

ABSTRACT
The Us11 protein of herpes simplex virus 1 (HSV-1) is an accessory factor with multiple functions. In virus-infected cells, it inhibits double-stranded RNA-dependent protein kinase (PKR), 2′,5′-oligoadenylate synthetase, RIG-I, and MDA-5. However, its precise role is incompletely defined. By screening a human cDNA library, we showed that the Us11 protein targets heat shock protein 90 (Hsp90), which inactivates TANK binding kinase 1 (TBK1) and antiviral immunity. When ectopically expressed, HSV-1 Us11 precludes TBK1 from access to Hsp90 and interferon (IFN) promoter activation. Consistently, the Us11 protein, upon HSV infection, suppresses the expression of beta interferon (IFN-β), RANTES, and interferon-stimulated genes. This is mirrored by a blockade in the phosphorylation of interferon regulatory factor 3. Mechanistically, the Us11 protein associates with endogenous Hsp90 to disrupt the Hsp90-TBK1 complex. Furthermore, Us11 induces destabilization of TBK1 through a proteasome-dependent pathway. Accordingly, Us11 expression facilitates HSV growth. In contrast, TBK1 expression restricts viral replication. These results suggest that control of TBK1 by Us11 promotes HSV-1 infection.
IMPORTANCE TANK binding kinase 1 plays a key role in antiviral immunity. Although multiple factors are thought to participate in this process, the picture is obscure in herpes simplex virus infection. We demonstrated that the Us11 protein of HSV-1 forms a complex with heat shock protein 90, which inactivates TANK binding kinase 1 and IFN induction. As a result, expression of the Us11 protein promotes HSV replication. These experimental data provide a new insight into the molecular network of virus-host interactions.
KEYWORDS: TANK binding kinase 1, herpes simplex virus, interferons, viral replication, virus-host interactions
INTRODUCTION
Herpes simplex virus 1 (HSV-1) infection proceeds in a temporal manner in which the virus undergoes gene expression, DNA replication, assembly, and egress (1). In this process, HSV-1 instigates a number of antiviral programs that operate coordinately (2). For example, Toll-like receptor 3 (TLR3) senses double-stranded RNA (dsRNA) from HSV-1 and subsequently stimulates interferon (IFN) induction (3). In the cytoplasm, RNA helicases, retinoid acid-inducible gene I (RIG-I), or melanoma differentiation-associated gene 5 (MDA5) recognizes host RNA induced by HSV-1 replication (4). In addition, DNA sensors, including cyclic GMP-AMP (GAMP) synthase, interferon-inducible protein 16 (IFI16), and DEAD box helicase 41 (DDX41), detect HSV-1 (5–8). Although they recognize distinct molecular patterns, these receptors transmit danger signals to TANK binding kinase 1 (TBK1), which phosphorylates interferon regulatory factor 3 (IRF3), leading to the induction of IFN-α/β, chemokines, and interferon-stimulated genes (ISG), such as dsRNA-dependent protein kinase (PKR) (2, 9).
Several lines of evidence suggest that HSV-1 perturbs innate immunity, which relies on an array of viral proteins (9). It has been demonstrated that an early (α) protein, ICP0, negatively regulates the expression of IFN-β and ISGs in human fibroblasts (10–12). This is ascribed to its capacity to mediate IFI16 degradation (13). In macrophages, another early protein, ICP27, dampens STING pathway activation and reduces cytokine expression (14). On the other hand, HSV-1 Us3 compromises TLR3 pathway signaling in monocytes (15). Mechanistically, Us3 is reported to phosphorylate and inactivate IRF3 (16). While the aforementioned HSV-1 proteins modulate the induction of cytokines, the γ134.5 protein is able to neutralize PKR as well (17). It is well established that onset of viral DNA replication activates PKR, which arrests protein synthesis by phosphorylation of the α subunit of eukaryotic translation initiation factor eIF-2 (eIF-2α) (17, 18). As a countermeasure, HSV-1 γ134.5 recruits protein phosphatase 1 (PP1) to dephosphorylate eIF-2α (19, 20). Therefore, a deletion or a site-specific mutation in the PP1 site of γ134.5 abrogates viral replication (19, 20). Intriguingly, a secondary mutation with early expression in the Us11 gene compensates for such a defect in HSV-infected cells (21, 22).
HSV-1 Us11 is a late (γ2) protein that regulates the accumulation of RNA species (23, 24). In HSV-infected cells, it cooperates with the γ134.5 protein to confer HSV-1 resistance to IFN-α (25). When expressed as an early (α) protein, Us11 expression precludes the shutoff viral protein synthesis mediated by PKR in the absence of γ134.5 (21, 26, 27). This requires its RNA binding domain to interact with PKR, leading to the prevention of eIF2α phosphorylation (28). Indeed, HSV-1 Us11 associates with and prevents PKR activation by a cellular protein, PACT (29). Moreover, Us11 negatively modulates 2′,5′-oligoadenylate synthetase (30). Recent work showed that HSV-1 Us11 binds to RIG-I or MDA5 when ectopically expressed. In correlation, HSV-1 Us11 downregulates the expression of IFN-α/β (31). In particular, the Us11 protein suppresses RIG-I activation by PACT, which coincides with a reduction in IFN-α/β expression in virus-infected cells (32). HSV-1 Us11 has also been suggested to modulate apoptosis and autophagy (33, 34). However, its role in HSV infection remains incompletely deciphered.
