absence of calcium resulted in only small changes in ellipticity, and clear effects were limited to the highest Aβ42/S100B ratios tested (Fig. 1C and fig. S3). Overall, the CD data support the possibility of a protein-protein interaction, and the observed increase in α-helical content of the complex might arise from an α helix formed in Aβ42, presumably on a longer time scale upon interaction with Ca2+-S100B. Note that upon interaction with Ca2+-S100B, the natively disordered p53 regulatory domain protein also acquires an α-helical conformation (23). Consistent with these findings, the Aβ peptide is known to undergo conformational switching (24) and to adopt α-helical conformations in hydrophobic environments (25).
To further characterize the structural details of this interaction, we used solution nuclear magnetic resonance (NMR) spectroscopy. First, unlabeled S100B was titrated into a 15N-labeled sample of Aβ42. In the absence of calcium, no interaction was observed at a molar ratio of 1:0.2 (Fig. 1D), or up to 1:1. However, with the addition of 0.1 equivalent of Ca2+-S100B, many Aβ42 peaks disappeared from the spectrum, and at 0.2 equivalent, all peaks except those corresponding to the Aβ42 termini had broadened beyond detection (Fig. 1E). Thus, it could not be determined whether Aβ42 forms an α-helical conformation upon binding. However, this result is indicative of a global interaction of Aβ42 with S100B, in agreement with previous experiments. We further speculated that the peak disappearances might also be caused either by the formation of a multimeric complex that is too large to be detected in the NMR experiments (although no precipitation was visible in the NMR titration experiments) or, alternatively, by an exchange rate that is in the intermediate regime on the NMR chemical shift time scale, which leads to peak broadening (26). Subsequent experiments have ruled out both possibilities. First, to investigate whether aggregates or a large complex is formed, small-angle x-ray scattering (SAXS) experiments were performed on S100B in the presence and absence of Aβ42. The scattering curves and pair-distance distribution plots clearly showed that S100B remains homodimeric in the presence of Aβ42 (fig. S4). We calculated the molecular weight of Ca2+-S100B to be 22,900 Da, which is in good agreement with its mass of 21,600 Da. The addition of the Aβ42 peptide leads to a shift of the pair-distance distribution function that corresponds to an increase of the molecular weight to 24,300 Da, which is in excellent agreement with an Aβ42/S100B complex. Larger S100B oligomers or aggregates were not detected. Since the maximum distance (Dmax) does not change, we conclude that the binding of the peptide occurs around the center of the S100B dimer, rather than at the outer edges of the structure. Aβ42 was not added in excess for the SAXS measurements to prevent formation of Aβ42 aggregates, which would complicate the SAXS data analysis. The SAXS data show that the Aβ42/S100B complex is in intermediate exchange and no higher–molecular weight species is formed, in agreement with the BLI binding kinetics data. Second, we attempted to observe the complex by NMR spectroscopy at other chemical exchange regimes by changing field strength (500 to 900 MHz) and by varying the temperature (0° to 45°C), but no improvement in the spectra of the complex was obtained.
Aβ42 binds S100B at a promiscuous peptide-binding region within the interfacial cleft
To investigate the binding of Aβ42 to S100B, we performed NMR HSQC titrations by addition of unlabeled Aβ42 to 15N-labeled S100B. In the absence of calcium, no change was observed in the S100B spectra even when a twofold excess of Aβ42 was added (Fig. 2A). Titration in the presence of calcium was consistent with the reverse NMR titration, in that many peaks disappeared with the addition of Aβ42 in the range of 0.2 to 2 molar equivalents (Fig. 2B). Assignment of the S100B HSQC spectrum was achieved by transfer of chemical shift values from the Biological Magnetic Resonance Bank data set 18995, and confirmed using a three-dimensional, 15N-separated NOESY, allowing assignment of approximately 92% of the backbone amides (27). The peak intensity ratios in the presence and absence of twofold Aβ42 were plotted and are marked as a heat map on the S100B structure (Fig. 2C). Most of the residues which undergo large variations upon Aβ42 binding occur in helix IV, where a previously described peptide-binding region is located (20) and which also corresponds to a segment with a higher aggregation score (9). Changes in residues in other parts of the structure, such as at the N terminus, indicate that minor overall structural changes in S100B may take place upon Aβ42 binding. However, SAXS analysis confirms that no major conformational changes in the S100B occur upon binding of Aβ42 (Fig. 2D). The binding region is also supported by the fact that there is a weaker interaction in the absence of calcium, as the binding of calcium causes the peptide-binding pocket to become accessible in the S100B structure. Therefore, our data show that Aβ42 binds to S100B in a calcium-facilitated fashion, at a promiscuous peptide-binding region located within the interfacial cleft.
Fig. 2. Mapping the interaction of Aβ42 with S100B.
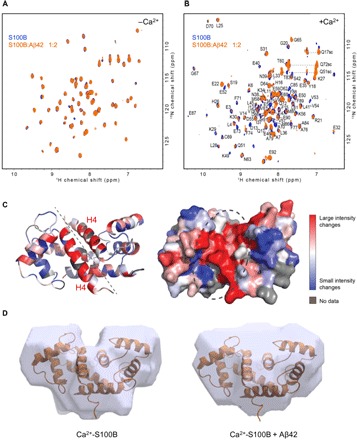
(A) HSQC spectra of 100 μM 15N-S100B in the absence (blue) and in the presence (orange) of 50 μM Aβ42. (B) HSQC spectra of 100 μM 15N-labeled S100B in the absence (blue) and in the presence (orange) of 50 μM Aβ42 with 10 mM CaCl2. (C) Structure of Ca2+-S100B [Protein Data Bank (PDB) code: 2H61], color-coded by the degree of difference in peak intensities in the absence or presence of Aβ42. Unassigned residues are colored gray, while residues that correspond to peaks that undergo large intensity changes are colored red, and residues that correspond to peaks that change little or not at all are colored blue, with a gradient for values in between. Left: Ribbon representation. Dashed line indicates the interfacial cleft. Right: Surface representation at the same orientation. Dashed circle highlights the putative Aβ42 binding region. (D) SAXS-based structural model of S100B in the absence (left) and in the presence (right) of Aβ42, both in the presence of calcium. The surface represents the best SAXS models based on the fit between the experimental data and the back-calculated SAXS data and have been overlapped with the experimental NMR structure (PDB code: 2K7O).
