

Abstract
The application of population genetic methods in combination with gene mapping strategies can help to identify genes and mutations selected during the evolution from wild plants to crops and to explore the considerable genetic variation still maintained in natural populations. We genotyped a grapevine germplasm collection of 44 wild (Vitis vinifera subsp. sylvestris) and 48 cultivated (V. vinifera subsp. sativa) accessions at 54 K single-nucleotide polymorphisms (SNPs) to perform a whole-genome comparison of the main population genetic statistics. The analysis of Wright Fixation Index (FST) along the whole genome allowed us to identify several putative “signatures of selection” spanning over two thousand SNPs significantly differentiated between sativa and sylvestris. Many of these genomic regions included genes involved in the adaptation to environmental changes. An overall reduction of nucleotide diversity was observed across the whole genome within sylvestris, supporting a small effective population size of the wild grapevine. Tajima’s D resulted positive in both wild and cultivated subgroups, which may indicate an ongoing balancing selection. Association mapping for six domestication-related traits was performed in combination with population genetics, providing further evidence of different perception and response to environmental stresses between sativa and sylvestris.
Grapes: Wild and cultivated grapevines adapted to different stresses
Wild and domesticated grapevine populations display genomic differences that likely arose as a result of evolutionary responses to different environmental pressures. Annarita Marrano of the Edmund Mach Foundation in San Michele All’adige, Italy, and colleagues characterized the genetic diversity found in 44 wild and 48 cultivated accessions of the Eurasian wine grape, Vitis vinifera. They analyzed 54,000 single DNA letters spread across the genome and found evidence that natural wild grapevine populations are on the decline. They also identified several genomic regions related to stress responses that evolved differently in grapes bred to live in vineyards and their wild relatives. The findings suggest the need to conserve wild grapevines, which may harbor genes encoding resilience factors that could aid in the development of hardier wine-producing crops in the future.
Introduction
The Eurasian grape (Vitis vinifera L.) is one of the most important crops worldwide for its global distribution and economic value1. V. vinifera L. exists as the cultivated form V. vinifera subsp. sativa (or vinifera; hereafter called sativa) and the wild-form V. vinifera subsp sylvestris (hereafter called sylvestris), which is assumed to be the ancestor of modern cultivars. The two subspecies exhibit several phenotypical differences, notably in flower sex, seed shape, bunch and berry size, and leaf morphology2. In particular, sylvestris is dioecious with separate male and female individuals, and in general produces few bunches with small, black and juiceless berries. On the contrary, sativa presents hermaphroditic flowers and enormous phenotypic variability regarding number, size, taste, and colour of the fruit. Previous surveys in grapevine collections have also outlined weak but clear genetic differentiation among cultivated and ex situ wild accessions3–5. The cultivated grapevine has broad genetic variation, which is likely the result of sexual reproduction, vegetative propagation, and somatic mutations occurring during the long history of grapevine cultivation1. In contrast, sylvestris is less diverse than the domesticated form6. The present distribution of the wild vinifera is fragmented in relict populations with very few individuals. The decline of sylvestris has drastically increased over the last two centuries because of the introduction of pests and diseases (phylloxera, downy and powdery mildew) from North America, and the anthropic impact on wild grapevine habitats7. Also, gene flow between cultivated and wild grapevines might lead to domestic introgression and genetic loss in the small relict populations of sylvestris8. Recently, several efforts have been devoted to the study of biotic and abiotic stresses-response in wild V. vinifera, revealing accessions tolerant to salt stress9, lime-induced chlorosis10 and downy mildew (Plasmopara viticola)11. These findings shift sylvestris into the center of attention as a valuable genetic resource for grapevine resilience breeding, which has been so far based on the exploitation of the innate resistance/tolerance of other Vitis species. The identification of genetic resistance in sylvestris may allow preserving the vinifera genomic background of high fruit quality in future breeding cycles. The need to investigate the genetic diversity of sylvestris becomes more significant considering the potential increased vulnerability to environmental changes and the appearance of new pests and diseases since the limited number of grape cultivars grown worldwide.
The morphological and genetic divergence between the two vinifera subspecies may be the result of different evolutionary forces acting on sativa in the vineyards and on sylvestris in floodplain forests. Whole-genome comparison of genetic diversity is a feasible strategy to discover the genes involved in this progressive and subtle differentiation between sativa and sylvestris12. Genome scanning for signatures of selection has been reported for several crops such as tomato13, maize14, rice15 and barrel medic16. In grapevine, Myles et al.4 used 9 K SNPs to compare the haplotype diversity between sativa and sylvestris, identifying a 5-Mb putative signature of selection on chromosome 17. Association mapping (AM) is an alternative to population genetics to uncover the genomic regions responsible for the phenotypic variation observed in V. vinifera. However, few studies of AM in fruit species have been reported so far, due to the difficulties in building up an ideal large association panel without an intricate pattern of population stratification and familial relatedness17. Chitwood et al.18 performed a genome-wide association scan (GWAS) to map the genetic basis of leaf morphology in grapevine, identifying a handful of SNPs associated with just four of the 13 phenotyped traits. This GWA study underlined the limited power of AM in grapevine, which can mainly be attributed to its rapid linkage disequilibrium (LD) decay19. More recently, Migicovsky et al.20 combined GWAS with selective sweep mapping to deal with the fast LD decay and increase the power of detecting loci targeted during the domestication and breeding of wine and table grapes.
In the present research, we assess the distribution and magnitude of the genomic differences between two populations of wild and domesticated grapevine. The study is organized into the following two main milestones, (i) the development of high-density SNP-based genotyping for a germplasm collection, including many authentic wild V. vinifera; (ii) the whole-genome scan for signatures of selection by using a combination of population genetics methods. Our results provide evidence of genetic differentiation between sativa and sylvestris individuals at genomic regions mainly involved in response to environmental stimuli. These findings draw attention to wild grapevines as a valuable source of resilience factors, whose re-discovery might be fundamental for sustainable agriculture in the future.
