

Abstract
Non-resolving inflammation drives the development of clinically dangerous atherosclerotic lesions by promoting sustained plaque inflammation, large necrotic cores, thin fibrous caps, and thrombosis. Resolution of inflammation is not merely a passive return to homeostasis, but rather an active process mediated by specific molecules, including fatty acid–derived specialized pro-resolving mediators (SPMs). In advanced atherosclerosis, there is an imbalance between levels of SPMs and proinflammatory lipid mediators, which results in sustained leukocyte influx into lesions, inflammatory macrophage polarization, and impaired efferocytosis. In animal models of advanced atherosclerosis, restoration of SPMs limits plaque progression by suppressing inflammation, enhancing efferocytosis, and promoting an increase in collagen cap thickness. This Review discusses the roles of non-resolving inflammation in atherosclerosis and highlights the unique therapeutic potential of SPMs in blocking the progression of clinically dangerous plaques.
Introduction
Atherosclerosis is a lipoprotein-driven, non-resolving chronic inflammatory disease of the arterial subendothelial space (intima) and involves activation of both the innate and adaptive branches of the host immune system. This disease process is the underlying cause of fatal heart attacks and strokes, which are responsible for the majority of deaths worldwide (1). Despite the availability of safe and effective LDL-lowering therapy, the incidence of cardiovascular disease has been increasing in the United States and other developed countries (2). A deeper understanding of the cellular mechanisms that promote atherosclerotic lesions, particularly the unique subset of advanced lesions that trigger vascular disease, may identify novel therapeutic targets to help stem this tide.
Atherosclerosis begins with the retention of apolipoprotein B–containing lipoproteins within the subendothelial intima of arteries, which leads to the activation of endothelial cells; recruitment of leukocytes, particularly Ly6chi monocytes; and accumulation of cells, extracellular matrix, and lipids (3). Monocytes differentiate into macrophages, the major leukocyte subset in the subendothelial region, and these macrophages then become foam cells through their ingestion of retained lipoproteins. The interaction of macrophages with retained apolipoprotein B–containing lipoproteins, some of which become modified by oxidation and other processes, triggers proinflammatory responses (4). Eventually, myofibroblasts derived from medial smooth muscle cells and possibly other sources enter the intima and lead to the formation of a collagenous fibrous cap that overlies the lipid-rich plaque core (1). Cells found in the atherosclerotic lesion, including macrophages, T cells, and dendritic cells, promote expression of proinflammatory cytokines and eicosanoids that sustain proinflammatory responses (5, 6). Plaque necrosis develops in the setting of continued inflammatory stimuli, defective clearance of dead cells, and primary necroptosis, and then plaque necrosis further amplifies the inflammatory response (7–9). Over time, the integrity of the fibrous cap may break down, leading to plaque rupture, which can trigger acute occlusive thrombosis and the associated clinical sequelae associated with tissue infarction (9–11). While the role of persistent inflammation has long been recognized in the progression of atherosclerosis, recent work by a number of groups has also identified defects in inflammation resolution as a key causal factor (12–15). Here, we present an overview of how impaired inflammation resolution drives atherosclerosis and discuss the therapeutic implications of this concept.
Resolution of inflammation and specialized pro-resolving mediators
Acute inflammation evolved as a physiological host defense response against both microbial and sterile pathogens (16). When acute inflammation cannot be controlled or resolved, chronic inflammation develops, which can result in organ dysfunction (17). Examples include autoimmune disease, asthma, Alzheimer’s disease, diabetes, obesity, and cardiovascular disease (12, 18–22). The acute inflammatory response is divided into two phases: initiation and resolution. During the initiation phase, chemical messengers including cytokines, chemokines, and lipid-derived mediators, e.g., prostaglandins and leukotrienes, recruit immune cells to the site of injury (17). Polymorphonuclear neutrophils (PMNs) arrive first and begin to phagocytose and neutralize pathogens. Simultaneously, monocytes are recruited to the area of inflammation and differentiate into macrophages that help to further clear pathogens as well as begin to orchestrate the next stage, the resolution of inflammation (23). This stage is characterized by quelling of the inflammatory response but with the continued clearance of pathogens; phagocytic removal of apoptotic cells in a process known as efferocytosis; and repair of damaged tissue. Resolution is characterized by a switch in the production of lipid mediators from proinflammatory mediators, e.g., leukotrienes, toward specialized pro-resolving mediators (SPMs), e.g., resolvins and lipoxins (24–26).
Proinflammatory lipid mediators, consisting of prostaglandins and leukotrienes, are formed from the n-6 fatty acid precursor arachidonic acid (AA). These mediators increase blood flow, endothelial cell permeability, and extravasation of PMNs (27). Lipid mediators that participate in resolution were identified using liquid chromatography–tandem mass spectrometry–based analysis of resolving inflammatory exudates from in vivo animal models and human cell culture systems (28). SPMs consist of at least four distinct families: lipoxins, resolvins, protectins, and maresins (25, 26, 29, 30). Like prostaglandins and leukotrienes, lipoxins are produced from n-6 fatty acid–derived AA (26). The other families of SPMs, including resolvins, protectins, and maresins, are generated from n-3 fatty acid–derived eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), or docosapentaenoic acid (DPA) (25, 29). Interestingly proinflammatory lipid mediators are able to trigger the synthesis of the SPMs, suggesting a direct cell biological link between the inflammatory and resolving phases of the inflammatory response (24).
In response to an inflammatory stimulus, polyunsaturated fatty acids including AA, EPA, DHA, and DPA are released from phospholipids by phospholipase enzymes and then catalyzed by a series of lipoxygenases, namely, 5-lipoxygenase (5-LOX), 12-lipoxygenase (12-LOX), and 15-lipoxygenase (15-LOX), to produce lipid mediators of inflammation and resolution (27). Proinflammatory leukotrienes such as leukotriene B4 (LTB4) and leukotriene C4 (LTC4) are derived from AA by 5-LOX and act on GPCRs such as leukotriene B4 receptor 1/2 (BLT1/2) and cysteinyl leukotriene receptor 1/2 (CysLT1/2). Activation of these receptors leads to increased expression of inflammatory pathway enzymes, e.g., 5-LOX and cyclooxygenase-2 (COX-2), and inflammatory cytokines, e.g., IL-6 and IL-8, which then promote the recruitment of neutrophils. Similarly to leukotrienes, SPMs also bind and act on their cognate GPCRs, which leads to the induction of pro-resolving cellular processes (31). These processes include apoptosis and decreased recruitment of leukocytes; efferocytosis; macrophage phenotypic shift from proinflammatory to pro-resolving; and tissue repair processes, including reconstitution of cells, matrix, and blood and lymph vessels (32, 33).
Imbalance of inflammatory and resolving mediators drives atherosclerosis progression
An imbalance of pro-resolving to proinflammatory lipid mediators has recently been identified as a key feature of advanced atherosclerosis in human and murine plaques (14, 15). Recent work has shown that regions of human atherosclerotic plaques characterized by large necrotic cores and thin fibrous caps had low levels of the pro-resolving mediator resolvin D1 (RvD1) and high levels of the proinflammatory mediator LTB4 (14). Conversely, plaque regions containing less necrosis and thicker fibrous caps demonstrated high levels of RvD1 and relatively lower levels of LTB4 (14). Thus, the ratio of RvD1 to LTB4 correlated strongly with plaque severity. Consistent with these findings, advanced aortic root plaques of Ldlr–/– mice fed a high-fat diet for 17 weeks showed a lower ratio of lesional RvD1 to LTB4 than did early atherosclerotic lesions from Ldlr–/– mice fed a high-fat diet for 8 weeks (14). Another study showed that the ratio of the SPMs maresin-1 (MaR1) and RvD2 to the proinflammatory mediators LTB4, prostaglandin A1, prostaglandin D2, and prostaglandin E2 was lower in Apoe–/– mice fed a high-fat diet for 16 weeks as compared with mice fed the diet for 4 or 8 weeks (15). An important area of future research will be understanding how the plaque environment affects mediator synthesis and/or degradation. Nonetheless, as reviewed in the following sections, evidence is now accumulating that this imbalance is causal in the development of features of clinically significant plaques, including continuous Ly6chi monocyte influx; inflammatory macrophage polarization; defective efferocytosis; and thinning of the stabilizing collagenous fibrous cap overlying lesions (Figure 1).