S100B delays Aβ42 fibrillation through modulation of primary and secondary nucleation
We then hypothesized that the interaction between S100B and Aβ42 would influence Aβ42 aggregation. To test this possibility, we carried out a quantitative global kinetic analysis of the effects of S100B on Aβ42 aggregation. We performed extensive Thioflavin-T (ThT) monitored kinetic analysis of Aβ42 aggregation (5 μM) at a range of S100B concentrations (1 to 200 μM) under quiescent conditions at physiological pH (pH 7.4), in the presence and absence of calcium (Fig. 3, A and B). We found that S100B decreased the rate of Aβ42 aggregation, under both the absence and presence of calcium. The Aβ42 aggregation reaction involves primary processes with nucleation of monomers followed by secondary processes, whereby the surface of formed Aβ42 fibrils serves as a catalyst for the subsequent generation of toxic oligomeric Aβ42 species, which then mature and convert into additional fibrils (Fig. 3C) (28). The kinetic analysis of these pathways and determination of the Aβ42 aggregation microscopic reaction rates (kn, primary nucleation; k+, elongation; and k2, secondary nucleation) in the presence of S100B allow for elucidation of how the interaction between Aβ42 and S100B affects the aggregation of Aβ42.
Fig. 3. S100B modulates Aβ42 aggregation.
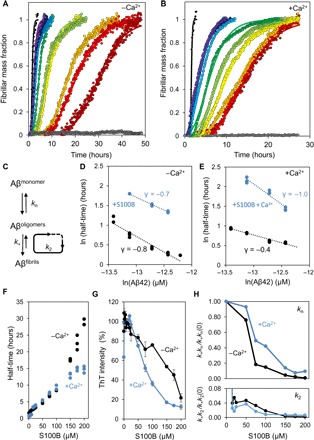
(A) Fibril formation of 5 μM Aβ42 (black) in 50 mM Hepes (pH 7.4) at 37°C under quiescent conditions in the presence of 10 μM (purple), 15 μM (dark blue), 20 μM (blue), 25 μM (light blue), 50 μM (dark green), 75 μM (light green), 100 μM (yellow), 150 μM (orange), 175 μM (red), and 200 μM (dark red) S100B. S100B (200 μM) alone, as control, is represented in gray. Plots represent averaged normalized intensity curves obtained from three independent replicates for each of the tested conditions. (B) Fibril formation of 10 μM Aβ42 (black) with 1.1 mM CaCl2 in 50 mM Hepes (pH 7.4) at 37°C under quiescent conditions in the presence of 10 μM (purple), 15 μM (dark blue), 20 μM (blue), 25 μM (light blue), 50 μM (dark green), 75 μM (light green), 100 μM (yellow), 150 μM (orange), 175 μM (red), and 200 μM (dark red) S100B. S100B (200 μM) alone, as control, is represented in gray. Plots represent averaged normalized intensity curves obtained from three independent replicates for each of the tested conditions. (C) Representative scheme of microscopic events occurring during Aβ42 aggregation: primary nucleation (kn) starting from monomers, elongation (k+) by monomer addiction to existing aggregates, and secondary nucleation (k2) from addition of monomers to fibril surface. (D) Log-log plot of the half-time of the Aβ42 aggregation reaction as a function of the initial Aβ42 monomer concentration in the absence (black) or in the presence (blue) of 15-fold excess of S100B. (E) Log-log plot of the half-time of the Aβ42 aggregation reaction as a function of the initial Aβ42 monomer concentration with 1.1 mM CaCl2 in the absence (black) or presence (blue) of 15-fold excess of S100B. (F) Half-times as function of increasing concentrations of S100B in the presence of Aβ42 with (blue) and without (black) 1.1 mM CaCl2. Dots represent values obtained from three independent replicates for each of the tested conditions. (G) Plot of ThT intensity of the end point of the aggregation kinetics in the presence of Aβ42 with increasing concentrations of S100B in the absence (black) and in the presence (blue) of 1.1 mM CaCl2. Dots represent averaged values obtained from three independent replicates for each of the tested conditions. (H) Plot of S100B-dependent change in microscopic rates of Aβ42 aggregation. The top panel represents the ratio of combination of kinetic rates describing primary nucleation (k+kn) with and without S100B in the absence (black) and in the presence (blue) of 1.1 mM CaCl2. The bottom panel represents the ratio of combination of kinetic rates describing secondary nucleation (k+k2) with and without S100B in the absence (black) and in the presence (blue) of 1.1 mM CaCl2. Microscopic rates were determined by individual fitting with the secondary nucleation model and with combined rate constants k+kn and k+k2 as free fitting parameters. Plots represent averaged values obtained from three independent replicates for each of the tested conditions.
To determine whether the effect of S100B implies a change of the Aβ42 aggregation mechanism, we determined the Aβ42 aggregation kinetics in a range of Aβ42 concentrations (1.5 to 5 μM) at pH 7.4, under quiescent conditions, and in the absence and presence of calcium and S100B (Fig. 3, D and E). The dependence of the reaction half-time (t1/2) on the initial concentration of monomeric Aβ42, represented in double-logarithmic plots, allowed the inference of the dominant reaction mechanism and the determination of the scaling exponent (γ), which is suggestive of the aggregation pathway involved (29).
Using this approach, we determined a scaling exponent γ = −0.8 for Aβ42 aggregation in the absence of S100B (Fig. 3D), which indicates that Aβ42 aggregation proceeds via a mechanism where secondary events dominate the generation of new fibrils, consistent with previously reported ranges of scaling exponents obtained under identical experimental conditions (28). In the presence of S100B, an increase in t1/2 is observed, but a linear plot is also obtained and the scaling exponent is unchanged, indicating that S100B delays Aβ42 aggregation without altering the dominant aggregation mechanism (Fig. 3D). To determine the effects of Ca2+-S100B on the process, we performed identical experiments in the presence of calcium. We carried out a control experiment in which we analyzed the influence of calcium on Aβ42 aggregation (Fig. 3E). We found that the scaling exponent increases (from γ = −0.7 to −0.4), indicating that calcium causes Aβ42 aggregation to be less dominated by secondary nucleation events. Under these conditions, the presence of S100B delays Aβ42 aggregation and imposes a significant decrease in the scaling exponent (from γ = −0.4 to −1.0), suggesting that Ca2+-S100B substantially affects secondary nucleation.
Analysis of the kinetic data revealed that, under both conditions, increasing concentrations of S100B result in a progressive delay of Aβ42 aggregation, with a corresponding increase in the t1/2 of the aggregation reaction of up to 30 times (Fig. 3F). Correspondingly, the presence of S100B results in a progressive decrease in the Aβ42 aggregation load, as inferred from decreased binding of ThT by the formed aggregates, which suggests an overall decrease in fibril mass. This effect on formed fibrils is enhanced by Ca2+-S100B, as under these conditions there is a substantial decrease in the amount of ThT-binding aggregates formed (Fig. 3G). A part of this effect may, however, be due to the fact that, since in the presence of calcium the Aβ42 aggregation is less dominated by secondary nucleation, there might be a decrease in the amount of formed ThT-binding fibrils.