This study was designed to identify a novel cellular target(s) of Us11. We report that the Us11 protein facilitates HSV-1 replication by binding to heat shock protein 90 (Hsp90), which disrupts the Hsp90 machinery. We provide evidence that HSV-1 Us11 associates with Hsp90. Upon displacement of TBK1 from Hsp90, Us11 induces destabilization of TBK1 via a proteasome-dependent pathway. We also demonstrate that HSV-1 Us11 suppresses IRF3 phosphorylation and IFN responses. Consequently, inhibition of TBK1 by Us11 promotes HSV growth. These results reveal a previously undocumented HSV mechanism in infected cells.
RESULTS
Heat shock protein 90 is a cellular target of the Us11 protein encoded by HSV-1.
To better understand the function of HSV-1 Us11, we sought to screen human cDNA library by the use of the yeast two-hybrid system. This approach identified heat shock protein 90 (Hsp90) as an interacting factor for HSV-1 Us11 (not shown). To test whether HSV Us11 binds to Hsp90 in mammalian cells, 293T cells were transfected with Flag-Us11 along with Myc-Hsp90, Myc-EGFP (Myc-enhanced green fluorescent protein), or a vector plasmid. Lysates of cells were then subjected to immunoprecipitation with antibody against Flag-Us11. Figure 1A shows that Hsp90 was coimmunoprecipitated with HSV Us11 but not with the vector or EGFP. To further verify the interaction between HSV-1 Us11 and Hsp90, we carried out a reverse experiment. As shown in Fig. 1B, HSV-1 Us11 was pulled down by Hsp90. However, no such interaction was detectable with a vector or EGFP. These experimental data suggest that Hsp90 is a host-interacting factor of the Us11 protein.
FIG 1.
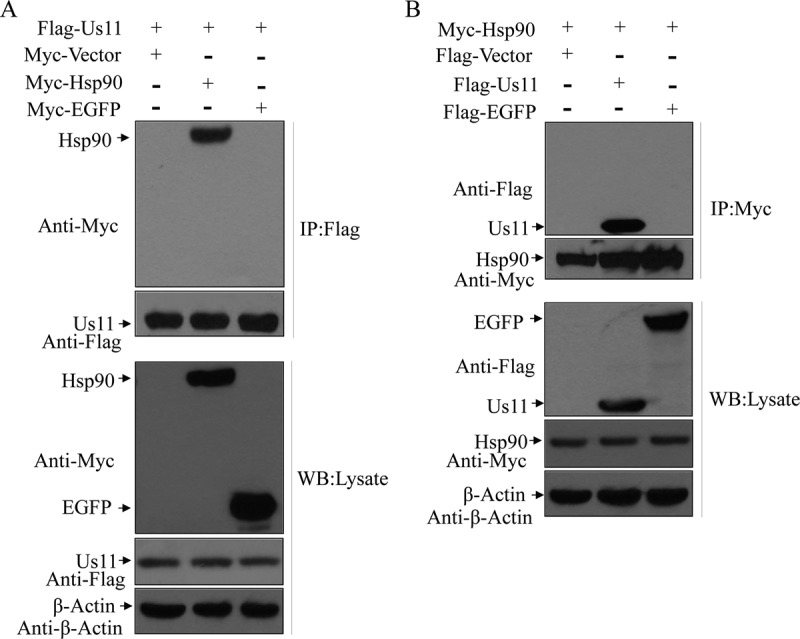
HSV Us11 interacts with Hsp90 in mammalian cells. (A) Monolayers of 293T cells were transfected with Flag-Us11 along with Myc-Hsp90, Myc-EGFP, or vector plasmid. At 36 h after transfection, cells were processed for immunoprecipitation (IP) with anti-Flag antibody. Precipitated proteins and whole-cell lysates were probed with antibodies against Flag, Myc, and β-actin as described in Materials and Methods. (B) 293T cells were transfected with Myc-Hsp90 along with Flag-Us11, Flag-EGFP, or vector plasmid as described for panel A. Cell lysates were subjected to immunoprecipitation and Western blot (WB) analysis with antibodies against Myc, Flag, and β-actin. The data are representative of results from three independent experiments.
HSV-1 Us11 disrupts the Hsp90-TBK1 complex in the absence of other viral proteins.