Figure 1. Imbalance of SPMs and leukotrienes (LTs) promotes instability of atherosclerotic plaques.

When the SPM/LT ratio is low (A), resolution of inflammation is impaired, leading to sustained inflammatory Ly6chi monocyte influx, platelet aggregation, proinflammatory macrophage polarization, impaired efferocytosis, large necrotic cores, and thin fibrous caps. Conversely, an increase in the SPM/LT ratio (B) promotes resolution, characterized by attenuated Ly6hi monocyte influx, pro-resolving macrophage polarization, enhanced efferocytosis, decreased necrotic core formation, and thicker fibrous caps.
Lipoxygenases play a critical role in the biosynthesis of both proinflammatory and pro-resolving mediators and thus can be critical regulators of their balance. Two key enzymes, 5-LOX and 12/15-LOX, have been studied in this context. Notably, humans express three related forms of this enzyme, 12-LOX, 15-LOX type 1, and 15-LOX type 2, while mice express only one form, which is usually referred to as 12/15-LOX because it catalyzes both 12- and 15-LOX reactions (34). 5-LOX plays a role in the formation of both proinflammatory leukotrienes and pro-resolving lipoxins, and the determination of which product is generated in a given setting is dependent on 5-LOX subcellular localization (34). Nuclear 5-LOX is located near leukotriene A4 (LTA4) hydrolase, which converts the product of 5-LOX, LTA4, to LTB4. In contrast, when 5-LOX is cytoplasmic, LTA4 is converted to lipoxin A4 (LXA4) by cytoplasmic 12/15-LOX. In macrophages, the nuclear localization of 5-LOX is mediated by MAPKAPK2-mediated (MK2-mediated) 5-LOX phosphorylation, and thus when MK2 is activated in macrophages, the inflammation/resolution balance is shifted toward inflammation (35–37).
Expression analysis of lipoxygenases has demonstrated that 5-LOX is significantly higher in human carotid atherosclerotic lesions compared with healthy arteries, where it has been found to have a predominantly nuclear localization (38, 39). Moreover, 5-LOX expression in atherosclerotic plaques correlated with patients’ clinical symptoms, including transient ischemic attack, stroke, and amaurosis fugax (38). The concept that 5-LOX is involved in atherosclerosis is also supported by human genetic findings. In a cohort of patients from the Los Angeles Atherosclerosis Study, Dwyer and colleagues identified a variant allele of ALOX5 that was associated with increased carotid intima-media thickness. Further, they observed that increased dietary intake of AA enhanced the atherogenic effect of the variant genotype, whereas increased dietary intake of n-3 fatty acids diminished its effect (40). These data are consistent with the concept that an increase in the ratio of AA-derived LTB4 to n-3 fatty acid–derived LXA4 promotes plaque progression. Supportive of a proinflammatory role for 5-LOX in atherogenesis, murine studies have demonstrated that the absence of 5-LOX in Apoe–/– mice dramatically decreased atherosclerotic lesion size (41). In contrast to 5-LOX, human studies have suggested that 15-LOX may be atheroprotective. In a case-control study, a polymorphism in ALOX15 that was associated with higher enzyme activity was enriched in a population of control subjects without coronary artery disease (CAD) versus patients with CAD (42). Consistent with this finding, the Coronary Artery Risk Development in Young Adults (CARDIA) study found that a SNP in the coding region of ALOX15 was associated with both a 20-fold reduction in 15-LOX activity in vitro and a 1.6-fold increased risk of CAD (43). In a hypercholesterolemic rabbit model, overexpression of 15-LOX in macrophages was protective against early atherogenesis (44). Similarly, transgenic mice overexpressing 12/15-LOX on the Apoe–/– background and fed a chow diet showed attenuated atherosclerosis progression, which was thought to be due to increased 12/15-LOX–mediated synthesis of LXA4 (12). However, Merched and colleagues demonstrated that the role of 12/15-LOX in atherosclerosis is not straightforward. These authors found that although 12/15-LOX–transgenic Apoe–/– mice on a chow diet had reduced atherosclerotic lesion area compared with control Apoe–/– mice, lesion size was similar between 12/15-LOX–transgenic Apoe–/– and control Apoe–/– mice when Western diet feeding was used (12, 45). Consistent with these results, they showed that hyperlipidemia decreased 12/15-LOX–derived LXA4 in the plasma of Western diet–fed 12/15-LOX–transgenic Apoe–/– mice and that LDL-supplemented medium blocked LXA4-induced efferocytosis in cultured macrophages. The mechanisms linking high-fat diet or LDL to decreased SPM plasma level and impaired SPM-mediated functions remain to be elucidated (45).
To identify a potential direct effect of SPMs on the progression of atherosclerosis, recent studies examined the effect of treating established lesions in atherosclerosis-prone mice with lipid mediators, including RvE1, RvD1, RvD2, RvD5n-3 DPA, MaR1, or aspirin-triggered LXA4 (ATL) (Table 1). Administration of RvE1, an E-series resolvin derived from EPA, to ApoE*3 Leiden transgenic mice fed a high-fat/high-cholesterol diet showed a significant reduction in lesion progression, accompanied by decreased expression of genes involved in atherogenesis, including those encoding ADAM17, TNF, and IFN-γ (46). The ADAM17 effect is particularly interesting given that Mer proto-oncogene tyrosine kinase (MerTK), a key macrophage efferocytosis receptor in atherosclerotic lesions, is cleaved by this protease, resulting in defective lesional efferocytosis (47). Additionally, because TNF-α and IFN-γ modulate macrophage polarization toward a more proinflammatory phenotype, a decrease in the expression of these cytokines may promote a pro-resolving macrophage phenotype and thereby favorably affect the resolution process (48). In cholesterol-fed rabbits with periodontitis, administration of RvE1 to the gums led to decreases in periodontal inflammation, atherosclerotic lesional inflammation, and necrotic core area (49). The D-series resolvin RvD1 led to smaller necrotic cores, enhanced rates of lesional efferocytosis, and thicker collagen caps when administered to high-fat-fed Ldlr–/– mice (14). Similarly, administration of RvD5n-3 DPA (derived from n-3 docosapentaenoic acid) to high-fat-fed Apoe–/– mice decreased atherosclerotic lesion size and reduced leukocyte and platelet activation (50). Treatment of Apoe–/– mice with RvD2 and MaR1 also promoted plaque stability by decreasing lesional macrophage numbers, halting expansion of necrotic cores, and thickening collagen caps (15). ATL treatment was shown to block atherosclerosis progression in high-fat-fed Apoe–/– mice by reducing lesional macrophage infiltration and apoptotic cell accumulation, and this protective effect required its cognate receptor, formyl peptide receptor 2 (FPR2/ALX) (51). In addition to SPMs, the proteins annexin A1 and IL-10 have shown pro-resolving effects in the setting of experimental inflammation (52–56). Drechsler et al. demonstrated that the absence of annexin A1 in high-fat-fed Apoe–/– mice led to increased arterial myeloid cell recruitment and atherosclerotic lesion size. Further, the intravenous delivery of the annexin A1–derived peptide fragment Ac2-26 to Apoe–/– mice led to a reduction in arterial macrophage accumulation and plaque size (57). Moreover, when Western diet–fed Ldlr–/– mice with pre-established atherosclerotic lesions were treated with atherosclerosis-targeting nanoparticles containing Ac2-26, the plaques showed decreases in overall plaque area, necrotic core size, and lesional oxidative stress and increases in fibrous cap thickness and IL-10 expression (58). In another study, treatment of Ldlr–/– mice with nanoparticles containing IL-10 led to similar improvements in advanced plaques (59). Together, these studies demonstrate that atherosclerosis progression and vulnerable plaque features can be ameliorated by treatment with exogenous SPMs or pro-resolving proteins. Although the these pro-resolving mediators act on distinct receptors, they appear to induce similar atheroprotective mechanisms, including enhanced efferocytosis, pro-resolving macrophage polarization, decreased necrotic core formation, and increased collagen synthesis with thickened fibrous cap development. Despite these important studies, the precise mechanisms of the atheroprotective effects of SPMs remain incompletely understood.