Materials and methods
SNP genotyping of a grapevine germplasm population
A germplasm collection of 48 cultivated (Vitis vinifera spp. sativa) and 44 wild female (Vitis vinifera spp. sylvestris) grapevines (Supplementary Table S1) was sorted at the FEM grape repository (ITA362) as described by Marrano et al.6. All samples were grafted on the rootstock Kober 5BB, and uniformly pruned and trained according to the Guyot system. DNA extraction was performed from young leaf tissue of one field grown plant per accession using the DNeasy 96 plant mini kit (QIAGEN, Germany). Both the Synergy HT Multi-Mode Microplate Reader (BioTek) and the NanoDrop 8000 UV-Vis Spectrophotometers (Thermo Scientific) were used to inspect DNA concentration and purity. DNA samples were adjusted to a minimum concentration of 100 ng/µL in 10 µL aliquots. The commercial GrapeReseq 20 K SNPs array (http://urgi.versailles.inra.fr/Species/Vitis/GrapeReSeq_Illumina_20K)21 was used to genotype the whole population with the Infinium technology according to the Illumina protocol (Illumina, Inc., San Diego, CA, USA). The genomic DNA of the Pinot Noir cultivar was used as a control. SNPs genotypes were scored using the Genotyping Module v1.9 of the Illumina GenomeStudio Data Analysis software. SNPs with a Call Freq score 0 and a GenTrain < 0.6 were filtered out. Markers with a Cluster Sep score < 0.4 were visually inspected for accuracy of the SNP calling. SNPs with R mean score > 0.3 and with clusters not overlapped were retained. The obtained high-quality SNPs were merged in a unique panel with 37 K SNPs from a RAD-seq data set previously generated6. For the SNPs shared between the two experiments, only the SNP profiles from the 20 K Illumina array were retained. Samples and SNP loci with a call rate < 0.8 were filtered out. Genotype imputation was performed to fill in missing data using LinkImpute v1.1.1 software, which is based on a k-nearest neighbour genotype imputation method (LD-kNNi) designed to work with unordered markers22. Finally, SNPs with a minor allele frequency (MAF) < 0.05 were removed using Plink v1.9 software23.
Analysis of population structure
The genetic structure of the germplasm population was analysed with fastSTRUCTURE v1.024. A number of ancestral genetic groups (K), ranging from 1 to 10, was tested by ten independent iterations for each K. The most likely K value was chosen running the algorithm for multiple choices of K and by plotting the marginal likelihood of the data. The software CLUMPP v1.1.225 was used to find optimal alignments of the independent runs and the output was used directly as input into the program for cluster visualisation DISTRUCT v1.126. Moreover, a Principal Component Analysis (PCA) was performed as implemented in ‘adegenet’27 R package for the multivariate analysis of genetic markers.
LD decay
Linkage disequilibrium (LD) was estimated between all SNPs with a MAF > 5% in the whole germplasm population and within sativa and sylvestris subgroups separately by using Plink v1.9 software23. The classical r2 estimate of the correlation between genotypes was used. LD decay was explored by plotting the median r2 in sequential bins of 10 Kb against the physical position. Moreover, LD landscape of each chromosome was also inspected through heat-map visualisation with the software Haploview v4.128.
Genomic differentiation between sativa and sylvestris genotypes
Fixation index (FST) was measured between sativa and sylvestris accessions with VCFtools v0.1.1329, setting a sliding window of 100 kb with a step size of 10 kb. Genomic windows with the top 5% of FST values were selected as candidate regions for further analysis. To verify the empirical cutoff with low false discovery rate, we performed whole-genome permutation tests to ascertain the thresholds for identifying genomic regions highly differentiated between the two grapevine subgroups. In more detail, all the genotypes of sativa and sylvestris were shuffled, and the FST analysis was performed with the same parameters 1000 times. Nucleotide diversity (π) and Tajima’s D30 were estimated along the whole genome in 100-kb windows with a step size of 10 kb using VCFtools, to interpret better the results gained with the FST analysis and clarify how sativa and sylvestris genotypes differentiated. The grape gene annotation v2.1 hosted on http://genomes.cribi.unipd.it/grape/31 was used to investigate the putative gene functions of the genomic regions with the top 5% of FST values. In particular, the distribution of the identified genes into different biological processes was evaluated using the weight01 method provided by the R package topGO32. The Kolmogorov–Smirnov-like test was performed to assess the significance of over-representation of GO categories compared with all genes in the grapevine gene prediction. Also, differentiation in the genomic regions reported in the literature associated with flower and fruit traits was checked.
Association mapping for domestication-related traits
Genotype-phenotype associations were tested for up to six domestication-related traits (single bunch weight (SBCW), single berry weight (SBW), yield, number of bunches per plant (NBCs), total soluble solids (Brix°) and pH; see Supplementary Note S1), using both genotypic best linear unbiased predictors (BLUPs) and the average performance of each sample in each year separately. Also, GWAS was run for the “Species” trait coded as a binary phenotype assigning 1 to sativa accessions and 0 to sylvestris samples. GWAS was carried out by applying three models, which account for different confounding factors to avoid spurious marker-trait associations (Supplementary Note S1). All three models are implemented in TASSEL v5.0 software33. A quantile–quantile (Q–Q) plot was used to choose the model that better fit population structure and familial relatedness in the marker-trait association (Supplementary Note S1). P values adjustment for multiple testing was performed, and the Bonferroni-corrected critical p values and False Discovery Rate (FDR) were used to identify significant marker-trait associations. Manhattan plots were displayed accordingly using the ‘qqman’ v0.1.3 R package34. The positions of markers significantly associated with phenotypes were used to investigate the grapevine gene annotation v2.1. With regard to the extent of LD, windows of 10 kb upstream and downstream, the SNPs of interest were used to identify candidate genes. If the markers fell within long LD blocks, the entire genomic region located between the extreme SNPs was explored.
Results and discussion
SNP genotyping of a grapevine germplasm population
A total of 92 wild and domesticated grapevine accessions were genotyped using the custom Illumina Infinium Vitis20K SNP array and a novel RAD-seq approach6. We merged the two SNPs matrices in a unique panel since the distribution of allele frequencies within the sativa and sylvestris subgroups showed the same trend (Supplementary Figure S1). The merged data set included totally 54,157 SNPs (Table 1). We first filtered for a missing rate > 0.2, removing six samples and 22,258 markers (Supplementary Table S1). As shown in Table 1, most of the SNPs filtered out due to high missing rate came from RAD-seq. This result is a common issue of all methods of reduce representation sequencing, where several technical factors led all the sequenced regions not to be evenly covered in all the individuals of the population35. After imputing the remaining missing genotypes, SNPs with a minor allele frequency (MAF) < 0.05 were removed. Most of the SNPs with a low MAF came from the Vitis20K array (Table 1), and they probably resulted from errors in the genotype calling. The final panel counted 86 samples and 26,893 SNPs (hereafter called 26 K SNPs) with an average of 1.3 K SNPs per chromosome. In particular, the SNP density ranged from one SNP every 15 kb on chr8 to one SNP every 21 kb on chr19.
Table 1.