As a chaperone in host cells, Hsp90 has been reported to interact with TBK1, which potentiates the induction of type I IFN (35). As such, we first evaluated the interaction of Hsp90 and TBK1 by immunoprecipitation. As expected, Hsp90, when ectopically expressed, precipitated TBK1 but not EGFP or the vector control (Fig. 2A). In contrast, TBK1 specifically pulled down Hsp90 (Fig. 2B). To assess whether HSV-1 Us11 has any impact on the Hsp90-TBK1 complex, 293T cells were transiently transfected with Us11, along with Hsp90, TBK1, EGFP, or a vector plasmid. Lysates of cells were subjected to immunoprecipitation. As illustrated in Fig. 2C, HSV-1 Us11 formed a complex with Hsp90. Under this experimental condition, it failed to associate with TBK1 or EGFP, suggesting that Hsp90 but not TBK1 is a direct target. Intriguingly, while Hsp90 associated with TBK1 in 293T cells, such interactions were abrogated in the presence of HSV-1 Us11 (Fig. 2D). Thus, HSV-1 Us11, when expressed in mammalian cells, precludes formation of the Hsp90-TBK1 complex.
FIG 2.

(A) Hsp90 associates with TBK1. Monolayers of 293T cells were transfected with Myc-Hsp90 along with Flag-TBK1, Flag-EGFP, or vector plasmid. At 36 h after transfection, cells were processed for immunoprecipitation with anti-Flag antibody. Precipitated proteins and whole-cell lysates were probed with antibodies against Myc, Flag, and β-actin. (B) TBK1 binds to Hsp90. 293T cells were transfected with Flag-TBK1 along with Myc-Hsp90, Myc-EGFP, or vector plasmid as described for panel A. Cell lysates were processed for immunoprecipitation and Western blot analysis with antibodies against Flag, Myc, and β-actin. (C) HSV Us11 interacts with Hsp90. Monolayers of 293T cells were transfected with Myc-Hsp90, Myc-TBK1, Myc-EGFP, or vector plasmid along with Flag-Us11. At 36 h after transfection, lysates of cells were processed for immunoprecipitation with anti-Flag antibody. Precipitated proteins and whole-cell lysates were probed with antibodies against Flag, Myc, and β-actin. (D) HSV Us11 disrupts the interaction of Hsp90 and TBK1. 293T cells were transfected with Myc-Hsp90 along with Flag-Us11, Flag-TBK1, or vector plasmid. At 36 h after transfection, cells were processed for immunoprecipitation and Western blot analysis with antibodies against Flag, Myc, and β-actin. The data are representative of results from three independent experiments.
The Us11 protein suppresses IFN promoter activation potentiated by Hsp90.
To investigate whether the Us11 protein functionally modulates Hsp90, we performed luciferase reporter assays. Figure 3A shows that Hsp90 itself, when ectopically expressed, failed to activate the IFN promoter. TBK1 modestly stimulated IFN-β promoter activation in 293T cells (Fig. 3B). Addition of Hsp90 enhanced TBK1-mediated gene expression in a dose-dependent manner. However, coexpression of HSV-1 Us11 reduced the stimulatory effect exerted by Hsp90, which paralleled the amount of Us11 transfected into 293T cells (Fig. 3C). Notably, the Us11 protein reduced the IFN promoter activation only in the presence of Hsp90 (Fig. 3D). These results suggest that the Us11 protein negatively modulates IFN promoter activation by TBK1 through Hsp90.
FIG 3.

(A) Hsp90 by itself does not activate the IFN-β promoter. 293T cells were transfected with an empty vector or different amounts of Myc-Hsp90 along with an IFN-β reporter. At 36 h after transfection, cells were harvested for luciferase assays. (B) Hsp90 enhances IFN-β promoter activation by TBK1. 293T cells were transfected with an empty vector, Flag-TBK1, or Myc-Hsp90 along with an IFN-β reporter for 36 h. Cells were harvested for luciferase assays. (C) Us11 inhibits IFN-β promoter activation in a dose-dependent manner. 293T cells were transfected with increasing doses of Flag-Us11 along with Flag-TBK1, Myc-Hsp90, and an IFN-β reporter. Cells were harvested for luciferase assays at 36 h after transfection. (D) Us11 inhibits IFN-β promoter activation potentiated by Hsp90. 293T cells were transfected with an empty vector, Flag-Us11, Flag-TBK1, or Myc-Hsp90 along with an IFN-β reporter. Cells were harvested for luciferase assays at 36 h after transfection. Results are representative of three experiments among triplicate samples and assessed by a two-tailed Student's t test (**, P < 0.01).
HSV-1 Us11 suppresses IRF3 phosphorylation and IFN induction in virus-infected cells.