Table 1. The effect of lipoxygenase and SPM receptor genetic engineering and SPM and pro-resolving protein administration on atherosclerosis in animal models of atherosclerosis.

Recent studies have also shown that the biosynthesis of SPMs is regulated by circadian rhythm. Using lipid mediator profiling of healthy volunteers, Colas et al. demonstrated that plasma concentrations of the DPA-derived D-series resolvins (RvDn-3 DPA) are diurnally regulated. Further, the pattern of regulation was significantly impaired in patients with cardiovascular disease (50). These patients were also found to have increased leukocyte activation and higher levels of circulating platelet-leukocyte aggregates, which have been shown to be a marker of inflammation (50). These findings are particularly interesting given previous work showing that proinflammatory Ly6chi monocytes and the proinflammatory cytokines IL-6 and TNF-α are also regulated by circadian rhythm (60, 61). These mechanisms may contribute to the development of increased atherosclerosis in mice with disruptions in components of the circadian clock (62, 63). Together, these findings suggest that diurnal regulation of SPM production may be important in their cardioprotective effects.
Along with the lipid mediators themselves, the role of their receptors in atherosclerosis has also been examined, though progress has been hindered by the complex and sometimes nonspecific relationship between resolving mediators and their receptors (Table 2). In addition, mechanistic studies have been limited in some cases by the lack of a murine homolog for human receptors. For example, human GPR32, an RvD1 receptor, does not exist in mice, which instead appear to largely utilize the FPR2/ALX receptor for RvD1 as well as for LXA4 (64). Interestingly, although genetic targeting of the FPR2/ALX receptor in mice prevented atherosclerosis progression as predicted, it actually decreased plaque stability in high-fat-fed Ldlr–/– mice, which may be due to non-SPM ligands for FPR2/ALX, such as serum amyloid A, that are proinflammatory (65). Notably, the targeting strategy used by these authors led to the deletion of both Fpr2 and Fpr3, which are the murine homologs of human FPR2/ALX (66, 67). The findings from this study are consistent with those of prior studies showing that the FPR2/ALX receptor induces both proinflammatory and pro-resolution actions depending on the activating ligand. For example, activation of FPR2/ALX by prokaryotic peptides induces leukocyte chemotaxis, while annexin 1 and its related N-terminal peptides cause a decrease in neutrophil migration (67–69). Deletion of BLT1, the receptor for LTB4, decreased formation of early lesions in Apoe–/– mice fed the Western diet for 4–8 weeks, but it did not lead to any change in lesion size after 19 weeks of high-fat-diet feeding (70). In a related study, treatment of short-term Western diet–fed Ldlr–/– mice or chow diet–fed Apoe–/– mice with a BLT1 antagonist led to reduced lesion area and decreased lesional CD11b expression (71). These combined data suggest that BLT1 signaling is important in early atherogenesis but that other proatherogenic processes become dominant in more advanced atherosclerosis. Finally, a recent study demonstrated that deficiency of the receptor for RvE1, ChemR23/ERV1, in Apoe–/– mice or in the hematopoietic cells of Ldlr–/– mice leads to increased lesion size and larger necrotic cores (72). Further, the loss of ChemR23/ERV1 was associated with decreased phagocytosis in vitro, decreased expression of genes involved in phagocytosis, and increased expression of genes relevant to macrophage lipoprotein uptake. Together, these data suggest an atheroprotective role for ChemR23/ERV1.
Table 2. SPM receptors and their atherorelevant downstream effects.

Macrophages show a high degree of plasticity and can perform either proatherogenic or antiatherogenic activities in response to environmental stimuli (73). As mentioned above, proinflammatory macrophages are prominent in unstable regions of plaques, whereas abundant pro-resolving macrophages are present in stable regions of plaques (74). Further, atherosclerotic plaques undergoing regression demonstrate increased numbers of pro-resolving macrophages, which presumably promote tissue repair. In this context, SPMs are capable of promoting phenotypic skewing in macrophages. For example, the administration of RvD2 and MaR1 to atheroprone mice promotes polarization of macrophages toward a pro-resolving phenotype (15). Interestingly, pro-resolving macrophages have been shown to synthesize higher levels of SPMs than proinflammatory macrophages (75), suggesting a resolution-amplification cycle. Pro-resolving macrophages have also been shown to have a higher efferocytic capacity than proinflammatory macrophages. Moreover, the pro-resolving protein IL-10 also induces polarization of macrophages toward a pro-resolving phenotype with enhanced efferocytosis (76, 77).
Role of SPMs in atherosclerotic lesional efferocytosis
Efferocytosis, the receptor-mediated clearance of apoptotic cells, has a critically important role in successful resolution responses, and its impairment has been highlighted in many non-resolving chronic inflammatory diseases, including atherosclerosis. Failure to clear apoptotic cells from atherosclerotic plaques in a timely fashion leads to secondary necrosis, which generates heightened inflammation owing to the release of inflammatory damage-associated molecular patterns (DAMPs) from the necrotic cells (4, 78–80). Therefore, efferocytosis is a crucial step in the resolution of lesional inflammation in chronic non-resolving inflammatory diseases. Interestingly, SPMs are able to increase the efferocytic capacity of macrophages (25, 81–83), and efferocytosis, in turn, promotes SPM production by macrophages, including LXA4, RvE1, and PD1 (75, 81, 84). SPMs, in particular the lipoxins, also act as chemoattractants that recruit monocytes that, after differentiating into macrophages, enhance the overall efferocytic capacity of resolving lesions (82, 85, 86). Thus, similar to the situation mentioned above with resolving macrophages, there is a positive resolution feedback cycle involving SPMs and efferocytosis.
Specific phagocytic receptors binding to phosphatidylserine on apoptotic cells have been implicated in the regulation of efferocytosis in atherosclerotic lesions. For example, genetic targeting of Mertk in fat-fed Apoe–/– or Ldlr–/– mice led to impaired efferocytosis, accumulation of apoptotic cells, and enlarged necrotic cores (87, 88). MerTK deficiency also leads to impaired SPM production by macrophages, exemplifying the aforementioned efferocytosis/resolution–amplification loop that, when disrupted, can cause devastating consequences (89). As mentioned above, the MerTK receptor can be inactivated by the metalloproteinase ADAM17, which cleaves the receptor and causes it to shed its soluble extracellular domain (47). Interestingly, high levels of soluble MerTK have been detected in both human and murine atherosclerotic lesions, suggesting that in advanced atherosclerosis, inactivation of the MerTK receptor in this fashion is one mechanism by which efferocytosis is impaired (89). In further support of this idea, analysis of human carotid plaques revealed that expression of ADAM17 is increased while that of MerTK is decreased in macrophages surrounding the necrotic cores as compared with macrophages in peripheral regions of the plaques (90). In addition, extracts obtained from the regions around necrotic cores induced cleavage of MerTK and upregulated expression of ADAM17 in cultured THP-1 macrophages (90).