Since the decrease in the slopes of the sigmoid transitions of Aβ42 aggregation in the presence of S100B and Ca2+- S100B (Fig. 3, A and B) is indicative of a delaying effect on the macroscopic aggregation rate, we then carried out a global fitting analysis to determine effects on the microscopic events during Aβ42 aggregation. To carry out this analysis, we determined the combination of kinetic constants describing the primary (knk+) and secondary (knk2) pathways from fits of Aβ42 aggregation kinetics to a secondary nucleation model (Fig. 3H). We observe that, irrespective of the presence of calcium, an overall decrease in microscopic rate constants takes place and that this decrease occurs mostly in secondary processes, whose rates decrease up to 160 times in the presence of S100B (Fig. 3H, bottom panel). The decrease in both microscopic events indicates a sequestration of Aβ42 monomers by S100B (30), in agreement with our results showing an interaction between Aβ42 and S100B. Furthermore, the observed effect on secondary nucleation processes consequently inhibits the catalytic cycle that leads to the formation of toxic oligomers, therefore potentially limiting Aβ42 toxicity (31).
We then investigated whether S100B has an inhibitory effect when Aβ42 aggregation proceeds via a mechanistic regime different from one dominated by secondary nucleation, as is observed under quiescent conditions. We therefore performed identical experiments under agitation, where the process of Aβ42 aggregation is known to be mechanistically dominated by fragmentation, which is a monomer-independent process, in which the rate depends on the concentration of existing fibrils (28). We observed that under these conditions, the presence of S100B also delays Aβ42 aggregation, decreases fibrillar mass, and likely affects secondary reactions presumably through binding to fibrils, as inferred from the observed increase in the scaling exponent (fig. S5). The effect of Ca2+-S100B is even more dramatic with a substantial suppression of formed fibril mass at a Aβ42/S100B molar ratio of 4:1 (fig. S6). These results confirm that S100B inhibit secondary processes during Aβ42 aggregation, irrespective of the aggregation pathway (secondary nucleation or fragmentation dominated). In addition, we also analyzed the impact of S100B on a shorter Aβ peptide fragment Aβ25–35, which is also present in the senile plaques, retains toxicity, and is also prone to β-aggregation. We observed that the presence of S100B results in a decrease in the mass of fibrils formed during the aggregation of Aβ25–35, as inferred from a decrease in the formation of ThT-reactive species (fig. S7).
S100B inhibits Aβ42 surface-catalyzed secondary nucleation and decreases toxicity
To further investigate the inhibition of secondary processes in Aβ42 aggregation by S100B, we performed seeding experiments under various conditions. To establish that S100B acts on Aβ42 fibrils, we performed experiments in which newly formed Aβ42 fibrils, produced in the presence and absence of S100B, were used to seed Aβ42 aggregation (Fig. 4). Pristine Aβ42 fibrils enhance secondary nucleation processes and increase aggregation rate of monomeric Aβ42 (Fig. 4A) (28, 31). However, when Aβ42 aggregation is measured in the presence of both Aβ42 fibril seeds and S100B, the seeding effect of these preformed fibrils is abolished and aggregation is delayed in proportion to S100B concentration (Fig. 4B). This finding suggests that S100B binds to preformed Aβ42 fibrils, as well as to Aβ42 monomers. To verify direct S100B binding to Aβ42 fibrils, we generated Aβ42 fibril seeds in the presence of S100B; when Aβ42 aggregation was measured in the presence of these seeds, a delay in aggregation was observed (Fig. 4C). This suppressing effect is further enhanced when S100B is added at both stages: in the production of Aβ42 fibril seeds and directly to Aβ42 monomers (Fig. 4D). These results suggest that the surface catalytic activity of Aβ42 fibrils is decreased when S100B is present, thus demonstrating the ability of S100B to arrest even ongoing Aβ42 fibrillation reactions. To obtain additional evidence for the interaction between S100B and Aβ42 fibrillar and oligomeric species, we used transmission electron microscopy (TEM) imaging with nanogold-conjugated anti-S100B and anti-Aβ42 antibodies in samples incubated for 24 hours at 37°C under quiescent conditions (Fig. 4E). Using this approach, we could identify S100B along the surface of Aβ42 fibrils as well as on immature Aβ42 oligomers (Fig. 4E, arrows), as predicted by our mechanistic observations of the effect of S100B over Aβ42 aggregation. Furthermore, we observed that, in the presence of S100B, Aβ42 fibrils were less abundant and have simpler structures. Although we detected long fibrils, these structures displayed no apparent twists or lateral assembly, and seemed to be formed by a single filament. To confirm the decrease in amyloid fibrils in the presence of S100B, we performed dot blot analysis with the conformational antibody OC that recognizes amyloid fibers (fig. S8). Addition of S100B leads to a decreased reactivity with antibody OC, supporting lower fibril content. With the A11 antibody, which detects prefibrillar oligomers, we have observed no signals in the dot blots, suggesting that these structures may be underrepresented at the reaction end points.
Fig. 4. S100B inhibits surface-catalyzed secondary nucleation of Aβ42 oligomers.
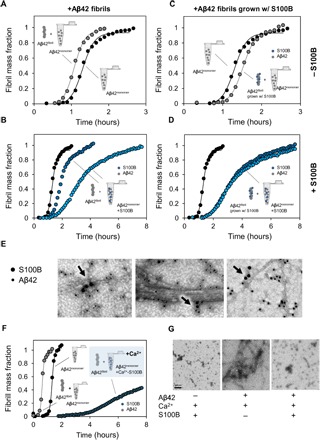
(A) Aggregation kinetics of 5 μM Aβ42 (black) seeded by the addition of 0.1 μM preformed Aβ42 fibrils (gray). (B) Aggregation kinetics of 5 μM Aβ42 (black) seeded by the addition of 0.1 μM preformed Aβ42 fibrils in the presence of 50 μM S100B (dark blue) and 150 μM S100B (light blue) in solution. (C) Aggregation kinetics of 5 μM Aβ42 (black) seeded by the addition of 0.1 μM preformed Aβ42 fibrils grown with S100B (gray). (D) Aggregation kinetics of 5 μM Aβ42 (black) seeded by the addition of 0.1 μM preformed Aβ42 fibrils grown with S100B in the presence of 50 μM S100B (dark blue) and 150 μM S100B (light blue) in solution. (E) TEM images with a nanogold-conjugated secondary antibody against S100B (15 nm) and Aβ42 (10 nm), showing binding of S100B to fibrils and oligomers. (F) Aggregation kinetics of 10 μM Aβ42 with and without the addition of 0.1 μM preformed Aβ42 fibrils in the presence of 50 μM Ca2+-S100B (dark blue) in solution. (G) TEM images of end points of ThT aggregation kinetics of 150 μM S100B, 10 μM Aβ42, and 10 μM Aβ42 + 150 μM S100B, with 1.1 mM CaCl2.
