

Abstract
Growth hormone (GH) has been shown to act directly on multiple tissues throughout the body. Historically, it was believed that GH acted directly in the liver and only indirectly in other tissues via insulin‐like growth hormone 1 (IGF‐1). Despite extensive work to describe GH action in individual tissues, a comparative analysis of acute GH signaling in key metabolic tissues has not been performed. Herein, we address this knowledge gap. Acute tissue response to human recombinant GH was assessed in mice by measuring signaling via phospho‐STAT5 immunoblotting. STAT5 activation is an easily and reliably detected early marker of GH receptor engagement. We found differential tissue sensitivities; liver and kidney were equally GH‐sensitive and more sensitive than white adipose tissue, heart, and muscle (gastrocnemius). Gastrocnemius had the greatest maximal response compared to heart, liver, white adipose tissue, and whole kidney. Differences in maximum responsiveness were positively correlated with tissue STAT5 abundance, while differences in sensitivity were not explained by differences in GH receptor levels. Thus, GH sensitivity and responsiveness of distinct metabolic tissues differ and may impact physiology and disease.
Keywords: GH responsiveness, GH sensitivity, tissues
Abbreviations
- eWAT
epididymal white adipose tissue
- gastroc
gastrocnemius
- GH
growth hormone
- IGF‐1
insulin‐like growth factor 1
- JAK2
Janus kinase 2
- MGF
mechano growth factor
- PRLR
prolactin receptor
- STAT5
signal transducer and activator of transcription 5
Growth hormone (GH), produced in the anterior pituitary, plays a major role in both longitudinal growth and metabolism 1, 2. Dysregulation in GH signaling, either increased in acromegaly and gigantism 3 or decreased in short stature or dwarfism, has profound consequences on growth and development 4, 5. GH also impacts life span; GH excess is associated with increased morbidity and premature mortality 6, while GH deficiency promotes longevity 7. GH binds cell surface receptors (GH receptor; GHR) on target cells, resulting in GHR‐associated Janus kinase 2 (JAK2) autophosphorylation and subsequent phosphorylation of GHR intracellular domain tyrosine residues 8, 9, 10, 11, 12, 13. Signal transducer and activator of transcription 5 (STAT5) docks at the phosphorylated GHR and is phosphorylated by JAK2. pSTAT5 dimers translocate to the nucleus to influence transcription of genes including insulin‐like growth factor (IGF)‐1 14, 15, 16; GH's metabolic and somatogenic effects are related to its influence on target cell gene expression.
Assessing acute GH effects in different key metabolic tissues may have once been considered an irrelevant question. Classically, GH was thought to exclusively target the liver, which would then produce IGF‐1 (aka somatomedin C) 17. IGF‐1 would subsequently act in an endocrine manner, modulating growth/metabolism in extrahepatic tissues. This is the somatomedin hypothesis of GH action 18. Later, D'Ercole et al. 19 showed that IGF‐1 is also produced locally by extrahepatic tissues in response to GH and that the level of IGF‐1 produced after GH administration differs between tissues. Further, Skottner et al. 20 demonstrated that administration of IGF‐1 did not affect longitudinal growth in hypophysectomized rats, except at very high concentrations, whereas GH administration induced significant growth. These pioneering studies suggested that IGF‐1 might be produced and act locally within target tissues, in contrast to the somatomedin hypothesis. Consistent with these observations, liver‐specific IGF‐1 knockout mice grow and develop normally, despite diminished circulating IGF‐1 21, 22, 23. As such, a revised hypothesis suggests that circulating (hepatic‐derived) IGF‐1 is responsible for negatively regulating GH secretion, whereas local (extrahepatic) IGF‐1 plays a primary role in longitudinal growth 24.
Despite interest in extrahepatic actions of GH and IGF‐1, little information is available that compares GH signaling among organs in intact animals. Because of the distinct roles of GH signaling in the liver compared to other metabolic tissues, we hypothesized that GH sensitivity and responsiveness would differ in hepatic versus extrahepatic tissues. Herein, we compare acute in vivo sensitivity and MAX responsiveness to exogenously administered GH in mice among liver, heart, kidney, skeletal muscle (gastrocnemius; gastroc), and epididymal white adipose tissue (eWAT). Our results indicate substantial differences between tissues that may be important for understanding tissue‐specific metabolic and growth‐promoting effects of GH.
Materials and methods
Unless otherwise stated, reagents were obtained from Sigma (St. Louis, MO).
Animals
All animal husbandry and experimental protocols were carried out according to the Guide for the Care and Use of Laboratory Animals [1996 (7th ed.), Washington, DC: National Research Council, National Academies Press] and in compliance with the local IACUC standards. At 15 weeks of age (± 3 days), male C57B6J mice (Jackson Laboratories; Cat. # 000664) were individually housed in standard conditions under a 12‐h : 12‐h light:dark cycle and had ad libitum access to standard rodent chow and water. After acclimatization to single housing, mice were placed in wire‐bottom cages without food at the beginning of the light cycle. Growth hormone challenge was performed in 6‐h fasted mice in a manner that is essentially identical to that described previously 25. Briefly, either saline (control) or human recombinant GH (2, 4, 8, 12.5, 20, 50, 80, 120, 200 ng/gbw; gift from Eli Lilly Co, Indianapolis, IN) was injected (i.v.) in anesthetized mice; 5 min thereafter, heart, liver, kidney, eWAT, and gastroc were rapidly excised in that order and flash‐frozen in liquid nitrogen prior to biochemical analysis. Total time of tissue extraction for each animal was 3–4 min. The duration of GH exposure was selected so as to capture only acute (and not secondary) effects of GH stimulation and thus most cleanly address the question of GH sensitivity.