Our group has demonstrated recently that intact MerTK controls the subcellular localization of 5-LOX, which, as mentioned in the previous section, favors production of SPMs (91). Under inflammatory conditions, 5-LOX is phosphorylated by MK2 and translocated to the nucleus, where it promotes the generation of LTB4 (36, 37). MerTK signaling inhibits MK2 and thereby leads to cytosolic localization of 5-LOX and the generation of LXA4 in macrophages. When MerTK is cleaved or genetically deleted, 5-LOX localizes to the nuclear membrane, and there is an increase in leukotrienes (91). Moreover, the atherosclerotic lesions of mice engineered to express a cleavage-resistant version of MerTK (MerTKCR) and crossed onto the Ldlr–/– background demonstrate attenuated nuclear localization of 5-LOX in macrophages, enhanced SPMs, increased rates of efferocytosis, smaller necrotic cores, and thicker fibrous caps compared with the lesions of Ldlr–/– mice with wild-type MerTK (89, 91). SPMs themselves are able to augment and therefore amplify this pathway. For example, RvD1, which is also induced by successful efferocytosis, can promote cytosolic localization of 5-LOX to favor the generation of LXA4 (37).
The mechanism by which MerTK signaling and RvD1 promote cytoplasmic 5-LOX and subsequent SPM generation in macrophages has recently been revealed (Figure 2). RvD1, by activating its receptor FPR2/ALX, lowers cytosolic calcium, which suppresses activity of the calcium-sensitive kinase calcium/calmodulin-dependent protein kinase II-γ (CaMKIIγ). CaMKII, which is activated in macrophages of advanced, but not early, mouse and human atherosclerotic lesions (92), activates MK2. Thus, the RvD1-mediated decrease in CaMKII activity lowers MK2 activity, which in turn leads to decreased phosphorylation and increased cytoplasmic localization of 5-LOX (68). Activation of the MerTK receptor by its ligands Gas6 and protein S or by apoptotic cells that have Gas6 or protein S on their surface also suppresses MK2 activity; decreases nuclear phospho–5-LOX, which is the form that leads to LTB4 synthesis; and increases SPMs (91). Recent evidence suggests that suppression of CaMKII activity is upstream of this effect, as it is with the RvD1–FPR2/ALX pathway. Finally, as yet another example of feedback and amplification in the resolution response, suppression of CaMKII activity has another pro-resolving role in macrophages, i.e., in addition to increasing SPM biosynthesis: its inactivation leads to an increase in Mertk transcription and MerTK-mediated efferocytosis via a pathway involving the transcription factors ATF6 and LXRα (92). In vivo, genetic deletion of CaMKIIγ from myeloid cells in Ldlr–/– mice enhances the efferocytic capacity of macrophages in atherosclerotic plaques and decreases plaque necrosis and inflammation (92). Thus, CaMKII activity in lesional macrophages represents an important hub orchestrating defective efferocytosis and impaired resolution. For this reason, its therapeutic inhibition may be considered as a new strategy to block advanced plaque progression.
Figure 2. RvD1 and MerTK signaling promote pro-resolving pathways in macrophages.
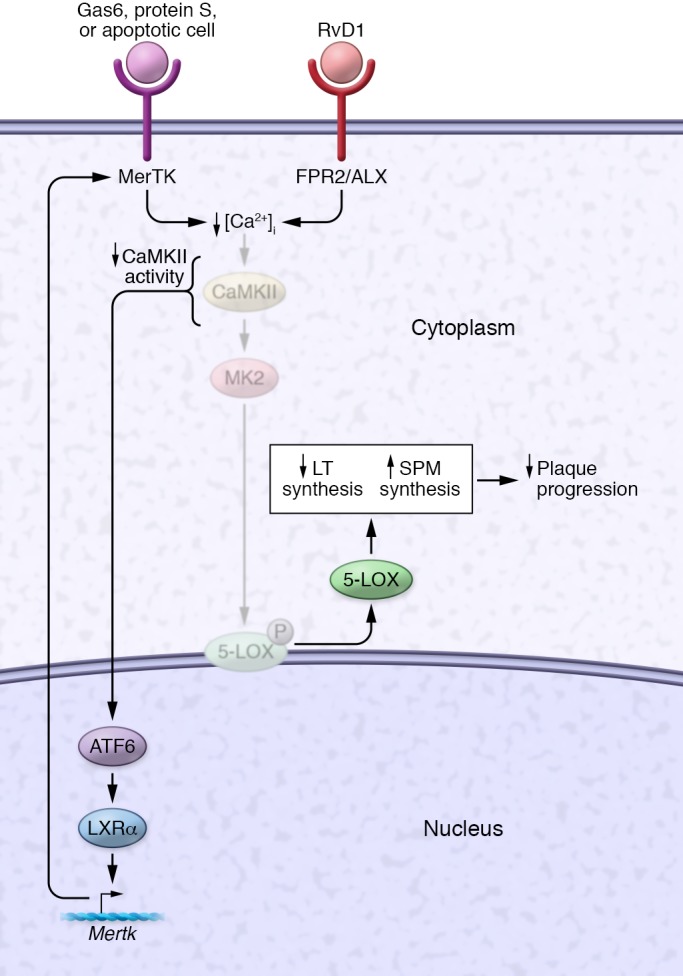
Activation of signaling through the FPR2/ALX receptor by RvD1 or through the MerTK receptor by Gas6, protein S, or apoptotic cells (with Gas6 or protein S on the surface of the apoptotic cells) lowers cytoplasmic calcium and thereby decreases the activity of CaMKII. The decrease in CaMKII activity triggers at least two pro-resolving processes. First, it lowers MK2 activity, which increases nonphosphorylated 5-LOX and promotes its nuclear-to-cytoplasmic translocation. In the cytoplasm, 5-LOX mediates the synthesis of SPMs, e.g., LTB4 in the presence of AA and RvD1 in the presence of DHA, and in the nucleus, 5-LOX causes a concomitant decrease in LT synthesis. The resulting increase in the SPM/LT ratio suppresses plaque progression. Second, suppression of CaMKII activity induces an ATF6-LXRα-MerTK pathway that increases the synthesis of MerTK and thus amplifies the pro-resolving program. Not shown here is that ADAM17-mediated MerTK cleavage, which occurs in advanced lesional macrophages, suppresses both efferocytosis and resolution in macrophages in vitro and in advanced atherosclerotic lesions.
In addition to efferocytosis, SPMs play a role in the apoptosis of neutrophils after they have engulfed pathogens, which is a critical step in resolution. For example, in a model of acute lung injury, RvE1, through its receptor BLT1, promoted apoptosis of neutrophils after their ingestion of opsonized E. coli by increasing caspase-3 and caspase-8 activation and by suppressing ERK- and Akt-dependent prosurvival signaling pathways (93). Similarly, aspirin-triggered lipoxins signal via FPR2/ALX, ERK, and Akt to block prosurvival signaling in neutrophils (94). Further, annexin A1 has also been shown to promote neutrophil apoptosis by a pathway involving L-selectin shedding from neutrophils, inhibition of PMN adhesion to endothelial cells, and activation of caspase-3. This pathway also leads to the externalization of annexin A1 on the PMNs, which promotes efficient efferocytosis of the cells (95–97). Finally, SPMs, including 15-epi-LXA4 and RvD1, promote autophagy and survival pathways in macrophages, which enhances efferocytosis and tissue resolution (98).