We also performed seeding experiments in the presence of calcium, as this condition evidenced a stronger suppression of Aβ42 aggregation. In agreement with experiments in the absence of calcium, when seeding is done in the presence of S100B and calcium, Aβ42 fibrils are also no longer able to seed Aβ42 aggregation, and the effect of Ca2+-S100B in suppressing fibrillation is even more pronounced than that observed in the absence of calcium (Fig. 4F). To confirm the effect of Ca2+-S100B in suppressing Aβ42 fibril formation, we performed TEM. We observed that incubation of Aβ42 with calcium results in the formation of typical oligomers and amyloid fibrils ca. 10 to 12 nm wide and several micrometers long, which do not differ from oligomers and fibrils formed in the absence of calcium (Fig. 4G and fig. S9). In agreement with its known aggregation propensity (32), Ca2+-S100B develops short protofilaments approximately 5 nm wide and oligomers of different sizes. However, no fibrillar aggregates are observed in a reaction mixture containing Aβ42, S100B, and calcium, thus corroborating a suppressing role of S100B on Aβ42 fibril formation (Fig. 4G). Overall, S100B suppresses secondary pathways of Aβ42 aggregation by engaging into noncovalent associations with growing aggregates and fibrils, a mechanism that has been described for canonical antiaggregation chaperones (31).
To gain insight into the possibility of a protective role of S100B, we next investigated whether S100B would rescue Aβ42-induced toxicity in neuronal cell cultures. Using neuronal SH-SY5Y cells, we combined the PrestoBlue assay to determine effects in cellular viability and the caspase-3/7 assay to measure effects on apoptotic processes. In these studies, differentiated SH-SY5Y cells were exposed to homogenous fractions of monomeric Aβ42 (7 μM) and cultured for 72 hours prior to analysis. The results of the control experiments reveal that after 72 hours, Ca2+-S100B (84 μM) does not significantly affect the cellular viability and the caspase-3/7 activity. In stark contrast, Aβ42 decreases cell viability by ~50% and induces a 3.5-fold increase in apoptotic enzymes, in agreement with its known toxicity. The presence of S100B results in a complete reversal of the toxic effects of Aβ42: Both viability and apoptosis levels of S100B-rescued cells match those of controls (Fig. 5, A and B). This result is in excellent agreement with the observed inhibitory effect of S100B over the Aβ42 microscopic reaction step that generates toxic oligomers and supports a chaperone-like role for S100B over Aβ42 aggregation.
Fig. 5. S100B protects SH-SY5Y cells against Aβ42 toxicity and apoptosis.
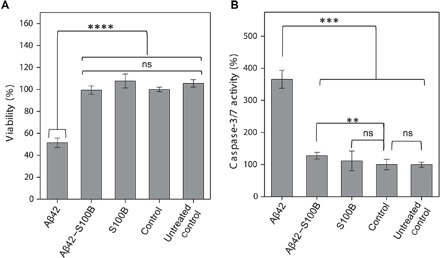
(A) Cell viability as measured by PrestoBlue reagent after 72 hours in differentiated SH-SY5Y cells for medium; buffer: 50 mM Hepes (pH 7.4) and 1.1 mM CaCl2, 7 μM Aβ42 with 1.1 mM CaCl2, and 7 μM Aβ42 + 84 μM S100B with 1.1 mM CaCl2. (B) Cell apoptosis as measured by caspase-3/7 activity after 72 hours in differentiated SH-SY5Y cells for medium; buffer: 50 mM Hepes (pH 7.4) and 1.1 mM CaCl2, 7 μM Aβ42 with 1.1 mM CaCl2, and 7 μM Aβ42 + 84 μM S100B with 1.1 mM CaCl2. Results represent mean and SD of two independent experiments (n = 8 and 5 wells per replicates per condition and plate). Statistically significant differences at the 95.0% confidence level using one-way analysis of variance (ANOVA) followed by Welch’s t test. ns, not significant; **P < 0.01, ***P < 0.001, ****P < 0.0001.
DISCUSSION
Recent research has uncovered a number of molecular chaperones capable of modulating Aβ42 aggregation through regulatory interactions that result in suppression of amyloid formation by acting on the microscopic mechanisms of amyloid formation reaction (31, 33). These molecular chaperones, such as the Brichos domain, underlie important regulatory processes in neuronal proteostasis through mechanisms that are now being uncovered (31, 33, 34). However, extracellular aggregation of Aβ42 in AD is a process that is also highly influenced by the biochemistry and composition of the synaptic environment in which the fibrillation reaction takes place, being influenced by metal ions, crowding effects, and extracellular proteins (35, 36). These phenomena, including regulatory interactions involving synaptic proteins and Aβ42, are particularly relevant when involving components of biological processes that are concurrently altered with the amyloid cascade. This is the case for the neuroinflammatory response, which involves the production and release of different proinflammatory cytokines, including S100B. Apart from regulating a number of intracellular functions, S100B is released by astrocytes and extracellular S100B has a dual effect, being neuroprotective and neurotrophic at nanomolar levels and neurotoxic at micromolar extracellular concentrations (37), as those found in AD and in the aging brain. Previous reports have described that, at nanomolar levels, S100B promotes survival of neuronal cells treated with neurotoxic amounts of Aβ (19). Immunohistochemical analysis of S100B expression in 5XFAD mice brains indeed confirms substantially elevated levels of S100B within plaques, in agreement with the higher extracellular levels of these two proteins in AD brains (Fig. 6A and fig. S10). However, the mechanisms by which S100B acts on modulating Aβ neurotoxicity remain unknown.
Fig. 6. S100B delays Aβ42 aggregation by suppressing primary and secondary nucleation.
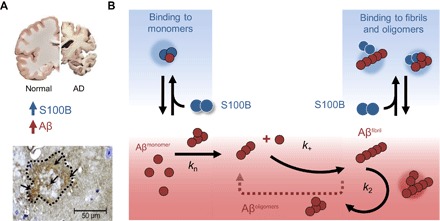
(A) In the AD brain, protein aggregation and exacerbated inflammation are well-established disease features, with elevated levels of Aβ42 and S100B. In AD animal models such as the 5XFAD mice, amyloid plaques (dotted contour) have intense staining for extracellular S100B (arrows) (see also fig. S7). (B) Extracellular S100B and Aβ42 engage in regulatory interactions, which are depicted in the scheme which summarizes the finding that S100B inhibits Aβ42 fibril formation mainly by affecting the lag phase and secondary nucleation, through interactions with monomeric and fibrillar Aβ42, suggesting a potential role of S100B as novel extracellular chaperone suppressing proteotoxicity in AD.