On the basis of the results described above, we reasoned that expression of Us11 might suppress the induction of type I IFN responses upon HSV infection. To test this, we analyzed the expression of antiviral genes by quantitative real-time PCR. Previous work suggested that wild-type virus inhibits the induction of IFN whereas the γ134.5 deletion mutant stimulates it (36). As Us11 works cooperatively with γ134.5 (25), we chose to assess its activity in the absence of γ134.5. For this purpose, we compared wild-type HSV-1, the γ134.5 null mutant (Δγ134.5), and the γ134.5 null mutant in which Us11 is driven by the α47 promoter (EUs11). As indicated in Fig. 4A, wild-type HSV-1 triggered a low level of IFN-β, ISG54, ISG56, and RANTES expression whereas the Δγ134.5 mutant stimulated robust responses, with a sharp increase in the expression of IFN-β ISG54, ISG56, and RANTES at 6 h postinfection. However, EUs11 reduced such responses to modest levels, indicative of a negative regulation of type I IFN responses by Us11 in HSV-infected cells.
FIG 4.

(A) Expression of Us11 inhibits the induction of antiviral genes. Mouse embryonic fibroblasts were either mock infected or infected with HSV-1, the Δγ134.5 mutant, or EUs11 (5 PFU/cell). At 6 h postinfection, total RNA extracted from cells was subjected to quantitative real-time PCR amplification for IFN-β, ISG56, ISG54, and RANTES. The data were normalized to 18S rRNA data and calculated as described in Materials and Methods. Results are expressed as fold activation with standard deviations among triplicate samples. The data were statistically analyzed by a two-tailed Student's t test (**, P < 0.01). (B) Cells were subjected to mock infection or were infected with the indicated viruses (5 PFU/cell). At 6 h postinfection, cell lysates were prepared and processed for Western blot analysis with antibodies against Hsp90, TBK1, IRF3, phosphorylated IRF3, Us11, ICP27, or β-actin. The data are representative of results from three independent experiments.
Since IRF3 phosphorylation by TBK1 drives the expression of antiviral genes (2), we asked whether HSV-1 Us11 perturbs IRF3 activation. Cells were subjected to mock infection or were infected with viruses. At 6 h postinfection, samples were processed for immunoblot analysis. As illustrated in Fig. 4B, viral infection had little effect on the expression of IRF3 or Hsp90 but reduced the level of TBK1, particularly in cells infected with EUs11, suggesting that Us11 downregulated TBK1. Like wild-type virus, EUs11 blocked phosphorylation of IRF3 whereas the Δγ134.5 mutant failed to do so. This was not due to the lack of virus infection, as the expression levels of ICP27 were comparable in virus-infected cells.
HSV-1 Us11 primes degradation of TBK1 via the proteasome-mediated pathway.
To further test the impact of Us11 on TBK1 expression, we performed time course analysis. Cells were subjected to mock infection or were infected with viruses. At 0, 3, and 6 h postinfection, lysates of cells were processed for Western blot analysis with anti-TBK1 antibody. As illustrated in Fig. 5A, expression of TBK1 remained virtually unchanged in the mock-infected cells. A marginal reduction in TBK1 expression was detected upon infection with wild-type HSV-1 or the Δγ134.5 mutant over the course of infection. While a similar phenotype was seen with EUs11, a pronounced reduction in TBK1 expression was detectable at 6 h postinfection, which paralleled the expression of Us11.
FIG 5.

(A) Expression of HSV Us11 induces TBK1 degradation. (A) Mouse embryonic fibroblasts (MEFs) were infected with wild-type HSV-1, the Δγ134.5 mutant, and EUs11 at 5 PFU/cell. At different time points postinfection, cell lysates were prepared and processed for Western blot analysis with antibodies against TBK1, Us11, Hsp90, and β-actin. (B and C) Effect of MG132 on TBK1 degradation. MEFs were subjected to mock infection or were infected with the indicated viruses (5 PFU/cell) without MG132 (B) or with MG132 (25 μM) (C). At 6 h postinfection, cell lysates were prepared and processed for Western blot analysis with antibodies against TBK1, Us11, ICP27, and β-actin. The data are representative of results from three independent experiments.
To assess the nature of TBK1 reduction, we measured the level of TBK1 in the presence or absence of MG132, which is an inhibitor of the proteasome degradation pathway. As shown in Fig. 5B, unlike the mock control, wild-type HSV-1, or Δγ134.5 mutant results, EUs11 induced a remarkable reduction in TBK1 expression in infected cells. However, treatment with MG312 restored TBK1 to levels comparable to those seen in the cells subjected to mock infection or infected with wild-type virus or the Δγ134.5 mutant (Fig. 5C). Here, the Us11 protein, upon infection, primed the degradation of TBK1 via a proteasome degradation pathway.
HSV-1 Us11 displaces TBK1 by formation of a complex with Hsp90 upon virus infection.
TBK1 activation is suggested to rely on the formation of a complex with Hsp90 (35). As such, we assessed the integrity of the Hsp90-TBK1 complex in virus-infected cells by immunoprecipitation. Cells were subjected to mock infection or were infected with viruses. At 6 h postinfection, samples were collected for analysis. As illustrated in Fig. 6A, Hsp90 formed a complex with TBK1 and IRF3 in the cells that had been subjected to mock infection. Similar results were seen in the cells infected with wild-type virus or the Δγ134.5 mutant, suggesting retention of the Hsp90 complex. However, in cells infected with EUs11, TBK1 was not detectable in the Hsp90 complex whereas the presence of Us11 was apparent in the Hsp90 complex. Interestingly, IRF3 remained associated with Hsp90. Western blot analysis confirmed protein expression in mock-infected or virus-infected cells.