Antiinflammation versus resolution therapy in atherosclerosis
In view of the evidence suggesting that chronic inflammation plays an important role in plaque progression, therapeutic targeting of inflammation has been considered as a complementary strategy to LDL reduction for lowering the risk of atherosclerotic vascular disease. This idea recently made a quantum leap in plausibility with the publication of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) trial, which examined approximately 10,000 post–myocardial infarction patients to determine whether inhibition of IL-1β with canakinumab, a monoclonal antibody directed against the proinflammatory cytokine IL-1β, could decrease cardiovascular events in patients with a history of myocardial infarction and elevated C-reactive protein (CRP) levels (99). More than 90% of the subjects were on concomitant statin therapy. In animal and preclinical models, IL-1β has been shown to promote inflammation in atherosclerosis by inducing the expression of leukocyte adhesion proteins, e.g., ICAM-1, E-selectin, and VCAM-1, that contribute to the accumulation of leukocytes in lesions (100, 101). The CANTOS investigators demonstrated that 150 mg of canakinumab administered every 3 months led to a statistically significant reduction in the primary composite endpoint of nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death (hazard ratio 0.85 vs. placebo, P = 0.021) (102). This effect was associated with a decrease in CRP but no significant changes in LDL or HDL in the plasma. Thus, the CANTOS trial showed a beneficial effect of blocking inflammation independently of the role of lipids, highlighting the promise of therapy directed toward inflammation in cardiovascular disease. A second trial exploring the potential benefit of low-dose methotrexate for cardiovascular disease is the Cardiovascular Inflammation Reduction Trial, whose results are expected in 2020 (103). Methotrexate, which has long been a mainstay of therapy for autoimmune disease, including rheumatoid arthritis, psoriasis, and psoriatic arthritis, has been shown to attenuate levels of the proinflammatory cytokines IL-6 and TNF-α as well as CRP in the plasma of these patients (104).
A concern with the use of antiinflammatory drug therapy is the potential to compromise of host defense (105–107), and in CANTOS there was a significant increase in the number of fatal infections associated with canakinumab therapy (incidence rate 0.31 vs. 0.18 events per 100 person-years, combined dose groups vs. placebo, P = 0.02) (102). In this context, translational cardiovascular researchers are beginning to consider the alternative strategy of boosting resolution to lower risk of cardiovascular disease, because this strategy would attack one of the root causes of advanced plaque progression while having the capacity to actually boost host defense rather than suppress it (108, 109). One possibility would be to increase the precursors of SPMs through dietary supplementation of n-3 fatty acids. This idea would be valid only if these precursors were rate-limiting for SPM biosynthesis in a manner that could increase the level of lesional SPMs. Conversely, if a major cause of low SPMs in lesions were a defect in a biosynthetic pathway distal to the step where n-3 fatty acids enter the pathway, e.g., excessive phosphorylation of 5-LOX (above), this strategy would not be expected to be effective. Indeed, when the outcomes of a number of n-3 fatty acid trials are considered together, the results have been rather disappointing. While observational population-based studies have suggested that a n-3 fatty acid–rich diet is associated with lower incidence of cardiovascular disease (110, 111), subsequent randomized clinical trials of n-3 fatty acid supplementation yielded mixed results, with only some suggesting a beneficial effect in preventing cardiovascular events (112–115). There have been questions related to the dose and types of n-3 fatty acids used in these trials, and two new cardiovascular outcome studies comparing statin therapy alone with statins plus either high-dose EPA or combined DHA/EPA are currently under way.
In view of the imbalance in SPMs versus leukotrienes in advanced atherosclerotic plaques (116, 117), investigators have tested the effect of atreleuton, a 5-LOX inhibitor, to lower leukotrienes. However, this enzyme is also involved in the synthesis of lipoxins and resolvins (above). Phase II studies conducted in patients with acute coronary syndromes demonstrated that while leukotriene production was decreased in patients given atreleuton, vascular inflammation within the ascending aorta and carotids as measured by fludeoxyglucose (FDG) PET did not show any significant change (118, 119). This lack of efficacy in this early attempt at resolution-targeting therapy may have been due to the dual role of this enzyme in SPM synthesis.
The most direct way to implement resolution mediator therapy is to administer exogenous mediators. Examples summarized in Table 1 and discussed above illustrate the concept that exogenous administration of SPMs provides a protective effect in animal models of atherosclerosis. These preclinical studies, together with the theoretical advantages of pro-resolution versus antiinflammatory therapy outlined above; the fact that SPMs are already being tested in clinical trials for other chronic inflammatory diseases (ClinicalTrials.gov, NCT00799552 and NCT00941018); and the new opportunities for plaque therapy in humans in the wake of CANTOS support further research into using resolution mediators to block plaque progression in humans.
Conclusions and future directions
Despite years of steady progress in the treatment and prevention of cardiovascular disease, death rates attributed to heart attack and stroke are again rising in the United States, presumably owing to the epidemic of obesity and diabetes (2). Aggressive lipid lowering remains the mainstay of therapy for atherosclerosis, but there is room for the development of complementary therapeutics. Although the recent CANTOS trial has demonstrated that targeting inflammation can successfully reduce cardiac mortality in high-risk populations, it has also confirmed that antiinflammatory therapies run the risk of compromising host defense. Our understanding of the etiology of atherosclerosis has recently evolved to include failed resolution as a prominent causal feature for the most clinically relevant advanced plaques, opening up the exciting possibility that clinical plaque progression could be targeted in a manner that would lessen immunosuppression. Moreover, in the context that obesity and type 2 diabetes are major risk factors for atherosclerotic cardiovascular disease — and a major reason why cardiovascular disease remains the leading cause of death (2) — a role for chronic inflammation and failed resolution in metabolic diseases is also emerging (120). Thus, resolution therapy may represent a novel avenue for the integrated treatment of cardiometabolic disease. Although we know that there is an imbalance of proinflammatory versus pro-resolving mediators in advanced atherosclerosis, we do not yet fully understand why the resolution process fails in this setting. On the one hand, we can consider the direct approach of administering exogenous SPMs, as discussed above. However, more mechanistic strategies must also be considered, and this will require a deeper understanding of how SPMs are synthesized and catabolized and how they work on effector cells to carry out various resolution processes. Finally, with the increasing availability of large genetic data sets, incorporating a functional genomic approach to the identification of gene signatures associated with defective inflammation resolution may help identify new targets as well as provide a strategy for the tailoring of therapy to specific individuals.
Acknowledgments
This work was supported by American Heart Association Fellow-to-Faculty award 17FTF33660643 (to ACD) and Post-Doctoral Fellowship grant 15POST25620024 (to BC) as well as by NIH grants T32 HL120826-05 (to CK) and HL075662, HL127464, and HL132412 (to IT). The authors thank Gabrielle Fredman for her inspiration and insight related to many topics covered in this Review.
Version 1. 07/02/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(7):2713–2723.https://doi.org/10.1172/JCI97950.
Contributor Information
Canan Kasikara, Email: Ck2894@cumc.columbia.edu.
Bishuang Cai, Email: bc2586@columbia.edu.
Ira Tabas, Email: iat1@columbia.edu.