Here, we investigated the relationship between the aggregation and inflammatory cascades in AD, taking the premise that the extracellular accumulation of S100B at micromolar levels along with Aβ42 might result in regulatory functions of this proinflammatory cytokine. Using a combination of structural biophysical methods, we have monitored the progression of changes in a range of Aβ42/S100B ratios between 0.2 and 2. We established that S100B undergoes a dynamic interaction with monomeric Aβ42 at a binding affinity (Kd ≈ 0.6 μM) that is compatible with the physiological range of concentrations of the two proteins in the extracellular space. Moreover, we observed that the interaction is preferential with Ca2+-S100B, suggesting that calcium-induced conformational changes promote interactions within the S100B dimer interfacial cleft, in what seems to be a molecular mechanism common to other S100B-target protein interactions (38). NMR analysis allowed the mapping of Aβ42 binding to a promiscuous peptide-binding region of the S100B interfacial cleft. The fact that we observe that most Aβ42 NMR peaks disappear in the presence of Ca2+-S100B with the exception of those at the termini regions, supports the possibility that other pathophysiological Aβ fragments may engage into similar regulatory interactions. Far UV-CD studies suggested that Aβ42 undergoes a conformational change into an α-helical conformation upon interaction with S100B, which could not be confirmed by NMR due to fast relaxation. Nevertheless, this is, however, a rather plausible possibility considering that the adoption of a helical conformation by a disordered region in p53 has also been reported upon its binding to S100B (23). The interconversion of Aβ42 into a helical conformer has been associated with a decreased propensity for amyloid formation (39), and a β strand to α helical transition by Aβ42 upon dynamic interaction with S100B could account for the observed effect on delaying fibrillation.
The results of this study show that S100B targets the molecular mechanism of Aβ42 aggregation reaction due to complexation with monomeric and oligomeric Aβ42 (Fig. 6B). We find that S100B delays Aβ42 fibril formation in a concentration-dependent manner and completely inhibits fibrillation upon Aβ42 interaction at the S100B interfacial cleft. We note that brain S100 proteins such as S100A9 and S100A12 also affect Aβ42 aggregation, suggesting similar regulatory effects mediated through interactions within the interfacial cleft, which is present across the S100 family. Regarding S100B, we showed that S100B targets highly specific molecular processes in the aggregation pathway of Aβ42. When calcium is bound to S100B, secondary nucleation is mostly affected, which is predominantly responsible for the generation of toxic oligomers. However, since in the presence of calcium the Aβ42 aggregation is less dominated by secondary nucleation, this additional contribution for the overall observed effect must also be taken into account. The reduction of the population of oligomers produced through surface-catalyzed secondary nucleation and complexation with free monomers of Aβ42 defines a new potential role of S100B as a molecular chaperone. S100B both retards de novo aggregation and delays aggregation in the presence of preformed aggregates, representing an effective mechanism for reduction of toxicity associated with Aβ42 fibrillation at any step of the reaction. In agreement, the presence of Ca2+-S100B counteracts Aβ42 toxicity in neuronal cell models by restoring cellular viability and decreasing apoptosis. As suggested by others, the up-regulation of S100B expression after the administration of Aβ to cell cultures may be the result of a protective response toward the Aβ insult (19), and this may involve mechanisms like the ones reported here. In conclusion, our findings show that S100B inhibits Aβ42 fibril formation mainly by affecting the lag phase and secondary nucleation, suggesting a potential role of S100B as novel extracellular chaperone suppressing proteotoxicity in AD. Given the importance of S100 proteins in physiopathological conditions, it remains to be explored if other S100 proteins also play roles in cellular proteostasis processes.
MATERIALS AND METHODS
Materials
All reagents were of the highest grade commercially available. ThT was obtained from Sigma. A Chelex resin (Bio-Rad) was used to remove contaminant trace metals from all solutions. S100B was expressed in Escherichia coli and purified to homogeneity using a previously established protocol. Expression of 15N-labeled S100B was performed with minimal medium, and purification was performed with the same protocol for S100B (11). Apo S100B was prepared by incubation at 37°C for 2 hours with a 300-fold excess of dithiothreitol (DTT) and 0.5 mM EDTA and eluted in a Superdex S75 (GE Healthcare). Human Aβ42 expression plasmid, as in (40), was a gift from J. Presto (Karolinska Institutet, Sweden). Recombinant Aβ42 was expressed in E. coli and purified according to (40). To obtain the monomeric form, 1 mg of Aβ42 was dissolved in 7 M guanidine hydrochloride and eluted in a Superdex S75 (GE Healthcare) with 50 mM Hepes (pH 7.4). Amyloid fibrils of Aβ42 were prepared by incubation of 80 μM Aβ42 at room temperature for 1 week. 15N-labeled Aβ42 was expressed in E. coli (BL21 DE3) using the pET28a vector (Novagen) in minimal medium. Cells were harvested after 3 to 4 hours by centrifugation. The pellet was resuspended in 20 mM tris (pH 8.0) and lysed by sonication. Inclusion bodies were isolated by centrifugation and then washed (resuspended, sonicated, and centrifuged) three times, the first time in 20 mM tris (pH 8.0), 0.4% Triton X-100, and the second and third times in 20 mM tris (pH 8.0). Inclusion bodies were solubilized in 8 M guanidine hydrochloride and 20 mM tris (pH 8.0) with sonication. The solubilized Aβ42 was finally purified by reversed-phase high-performance liquid chromatography using a SOURCE 30RPC column (GE Healthcare), with a linear gradient of 10 mM NH4OH into 80% acetonitrile, 0.3% trifluoroacetic acid. Fractions containing Aβ42 were combined, lyophilized, and stored at −80°C. To prepare monomeric Aβ42 for NMR or SAXS measurements, the lyophilized peptide was dissolved in 10 mM NaOH, briefly sonicated, and centrifuged or filtered to remove preformed aggregates before dilution in buffer at a molar ratio of 1:1. Low-bind tubes (Axygen Scientific, Corning) were used for all manipulation of Aβ42.
CD spectroscopy
CD measurements were performed on a Jasco J-1500 spectropolarimeter equipped with a Peltier-controlled thermostated cell support. Far UV-CD spectra (200 to 260 nm) were recorded at 4 μM S100B with or without 80 μM CaCl2 in 50 mM tris (pH 7.4). Monomeric Aβ42 was added to individual solutions with S100B and incubated overnight at 4°C.
NMR spectroscopy
NMR spectra were acquired at 4°C on a Bruker Avance III 500-MHz spectrometer, equipped with a 5-mm triple-resonance TCI cryogenic probe head. Spectra were processed using Topspin (Bruker Biospin Ltd.) and analyzed with CCPN (Collaborative Computing Project for NMR) analysis. The NOESY spectrum used a NOESY mixing time of 100 ms. Samples were prepared at a concentration of 430 μM (NOESY) or 100 μM (HSQC titrations of S100B or Aβ42), in 50 mM Hepes (pH 7.3), 5 mM DTT, 10% D2O, with or without 10 mM CaCl2. The chemical shift assignments for S100B under these conditions have been deposited in the BioMagResBank under the accession number 27199.