Liver samples for PRLR mRNA positive control were harvested from female C56Bl6/J mice that were ad libitum fed and age‐matched, age 2–3 months. Pregnant samples were harvested at gestational day 16.5.
This study protocol was approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee.
Immunoblotting
Protein lysates were prepared from tissues crushed to powder under liquid nitrogen (~ 20 mg) using 300 μL of tissue lysis buffer (50 mm Tris 7.3, 150 mm NaCl, 1 mm EDTA pH 8.1, 1.5 mm MgCl2, 10% glycerol, 1% Triton X‐100, 10 mm Na4P2O7, 100 mm NaF, 1 mm Na3VO4, 1 mm phenylmethanesulfonyl fluoride, 5 μg·mL−1 aprotinin, and 5 μg·mL−1 leupeptin). Lysates were resolved under reducing conditions by SDS/PAGE and transferred to nitrocellulose membranes (Amersham Biosciences), followed by blocking with 2% BSA. Membranes were immunoblotted (Table 1) with anti‐phospho‐STAT5 antibody (Y694; Cell Signaling; 9351L) (1 : 1000), which reacts with both phosphorylated Y694 in STAT5A and Y699 in STAT5B; anti‐STAT5 antibody (Santa Cruz Biotechnology; sc‐835) (1 : 1000); anti‐GHR (polyclonal anti‐GHRcytAL‐47; against the intracellular domain of GHR) 26 (1 : 1000); anti‐PRLR (anti‐PRLRcytAL‐84; against the human PRLR ICD) 27 (1 : 1000); anti‐PRL‐R (H‐300) (Santa Cruz Biotechnology; sc‐20992); and anti‐JAK2 (anti‐JAK2AL‐33) 28 (1 : 1000). Densitometry was performed using UVP Software 8.0.
Table 1.
Antibody table
| Antigen sequence (if known) | Name of antibody | Manufacturer, catalog #, and/or name of individual providing the antibody | Species raised in; monoclonal or polyclonal | Dilution used | RRID |
|---|---|---|---|---|---|
| Y694, mouse, rat, bovine, human | Anti‐pSTAT5 | Cell Signaling, 9351L | Rabbit; polyclonal | 1 : 1000 | AB_331594 |
| Stat5 (C‐17) human, mouse, rat | STAT5 (C‐17) | Santa Cruz, Cat. # sc‐836 | Rabbit; polyclonal | 1 : 1000 | AB_632446 |
| Residues 271‐620 | Anti‐GHRcytAL‐47 | Stuart J. Frank | Rabbit; polyclonal | 1 : 1000 | AB_2713931 |
| PRLR intracellular domain | Anti‐PRLRcytAL‐48 | Stuart J. Frank | Rabbit; polyclonal | 1 : 1000 | AB_2665406 |
| Residues 323‐622 | PRL‐R (H‐300) | Santa Cruz, Cat. # sc‐20992 | Rabbit; polyclonal | 1 : 1000 | AB_2237692 |
| Residues 746‐1129 | Anti‐JAK2AL‐33 | Stuart J. Frank | Rabbit; polyclonal | 1 : 1000 | AB_2665398 |
Curve fitting and statistical analysis
Dose–response curve data were fit to the sigmoidal dose–response curve (with variable slope): [Y = BOTTOM + (TOP‐BOTTOM)/(1 + 10^((LogEC50‐X)*HillSlope))]; Y = response; X = log[dose]; HillSlope = slope of linear section of the dose–response curve; TOP = point in the dose–response curve at which an increase in ‘X’ yields little to no increase in ‘Y’; and EC50 = the effective concentration (or dose) at which 50% of the MAX response is achieved 29. This analysis was performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, California, USA, http://www.graphpad.com). Sensitivity was defined by the value EC50. Responsiveness was defined by the TOP value, herein referred to as the MAX response. During the constrained fit, the TOP and BOTTOM parameters were fixed at 100 and 0, respectively. Tissue‐specific differences in protein abundances were assessed via one‐way ANOVA using SPSS followed by post hoc analysis via Tukey's test. Regression analysis to assess the correlation between STAT5 abundance and MAX response was performed using Excel.
Gene analysis
mRNA was isolated from mouse tissues using either a QIAGEN RNeasy Mini Kit (Cat. No. 74104) or TRIzol RNA isolation reagent according to the manufacturer's recommended protocol for RNA isolation. Reverse transcription was performed using the High‐Capacity cDNA RT Kit (Cat. No. 4368814) from Thermo Fisher. qPCR measurements were carried out using the mPRLR TaqMan Gene Exp. Assay (Assay ID Mm04336676_m1, Cat. No. 4351372) from Thermo Fisher.