Summary of SNPs filtering after the population genotyping assays with the Vitis20K Illumina chip and RAD-seq approaches
| Genotyping technology | Initial No. of SNPs | No of SNPs removed for a missing rate > 0.2 | No of SNPs removed for a MAF < 0.05 | Final No of SNPs |
|---|---|---|---|---|
| Vitis20K | 16,563 | 338 | 3600 | 12,625 |
| RAD-seq | 37,594 | 21,920 | 1330 | 14,268 |
| Total | 54,157 | 22,258 | 4930 | 26,893 |
Analysis of population structure
We used the 26 K SNPs panel to investigate the population structure and visualise the relationships among individual accessions using two different approaches: Principal Component Analysis (PCA) and model-based clustering. Figure 1 shows the first two principal components (PCs), which accounted for the 21% of the total variance. PC1 differentiates sylvestris genotypes from the cultivated varieties, whereas PC2 reflects the variability among sativa accessions.
Fig. 1. Visualisation of the genetic relationships among wild and cultivated vinifera by their projection onto the first two Principal Component axes.

Along each axis, the proportion of the total variance accounted by each PC is shown in parentheses
As a second approach, we used the clustering algorithm implemented in fastSTRUCTURE software (Fig. 2). The optimal number of subgroups was three, with 81% of the individuals showing a clear assignment to a cluster (membership likelihood > 0.75; Supplementary Table S2). The two principal groups included 28 sativa accessions and 36 sylvestris individuals, respectively, while Pinot Noir, Gewürtztraminer (an aromatic mutation of Traminer Rot) and Mornan Noir cultivars clustered together in a third separate group. Previous studies with microsatellite markers (SSRs)36 have already suggested the first-degree relationship of Pinot Noir and Traminer. Moreover, Pinot Noir and Traminer have presumably ancient origins, and many modern cultivars are their first-degree relatives37. Indeed, many of the 19 genotypes (13 sativa and six sylvestris) not assigned to a defined group by fastSTRUCTURE exhibited admixture with this small cluster (K2, Supplementary Table S2). However, the analysis of the population structure highlighted how sativa and sylvestris individuals were well distinguished as two separated groups with a low level of admixture. This result is consistent with previous reports based on SSR and SNP genetic profiles, which showed a clear distinction between wild and cultivated individuals5. However, we used sylvestris individuals previously clustered through a hierarchical STRUCTURE analysis, and sativa accessions selected from a core collection that maximises the genetic diversity present in the whole germplasm collection6. Therefore, biases in allele frequencies may have been introduced, leading to an underestimation of the real level of admixture between the two subspecies.
Fig. 2. Barplot of admixture proportions of wild and cultivated subpopulations, as measured by fastSTRUCTURE at K = 3.
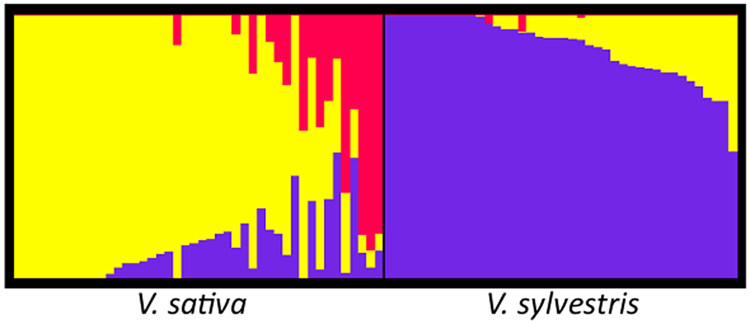
Each individual is represented by a vertical bar, reflecting assignment probabilities to each of the three groups. K1: purple bars; K2: red bars; K3: yellow bars
Estimation of linkage disequilibrium
We used the 26 K SNPs data set to estimate and validate the level of LD along the whole genome within the investigated germplasm collection of grapevine6. The LD, as measured by the standard r2 correlation coefficient, decayed below 0.2 within 10 kb (Fig. 3). Such rapid LD decay is consistent with the results of Myles et al.4, which detected a low level of LD (r2 < 0.2) at short physical distances using the Vitis9K SNP array. Lijavetzky et al38. observed an even lower level of LD, which decayed within 100–200 bp in more than 200 gene sequences. On the other hand, Nicolas et al.17 found that the decay of LD down to 0.2 ranged from 9 to 458 kb. These discrepancies may be related to the low number of genomic regions investigated in both LD surveys compared to our genome-wide analysis of LD. We confirmed the evidence of a rapid LD decay in grapevine, which is in agreement with the high polymorphic rate of the grapevine genome39.
Fig. 3. Decay of LD in sativa and sylvestris separately.
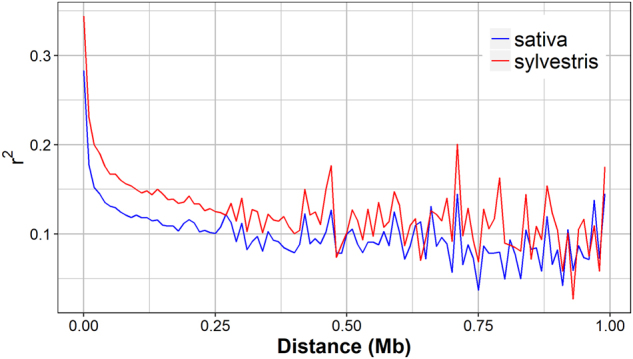
Each point represents the median r2 value in sequential bins of 10 Kb against the physical position
When analysed separately in the two subspecies, the decay of LD appeared slower within the sylvestris group, where r2 reached values below 0.2 within 20 kb. Therefore, the increase in SNPs density throughout the genome did not have any significant contribution towards shaping the LD patterns within the two grapevine subpopulations. The longer extent of LD observed in the sylvestris subgroup can be related to an elevated level of inbreeding as well as to the common Italian origin of most of our wild accessions6. The differences in LD extent between sativa and sylvestris accessions were more evident by the comparison of the LD patterns on each chromosome. In particular, long-range LD (LRLD) between widely separated loci on the chromosome (distance > 1 Mb) was observed for almost all chromosomes in the sylvestris group (Supplementary Figure S3). The presence of LRLD suggested the action of some forces, such as genetic drift, hitchhiking with positive-selected mutation or structural variation in chromosomes40. Blocks of short-range LD were also observed within sativa on chromosomes 2, 6, 17 and 18 (Supplementary Figure S2). Here major QTLs associated with important traits in grapevine have been identified, such as those for flower sex and berry skin colour on chr241, and for fruit weight on chr17 and chr1842.