FIG 6.

(A) HSV Us11 precludes Hsp90-TBK1 complex from formation in infected cells. MEFs were subjected to mock infection or were infected with the indicated viruses (5 PFU/cell). At 6 h postinfection, cells were processed for immunoprecipitation with anti-Hsp90 antibody. Precipitated proteins and whole-cell lysates were probed with antibodies against Us11, TBK1, IRF3, Hsp90, ICP27, or β-actin. (B) Us11 does not affect the binding of Cdc37 to Hsp90. Cell were infected and processed as described for panel A. Samples were probed with antibodies against Us11, Cdc37, Hsp90, ICP27, and β-actin. The data are representative of results from three independent experiments.
To further explore the way in which Us11 functions, we analyzed the Cdc37 cochaperone, which participates in the recruitment of client kinases by Hsp90 (37). Figure 6B shows that Hsp90 formed a complex with Cdc37 in mock-infected cells as measured by coimmunoprecipitation. This interaction was not affected in cells infected with wild-type HSV-1, the Δγ134.5 mutant, or EUs11. Although present in the Hsp90 machinery, Us11 failed to displace Cdc37. We conclude that Us11 is able to complete with TBK1 for Hsp90, precluding IRF3 phosphorylation upon HSV infection.
Inhibition of TBK1 by the Us11 protein facilitates viral replication.
To evaluate the significance of Us11-TBK1 interactions, we performed viral growth assays. As shown in Fig. 7A, wild-type HSV-1 replicated efficiently in both TBK1+/+ and TBK1−/− cells, reaching titers of 2 × 106 and 2 × 107 PFU/ml, respectively. In contrast, the Δγ134.5 mutant barely grew in TBK1+/+ cells but replicated to a titer of 5 × 104 PFU/ml in TBK1−/− cells. EUs11 exhibited a different growth pattern. In TBK1+/+ cells, it replicated to an intermediate level, with a titer of 8 × 104 PFU/ml. In TBK1−/− cells, EUs11 replicated almost like the wild-type virus, with a titer of 7 × 106 PFU/ml. Hence, the ability of EUs11 to neutralize TBK1 restriction is attributable to the Us11 protein.
FIG 7.

The Us11-TBK1 interaction influences viral replication. (A) Viral replication in TBK1+/+ or TBK1−/− cells. Cells were infected with HSV-1, the Δγ134.5 mutant, or EUs11 at 0.01 PFU/cell. At 48 h postinfection, cells were harvested and the total virus yields were determined on Vero cells. (B) Kinetics of viral growth in TBK1+/+ cells. MEFs were infected with the indicated viruses as described for panel A. At different time points postinfection, viral yields were determined on Vero cells. (C) Kinetics of viral growth in TBK1−/− MEFs. Viral infection was performed as described for panel B. The data are representative of results from three experiments with triplicate samples. Differences between the selected groups were statistically assessed by a two-tailed Student's t test (*, P < 0.05; **, P < 0.01).
Next, we examined the kinetics of viral growth. As shown Fig. 7B, in TBK1+/+ cells, wild-type HSV-1 grew steadily at 0, 12, 24, 36, and 48 h postinfection, with a titer increasing to 3 × 106 PFU/ml at 48 h postinfection. Under this experimental condition, EUs11 replicated with a similar trend but at a lower level, with a titer of 1 × 105 PFU at 48 h postinfection. The γ134.5 null mutant replicated poorly throughout infection, with a titer of <1 × 102 PFU/ml. In TBK1−/− cells (Fig. 7C), viruses replicated more efficiently at each time point examined, with faster growth kinetics in the absence of a TBK1 blockade. Taken in combination, these results suggest that the Us11-TBK1 interaction determines the outcome of HSV infection.
DISCUSSION
Previous studies have demonstrated that HSV-1 Us11 facilitates viral replication through inhibition of dsRNA-dependent protein kinase PKR (21, 25–28). Although not fully deciphered, these results partly explain why HSV downregulates type I IFN responses. In the present study, we showed that the Us11 protein of HSV-1 targets heat shock 90 protein. By engagement with Hsp90, HSV-1 Us11 destabilizes TNAK binding kinase 1, which subsequently suppresses the activation of IRF3 and IFN expression. These data unveil a regulatory interface where interplay of HSV-1 Us11 and TBK1 influences HSV infection.