References
- 1.Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Behn A, Ur E. The obesity epidemic and its cardiovascular consequences. Curr Opin Cardiol. 2006;21(4):353–360. doi: 10.1097/01.hco.0000231406.84554.96. [DOI] [PubMed] [Google Scholar]
- 3.Williams KJ, Tabas I. The response-to-retention hypothesis of atherogenesis reinforced. Curr Opin Lipidol. 1998;9(5):471–474. doi: 10.1097/00041433-199810000-00012. [DOI] [PubMed] [Google Scholar]
- 4.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38(6):1092–1104. doi: 10.1016/j.immuni.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 7.Kolodgie FD, et al. Pathologic assessment of the vulnerable human coronary plaque. Heart. 2004;90(12):1385–1391. doi: 10.1136/hrt.2004.041798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ball RY, Stowers EC, Burton JH, Cary NR, Skepper JN, Mitchinson MJ. Evidence that the death of macrophage foam cells contributes to the lipid core of atheroma. Atherosclerosis. 1995;114(1):45–54. doi: 10.1016/0021-9150(94)05463-S. [DOI] [PubMed] [Google Scholar]
- 9.Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278(5):483–493. doi: 10.1111/joim.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114(12):1852–1866. doi: 10.1161/CIRCRESAHA.114.302721. [DOI] [PubMed] [Google Scholar]
- 11.Libby P. The molecular mechanisms of the thrombotic complications of atherosclerosis. J Intern Med. 2008;263(5):517–527. doi: 10.1111/j.1365-2796.2008.01965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008;22(10):3595–3606. doi: 10.1096/fj.08-112201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10(1):36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fredman G, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859. doi: 10.1038/ncomms12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viola JR, et al. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res. 2016;119(9):1030–1038. doi: 10.1161/CIRCRESAHA.116.309492. [DOI] [PubMed] [Google Scholar]
- 16.Geering B, Stoeckle C, Conus S, Simon HU. Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol. 2013;34(8):398–409. doi: 10.1016/j.it.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol. 2010;177(4):1576–1591. doi: 10.2353/ajpath.2010.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White PJ, Arita M, Taguchi R, Kang JX, Marette A. Transgenic restoration of long-chain n-3 fatty acids in insulin target tissues improves resolution capacity and alleviates obesity-linked inflammation and insulin resistance in high-fat-fed mice. Diabetes. 2010;59(12):3066–3073. doi: 10.2337/db10-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Titos E, et al. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol. 2011;187(10):5408–5418. doi: 10.4049/jimmunol.1100225. [DOI] [PubMed] [Google Scholar]
- 20.Medeiros R, et al. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am J Pathol. 2013;182(5):1780–1789. doi: 10.1016/j.ajpath.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Planagumà A, et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am J Respir Crit Care Med. 2008;178(6):574–582. doi: 10.1164/rccm.200801-061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giera M, et al. Lipid and lipid mediator profiling of human synovial fluid in rheumatoid arthritis patients by means of LC-MS/MS. Biochim Biophys Acta. 2012;1821(11):1415–1424. doi: 10.1016/j.bbalip.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 24.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2(7):612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 25.Serhan CN, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196(8):1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clària J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci U S A. 1995;92(21):9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serhan CN, Chiang N, Dalli J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin Immunol. 2015;27(3):200–215. doi: 10.1016/j.smim.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000;192(8):1197–1204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Serhan CN, et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med. 2009;206(1):15–23. doi: 10.1084/jem.20081880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bannenberg GL, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. 2005;174(7):4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 31.Serhan CN, Krishnamoorthy S, Recchiuti A, Chiang N. Novel anti-inflammatory–pro-resolving mediators and their receptors. Curr Top Med Chem. 2011;11(6):629–647. doi: 10.2174/1568026611109060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalli J, et al. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J. 2013;27(7):2573–2583. doi: 10.1096/fj.13-227728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell S, et al. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J Am Soc Nephrol. 2002;13(10):2497–2507. doi: 10.1097/01.ASN.0000032417.73640.72. [DOI] [PubMed] [Google Scholar]
- 34.Haeggström JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111(10):5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 35.Wong A, Cook MN, Foley JJ, Sarau HM, Marshall P, Hwang SM. Influx of extracellular calcium is required for the membrane translocation of 5-lipoxygenase and leukotriene synthesis. Biochemistry. 1991;30(38):9346–9354. doi: 10.1021/bi00102a030. [DOI] [PubMed] [Google Scholar]
- 36.Woods JW, et al. 5-Lipoxygenase and 5-lipoxygenase-activating protein are localized in the nuclear envelope of activated human leukocytes. J Exp Med. 1993;178(6):1935–1946. doi: 10.1084/jem.178.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fredman G, et al. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc Natl Acad Sci U S A. 2014;111(40):14530–14535. doi: 10.1073/pnas.1410851111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qiu H, et al. Expression of 5-lipoxygenase and leukotriene A4 hydrolase in human atherosclerotic lesions correlates with symptoms of plaque instability. Proc Natl Acad Sci U S A. 2006;103(21):8161–8166. doi: 10.1073/pnas.0602414103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spanbroek R, et al. Expanding expression of the 5-lipoxygenase pathway within the arterial wall during human atherogenesis. Proc Natl Acad Sci U S A. 2003;100(3):1238–1243. doi: 10.1073/pnas.242716099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dwyer JH, et al. Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N Engl J Med. 2004;350(1):29–37. doi: 10.1056/NEJMoa025079. [DOI] [PubMed] [Google Scholar]
- 41.Mehrabian M, et al. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ Res. 2002;91(2):120–126. doi: 10.1161/01.RES.0000028008.99774.7F. [DOI] [PubMed] [Google Scholar]
- 42.Wittwer J, Bayer M, Mosandl A, Muntwyler J, Hersberger M. The c.-292C>T promoter polymorphism increases reticulocyte-type 15-lipoxygenase-1 activity and could be atheroprotective. Clin Chem Lab Med. 2007;45(4):487–492. doi: 10.1515/CCLM.2007.103. [DOI] [PubMed] [Google Scholar]
- 43.Assimes TL, et al. A near null variant of 12/15-LOX encoded by a novel SNP in ALOX15 and the risk of coronary artery disease. Atherosclerosis. 2008;198(1):136–144. doi: 10.1016/j.atherosclerosis.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen J, et al. Macrophage-mediated 15-lipoxygenase expression protects against atherosclerosis development. J Clin Invest. 1996;98(10):2201–2208. doi: 10.1172/JCI119029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merched AJ, Serhan CN, Chan L. Nutrigenetic disruption of inflammation-resolution homeostasis and atherogenesis. J Nutrigenet Nutrigenomics. 2011;4(1):12–24. doi: 10.1159/000326890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salic K, et al. Resolvin E1 attenuates atherosclerosis in absence of cholesterol-lowering effects and on top of atorvastatin. Atherosclerosis. 2016;250:158–165. doi: 10.1016/j.atherosclerosis.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 47.Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cδ, and p38 mitogen-activated protein kinase (MAPK) J Biol Chem. 2011;286(38):33335–33344. doi: 10.1074/jbc.M111.263020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hasturk H, et al. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler Thromb Vasc Biol. 2015;35(5):1123–1133. doi: 10.1161/ATVBAHA.115.305324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colas RA, et al. Impaired production and diurnal regulation of vascular RvDn-3 DPA increase systemic inflammation and cardiovascular disease. Circ Res. 2018;122(6):855–863. doi: 10.1161/CIRCRESAHA.117.312472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petri MH, et al. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E–/– mice. Br J Pharmacol. 2017;174(22):4043–4054. doi: 10.1111/bph.13707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chatterjee BE, et al. Annexin 1-deficient neutrophils exhibit enhanced transmigration in vivo and increased responsiveness in vitro. J Leukoc Biol. 2005;78(3):639–646. doi: 10.1189/jlb.0405206. [DOI] [PubMed] [Google Scholar]