Genomic differentiation between sativa and sylvestris genotypes
Since the analysis of population structure underlined a clear separation between sativa and sylvestris accessions, we computed population differentiation statistic (FST) across the grapevine genome to identify genomic regions with altered allele frequency among the two V. vinifera subspecies. The overall level of genetic differentiation between cultivated and wild grapes was moderate (FST = 0.12). A similar genetic divergence was reported among Western European cultivars and wild genotypes4 as well as among grapevine accessions of sativa and sylvestris from Spain43 and Morocco44. This low level of genetic differentiation suggests gene flow between cultivated and wild individuals. However, we observed a non-random distribution of divergent sites along the whole genome: the 95th percentile of the FST empirical distribution was > 0.27, and no positive signals were found to pass this empirical cutoff after the permutation test (Supplementary Figure S4). Sativa and sylvestris individuals differed significantly at 2461 SNPs included in 2001 windows. More than half (63.8%) of those variants belonged to intergenic or UTR/intron regions, whereas the remaining 26.8% and 9.4 % were synonymous and nonsynonymous, respectively. All 19 chromosomes of the grapevine genome showed divergent sites, ranging from 14 to 382 bins on chr12 and chr4, respectively (Fig. 4a).
Fig. 4.

a Manhattan plot of FST values for all SNP sites between cultivars and wild grapevines. The horizontal blue and red lines indicate, respectively the 95th (FST = 0.27) and the 99th (FST = 0.37) percentiles of the FST empirical distribution. Circles show the putative functions and the related metabolic processes of the genes with the highest FST values in the enriched functional classes (i.e., the ERF2 and RAP2 genes on chr15 and chr18, respectively). b Reduction in nucleotide diversity in the comparison of sylvestris and sativa accessions (πsylvestris/πsativa) across the genome
A shift in the allele distribution within populations may result from a sweep toward fixation of a selected locus and its nearby hitchhikers45. This sweep causes a population-wide reduction in the genetic diversity around the selected locus. Therefore, nucleotide diversity (π)46 was evaluated across the grapevine genome in sativa and sylvestris groups separately. As shown in Fig. 4b, the average value of the ratio πsylvestris/πsativa was 0.89, suggesting that π is slightly higher in the cultivated grapevine for most of the investigated genomic regions. Several surveys in grapevine germplasm collections consisting of both cultivated and wild V. vinifera accessions reported this overall lower genetic diversity in the wild gene pool compared to the cultivated panel47. The wild relatives are today present in low number in isolated populations, most likely due to the anthropogenic pressure on their natural habitats and the introduction of disease-causing agents from North America at the end of the 19th century7. On the other hand, the cultivated grape has a bigger effective population size present in multiple locations, where sexual crossing and somatic mutations coupled with a massive vegetative propagation have occurred, thus accumulating and increasing genetic variability. Nevertheless, our selection of sativa accessions from a core collection may overestimate the real level of nucleotide diversity in cultivated grapevines. A drastic reduction in nucleotide diversity of sylvestris individuals (πsylvestris/πsativa = 0) was observed on chromosomes 5, 14 and 15 at genomic regions with a total of 6 SNPs monomorphic in sylvestris. At the same time, sativa showed a reduction in nucleotide diversity on chromosomes 5, 12, and 19, where πsylvestris/πsativa had values higher than 10. However, except for the reduction of genetic diversity in sativa on chr19 (FST = 0.32), no divergence in allele frequencies was observed for the other genomic regions with extreme values of πsylvestris/πsativa. Both cultivated and wild individuals showed low minor allele frequency at those loci (MAF < 0.1). This reduction in nucleotide diversity in both subspecies may indicate reciprocal introgressions between wild and cultivated grapes or could reflect the occurrence of purifying selections, affecting diversity in both populations14.
Another common test used to detect signals of selection as a distortion of allele frequency and nucleotide diversity is the Tajima’s D, which compares the number of pairwise differences between individuals with the total number of segregating polymorphisms30. We observed mostly positive values of Tajima’s D in both wild (D: ~0.89) and cultivated (D: ~1.35) subgroups. As reported by Riahi et al.47, a positive value of Tajima’s D, especially for cultivated accessions, may indicate an excess of intermediate frequency alleles in these populations. Such configuration of allele frequencies may arise by balancing selection, which maintains both alleles at the selected loci48. According to Delph & Kelly49, this is the result of a heterozygote advantage or a spatial and temporal habitat heterogeneity. A balancing selection is in line with the high heterozygosity of grapevine genome and with the heterogeneity of uses and habitats to which V. vinifera is adapted.
Identification of biological functions underlying the selective sweeps
We looked at the new gene prediction v2.1 of the grapevine genome within windows of 20 kb around the SNPs detected as putatively under selection. Out of the 2032 predicted genes found in LD with the most significant SNPs, 1714 were annotated. Twelve functional classes were significantly enriched in the list of differentiated genes (Table 2), accounting for 109 of them (Supplementary Table S3). Most (69%) of these genes had predicted functions related to organic compound metabolism, mainly nitrogen and carbohydrate, while 24% was assigned to functional classes involved in perception, response, and adaptation to environmental stimuli.
Table 2.
Functional Classes significantly differentiated between sativa and sylvestris accessions
| GO ID | Term | Annotated genes | Significant genes | P value |
|---|---|---|---|---|
| GO:0071704 | Organic substance metabolic process |
1516 | 33 | 0.01596 |
| GO:0006807 | Nitrogen compound metabolic process |
604 | 32 | 0.01372 |
| GO:0005975 | Carbohydrate metabolic process | 148 | 10 | 0.00019 |
| GO:0055114 | Oxidation-reduction process | 143 | 9 | 0.00262 |
| GO:0009737 | Response to abscisic acid | 114 | 8 | 0.00232 |
| GO:0006952 | Defense response | 446 | 3 | 0.03388 |
| GO:0032259 | Methylation | 72 | 5 | 0.01715 |
| GO:0009607 | Response to biotic stimulus | 124 | 3 | 0.00045 |
| GO:0009651 | Response to salt stress | 50 | 2 | 0.01213 |
| GO:0010363 | Regulation of plant-type hypersensitive response | 20 | 2 | 0.0378 |
| GO:0010118 | Stomatal movement | 9 | 1 | 0.00899 |
| GO:0090305 | Nucleic acid phosphodiester bond hydrolysis | 11 | 1 | 0.03897 |
Out of the 109 genes in the enriched classes, 14 showed FST values > 0.37 (99th percentile of the FST empirical distribution; Supplementary Table S3). Therefore, understanding the putative functions and the related metabolic processes of these genes is of particular relevance in the genomic comparison between sativa and sylvestris (Fig. 4). At the top of the genes list with the highest value of FST, we identified the ‘RPL5B’ gene (VIT_204s0008g00050; Fig. 4 and Supplementary Table S3), which codifies the 60 S ribosomal protein L5-2. This gene could imply differences in organ development and expansion between the two subspecies. The angusta3 (ang3) mutant of A. thaliana for RPL5b gene displayed altered growth and development for several organs, notably leaves50. It is likely that balancing selection (Dsat = 1.13; Dsyl = 1.37) has acted to promote the high morphological variation observable nowadays in leaf shape and size within and between cultivated and wild grapevines51.