HSV-1 Us11 is a true late (γ2) gene whose expression depends on viral DNA replication (38). As γ134.5 expression precedes Us11 expression, the two proteins are thought to work sequentially at the discrete phase of HSV replication (25, 39). Accordingly, deletion of γ134.5 results in translation arrest of Us11 mRNA-mediated dsRNA-dependent protein kinase PKR (17). When expressed as an early (α) gene, Us11 is able to facilitate HSV replication in the absence of γ134.5 in infected cells (21). The observation that the Us11 protein neutralizes antiviral immunity by TBK1 suggests an additional layer of virus-host interactions. This notion is suggested by three lines of evidence. First, yeast two-hybrid screen revealed Hsp90 as a target of the Us11 protein. When expressed, Us11 interrupted the Hsp90-TBK1 interaction. Consistently, it suppressed IFN promoter activation potentiated by Hsp90. Second, in HSV-infected cells, Us11 disrupted the Hsp90-TBK1 complex, resulting in TBK1 destabilization. As a result, Us11 inhibited IRF3 phosphorylation and IFN induction. Third, inhibition of TBK1 by HSV Us11 partially restored viral growth in the absence of γ134.5.
The precise mechanism of Us11 action is unknown. It has been shown that the carboxyl-terminal domain of Us11 inhibits PKR, 2′,5′-oligoadenylate synthetase, RIG-I, and MDA-5 (28, 30–32). Our experimental data do not exclude the possibility that Us11 may perturb TBK1 through this domain. As PKR deficiency has no impact on IFN induction upon HSV infection (40), it is reasonable to believe that Us11 suppresses the expression of type I IFN independently of PKR. However, the observed phenotype may stem from cofounding effects on the block of 2′,5′-oligoadenylate synthetase, RIG-I, or MDA-5. Additional work is required to address this issue. We postulate that the Us11 protein, by coordinately modulating innate immune pathways, likely creates a favorable environment for HSV infection.
Hsp90 is best characterized as a chaperone that interacts with clients through its central domain (41). While the N-terminal domain of Hsp90 binds ATP, the C-terminal domain initiates dimerization. Because Hsp90 and Cdc37 are implicated in TBK1 activation in the RIG-I and cytosolic DNA recognition pathways (35, 42), an issue arises as to how HSV-1 Us11 interacts with the Hsp90 machinery. We noted that Us11, when expressed, disrupted the Hsp90-TBK1 interaction. This was also seen in HSV-infected cells, where Us11 formed a complex with endogenous Hsp90. Congruently, Us11 expression paralleled reductions in IRF3 phosphorylation and cytokine expression. A plausible explanation is that association of Us11 with Hsp90 may create a physical barrier that masks the TBK1 binding site on Hsp90. As an alternative, HSV Us11 may mediate a gross change in Hsp90 conformation which abrogates the Hsp90-TBK1 complex. Since the interactions among IRF3, Cdc37, and Hsp90 were unaltered upon Us11 expression, we favor the model that HSV-1 Us11 may compete with TBK1 for Hsp90 binding.
Our work suggests that the Us11 protein of HSV-1 destabilizes TBK1. Us11, when expressed in HSV-infected mouse embryonic fibroblast (MEF) cells, primed a reduction in the steady-state level of TBK1. The observed effect was evident with TBK1 but not IRF3. Remarkably, TBK1 degradation appeared to rely on the proteasome-dependent pathway, as it was blunted in the presence of a proteasome inhibitor. The details of the mechanism are to be established. In this context, it is noteworthy that at least two separate degradation pathways exist for Hsp90 client kinases (43). The first pathway involves CUL5, elongin B, and elongin C, while the second one requires the E3 HECTD3 ligase. We speculate that HSV Us11 may destabilize TBK1 via one of these two pathways. Work is in progress to test this hypothesis.
TBK1 sits at the center of innate immune pathways leading to type I IFN production (2, 44). In response to TLR, RIG-I/MDA5, and DNA sensor pathways, it is activated in a signal-dependent manner. As such, it is not surprising that HSV has evolved various strategies to dampen or evade host antiviral immunity. For example, as an immediate early protein, ICP27 associates with and inhibits TBK1 and STING in macrophages (14). Similarly, UL46 targets TBK1 and STING (45). In addition, γ134.5 negatively regulates TBK1 early in HSV infection (36, 40). The results of the present work suggest that Us11 inhibits TBK1 through its interaction with Hsp90. As HSV triggers innate immune responses in a cell type- and time-dependent manner (46), these viral proteins may act cooperatively to facilitate HSV replication.
MATERIALS AND METHODS
Cells and viruses.
Vero and 293T cells were obtained from the American Type Culture Collection. TBK1+/+ and TBK1−/− mouse embryonic fibroblasts (MEFs) have been described previously (36). All cells were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. HSV-1(F) (47) is a prototype HSV-1 strain used in this study. In recombinant Δγ134.5 virus, a 1-kb fragment from the coding region of the γ134.5 gene was deleted (48). In EUs11 virus, the promoter of Us11 was deleted where its expression is driven by the α47 promoter. This was constructed via insertion of EGFP into the Us12 locus in the Δγ134.5 mutant, which was subsequently replaced with Us11 by transfection with plasmid pRB4028 (27). The virus construct was plaque purified and verified by PCR and DNA sequencing. The expression of Us11 was confirmed by Western analysis using anti-Us11 antibody (24). Preparation of viral stock and titration of infectivity were carried out with Vero cells.