Small-angle x-ray scattering
SAXS data for solutions of S100B and Aβ42 were recorded on an in-house SAXS instrument (SAXSess mc2, Anton Paar) equipped with a Kratky camera, a sealed X-ray tube source, and a two-dimensional Princeton Instrument PI•SCX:4300 charge-coupled device detector (Roper Scientific). The scattering patterns were measured with a 180-min exposure time (1080 frames of each 10 s); radiation damage was excluded by comparison of individual frames at different time points of the measurement. Samples were prepared at a concentration of 100 μM S100B and Aβ42 in 50 mM Hepes (pH 7.3), 5 mM DTT, 10% D2O, with 10 mM CaCl2. The measurements were performed at 20°C. All SAXS data were analyzed with the package ATSAS (version 2.5). The data were processed with the SAXSQuant software (version 3.9) and desmeared using the program GIFT. The forward scattering, I(0), the radius of gyration, Rg, the maximum dimension, Dmax, and the interatomic distance distribution functions, P(R), were computed with the program GNOM. The masses of the solutes were evaluated by using the Porod volume. For ab initio model generation, a total of 50 models were calculated using the program DAMMIF with C2 symmetry and aligned and averaged using the program DAMCLUST. The generated models were aligned with the NMR structure of S100B (PDB code: 2K7O), and figures were generated using the best structures based on the fit to the experimental data.
Isothermal titration calorimetry
The interaction of S100B with Aβ42, with or without 1.5 mM CaCl2, was performed on a Microcal ITC 200 calorimeter. This study was performed at pH 7.4 and 25°C. Titrations were performed by injecting 2 μl of aliquots of 150 μM S100B into 15 μM Aβ42 in the sample cell. Each injection was made with a 140-s spacing interval between subsequent 38 injections with stirring at 400 rpm. S100B and Aβ42 were previously dialyzed against 50 mM Hepes (pH 7.4), 5 mM tris(2-carboxyethyl)phosphine (TCEP), with or without 1.5 mM CaCl2. The titration curves were corrected using the buffer with or without CaCl2 as a buffer control titration. Data were analyzed using the nonlinear regression analyses of one-binding site model.
Biolayer interferometry
Streptavidin optical sensors were used to record the capture kinetics of Aβ42 onto the biotinylated S100B (28 μg/ml)–coated surface of the sensor on a BLItz instrument (ForteBio) in advanced kinetics model. The assays were performed in 50 mM Hepes, 5 mM TCEP (pH 7.4) buffer in the absence and presence of 5 mM CaCl2 at 25°C and 1000 rpm. Since this suggested a submicromolar affinity constant (Kd), the determination of the equilibrium dissociation constant was determined from the analysis of sensorgram traces obtained for interactions with S100B ranging from 0.1 to 1.5 μM in the presence of Ca2+, and from 0.5 to 3 μM with no Ca2+ present.
Aβ42 aggregation kinetics
Aggregation kinetics were performed by recording the ThT fluorescence intensity as a function of time in a plate reader (FLUOstar OPTIMA, BMG Labtech) with a 440-nm excitation filter and a 480-nm emission filter. The fluorescence was recorded using bottom optics in half-area 96-well polyethylene glycol-coated black polystyrene plates with a clear bottom (Corning, 3881). Aβ42 monomer was isolated by gel filtration (Tricorn Superdex75 column, GE Healthcare) in 50 mM Hepes (pH 7.4) and diluted in the same buffer with 0.5 mM EDTA or 1.1 mM CaCl2. ThT (10 μM) was added to each condition. The assays were performed at 37°C, without agitation with fluorescence read every 400 s. Aggregation kinetic data were analyzed using the AmyloFit platform, which implements the master equations derived from basin-hopping algorithm that describe the evolution of total fibril mass in the presence of primary and secondary nucleation events, and from which microscopic processes and reaction rates can be determined from global fitting (29). The kinetic curves were fitted using the secondary nucleation model. The normalized intensity curves and corresponding fits were extracted from the platform and presented in Fig. 3 (A and B) as fibrillar mass fraction.
TEM and immunogold labeling
For the analysis of structure and morphology, 5-μl sample aliquots were adsorbed to carbon-coated collodion film supported on 400-mesh copper grids and negatively stained with 1% uranyl acetate. For immunogold labeling, 5-μl sample aliquots were adsorbed to carbon-coated collodion film supported on 400-mesh nickel grids for 5 min. After washing with phosphate-buffered saline (PBS), the grids were blocked in 1% bovine serum albumin (BSA) in PBS for 10 min, and then incubated with each primary antibody: rabbit anti–amyloid-β (1:50; Sigma) or mouse anti-S100B (1:50), diluted in 1% BSA/PBS; for double labeling, grids were sequentially incubated with each primary antibody. Following washing with PBS, grids were then incubated with either anti-rabbit or anti-mouse secondary antibodies conjugated to 10- or 15-nm colloidal gold, respectively, or sequentially incubated with each one, for double labeling. Following washing with PBS, samples were negatively stained with 1% uranyl acetate. The grids were visualized with a JEOL JEM-1400 transmission electron microscope equipped with an Orious Sc1000 digital camera, and exhaustively observed. As a control, we performed the immunegold labeling, incubating only the secondary antibodies, and no colloidal gold was observed.
Cell culture
SH-SY5Y cells were cultured in Dulbecco’s modified essential medium/Nutrient Mix F12 (1:1 ratio). Medium was supplemented with 10 U penicillin/ml, 100 μM streptomycin (Sigma), and 10 % fetal bovine serum (Gibco). Cells were maintained at 37°C in a humidified incubator with 5% CO2/95% air. Differentiation of SH-SY5Y cells was induced by incubation with 10 μM retinoic acid (RA) (Sigma) for 5 to 6 days. Split cells were reseeded at a density of 25,000 cells per well in a white-walled, clear-bottomed 96-well plate, and after the cells attached, the medium was changed to that without sera and supplemented with brain-derived neutrophic factor human (BDNP) (25 ng/ml) (Sigma) and differentiation continued for an additional 5 days. After differentiation, the culture medium was replaced with fresh medium without sera. Samples previously filtered in 0.2-μm sterile filters were added at final concentrations of 7 μM Aβ42, 7 μM Aβ42 + 84 μM S100B, 84 μM S100B, and 50 mM Hepes (pH 7.4) in a 1:4 sample/media ratio; untreated control was maintained only with cell culture. Cells were incubated for 74 hours at 37°C in a humidified incubator with 5% CO2/95% air previous to viability and apoptosis assays.