Data display
Due to differences in normalizing proteins across tissues, densitometry data are normalized to total protein loaded on the gel except in Fig. 2 where pSTAT5 is normalized to STAT5 as a loading control; tissue differences in STAT5 abundance do not influence sensitivity. To reduce positional bias during the immunoblot transfer procedure, samples were loaded on gels in randomized order; where possible, n = 1 for each GH dose was included on each gel. Densitometry was performed on nonmanipulated blots. For clarity, representative blots presented were constructed as follows: A single gel was chosen for each tissue, after which lanes were rearranged such that GH doses were displayed in ascending order.
Figure 2.
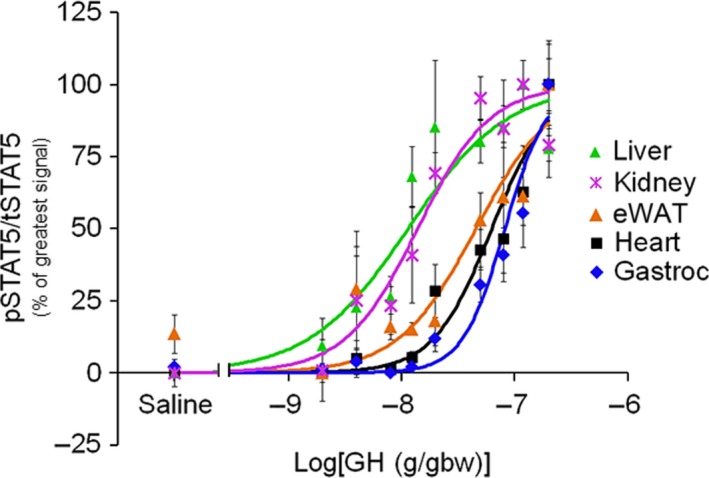
Tissue GH sensitivity (constrained fit). Dose–response data normalized such that the highest value within each tissue is 100 and lowest is 0, and fit to the Hill equation with the constraints: TOP = 100 and BOTTOM = 0.
Results
Murine peripheral tissues display differential sensitivity and MAX response to GH
Although they varied greatly in responsiveness, with kidney being the least maximally responsive, all tissues examined displayed dose‐dependent GH effects on STAT5 phosphorylation (Figs 1A and 2). Calculation of EC50 values (see the Methods section for details) revealed tissue‐specific differences in GH sensitivity (Fig. 1B, Table 2). EC50 values for liver and kidney did not differ significantly, although both were substantially lower (i.e., greater sensitivity) than eWAT, heart, and gastrocnemius. Differences were likewise observed in tissue responsiveness (Fig. 1B, Table 2). Gastrocnemius had the greatest (extrapolated) MAX GH response, followed by heart, liver, eWAT, and kidney (Fig. 1B, Table 2). There was large variability (i.e., confidence intervals) in the EC50 and MAX values for gastrocnemius, heart, and eWAT because the predicted MAX value was not defined by experimental data points (as predicted GH doses required for MAX response were too high). Therefore, as a secondary analysis we normalized the data for each curve such that the highest experimental data point was 100 while the lowest was 0, and fit the data to the sigmoidal dose–response curve using the constraints TOP = 100 and BOTTOM = 0 (Fig. 2). This analysis yielded similar EC50 calculations for liver and kidney, as well as similar R2 values for all curve fits. Furthermore, it confirmed, statistically, that liver and kidney have the same EC50 and that they are significantly more sensitive than eWAT, heart, and gastrocnemius (Fig. 2, Table 2).
Figure 1.
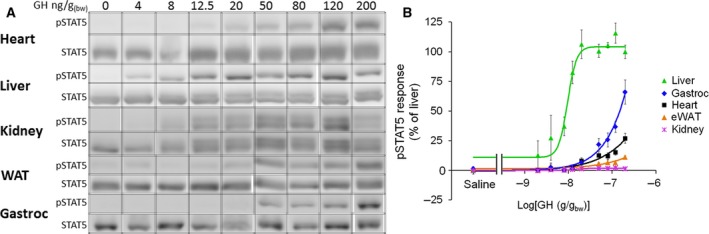
Tissue GH sensitivity and responsiveness (free fit). (A) Representative blots for dose‐dependent GH‐induced STAT5 phosphorylation in liver, kidney, eWAT, heart, and gastrocnemius muscle. The exposure of the blots was adjusted to be able to visualize dose dependencies of the different tissues, and as such, the intensities of bands may not be compared between tissues. (B) Dose–response data from liver, kidney, eWAT, heart, and gastrocnemius fit to the Hill equation without fit constraints (mean ± SEM; n = 3–10).
Table 2.