- 53.Babbin BA, et al. Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J Immunol. 2008;181(7):5035–5044. doi: 10.4049/jimmunol.181.7.5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ouyang N, et al. MC-12, an annexin A1-based peptide, is effective in the treatment of experimental colitis. PLoS One. 2012;7(7):e41585. doi: 10.1371/journal.pone.0041585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siqueira Mietto B, Kroner A, Girolami EI, Santos-Nogueira E, Zhang J, David S. Role of IL-10 in resolution of inflammation and functional recovery after peripheral nerve injury. J Neurosci. 2015;35(50):16431–16442. doi: 10.1523/JNEUROSCI.2119-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kearley J, Barker JE, Robinson DS, Lloyd CM. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J Exp Med. 2005;202(11):1539–1547. doi: 10.1084/jem.20051166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drechsler M, et al. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ Res. 2015;116(5):827–835. doi: 10.1161/CIRCRESAHA.116.305825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fredman G, et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med. 2015;7(275):275ra20. doi: 10.1126/scitranslmed.aaa1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kamaly N, et al. Targeted interleukin-10 nanotherapeutics developed with a microfluidic chip enhance resolution of inflammation in advanced atherosclerosis. ACS Nano. 2016;10(5):5280–5292. doi: 10.1021/acsnano.6b01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science. 2013;341(6153):1483–1488. doi: 10.1126/science.1240636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keller M, et al. A circadian clock in macrophages controls inflammatory immune responses. Proc Natl Acad Sci U S A. 2009;106(50):21407–21412. doi: 10.1073/pnas.0906361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang L, et al. Overexpression of CRY1 protects against the development of atherosclerosis via the TLR/NF-κB pathway. Int Immunopharmacol. 2015;28(1):525–530. doi: 10.1016/j.intimp.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 63.Pan X, Jiang XC, Hussain MM. Impaired cholesterol metabolism and enhanced atherosclerosis in clock mutant mice. Circulation. 2013;128(16):1758–1769. doi: 10.1161/CIRCULATIONAHA.113.002885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krishnamoorthy S, et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci U S A. 2010;107(4):1660–1665. doi: 10.1073/pnas.0907342107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petri MH, Laguna-Fernández A, Gonzalez-Diez M, Paulsson-Berne G, Hansson GK, Bäck M. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc Res. 2015;105(1):65–74. doi: 10.1093/cvr/cvu224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dufton N, et al. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184(5):2611–2619. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ye RD, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61(2):119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Norling LV, Dalli J, Flower RJ, Serhan CN, Perretti M. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: receptor-dependent actions. Arterioscler Thromb Vasc Biol. 2012;32(8):1970–1978. doi: 10.1161/ATVBAHA.112.249508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hayhoe RP, Kamal AM, Solito E, Flower RJ, Cooper D, Perretti M. Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: indication of distinct receptor involvement. Blood. 2006;107(5):2123–2130. doi: 10.1182/blood-2005-08-3099. [DOI] [PubMed] [Google Scholar]
- 70.Subbarao K, et al. Role of leukotriene B4 receptors in the development of atherosclerosis: potential mechanisms. Arterioscler Thromb Vasc Biol. 2004;24(2):369–375. doi: 10.1161/01.ATV.0000110503.16605.15. [DOI] [PubMed] [Google Scholar]
- 71.Aiello RJ, Bourassa PA, Lindsey S, Weng W, Freeman A, Showell HJ. Leukotriene B4 receptor antagonism reduces monocytic foam cells in mice. Arterioscler Thromb Vasc Biol. 2002;22(3):443–449. doi: 10.1161/hq0302.105593. [DOI] [PubMed] [Google Scholar]
- 72.Laguna-Fernandez A, et al. ERV1/ChemR23 signaling protects from atherosclerosis by modifying oxLDL uptake and phagocytosis in macrophages. Circulation. doi: 10.1161/CIRCULATIONAHA.117. [published online ahead of print May 8, 2018]. https://doi.org/10.1161/CIRCULATIONAHA.117.032801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li S, et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ Res. 2009;105(11):1072–1082. doi: 10.1161/CIRCRESAHA.109.199570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chistiakov DA, Bobryshev YV, Nikiforov NG, Elizova NV, Sobenin IA, Orekhov AN. Macrophage phenotypic plasticity in atherosclerosis: the associated features and the peculiarities of the expression of inflammatory genes. Int J Cardiol. 2015;184:436–445. doi: 10.1016/j.ijcard.2015.03.055. [DOI] [PubMed] [Google Scholar]
- 75.Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120(15):e60–e72. doi: 10.1182/blood-2012-04-423525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Paoli F, Staels B, Chinetti-Gbaguidi G. Macrophage phenotypes and their modulation in atherosclerosis. Circ J. 2014;78(8):1775–1781. doi: 10.1253/circj.CJ-14-0621. [DOI] [PubMed] [Google Scholar]
- 77.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 78.Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25(6):1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7. [DOI] [PubMed] [Google Scholar]
- 79.Linton MF, Babaev VR, Huang J, Linton EF, Tao H, Yancey PG. Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ J. 2016;80(11):2259–2268. doi: 10.1253/circj.CJ-16-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. 2009;86(5):1089–1095. doi: 10.1189/jlb.0209115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447(7146):869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. 2000;164(4):1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 83.Chiang N, Dalli J, Colas RA, Serhan CN. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J Exp Med. 2015;212(8):1203–1217. doi: 10.1084/jem.20150225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ohira T, Arita M, Omori K, Recchiuti A, Van Dyke TE, Serhan CN. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J Biol Chem. 2010;285(5):3451–3461. doi: 10.1074/jbc.M109.044131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maddox JF, Serhan CN. Lipoxin A4 and B4 are potent stimuli for human monocyte migration and adhesion: selective inactivation by dehydrogenation and reduction. J Exp Med. 1996;183(1):137–146. doi: 10.1084/jem.183.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maddox JF, Hachicha M, Takano T, Petasis NA, Fokin VV, Serhan CN. Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J Biol Chem. 1997;272(11):6972–6978. doi: 10.1074/jbc.272.11.6972. [DOI] [PubMed] [Google Scholar]
- 87.Ait-Oufella H, et al. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28(8):1429–1431. doi: 10.1161/ATVBAHA.108.169078. [DOI] [PubMed] [Google Scholar]
- 88.Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe–/– mice. Arterioscler Thromb Vasc Biol. 2008;28(8):1421–1428. doi: 10.1161/ATVBAHA.108.167197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cai B, et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest. 2017;127(2):564–568. doi: 10.1172/JCI90520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Garbin U, et al. Expansion of necrotic core and shedding of Mertk receptor in human carotid plaques: a role for oxidized polyunsaturated fatty acids? Cardiovasc Res. 2013;97(1):125–133. doi: 10.1093/cvr/cvs301. [DOI] [PubMed] [Google Scholar]