Cytotoxicity and cell viability assays
Caspase-3/7 activity was measured using Apo-ONE Homogeneous Caspase-3/7 assay (Promega). Caspase-3/7 substrate was diluted 1:100 in lysis buffer and added to the cell medium in a ratio of 1:1. The cell/reagent mix was then incubated for 1 hour before measuring the fluorescence at 499-nm excitation and 521-nm emission in a Gemini EM Microplate Reader. Cell viability was measured using the resazurin-based assay PrestoBlue Cell Viability Reagent (Invitrogen). The PrestoBlue reagent was added to the cell culture medium in a 1:10 ratio and incubated for 1 hour at 37°C with 5% CO2. Fluorescence was measured with λex = 560 nm and λem = 590 nm using a Gemini EM Microplate Reader. Reduction of viability was calculated as the percentage of fluorescence for each sample compared with buffer-treated cells. The background fluorescence was subtracted from all the values. Statistical analysis was performed with the GraphPad Prism 7.03 program. The significance of differences between the groups was evaluated using one-way ANOVA followed by Welch’s t test (ns indicates not significant; **P < 0.01, ***P < 0.001, ****P < 0.0001).
Supplementary Material
Acknowledgment
Funding: This work was supported by Fundação para a Ciência e a Tecnologia (Portugal) through grants PTDC/NEU-NMC/2138/2014 (to C.M.G.), POCI-01-0145-FEDER-007274 (to I.C.), IF/01046/2014 (to C.M.G.), UID/Multi/04046/2013 [to BioISI (Biosystems and Integrative Sciences Institute)], and PhD fellowship SFRH/BD/101171/2014 (to J.S.C.). Bial Foundation is acknowledged through grant PT/FB/BL-2014-343 (to C.M.G.). Norte Portugal Regional Operational Programme (NORTE 2020) is acknowledged through grant Norte-01-0145-FEDER-000008. ITQB/UNL (Instituto de Tecnologia Química e Biológica/Universidade Nova de Lisboa) is acknowledged for access to the ITC and BLI instruments. The German Research Foundation is acknowledged through grants FR1488/3 and FR1488/5 (to G.F.). V.M. is supported by the CJ Martin Early Career Fellowship from the National Health and Medical Research Council of Australia. T.M. is supported by the Integrative Metabolism Research Center Graz, the Austrian infrastructure program 2016/2017, BioTechMed/Graz, Omics Center Graz, the President’s International Fellowship Initiative of the Chinese Academy of Sciences (no. 2015VBB045), the National Natural Science Foundation of China (no. 31450110423), and the Austrian Science Fund (FWF: P28854 and W1226-B18). Author contributions: C.M.G. conceived and designed the study, analyzed the data, and wrote the manuscript with J.S.C. J.S.C. expressed and purified recombinant human S100B, N15-labeled S100B, and Aβ42, with assistance from R.D. J.S.C. performed CD experiments and aggregation kinetics and analysis. V.K.M. performed the NMR experiments and analyzed the data with B.R. I.C. and M.A. performed immunogold labeling and TEM imaging. S.S.L. performed the ITC and BLI experiments. J.M. performed the cell-based viability assays. H.M.B. performed the preliminary aggregation experiments. C.G. performed the SAXS experiments and analyzed the data with T.M. G.F. provided recombinant S100B, and K.K. and G.F. performed the analysis of S100B expression in 5XFAD mice. B.R. and T.M. provided access to biophysical instrumentation. All authors revised and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/6/eaaq1702/DC1
Supplementary Methods
fig. S1. Isothermal titration calorimetry analysis of the Aβ42/S100B interaction.
fig. S2. BLI analysis of the Aβ42/S100B interaction.
fig. S3. CD analysis of the Aβ42/S100B interaction in the absence of CaCl2.
fig. S4. SAXS analysis of the Aβ42/S100B complex.
fig. S5. Effect of apo-S100B over Aβ42 aggregation in a fragmentation-dominated regime.
fig. S6. Effect of Ca2+-S100B over Aβ42 aggregation in a fragmentation-dominated regime.
fig. S7. S100B inhibits Aβ25–35 aggregation.
fig. S8. Dot blot analysis of Aβ42 aggregates formed in the presence of S100B.
fig. S9. TEM images of Aβ aggregates formed in the presence and absence of S100B.
fig. S10. S100B accumulates at high levels around plaques in AD mice brains.
REFERENCES AND NOTES
- 1.Masters C. L., Bateman R., Blennow K., Rowe C. C., Sperling R. A., Cummings J. L., Alzheimer’s disease. Nat. Rev. Dis. Primers 1, 15056 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Kato S., Gondo T., Hoshii Y., Takahashi M., Yamada M., Ishihara T., Confocal observation of senile plaques in Alzheimer’s disease: Senile plaque morphology and relationship between senile plaques and astrocytes. Pathol. Int. 48, 332–340 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Li C., Zhao R., Gao K., Wei Z., Yin M. Y., Lau L. T., Chui D., Yu A. C., Astrocytes: Implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 8, 67–80 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Marshak D. R., Pesce S. A., Stanley L. C., Griffin W. S., Increased S100 beta neurotrophic activity in Alzheimer’s disease temporal lobe. Neurobiol. Aging 13, 1–7 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Sheng J. G., Mrak R. E., Griffin W. S., S100β protein expression in Alzheimer disease: Potential role in the pathogenesis of neuritic plaques. J. Neurosci. Res. 39, 398–404 (1994). [DOI] [PubMed] [Google Scholar]
- 6.Mrak R. E., Griffinbc W. S., The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 22, 915–922 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Sorci G., Bianchi R., Riuzzi F., Tubaro C., Arcuri C., Giambanco I., Donato R., S100B Protein, A Damage-Associated Molecular Pattern Protein in the Brain and Heart, and Beyond. Cardiovasc. Psychiatry Neurol. 2010, 656481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostendorp T., Diez J., Heizmann C. W., Fritz G., The crystal structures of human S100B in the zinc- and calcium-loaded state at three pH values reveal zinc ligand swapping. Biochim. Biophys. Acta 1813, 1083–1091 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Carvalho S. B., Botelho H. M., Leal S. S., Cardoso I., Fritz G., Gomes C. M., Intrinsically disordered and aggregation prone regions underlie β-aggregation in S100 proteins. PLOS ONE 8, e76629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fritz G., Botelho H. M., Morozova-Roche L. A., Gomes C. M., Natural and amyloid self-assembly of S100 proteins: Structural basis of functional diversity. FEBS J. 277, 4578–4590 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Botelho H. M., Fritz G., Gomes C. M., Analysis of S100 oligomers and amyloids. Methods Mol. Biol. 849, 373–386 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Rothermundt M., Peters M., Prehn J. H., Arolt V., S100B in brain damage and neurodegeneration. Microsc. Res. Tech. 60, 614–632 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Peña L. A., Brecher C. W., Marshak D. R., β-Amyloid regulates gene expression of glial trophic substance S100β in C6 glioma and primary astrocyte cultures. Brain Res. Mol. Brain Res. 34, 118–126 (1995). [DOI] [PubMed] [Google Scholar]