The list of genes with a significant differentiation between wild and cultivated grapevines was also particularly enriched in genes with a role in the carbohydrate metabolic processes (Table 2). For instance, the identification of the soluble starch synthase IV-1 gene (SS4; VIT_211s0065g00150; FST = 0.4; Fig. 4) highlighted differences between the two subspecies in starch and sucrose metabolism, which is relevant for berry development52. We also identified the nuclear transport factor 2 (NTF2) gene (VIT_217s0000g05240), which has a predicted role in response to abscisic acid (ABA), the main plant hormone promoting grape ripening53. NTF2 gene is located within the significant signature of selection on chr17, which includes candidate domestication-loci for berry size and development19. A reduction of nucleotide diversity in sativa accessions (πsylvestris/πsativa = 1.23) was observed at this locus, supporting evidence of a putative selection for berry composition and ripening traits in cultivated grapevines. Another diversified gene involved in the carbohydrate metabolism is the NADP-isocitrate dehydrogenase gene (cICDH; VIT_204s0079g00530), which catalyses the oxidative decarboxylation of isocitrate. An upregulation of the genes encoding isocitrate dehydrogenases in tobacco (Nicotiana tabacum cv Xanthi) and grape (V. vinifera cv Sultanina) accompanied increased aminating activity of glutamate dehydrogenase (GDH) under stress conditions, such as salinity54.
Several of the loci highly differentiated between sativa and sylvestris were involved in response to different environmental stimuli, in agreement with recent findings55. For instance, we identified the 10 kDa chaperonin gene (CPN10; VIT_208s0040g01150), the ‘LPA66’ gene (VIT_204s0008g00480), the rhomboid-like protein 11 gene (RBL11; VIT_204s0008g03830), the desacetoxyvindoline 4-hydroxylase gene (VIT_204s0008g01360), and the FATB gene (VIT_217s0000g01100). The latter has revealed a crucial role in seed development and viability as well as in the promotion of the hypersensitive response (HR) to pathogen attack in Arabidopsis56. We also observed differences in allele frequencies at the ERF2 transcription factor (VIT_215s0021g01590) and ‘RAP2’ (VIT_218s0001g05250) genes, which encode two members of the APETALA 2/ethylene-responsive element-binding factor (AP2/ERF) family. ERF proteins are ethylene-responsive element (GCC box)-binding proteins, and in tobacco, the GCC box has been found in the promoter of many defense genes57. Instead, RAP2 is a dehydration-responsive element-binding protein (DREB) with a role in plant abiotic stress responses such as high-salt stress, water deficit, and extreme temperatures58. Finally, we identified a splicing factor 3b subunit 1-like gene (VIT_208s0040g00270), supporting that alternative splicing may contribute to evolutionary adaptation through the assortment of different protein isoforms as a quick response to selective pressure31.
For almost all the stress-related genes identified, we observed a modest reduction in nucleotide diversity in sylvestris (πsylvestris/ πsativa ~0.95), associated with a positive value of the Tajima’s D (Dsylvestris = 1.41). These results imply that a balancing selection was likely acting in wild populations for adaptation to several environmental changes that may have occurred in their natural habitat along river banks. Our results are in line with recent studies on the tolerance of sylvestris genotypes to different stress conditions such as pathogen attack11 or calcareous soils10, which suggest that sylvestris grapevines represent valuable resources of resilience genes or alleles likely lost during the domestication process. This would have made cultivated grapevine dependent on agricultural means such as fertilisation, irrigation, weeding, and chemical plant protection. The CPN10 and RAP2 genes represent an exception to this trend. Indeed, lower genetic diversity (πsylvestris/πsativa CPN10 = 1.35; πsylvestris/πsativa RAP2 = 1.22, respectively) was observed at these loci in sativa accessions, suggesting a putative ongoing selection for adaptive mechanisms to salt stress in the cultivated grapevine.
In addition to the GO enrichment analysis, we looked for genes identified in previous QTL mapping studies as associated with main agronomic traits in grapevine, such as berry weight, berry skin colour and flower sex (Supplementary Table S4). We found several genes of those underlying berry weight QTLs42 such as the genes for the xyloglucan endotransglycosylase (XTH; VIT_201s0150g00460)59, the histone deacetylase 2 C (VvHD2C; VIT_206s0061g01240)60 and the cytochrome p450 78a3-like (CYP78A10; VIT_217s0000g05110), which has been found to regulate fruit size during tomato domestication61. Moreover, we found a signature of selection spanning from 4.7 to 5.0 Mb on chr2 (FST ~ 0.31), including 4 SNPs in LD with the APT, SNP4AC and Vvib23 markers for flower sex62. We also observed differences in allele frequency (FST = 0.36) between wild individuals, bearing colored fruits, and cultivated genotypes, composed by both colored and white varieties, at one of the MYB-type transcription factor genes on chr2 (MYB113; VIT_202s0033g00460)4 and within other candidate genes identified at berry skin color QTLs63 (Table S4).
Association mapping for six domestication-related traits in grapevine
The two vinifera subspecies exhibited an enormous phenotypic differentiation for six domestication-related traits, notably single berry weight (g; SBW), single bunch weight (g; SBCW) and berry flesh pH (Fig. 5a, b; Supplementary Note S1). For most traits, the cultivated individuals showed higher variability than the wild genotypes (Supplementary Figure S6 and Supplementary Table S5). In particular, sativa yielded on average numerous bunches with big and sweet berries, while sylvestris produced a few clusters with small, juiceless and acid fruits (Supplementary Note S1). These differences were more evident after estimating the Pearson’s correlation coefficient (R) between each pair of variables in the whole population and the two subgroups separately. While in the entire population yield was more correlated (R ~0,8) with both SBW and SBCW than with NBCs (R = 0.4), in sylvestris, it was highly correlated with both NBCs and SBCW rather than with SBW (Supplementary Table S6). This correlation suggests that the productivity of wild grapevine depends mainly on the number of clusters and the number of berries per bunch produced since fruit weight barely reached values higher than 1.5 g. Furthermore, we observed a significant inverse correlation (Supplementary Table S6) for total soluble solids (Brix°) with SBW and yield in both the whole population and the cultivated grapevines. This result can be explained by the shrinkage of berries, which occurs during véraison because of the loss of water by transpiration, as well as by the decrease of sugar concentration as berry size increases64.