- 91.Cai B, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U S A. 2016;113(23):6526–6531. doi: 10.1073/pnas.1524292113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Doran AC, et al. CAMKIIγ suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J Clin Invest. 2017;127(11):4075–4089. doi: 10.1172/JCI94735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.El Kebir D, Gjorstrup P, Filep JG. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci U S A. 2012;109(37):14983–14988. doi: 10.1073/pnas.1206641109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.El Kebir D, et al. Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: a novel mechanism for resolution of inflammation. J Immunol. 2007;179(1):616–622. doi: 10.4049/jimmunol.179.1.616. [DOI] [PubMed] [Google Scholar]
- 95.Zouki C, Ouellet S, Filep JG. The anti-inflammatory peptides, antiflammins, regulate the expression of adhesion molecules on human leukocytes and prevent neutrophil adhesion to endothelial cells. FASEB J. 2000;14(3):572–580. doi: 10.1096/fasebj.14.3.572. [DOI] [PubMed] [Google Scholar]
- 96.Watson RW, Rotstein OD, Nathens AB, Parodo J, Marshall JC. Neutrophil apoptosis is modulated by endothelial transmigration and adhesion molecule engagement. J Immunol. 1997;158(2):945–953. [PubMed] [Google Scholar]
- 97.Walther A, Riehemann K, Gerke V. A novel ligand of the formyl peptide receptor: annexin I regulates neutrophil extravasation by interacting with the FPR. Mol Cell. 2000;5(5):831–840. doi: 10.1016/S1097-2765(00)80323-8. [DOI] [PubMed] [Google Scholar]
- 98.Prieto P, et al. Activation of autophagy in macrophages by pro-resolving lipid mediators. Autophagy. 2015;11(10):1729–1744. doi: 10.1080/15548627.2015.1078958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162(4):597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 100.Qamar A, Rader DJ. Effect of interleukin 1β inhibition in cardiovascular disease. Curr Opin Lipidol. 2012;23(6):548–553. doi: 10.1097/MOL.0b013e328359b0a6. [DOI] [PubMed] [Google Scholar]
- 101.Welsh P, Grassia G, Botha S, Sattar N, Maffia P. Targeting inflammation to reduce cardiovascular disease risk: a realistic clinical prospect? Br J Pharmacol. 2017;174(22):3898–3913. doi: 10.1111/bph.13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ridker PM, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 103.Everett BM, et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166(2):199–207.e15. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chan ES, Cronstein BN. Molecular action of methotrexate in inflammatory diseases. Arthritis Res. 2002;4(4):266–273. doi: 10.1186/ar419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fallahi-Sichani M, Flynn JL, Linderman JJ, Kirschner DE. Differential risk of tuberculosis reactivation among anti-TNF therapies is due to drug binding kinetics and permeability. J Immunol. 2012;188(7):3169–3178. doi: 10.4049/jimmunol.1103298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.St Clair EW, et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: a randomized, controlled trial. Arthritis Rheum. 2004;50(11):3432–3443. doi: 10.1002/art.20568. [DOI] [PubMed] [Google Scholar]
- 107.Breedveld FC, et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54(1):26–37. doi: 10.1002/art.21519. [DOI] [PubMed] [Google Scholar]
- 108.Spite M, et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461(7268):1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chiang N, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484(7395):524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.de Oliveira Otto MC, et al. Circulating and dietary omega-3 and omega-6 polyunsaturated fatty acids and incidence of CVD in the Multi-Ethnic Study of Atherosclerosis. J Am Heart Assoc. 2013;2(6):e000506. doi: 10.1161/JAHA.113.000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sperling LS, Nelson JR. History and future of omega-3 fatty acids in cardiovascular disease. Curr Med Res Opin. 2016;32(2):301–311. doi: 10.1185/03007995.2015.1120190. [DOI] [PubMed] [Google Scholar]
- 112.No authors listed Dietary supplementation with n-3 polyunsaturated fatty acids vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet. 1999;354(9177):447–455. doi: 10.1016/S0140-6736(99)07072-5. [DOI] [PubMed] [Google Scholar]
- 113.Oikawa S, et al. Suppressive effect of EPA on the incidence of coronary events in hypercholesterolemia with impaired glucose metabolism: sub-analysis of the Japan EPA Lipid Intervention Study (JELIS) Atherosclerosis. 2009;206(2):535–539. doi: 10.1016/j.atherosclerosis.2009.03.029. [DOI] [PubMed] [Google Scholar]
- 114.ORIGIN Trial Investigators, et al. n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med. 2012;367(4):309–318. doi: 10.1056/NEJMoa1203859. [DOI] [PubMed] [Google Scholar]
- 115.Risk Prevention Study Collaborative Group. et al. n-3 fatty acids in patients with multiple cardiovascular risk factors. N Engl J Med. 2013;368(19):1800–1808. doi: 10.1056/NEJMoa1205409. [DOI] [PubMed] [Google Scholar]
- 116.De Caterina R, Zampolli A. From asthma to atherosclerosis — 5-lipoxygenase, leukotrienes, and inflammation. N Engl J Med. 2004;350(1):4–7. doi: 10.1056/NEJMp038190. [DOI] [PubMed] [Google Scholar]
- 117.Bäck M. Leukotriene signaling in atherosclerosis and ischemia. Cardiovasc Drugs Ther. 2009;23(1):41–48. doi: 10.1007/s10557-008-6140-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tardif JC, et al. Treatment with 5-lipoxygenase inhibitor VIA-2291 (Atreleuton) in patients with recent acute coronary syndrome. Circ Cardiovasc Imaging. 2010;3(3):298–307. doi: 10.1161/CIRCIMAGING.110.937169. [DOI] [PubMed] [Google Scholar]
- 119.Gaztanaga J, et al. A phase 2 randomized, double-blind, placebo-controlled study of the effect of VIA-2291, a 5-lipoxygenase inhibitor, on vascular inflammation in patients after an acute coronary syndrome. Atherosclerosis. 2015;240(1):53–60. doi: 10.1016/j.atherosclerosis.2015.02.027. [DOI] [PubMed] [Google Scholar]
- 120.Spite M, Clària J, Serhan CN. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2014;19(1):21–36. doi: 10.1016/j.cmet.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ariel A, Chiang N, Arita M, Petasis NA, Serhan CN. Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-α secretion from human T cells. J Immunol. 2003;170(12):6266–6272. doi: 10.4049/jimmunol.170.12.6266. [DOI] [PubMed] [Google Scholar]
- 122.Hachicha M, Pouliot M, Petasis NA, Serhan CN. Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor necrosis factor 1alpha-initiated neutrophil responses and trafficking: regulators of a cytokine-chemokine axis. J Exp Med. 1999;189(12):1923–1930. doi: 10.1084/jem.189.12.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chiang N, Fierro IM, Gronert K, Serhan CN. Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J Exp Med. 2000;191(7):1197–1208. doi: 10.1084/jem.191.7.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sun YP, et al. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem. 2007;282(13):9323–9334. doi: 10.1074/jbc.M609212200. [DOI] [PubMed] [Google Scholar]
- 125.Scannell M, et al. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. 2007;178(7):4595–4605. doi: 10.4049/jimmunol.178.7.4595. [DOI] [PubMed] [Google Scholar]
- 126.Arita M, Ohira T, Sun YP, Elangovan S, Chiang N, Serhan CN. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J Immunol. 2007;178(6):3912–3917. doi: 10.4049/jimmunol.178.6.3912. [DOI] [PubMed] [Google Scholar]
- 127.Arita M, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. 2005;201(5):713–722. doi: 10.1084/jem.20042031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Oh SF, Pillai PS, Recchiuti A, Yang R, Serhan CN. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J Clin Invest. 2011;121(2):569–581. doi: 10.1172/JCI42545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Oh SF, Dona M, Fredman G, Krishnamoorthy S, Irimia D, Serhan CN. Resolvin E2 formation and impact in inflammation resolution. J Immunol. 2012;188(9):4527–4534. doi: 10.4049/jimmunol.1103652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fredman G, Van Dyke TE, Serhan CN. Resolvin E1 regulates adenosine diphosphate activation of human platelets. Arterioscler Thromb Vasc Biol. 2010;30(10):2005–2013. doi: 10.1161/ATVBAHA.110.209908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9(1):62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- 132.Dalli J, et al. Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem Biol. 2013;20(2):188–201. doi: 10.1016/j.chembiol.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]