Curve fit parameters: from fitting dose–response data from Fig. 1B to the Hill equation without constraints (free fit), and from Fig. 2 using fit constraints (constrained fit)
| Liver | Kidney | eWAT | Heart | Gastroc | |
|---|---|---|---|---|---|
| Free fit | |||||
| EC50 (ng/gbw) | 10 | 14 | 1248 | 4901 | 1642 |
| MAX response (A.U.) | 104.3 | 1.8 | 53.6 | 296.1 | 615.2 |
| R2 | 0.8622 | 0.65 | 0.6343 | 0.6928 | 0.7379 |
| Constrained fit | |||||
| EC50 (ng/gbw) | 11 | 14 | 46 | 63 | 82 |
| EC50 (ng/gbw) 95% CI | 7.7–16.9 | 9.2–20.9 | 30.9–67.1 | 47.0–83.9 | 65.0–103.5 |
| R2 | 0.7344 | 0.6744 | 0.6288 | 0.722 | 0.7241 |
Differential abundance of GH signaling proteins among tissues
The factors that influence tissue sensitivity to a hormone often reside at the level of the receptor. Accordingly, we assessed GHR abundance by immunoblotting with an antibody against the GHR intracellular domain, which revealed highest abundance in eWAT (2.05 A.U.), followed by liver (1.00 A.U.), heart (0.61 A.U.), kidney (0.29 A.U.), and gastrocnemius (0.28 A.U.) (Fig. 3A,E). This study utilized human GH, which can also induce STAT5 phosphorylation via the prolactin receptor (PRLR) 30, 31, 32. We compared PRLR‐expressing MIN6 cells to the relevant mouse tissues by immunoblotting with two distinct anti‐PRLR sera (anti‐PRLRcytAL‐84 and anti‐PRL‐R (H‐300); Fig. 4A,B, respectively). No bands in common were detected by these sera in the mouse tissues, but a common PRLR band was detected by both in the MIN6 positive control. Analysis of prlr mRNA levels validated the conclusion that little or no expression was detected in the mouse tissues tested (Fig. 5). Thus, analyses of GHR and PRLR abundance did not readily explain observed tissue‐specific differences in GH sensitivity (although relatively high GHR expression in the liver may contribute to elevated GH sensitivity in this tissue).
Figure 3.
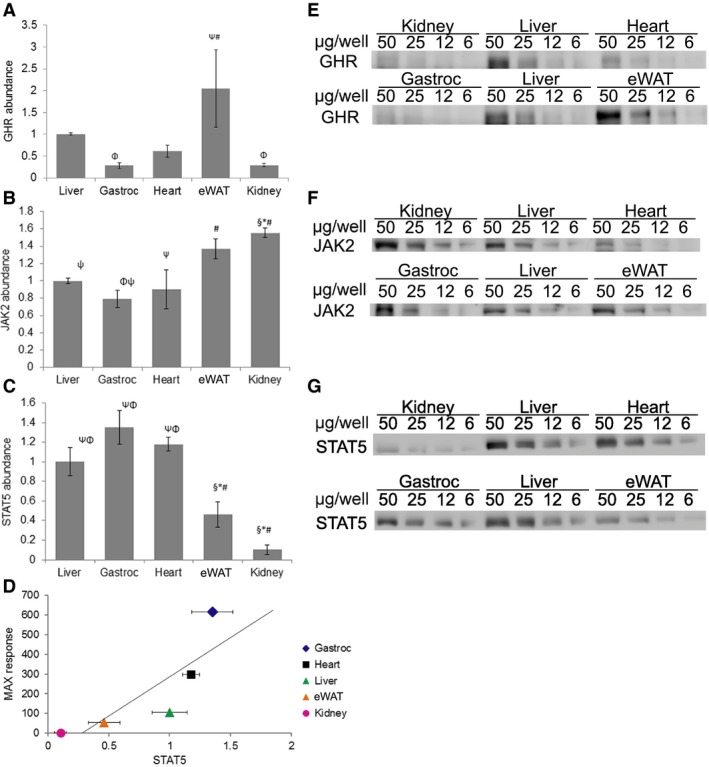
GH signaling components. Densitometry analysis and representative blots of tissue‐specific comparison of protein abundance for (A,E) GHR, (B,F) JAK2, and (C,G) STAT5 (relative to liver) (mean ± SEM; n = 5). Significance: All symbols represent P < 0.05 compared to: *, liver; #, gastrocnemius; §, heart; Φ, eWAT; and Ψ, kidney. (D) Linear regression analysis displaying the correlation between tissue‐specific MAX response and STAT5 (mean; n = 5) abundance (correlation coefficient: + 0.83; P = 0.082).
Figure 4.