Fig. 5. Comparison of phenotypic data between cultivated (in grey) and wild (in white) individuals in the two years of measurements.

(a) Box-plots of single bunch weight, single berry weight and number of bunches per plant. (b) Box-plots of yield, pH and total soluble solids.
We attempted an association mapping study to dissect the genetic basis of the phenotype variation observed between the wild and cultivated grapevines. The strong selection occurred during domestication may have extended the LD surrounding the target loci so that the SNP density required to map domestication-related traits may be lower than that required for unselected traits20. Also, aware of the limitations in size and population structure of our association panel, we combined GWAS and population genetics, displaying here only the marker-trait associations that were also identified as putative signatures of selection (Supplementary Figure S8-11). For instance, single berry weight (SBW) was significantly associated before p value correction with 2 SNPs on chr6 (Supplementary Table S7), which both fell within a genomic region significantly differentiated between sativa and sylvestris individuals (FST = 0.28). These SNPs, separated by 6.7 kb, are in LD with five genes, among which a Ca2+ transporting ATPase endoplasmic reticulum-type-like gene (Supplementary Tables S10). This finding supports the role of calcium ion in the development of grape berries65.
Significant values of FST were also observed for one SNP on chr15 associated with the number of bunches per plant (NBCs; FST = 0.28), and one marker on chr14 significantly correlated with the total soluble solids (Brix°; FST = 0.32; Supplementary Table S7). In particular, different values of Brix° at collection were observed among the three genotypes AA (0), AB (1) and BB (2) of the marker chr14_26697249 (Supplementary Figure S13), which belongs to a long LD block of 150 kb (Supplementary Table S8). In this region, we identified the cytochrome p450 724b1 gene implicated in the biosynthesis of brassinosteroids (BR), whose endogenous levels increase simultaneously with berry weight and soluble solids (Brix°) at the onset of berry ripening66.
We identified the highest number of marker-trait associations for the “Species,” a binary trait accounting for the level of genetic differentiation between cultivated and wild grapevine67. In particular, 34 SNPs were associated with the subspecies membership, out of which 3 SNPs on chr15 exhibited significant Bonferroni-corrected associations also with GLM-Q3 (see Supplementary Note S1; Supplementary Table S7 and Supplementary Figure S10). Notably, in LD with those three SNPs on chr15, we identified the nitrate transporter –like NRT1 gene, which has been correlated with the divergence in nitrate-use between the subspecies Oryza sativa L. indica and japonica68. According to the genome scan for signatures of selection, the GWAS test on ‘species’ trait led to identifying genes involved in response to environmental stresses (Supplementary Table S8). For instance, the salt overly sensitive 1 (SOS1) gene encodes a Na+/H+ antiporter, which is the downstream target of the Salt Overly Sensitive (SOS) signaling pathway, involved in controlling ion homoeostasis during salt stress69. We also identified the hypoxia up-regulated protein 1-like (HRE1) gene, which encodes an ERF transcription factor. HRE1 responds rapidly to oxygen deprivation by maintaining the expression of some anaerobic genes such as the alcohol dehydrogenase (ADH) gene70. Finally, the identification of the arginase gene, involved in the biosynthesis of polyamines, and the sugar transporter erd6-like 16-like gene, which encodes a monosaccharide transporter71, highlighted how sativa and sylvestris might present differences in the metabolisms of polyamines and sugars.
Conclusions
We displayed a whole-genome survey of the genetic differentiation between wild and cultivated grapevines by using population genetics approaches. An overall reduction of genetic diversity was observed within the wild panel, supporting the occurrence of an ongoing progressive decline of natural wild grapevine populations, and the necessity of developing new strategies for the characterization and conservation of V.v. sylvestris. Moreover, we identified several genomic regions with divergent allele frequencies between grapevine cultivars and their wild relatives. These genomic regions showed significant enrichment in functional gene classes related to responses to biotic and abiotic stresses, unraveling different putative mechanisms of adaptation to environmental changes. While grapevine cultivars are almost entirely dependent on human agricultural practices, the wild forms included in our study seem to have kept facing the permanent environmental alterations in their natural habitats. Future genome-scans using broader grapevine populations including sylvestris from their current whole worldwide distribution may confirm whether the differentiation in stress-related genomic regions is common evidence between all wild and cultivated vinifera populations, or if it is limited to the analyzed ex situ germplasm. Moreover, our study provides candidate genes for future functional genomics studies, to assess how the two forms of V. vinifera react under particular environmental stresses such as water deficit and pathogen attacks. In conclusion, our results support the large potential of sylvestris as a source of resilience factors in future breeding programs to deal with climate change and the increasing demand of sustainable viticulture.
Electronic supplementary material
GENOMIC SIGNATURES OF DIFFERENT ADAPTATIONS TO ENVIRONMENTAL STIMULI BETWEEN WILD AND CULTIVATED Vitis vinifera L
Acknowledgements
We are grateful to Giovanna Flaim for English supervision.
Authors’ contributions
A.M. conceived the study and elaborated its design, carried out the genotyping and phenotyping of the whole population, performed the analysis of LD decay, population genetics, GO enrichment and GWAS, and wrote the manuscript. D.M. participated in the study design and provided support in interpreting the results and writing the manuscript. S.L. provided lab assistance during the SNP genotyping and in the field during the trait phenotyping. D.N. provided support in interpreting the results of population genetics. M.S.G. supervised and coordinated the study, and drafted part of the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41438-018-0041-2).