Plasmids and reporter assays.
The Flag-Us11 plasmid was constructed by inserting a PCR-amplified fragment into the BamHI and EcoRI sites of pCMV-tag2B. The Flag-TBK1 plasmid was constructed by inserting a PCR-amplified fragment into the BamHI and SalI sites of pCMV-tag2B. The Flag-EGFP plasmid was constructed by inserting a PCR-amplified fragment into the BamHI and EcoRI sites of pCMV-tag2B. The Myc-Hsp90 plasmid was constructed by inserting a PCR-amplified fragment into the BamHI and XhoI sites of pCMV-Myc. The Myc-EGFP plasmid was constructed by inserting a PCR-amplified fragment into the BamHI and XhoI sites of pCMV-Myc. The pRB4028 plasmid contains the US10 gene and the α47 promoter juxtaposed with the US11 coding sequence (27). pTK-Luc and pIFN-β-Luc were described elsewhere (36). Reporter assays were carried out as described previously (36).
Immunoprecipitation analysis.
To examine protein interactions, immunoprecipitation was carried out as described previously (40). Briefly, cells were lysed with ice-cold immunoprecipitation buffer. After centrifugation, cell extracts were incubated with the indicated antibodies and agarose conjugated with protein A/G at 4°C. The immobilized protein beads were subjected to immunoblotting analysis.
Viral infection assay.
Cells were infected with viruses. At various time points, cells were harvested and freeze-thawed three times. Viral yields were determined on Vero cells at 37°C (36). For MG132 treatment, MG132 was added into the cell culture medium (25 μM) immediately after virus infection. At 6 h postinfection, the cells were harvested and lysed for immunoblotting analysis.
Immunoblotting analysis.
Cells were harvested, washed with phosphate-buffered saline (PBS), and lysed with ice-cold immune precipitation assay buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA, 1.0% Triton X-100, and protease inhibitor cocktail) for 30 min on ice. After centrifugation, supernatants were mixed with disruption buffer (50 mM Tris-HCl [pH 6.8], 2% SDS, 0.1% bromophenol blue, 10% glycerol, and 100 nM β-mercaptoethanol) and boiled. Samples were then subjected to electrophoresis on denaturing polyacrylamide gels, transferred to nitrocellulose membranes, blocked with 5% bovine serum albumin (BSA), and reacted with antibodies against TBK1 (catalog no. 3504; Cell Signaling Technology), IRF3 (catalog no. 4302; Cell Signaling Technology), phosphorylated IRF3 (catalog no. 4947; Cell Signaling Technology), Hsp90 (sc-13119; Santa Cruz), Cdc37 (catalog no. 4793; Cell Signaling Technology), β-actin (Sigma), Us11 (24), ICP27 (Virusys Inc.), anti-Flag–horseradish peroxidase (anti-Flag–HRP) (Sigma), or anti-Myc-HRP (Cell Signaling Technology). The membranes were rinsed in phosphate-buffered saline (PBS) and reacted with either donkey anti-rabbit (Santa Cruz) or donkey anti-mouse (Santa Cruz) immunoglobulin conjugated to horseradish peroxidase and developed with an enhanced chemiluminescence Western blot detection system kit (Amersham Pharmacia Biotechnology).
Quantitative real-time PCR assay.
Cells were subjected to mock infection or were infected with viruses. At the indicated time points, total RNA was harvested from cells using an RNeasy kit (Qiagen) and subjected to DNase I digestion (New England BioLabs). cDNA was synthesized using a High Capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCR was performed using an Applied Biosystems ABI Prism 7900HT instrument with ABI SYBR green master mix (Applied Biosystems). Gene expression levels were normalized to that of the endogenous control 18S rRNA gene. Relative gene expression levels were determined as described previously (49). Primers for each gene were chosen according to the recommendation of the qPrimerDepot database (12). Primer sequences were as follows: for mouse IFN-β, AAT TTC TCC AGC ACT GGG TG and AGT TGA GGA CAT CTC CCA CG; for mouse ISG54, GCA AGA TGC ACC AAG ATG AG and CAC TCT CCA GGC AAC CTC TT; for mouse ISG56, CAA GGC AGG TTT CTG AGG AG and AAG CAG ATT CTC CAT GAC CTG; for mouse RANTES, CTG CTG CTT TGC CTA CCT CT and CAC TTC TTC TCT GGG TTG GC; for 18S rRNA, CCT GCG GCT TAA TTT GAC TC and AAC CAG ACA AAT CGC TCC AC.
ACKNOWLEDGMENTS
We thank Bernard Roizman and Kevin Cassady for providing valuable reagents.
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases (AI112755 to B.H.).