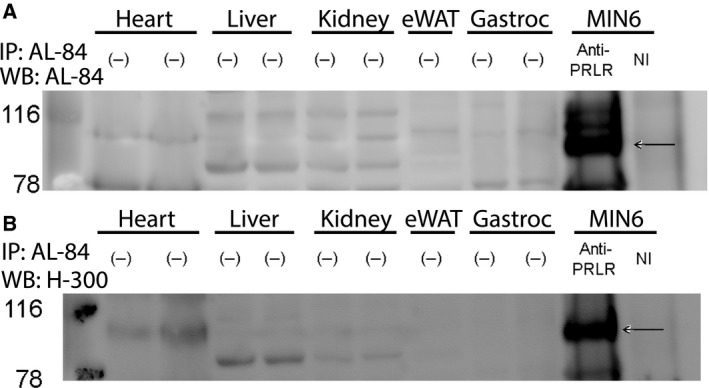
PRLR immunoblot of tissue lysates from various mouse tissues and in MIN6 mouse insulinoma cells (positive control). The black arrows denote the PRLR in the IP control. (−) denotes that no immunoprecipitation was performed. NI denotes nonimmune serum. Immunoprecipitation was performed with anti‐PRLRcytAL‐84, and resolved eluates were immunoblotted sequentially with (A) anti‐PRLRcytAL‐84 and (B) anti‐PRL‐R (H‐300).
Figure 5.
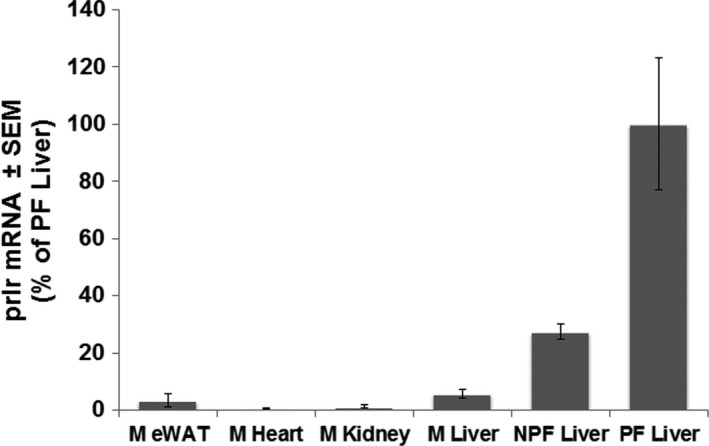
Comparison of PRLR mRNA expression between the male mouse tissues and with livers from pregnant and nonpregnant female mice. M, male; NPF, nonpregnant virgin female; PF, pregnant female. Data represented as mean ± SEM; n = 3‐5 for each condition.
In contrast to hormone sensitivity, the responsiveness of a tissue to a hormone is influenced by factors downstream of the receptor, including abundance of downstream signaling molecules. To this end, we assessed JAK2 and STAT5 abundance by immunoblotting. Relatively modest differences were observed in JAK2 abundance, with lowest levels in gastrocnemius and highest levels in kidney (Fig. 3B,F). STAT5 abundance did not differ between liver, gastrocnemius, and heart, but was significantly lower in eWAT and kidney (Fig. 3C,G). Regression analysis revealed a correlation (correlation coefficient: + 0.8296) between the STAT5 abundance in a tissue and its MAX response (P = 0.082) (Fig. 3D).
Discussion
The purpose of the current study was to define tissue‐specific differences in GH sensitivity and MAX responsiveness to GH. Here, we report that the order of GH sensitivity was liver = kidney > eWAT = heart = gastrocnemius, while the order of GH MAX responsiveness was gastrocnemius > heart > liver > eWAT > kidney and roughly correlated with STAT5 protein abundance. Such observations lead to questions with regard to physiologic significance. While the MAX response predicted from the free curve fitting in gastrocnemius and heart was much greater than in the other tissues, the levels of GH required to attain that MAX stimulation are far beyond physiologic levels. However, despite the error in these predicted values being large, the fact that there was a near‐significant correlation between STAT5 and MAX response supports the idea that these values are good estimates. The liver and kidney exhibit the highest level of GH sensitivity (relative to other tissues investigated), and the other three tissues were indistinguishable statistically. That this general relationship holds true regardless of whether constraints were used supports the idea that the liver and kidney respond to GH at much lower concentrations than eWAT, heart, and gastrocnemius.
Growth hormone plays a number of important roles in the liver, including generation of circulating IGF‐1 (which acts in a negative feedback manner on GH secretion) and hepatic metabolism. In the latter case, GH effects generally oppose those of insulin; specifically, these effects suppress glycolysis in favor of fatty acid oxidation and promote glycogenolysis and in prolonged fasting conditions promote gluconeogenesis 1, 33, 34, 35, 36. Thus, increased GH secretion during sleep likely plays an important role in maintenance of blood glucose levels via multiple mechanisms. Interestingly, the kidney is also a gluconeogenic tissue, contributing up to 50% of endogenous glucose production in the starved state 37. GH signaling in the kidney is also important for normal sodium and water retention; GH deficiency leads to renal insufficiency, while excess leads to hypertension, renal hypertrophy, and failure 37. Thus, our observation that the kidney is relatively GH‐sensitive (similar to the liver) is consistent with essential GH actions in this tissue. GH signaling is also important in eWAT, as this endocrine factor shifts metabolism from glucose utilization toward lipolysis and fatty acid oxidation, thereby minimizing reliance on muscle protein catabolism during periods of fasting (such as the sleep period) 1, 38, 39, 40. In contrast, GH signaling in the adult heart must be closely regulated, thus preventing excessive growth (e.g., in acromegaly) and subsequent contractile dysfunction 41. Similar to the heart, GH signaling in skeletal muscle mainly influences muscle size, but not contractile force 38, 40, 42. Our observation that skeletal muscle has decreased responsiveness to circulating GH may be explained by the existence of mechano growth factor (MGF), an alternative splice variant of the igf‐1 gene. MGF expression is increased in response to muscle stretch and exercise 43. Even hypophysectomized mice retain the ability to upregulate MGF in response to exercise 43, 44. The low GH sensitivity of gastrocnemius muscle may suggest that skeletal muscle growth in an adult mouse, in response to exercise, for example, may be through GH‐independent mechanisms.