References
- 1.This P, Lacombe T, Thomas MR. Historical origins and genetic diversity of wine grapes. Trends Genet. 2006;22:511–519. doi: 10.1016/j.tig.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Drori, E. et al. Collection and characterization of grapevine genetic resources (Vitis vinifera) in the Holy Land, towards the renewal of ancient winemaking practices. Sci. Rep. 7, 44463 (2017). [DOI] [PMC free article] [PubMed]
- 3.Emanuelli F, et al. Genetic diversity and population structure assessed by SSR and SNP markers in a large germplasm collection of grape. BMC Plant Biol. 2013;13:39. doi: 10.1186/1471-2229-13-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Myles S, et al. Genetic structure and domestication history of the grape. Proc. Natl Acad. Sci. USA. 2011;108:3530–3535. doi: 10.1073/pnas.1009363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Lorenzis G, Chipashvili R, Failla O, Maghradze D. Study of genetic variability in Vitis vinifera L. germplasm by high-throughput Vitis18kSNP array: the case of Georgian genetic resources. BMC Plant Biol. 2015;15:154. doi: 10.1186/s12870-015-0510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marrano A, et al. SNP-discovery by RAD-sequencing in a germplasm collection of wild and cultivated grapevines (V. vinifera L.) PLoS ONE. 2017;12:1–19. doi: 10.1371/journal.pone.0170655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arroyo-García, R. A. & Revilla, E. in The Mediterranean Genetic Code—Grapevine and Olive (ed Sladonja, B.) 51–72. London: Headquarters IntechOpen Limited, (2013).
- 8.Di Vecchi-Staraz M, et al. Low level of pollen-mediated gene flow from cultivated to wild grapevine: Consequences for the evolution of the endangered subspecies Vitis vinifera L. subsp. silvestris. J. Hered. 2009;100:66–75. doi: 10.1093/jhered/esn084. [DOI] [PubMed] [Google Scholar]
- 9.Doulati Baneh H, Hassani A, Abdollahi R. Growth and physiological responses of some wild grapevine (Vitis vinifera L. ssp. sylvestris) genotypes to salinity. Bulg. J. Agric. Sci. 2015;21:530–535. [Google Scholar]
- 10.Cambrollé J, García JL, Figueroa ME, Cantos M. Physiological responses to soil lime in wild grapevine (Vitis vinifera ssp. sylvestris) Environ. Exp. Bot. 2014;105:25–31. doi: 10.1016/j.envexpbot.2014.04.004. [DOI] [Google Scholar]
- 11.Duan D, et al. Genetic diversity of stilbene metabolism in Vitis sylvestris. J. Exp. Bot. 2015;66:3243–3257. doi: 10.1093/jxb/erv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen R, et al. Genomic scans for selective sweeps using SNP data Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin T, et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 2014;46:1220–1226. doi: 10.1038/ng.3117. [DOI] [PubMed] [Google Scholar]
- 14.Hufford, M. B. et al. The genomic signature of crop-wild introgression in maize. PLoS Genet. 9, e1003477 (2013). [DOI] [PMC free article] [PubMed]
- 15.Huang X, et al. A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012;490:497–501. doi: 10.1038/nature11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonhomme M, et al. Genomic signature of selective sweeps illuminates adaptation of Medicago truncatula to root-associated microorganisms. Mol. Biol. Evol. 2015;32:2097–2110. doi: 10.1093/molbev/msv092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicolas SD, et al. Genetic diversity, linkage disequilibrium and power of a large grapevine (Vitis vinifera L) diversity panel newly designed for association studies. BMC Plant Biol. 2016;16:74. doi: 10.1186/s12870-016-0754-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chitwood DH, et al. A modern ampelography: a genetic basis for leaf shape and venation patterning in grape. Plant Physiol. 2014;164:259–272. doi: 10.1104/pp.113.229708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Myles, S. et al. Rapid genomic characterization of the genus Vitis. PLoS ONE5, e8219 (2010). [DOI] [PMC free article] [PubMed]
- 20.Migicovsky Z, et al. Patterns of genomic and phenomic diversity in wine and table grapes. Hortic. Res. 2017;4:17035. doi: 10.1038/hortres.2017.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LePaslier, M.-C. et al. The GRAPERESEQ 18K Vitis genotyping chip. In IX International Symposium on Grapevine Physiology & Biotechnology 18. La Serena (CHL) (2013).
- 22.Money D, et al. LinkImpute: Fast and Accurate Genotype Imputation for Non-Model Organisms. G3. 2015;5:2383–2390. doi: 10.1534/g3.115.021667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raj A, Stephens M, Pritchard JK. FastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics. 2014;197:573–589. doi: 10.1534/genetics.114.164350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jakobsson M, Rosenberg NA. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- 26.Rosenberg NA. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes. 2004;4:137–138. doi: 10.1046/j.1471-8286.2003.00566.x. [DOI] [Google Scholar]
- 27.Jombart T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 28.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 29.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vitulo N, et al. A deep survey of alternative splicing in grape reveals changes in the splicing machinery related to tissue, stress condition and genotype. BMC Plant Biol. 2014;14:99. doi: 10.1186/1471-2229-14-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexa A, Rahnenführer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 33.Bradbury PJ, et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 34.Turner, S. D. qqman: an R package for visualizing GWAS results using Q–Q and manhattan plots. bioRxiv 1–2 (2014).
- 35.Deschamps S, Llaca V, May GD. Genotyping-by-sequencing in plants. Biology (Basel) 2012;1:460–483. doi: 10.3390/biology1030460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Regner F, Stadlbauer A, Eisenheld C, Kaserer H. Genetic relationships among Pinots and related cultivars. Am. J. Enol. Vitic. 2000;51:7–14. [Google Scholar]
- 37.Adam-Blondon, A., Martinez-Zapater, J. M. & Kole, C. Genetics, Genomics and Breeding of Grapes St. Helier, Jersey, British Channel Islands: Science Publishers (2011).