- 14.Sheng J. G., Mrak R. E., Bales K. R., Cordell B., Paul S. M., Jones R. A., Woodward S., Zhou X. Q., McGinness J. M., Griffin W. S. T., Overexpression of the neuritotrophic cytokine S100β precedes the appearance of neuritic β-amyloid plaques in APPV717F mice. J. Neurochem. 74, 295–301 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mori T., Koyama N., Arendash G. W., Horikoshi-Sakuraba Y., Tan J., Town T., Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer’s disease. Glia 58, 300–314 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Craft J. M., Watterson D. M., Marks A., Van Eldik L. J., Enhanced susceptibility of S-100B transgenic mice to neuroinflammation and neuronal dysfunction induced by intracerebroventricular infusion of human β-amyloid. Glia 51, 209–216 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Mori T., Town T., Tan J., Yada N., Horikoshi Y., Yamamoto J., Shimoda T., Kamanaka Y., Tateishi N., Asano T., Arundic acid ameliorates cerebral amyloidosis and gliosis in Alzheimer transgenic mice. J. Pharmacol. Exp. Ther. 318, 571–578 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Roltsch E., Holcomb L., Young K. A., Marks A., Zimmer D. B., PSAPP mice exhibit regionally selective reductions in gliosis and plaque deposition in response to S100B ablation. J. Neuroinflammation 7, 78 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Businaro R., Leone S., Fabrizi C., Sorci G., Donato R., Lauro G. M., Fumagalli L., S100B protects LAN-5 neuroblastoma cells against Aβ amyloid-induced neurotoxicity via RAGE engagement at low doses but increases Aβ amyloid neurotoxicity at high doses. J. Neurosci. Res. 83, 897–906 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Santamaria-Kisiel L., Rintala-Dempsey A. C., Shaw G. S., Calcium-dependent and -independent interactions of the S100 protein family. Biochem J. 396, 201–214 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jensen J. L., Indurthi V. S. K., Neau D. B., Vetter S. W., Colbert C. L., Structural insights into the binding of the human receptor for advanced glycation end products (RAGE) by S100B, as revealed by an S100B-RAGE-derived peptide complex. Acta Crystallogr. D Biol. Crystallogr. 71, 1176–1183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilder P. T., Lin J., Bair C. L., Charpentier T. H., Yang D., Liriano M., Varney K. M., Lee A., Oppenheim A. B., Adhya S., Carrier F., Weber D. J., Recognition of the tumor suppressor protein p53 and other protein targets by the calcium-binding protein S100B. Biochim. Biophys. Acta 1763, 1284–1297 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Rustandi R. R., Baldisseri D. M., Weber D. J., Structure of the negative regulatory domain of p53 bound to S100B(beta beta). Nat. Struct. Biol. 7, 570–574 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Serpell L. C., Alzheimer’s amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta 1502, 16–30 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Jarvet J., Danielsson J., Damberg P., Oleszczuk M., Gräslund A., Positioning of the Alzheimer Aβ(1–40) peptide in SDS micelles using NMR and paramagnetic probes. J. Biomol. NMR 39, 63–72 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Göbl C., Madl T., Simon B., Sattler M., NMR approaches for structural analysis of multidomain proteins and complexes in solution. Prog. Nucl. Magn. Reson. Spectrosc. 80, 26–63 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Gupta A. A., Chou R.-H., Li H., Yang L.-W., Yu C., Structural insights into the interaction of human S100B and basic fibroblast growth factor (FGF2): Effects on FGFR1 receptor signaling. Biochim. Biophys. Acta 1834, 2606–2619 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Cohen S. I. A., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., Knowles T. P. J., Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meisl G., Kirkegaard J. B., Arosio P., Michaels T. C. T., Vendruscolo M., Dobson C. M., Linse S., Knowles T. P. J., Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 11, 252–272 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Arosio P., Vendruscolo M., Dobson C. M., Knowles T. P., Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 35, 127–135 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Cohen S. I. A., Arosio P., Presto J., Kurudenkandy F. R., Biverstål H., Dolfe L., Dunning C., Yang X., Frohm B., Vendruscolo M., Johansson J., Dobson C. M., Fisahn A., Knowles T. P. J., Linse S., A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 22, 207–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.S. B. Carvalho, I. Cardoso, H. M. Botelho, K. Yanamandra, G. Fritz, C. M. Gomes, L. A. Morozova-Roche, Structural heterogeneity and bioimaging of S100 amyloid assemblies, in Bionanoimaging: Protein Misfolding and Aggregation, V. Uversky, Y. Lyubchenko, Eds. (Elsevier, 2013), vol. 18, pp. 197–212. [Google Scholar]
- 33.Arosio P., Michaels T. C. T., Linse S., Månsson C., Emanuelsson C., Presto J., Johansson J., Vendruscolo M., Dobson C. M., Knowles T. P. J., Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 7, 10948 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen G., Abelein A., Nilsson H. E., Leppert A., Andrade-Talavera Y., Tambaro S., Hemmingsson L., Roshan F., Landreh M., Biverstål H., Koeck P. J. B., Presto J., Hebert H., Fisahn A., Johansson J., Bri2 BRICHOS client specificity and chaperone activity are governed by assembly state. Nat. Commun. 8, 2081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leal S. S., Botelho H. M., Gomes C. M., Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev. 256, 2253–2270 (2012). [Google Scholar]
- 36.Cristovao J. S., Santos R., Gomes C. M., Metals and neuronal metal binding proteins implicated in Alzheimer’s disease. Oxid. Med. Cell. Longevity 2016, 9812178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Donato R., S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell Biol. 33, 637–668 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Zimmer D. B., Wright Sadosky P., Weber D. J., Molecular mechanisms of S100-target protein interactions. Microsc. Res. Tech. 60, 552–559 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Soto C., Castaño E. M., Frangione B., Inestrosa N. C., The α-helical to β-strand transition in the amino-terminal fragment of the amyloid β-peptide modulates amyloid formation. J. Biol. Chem. 270, 3063–3067 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Walsh D. M., Thulin E., Minogue A. M., Gustavsson N., Pang E., Teplow D. B., Linse S., A facile method for expression and purification of the Alzheimer’s disease-associated amyloid beta-peptide. FEBS J. 276, 1266–1281 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/6/eaaq1702/DC1
Supplementary Methods
fig. S1. Isothermal titration calorimetry analysis of the Aβ42/S100B interaction.
fig. S2. BLI analysis of the Aβ42/S100B interaction.
fig. S3. CD analysis of the Aβ42/S100B interaction in the absence of CaCl2.
fig. S4. SAXS analysis of the Aβ42/S100B complex.
fig. S5. Effect of apo-S100B over Aβ42 aggregation in a fragmentation-dominated regime.
fig. S6. Effect of Ca2+-S100B over Aβ42 aggregation in a fragmentation-dominated regime.
fig. S7. S100B inhibits Aβ25–35 aggregation.
fig. S8. Dot blot analysis of Aβ42 aggregates formed in the presence of S100B.
fig. S9. TEM images of Aβ aggregates formed in the presence and absence of S100B.
fig. S10. S100B accumulates at high levels around plaques in AD mice brains.