Subsequent interrogation of known GH signaling components provided potential mechanistic insights with regard to tissue‐specific differences in GH responsiveness/sensitivity. For example, STAT5 levels were correlated with MAX response in a given tissue. Additionally, GHR levels were relatively high in liver, consistent with high GH sensitivity. Our findings are consistent with those of Walker et al. 45, who reported that GHR mRNA in the rat kidney was roughly 33% that of liver. Additional studies are required to elucidate fully the mechanisms mediating tissue‐specific differences in GH sensitivity/responsiveness.
The current study focused on a particular acute signaling response of various tissues to exogenously administered GH (namely STAT5 phosphorylation). This approach has benefits and drawbacks. Although we did not assess the long‐term response to endogenous GH pulses, this approach allowed us to directly compare acute responses to GH in multiple tissues simultaneously. As STAT5 is a critical mediator of acute GH action, we were able to observe direct GH effects, rather than compensatory effects over longer periods. Nonetheless, we acknowledge that our studies do not discriminate between the STAT5A and STAT5B isoforms of STAT5. As different tissues may express varying ratios of these isoforms, our conclusions concerning maximum responsiveness based on STAT5 abundance should be interpreted with caution.
As noted above, GH stimulates glycogenolysis in liver and kidney during fasting 35, 36. The mice in this study were fasted for 6 h prior to GH treatment. Therefore, it is possible that we would have observed a different relationship among tissues of GH sensitivity in mice if food had not been withdrawn in the 6 h leading up to GH treatment. However, the period of fasting corresponded to the first 6 h of the rest phase, during which food consumption is generally reduced (relative fasting), compared to the active period 46. Thus, the relative physiologic effects of the strict fast are likely limited. We are mindful, however, that GH sensitivity and MAX response were only assessed at one time of day in our study. Because the circadian clock may control both secretion and sensitivity to hormones 47, it is possible that relative tissue sensitivity to GH may vary depending on the time of day.
In summary, the current study reveals a correlation between STAT5 abundance and the MAX GH response in these tissues, while GH sensitivity is not correlated with GHR. Thus, an important determinant of MAX GH response appears to be STAT5 abundance, while the determinants of in vivo GH sensitivity are more complex. We speculate that in pathological states, GH action may be influenced by alterations in GH sensitivity and/or responsiveness, not solely by changes in circulating GH levels. Our data from wild‐type mice will serve as a template for analyzing such changes in disease states.
Author contributions
RDB, MEY, and SJF conceived and designed the project, analyzed and interpreted the data, and wrote the manuscript. RDB, RRB, and GRM acquired the data.
Acknowledgements
The authors appreciate discussions with Drs. Y. Zhang, Y. Gan, Y. Liu, A. Buckels, A. Shalev, S.M. Bailey, S. Morrison, and V. Thannickal. This work was supported by a VA Merit Review Award (to SJF) and NIH R01‐DK107441 (to SJF).
References
- 1. Moller N and Jorgensen JO (2009) Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev 30, 152–177. [DOI] [PubMed] [Google Scholar]
- 2. Brooks AJ and Waters MJ (2010) The growth hormone receptor: mechanism of activation and clinical implications. Nat Rev Endocrinol 6, 515–525. [DOI] [PubMed] [Google Scholar]
- 3. Vilar L, Vilar CF, Lyra R, Lyra R and Naves LA (2016) Acromegaly: clinical features at diagnosis. Pituitary 20, 22–32. [DOI] [PubMed] [Google Scholar]
- 4. Alatzoglou KS, Webb EA, Le Tissier P and Dattani MT (2014) Isolated growth hormone deficiency (GHD) in childhood and adolescence: recent advances. Endocr Rev 35, 376–432. [DOI] [PubMed] [Google Scholar]
- 5. Laron Z (2004) Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958‐2003. J Clin Endocrinol Metab 89, 1031–1044. [DOI] [PubMed] [Google Scholar]
- 6. Dineen R, Stewart PM and Sherlock M (2016) Acromegaly. QJM. 1‐10 [DOI] [PubMed]
- 7. Laron Z (2008) The GH‐IGF1 axis and longevity. The paradigm of IGF‐1 deficiency. Hormones (Athens) 7, 24–27. [DOI] [PubMed] [Google Scholar]
- 8. Argetsinger LS, Campbell GS, Yang X, Witthuhn BA, Silvennoinen O, Ihle JN and Carter‐Su C (1993) Identification of JAK2 as a growth hormone receptor‐associated tyrosine kinase. Cell 74, 237–244. [DOI] [PubMed] [Google Scholar]