- 38.Lijavetzky D, Cabezas JA, Ibáñez A, Rodríguez V, Martínez-Zapater JM. High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology. BMC Genom. 2007;8:424. doi: 10.1186/1471-2164-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Velasco, R. et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE2, e1326 (2007). [DOI] [PMC free article] [PubMed]
- 40.Koch, E., Ristroph, M. & Kirkpatrick, M. Long range linkage disequilibrium across the human genome. PLoS ONE8, e80754 (2013). [DOI] [PMC free article] [PubMed]
- 41.Costantini L, Battilana J, Lamaj F, Fanizza G, Grando MS. Berry and phenology-related traits in grapevine (Vitis vinifera L.): from quantitative trait loci to underlying genes. BMC Plant Biol. 2008;8:38. doi: 10.1186/1471-2229-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doligez A, et al. New stable QTLs for berry weight do not colocalize with QTLs for seed traits in cultivated grapevine (Vitis vinifera L.) BMC Plant. Biol. 2013;13:217. doi: 10.1186/1471-2229-13-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Andrés MT, et al. Genetic diversity of wild grapevine populations in Spain and their genetic relationships with cultivated grapevines. Mol. Ecol. 2012;21:800–816. doi: 10.1111/j.1365-294X.2011.05395.x. [DOI] [PubMed] [Google Scholar]
- 44.Zinelabidine LH, Haddioui A, Bravo G, Arroyo-García R, Zapater JMM. Genetic origins of cultivated and wild grapevines from Morocco. Am. J. Enol. Vitic. 2010;61:83–90. [Google Scholar]
- 45.Vitti JJ, Grossman SR, Sabeti PC. Detecting natural selection in genomic data. Annu. Rev. Genet. 2013;47:97–120. doi: 10.1146/annurev-genet-111212-133526. [DOI] [PubMed] [Google Scholar]
- 46.Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riahi L, et al. Single nucleotide polymorphism and haplotype diversity of the gene NAC4 in grapevine. Ind. Crops Prod. 2013;43:718–724. doi: 10.1016/j.indcrop.2012.08.021. [DOI] [Google Scholar]
- 48.Charlesworth D. Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2006;2:379–384. doi: 10.1371/journal.pgen.0020064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delph LF, Kelly JK. On the importance of balancing selection in plants Lynda. New Phytol. 2013;100:130–134. doi: 10.1111/nph.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Devis D, Firth SM, Liang Z, Byrne ME. Dosage sensitivity of RPL9 and concerted evolution of ribosomal protein genes in plants. Front. Plant Sci. 2015;6:1–12. doi: 10.3389/fpls.2015.01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bodor P, Ladányi M, Grzeskowiak L, Grando MS, Bisztray GD. Ampelometric evaluation of wild grape (Vitis vinifera L. ssp. sylvestris (C.C. Gmel.) Hegi) accessions in the germplasm collection of FEM-IASMA, Italy. Vitis J. Grapevine Res. 2015;54:213–215. [Google Scholar]
- 52.Agudelo-Romero, P. et al. Search for transcriptional and metabolic markers of grape pre-ripening and ripening and insights into specific aroma development in three Portuguese cultivars. PLoS ONE8, e60422 (2013). [DOI] [PMC free article] [PubMed]
- 53.Fortes AM, Teixeira RT, Agudelo-Romero P. Complex interplay of hormonal signals during grape berry ripening. Molecules. 2015;20:9326–9343. doi: 10.3390/molecules20059326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skopelitis DS, et al. Abiotic stress generates ROS that signal expression of anionic glutamate dehydrogenases to form glutamate for proline synthesis in tobacco and grapevine. Plant Cell. 2006;18:2767–2781. doi: 10.1105/tpc.105.038323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou, Y., Massonnet, M., Sanjak, J. S., Cantu, D. & Gaut, B. S. Evolutionary genomics of grape (Vitis vinifera ssp. vinifera) domestication. Proc. Natl Acad. Sci.10.1073/pnas.1709257114 (2017). [DOI] [PMC free article] [PubMed]
- 56.Raffaele S, et al. A MYB transcription factor regulates very-long-chain fatty acid biosynthesis for activation of the hypersensitive cell death response in Arabidopsis. Plant Cell. 2008;20:752–767. doi: 10.1105/tpc.107.054858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakano T, Nishiuchi T, Suzuki K, Fujimura T, Shinshi H. Studies on transcriptional regulation of endogenous genes by ERF2 transcription factor in tobacco cells. Plant Cell Physiol. 2006;47:554–558. doi: 10.1093/pcp/pcj017. [DOI] [PubMed] [Google Scholar]
- 58.Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta. 2012;1819:86–96. doi: 10.1016/j.bbagrm.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 59.Chervin C, et al. Ethylene seems required for the berry development and ripening in grape, a non-climacteric fruit. Plant Sci. 2004;167:1301–1305. doi: 10.1016/j.plantsci.2004.06.026. [DOI] [Google Scholar]
- 60.Kaps ML, Cahoon Ga. Growth and fruiting of container-grown Seyval blanc grapevines modified by changes in crop level, leaf number and position, and light exposure. Am. J. Enol. Vitic. 1992;43:191–199. [Google Scholar]
- 61.Chakrabarti M, et al. A cytochrome P450 regulates a domestication trait in cultivated tomato. Proc. Natl Acad. Sci. USA. 2013;110:17125–17130. doi: 10.1073/pnas.1307313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fechter I, et al. Candidate genes within a 143 kb region of the flower sex locus in Vitis. Mol. Genet. Genom. 2012;287:247–259. doi: 10.1007/s00438-012-0674-z. [DOI] [PubMed] [Google Scholar]
- 63.Costantini L, et al. New candidate genes for the fine regulation of the colour of grapes. J. Exp. Bot. 2015;66:4427–4440. doi: 10.1093/jxb/erv159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Coombe BG, McCarthy MG. Dynamics of grape berry growth and physiology of ripening. Aust. J. Grape Wine Res. 2000;6:131–135. doi: 10.1111/j.1755-0238.2000.tb00171.x. [DOI] [Google Scholar]
- 65.Deluc LG, et al. Transcriptomic and metabolite analyses of Cabernet Sauvignon grape berry development. BMC Genome. 2007;8:429. doi: 10.1186/1471-2164-8-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Symons GM, et al. Grapes on steroids. brassinosteroids are involved in grape berry ripening. Plant. Physiol. 2006;140:150–158. doi: 10.1104/pp.105.070706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leforestier D, et al. Genomic basis of the differences between cider and dessert apple varieties. Evol. Appl. 2015;8:650–661. doi: 10.1111/eva.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu B, et al. Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies. Nat. Genet. 2015;47:834–838. doi: 10.1038/ng.3337. [DOI] [PubMed] [Google Scholar]
- 69.Ji H, et al. The salt overly sensitive (SOS) pathway: Established and emerging roles. Mol. Plant. 2013;6:275–286. doi: 10.1093/mp/sst017. [DOI] [PubMed] [Google Scholar]
- 70.Licausi F, et al. HRE1 and HRE2, two hypoxia-inducible ethylene response factors, affect anaerobic responses in Arabidopsis thaliana. Plant. J. 2010;62:302–315. doi: 10.1111/j.1365-313X.2010.04149.x. [DOI] [PubMed] [Google Scholar]
- 71.Afoufa-Bastien D, et al. The Vitis vinifera sugar transporter gene family: phylogenetic overview and macroarray expression profiling. Bmc. Plant. Biol. 2010;10:245. doi: 10.1186/1471-2229-10-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GENOMIC SIGNATURES OF DIFFERENT ADAPTATIONS TO ENVIRONMENTAL STIMULI BETWEEN WILD AND CULTIVATED Vitis vinifera L