- 9. Frank SJ, Gilliland G, Kraft AS and Arnold CS (1994) Interaction of the growth hormone receptor cytoplasmic domain with the JAK2 tyrosine kinase. Endocrinology 135, 2228–2239. [DOI] [PubMed] [Google Scholar]
- 10. Frank SJ (2002) Receptor dimerization in GH and erythropoietin action–it takes two to tango, but how? Endocrinology 143, 2–10. [DOI] [PubMed] [Google Scholar]
- 11. Gent J, van Kerkhof P, Roza M, Bu G and Strous GJ (2002) Ligand‐independent growth hormone receptor dimerization occurs in the endoplasmic reticulum and is required for ubiquitin system‐dependent endocytosis. Proc Natl Acad Sci USA 99, 9858–9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brooks AJ, Dai W, O'Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, Gardon O, Tunny KA, Blucher KM, Morton CJ et al (2014) Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 344, 1249783. [DOI] [PubMed] [Google Scholar]
- 13. Liu Y, Berry PA, Zhang Y, Jiang J, Lobie PE, Paulmurugan R, Langenheim JF, Chen WY, Zinn KR and Frank SJ (2014) Dynamic analysis of GH receptor conformational changes by split luciferase complementation. Mol Endocrinol 28, 1807–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lanning NJ and Carter‐Su C (2006) Recent advances in growth hormone signaling. Rev Endocr Metab Disord 7, 225–235. [DOI] [PubMed] [Google Scholar]
- 15. Frank SJ and Messina JL (2002) Growth hormone receptor In Cytokine Reference On‐Line (Oppenheim JJ. and Feldman M, eds), pp. 1–21. Academic Press, Harcourt, London, UK. [Google Scholar]
- 16. Woelfle J, Billiard J and Rotwein P (2003) Acute control of insulin‐like growth factor‐I gene transcription by growth hormone through Stat5b. J Biol Chem 278, 22696–22702. [DOI] [PubMed] [Google Scholar]
- 17. Salmon WD and Daughaday WH (1957) A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro . J Lab Clin Med 49, 825–836. [PubMed] [Google Scholar]
- 18. Daughaday WH, Hall K, Raben MS, Salmon WD Jr, van den Brande JL and van Wyk JJ (1972) Somatomedin: proposed designation for sulphation factor. Nature 235, 107. [DOI] [PubMed] [Google Scholar]
- 19. D'Ercole AJ, Stiles AD and Underwood LE (1984) Tissue concentrations of somatomedin C: further evidence for multiple sites of synthesis and paracrine or autocrine mechanisms of action. Proc Natl Acad Sci USA 81, 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Skottner A, Clark RG, Robinson IC and Fryklund L (1987) Recombinant human insulin‐like growth factor: testing the somatomedin hypothesis in hypophysectomized rats. J Endocrinol 112, 123–132. [DOI] [PubMed] [Google Scholar]
- 21. Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B and LeRoith D (1999) Normal growth and development in the absence of hepatic insulin‐like growth factor I. Proc Natl Acad Sci USA 96, 7324–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Tornell J, Isaksson OG, Jansson JO et al (1999) Liver‐derived insulin‐like growth factor I (IGF‐I) is the principal source of IGF‐I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci USA 96, 7088–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ueki I, Ooi GT, Tremblay ML, Hurst KR, Bach LA and Boisclair YR (2000) Inactivation of the acid labile subunit gene in mice results in mild retardation of postnatal growth despite profound disruptions in the circulating insulin‐like growth factor system. Proc Natl Acad Sci USA 97, 6868–6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. LeRoith D, Bondy C, Yakar S, Liu J and Butler A (2001) The somatomedin hypothesis: 2001. Endocr Rev 22, 53–74. [DOI] [PubMed] [Google Scholar]
- 25. McGinnis GR, Tang Y, Brewer RA, Brahma MK, Stanley HL, Shanmugam G, Rajasekaran NS, Rowe GC, Frank SJ, Wende AR et al (2017) Genetic disruption of the cardiomyocyte circadian clock differentially influences insulin‐mediated processes in the heart. J Mol Cell Cardiol 110, 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Y, Guan R, Jiang J, Kopchick JJ, Black RA, Baumann G and Frank SJ (2001) Growth hormone (GH)‐induced dimerization inhibits phorbol ester‐stimulated GH receptor proteolysis. J Biol Chem 276, 24565–24573. [DOI] [PubMed] [Google Scholar]
- 27. Chen H, Kleinberger JW, Takane KK, Salim F, Fiaschi‐Taesch N, Pappas K, Parsons R, Jiang J, Zhang Y, Liu H et al (2015) Augmented Stat5 signaling bypasses multiple impediments to lactogen‐mediated proliferation in human beta‐cells. Diabetes 64, 3784–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang J, Liang L, Kim SO, Zhang Y, Mandler R and Frank SJ (1998) Growth hormone‐dependent tyrosine phosphorylation of a GH receptor‐associated high molecular weight protein immunologically related to JAK2. Biochem Biophys Res Comm 253, 774–779. [DOI] [PubMed] [Google Scholar]
- 29. MIller JR (2003) GraphPad Prism Version 4.0 Step‐by‐Step Examples. GraphPad Software Inc, San Diego, CA. [Google Scholar]
- 30. Fu YK, Arkins S, Fuh G, Cunningham BC, Wells JA, Fong S, Cronin MJ, Dantzer R and Kelley KW (1992) Growth hormone augments superoxide anion secretion of human neutrophils by binding to the prolactin receptor. J Clin Invest 89, 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cunningham BC, Bass S, Fuh G and Wells JA (1990) Zinc mediation of the binding of human growth hormone to the human prolactin receptor. Science 250, 1709–1712. [DOI] [PubMed] [Google Scholar]
- 32. Somers W, Ultsch M, De Vos AM and Kossiakoff AA (1994) The X‐ray structure of a growth hormone‐prolactin receptor complex. Nature 372, 478–481. [DOI] [PubMed] [Google Scholar]
- 33. Kim YD, Li T, Ahn SW, Kim DK, Lee JM, Hwang SL, Kim YH, Lee CH, Lee IK, Chiang JY et al (2012) Orphan nuclear receptor small heterodimer partner negatively regulates growth hormone‐mediated induction of hepatic gluconeogenesis through inhibition of signal transducer and activator of transcription 5 (STAT5) transactivation. J Biol Chem 287, 37098–37108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cordoba‐Chacon J, Majumdar N, List EO, Diaz‐Ruiz A, Frank SJ, Manzano A, Bartrons R, Puchowicz M, Kopchick JJ and Kineman RD (2015) Growth hormone inhibits hepatic de novo lipogenesis in adult mice. Diabetes 64, 3093–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ghanaat F and Tayek JA (2005) Growth hormone administration increases glucose production by preventing the expected decrease in glycogenolysis seen with fasting in healthy volunteers. Metabolism 54, 604–609. [DOI] [PubMed] [Google Scholar]
- 36. Hoybye C, Chandramouli V, Efendic S, Hulting AL, Landau BR, Schumann WC and Wajngot A (2008) Contribution of gluconeogenesis and glycogenolysis to hepatic glucose production in acromegaly before and after pituitary microsurgery. Horm Metab Res 40, 498–501. [DOI] [PubMed] [Google Scholar]
- 37. Kamenicky P, Mazziotti G, Lombes M, Giustina A and Chanson P (2014) Growth hormone, insulin‐like growth factor‐1, and the kidney: pathophysiological and clinical implications. Endocr Rev 35, 234–281. [DOI] [PubMed] [Google Scholar]
- 38. Jorgensen JO, Rubeck KZ, Nielsen TS, Clasen BF, Vendelboe M, Hafstrom TK, Madsen M and Lund S (2010) Effects of GH in human muscle and fat. Pediatr Nephrol 25, 705–709. [DOI] [PubMed] [Google Scholar]
- 39. Nielsen S, Moller N, Christiansen JS and Jorgensen JO (2001) Pharmacological antilipolysis restores insulin sensitivity during growth hormone exposure. Diabetes 50, 2301–2308. [DOI] [PubMed] [Google Scholar]
- 40. Sun LY and Bartke A (2014) Tissue‐specific GHR knockout mice: metabolic phenotypes. Front Endocrinol (Lausanne) 5, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palmeiro CR, Anand R, Dardi IK, Balasubramaniyam N, Schwarcz MD and Weiss IA (2012) Growth hormone and the cardiovascular system. Cardiol Rev 20, 197–207. [DOI] [PubMed] [Google Scholar]
- 42. Mavalli MD, DiGirolamo DJ, Fan Y, Riddle RC, Campbell KS, van Groen T, Frank SJ, Sperling MA, Esser KA, Bamman MM et al (2010) Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J Clin Invest 120, 4007–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goldspink G and Yang SY (2001) Effects of activity on growth factor expression. Int J Sport Nutr Exerc Metab 11 (Suppl), S21–S27. [DOI] [PubMed] [Google Scholar]
- 44. Frost RA and Lang CH (2003) Regulation of insulin‐like growth factor‐I in skeletal muscle and muscle cells. Minerva Endocrinol 28, 53–73. [PubMed] [Google Scholar]
- 45. Walker JL, Moats‐Staats BM, Stiles AD and Underwood LE (1992) Tissue‐specific developmental regulation of the messenger ribonucleic acids encoding the growth hormone receptor and the growth hormone binding protein in rat fetal and postnatal tissues. Pediatr Res 31, 335–339. [DOI] [PubMed] [Google Scholar]
- 46. Shieh KR, Lee HJ and Yang SC (2008) Different patterns of food consumption and locomotor activity among taiwanese native rodents, formosan wood mice (apodemus semotus), and common laboratory mice, C57BL/6 (Mus musculus). Chin J Physiol 51, 129–135. [PubMed] [Google Scholar]
- 47. Gamble KL, Berry R, Frank SJ and Young ME (2014) Circadian clock control of endocrine factors. Nat Rev Endocrinol 10, 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]