REFERENCES
- 1.Whitley RJ, Roizman B. 2001. Herpes simplex virus infections. Lancet 357:1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- 2.Ma Y, He B. 2014. Recognition of herpes simplex viruses: Toll-like receptors and beyond. J Mol Biol 426:1133–1147. doi: 10.1016/j.jmb.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Heron B, Vallee L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL. 2007. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 4.Chiang JJ, Sparrer KMJ, van Gent M, Lassig C, Huang T, Osterrieder N, Hopfner KP, Gack MU. 2018. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat Immunol 19:53–62. doi: 10.1038/s41590-017-0005-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. 2011. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM. 2015. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112:E1773–E1781. doi: 10.1073/pnas.1424637112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paladino P, Mossman KL. 2009. Mechanisms employed by herpes simplex virus 1 to inhibit the interferon response. J Interferon Cytokine Res 29:599–607. doi: 10.1089/jir.2009.0074. [DOI] [PubMed] [Google Scholar]
- 10.Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. 2004. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol 78:1675–1684. doi: 10.1128/JVI.78.4.1675-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melroe GT, Silva L, Schaffer PA, Knipe DM. 2007. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: potential role in blocking IFN-beta induction. Virology 360:305–321. doi: 10.1016/j.virol.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eidson KM, Hobbs WE, Manning BJ, Carlson P, DeLuca NA. 2002. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J Virol 76:2180–2191. doi: 10.1128/jvi.76.5.2180-2191.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, Mettenleiter T, Chen ZJ, Knipe DM, Sandri-Goldin RM, Enquist LW, Hartmann R, Mogensen TH, Rice SA, Nyman TA, Matikainen S, Paludan SR. 2016. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J 35:1385–1399. doi: 10.15252/embj.201593458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peri P, Mattila RK, Kantola H, Broberg E, Karttunen HS, Waris M, Vuorinen T, Hukkanen V. 2008. Herpes simplex virus type 1 Us3 gene deletion influences Toll-like receptor responses in cultured monocytic cells. Virol J 5:140. doi: 10.1186/1743-422X-5-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S, Wang K, Lin R, Zheng C. 2013. Herpes simplex virus 1 serine/threonine kinase US3 hyperphosphorylates IRF3 and inhibits beta interferon production. J Virol 87:12814–12827. doi: 10.1128/JVI.02355-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou J, Roizman B. 1992. The γ134.5 gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc Natl Acad Sci U S A 89:3266–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou J, Chen JJ, Gross M, Roizman B. 1995. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with γ134.5-mutants of herpes simplex virus 1. Proc Natl Acad Sci U S A 92:10516–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He B, Gross M, Roizman B. 1997. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 94:843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He B, Gross M, Roizman B. 1998. The γ134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J Biol Chem 273:20737–20743. doi: 10.1074/jbc.273.33.20737. [DOI] [PubMed] [Google Scholar]
- 21.Mohr I, Gluzman Y. 1996. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J 15:4759–4766. [PMC free article] [PubMed] [Google Scholar]
- 22.He B, Chou J, Brandimarti R, Mohr I, Gluzman Y, Roizman B. 1997. Suppression of the phenotype of γ134.5-herpes simplex virus 1: failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J Virol 71:6049–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roller RJ, Roizman B. 1990. The herpes simplex virus Us11 open reading frame encodes a sequence-specific RNA-binding protein. J Virol 64:3463–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roller RJ, Roizman B. 1992. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J Virol 66:3624–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mulvey M, Camarena V, Mohr I. 2004. Full resistance of herpes simplex virus type 1-infected primary human cells to alpha interferon requires both the Us11 and gamma(1)34.5 gene products. J Virol 78:10193–10196. doi: 10.1128/JVI.78.18.10193-10196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cassady KA, Gross M, Roizman B. 1998. The herpes simplex virus US11 protein effectively compensates for the γ134.5 gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J Virol 72:8620–8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cassady KA, Gross M, Roizman B. 1998. The second-site mutation in the herpes simplex virus recombinants lacking the γ134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J Virol 72:7005–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poppers J, Mulvey M, Khoo D, Mohr I. 2000. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J Virol 74:11215–11221. doi: 10.1128/JVI.74.23.11215-11221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters GA, Khoo D, Mohr I, Sen GC. 2002. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J Virol 76:11054–11064. doi: 10.1128/JVI.76.21.11054-11064.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sànchez R, Mohr I. 2007. Inhibition of cellular 2′-5′ oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J Virol 81:3455–3464. doi: 10.1128/JVI.02520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xing J, Wang S, Lin R, Mossman KL, Zheng C. 2012. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol 86:3528–3540. doi: 10.1128/JVI.06713-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kew C, Lui PY, Chan CP, Liu X, Au SW, Mohr I, Jin DY, Kok KH. 2013. Suppression of PACT-induced type I interferon production by herpes simplex virus 1 Us11 protein. J Virol 87:13141–13149. doi: 10.1128/JVI.02564-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Javouhey E, Gibert B, Arrigo AP, Diaz JJ, Diaz-Latoud C. 2008. Protection against heat and staurosporine mediated apoptosis by the HSV-1 US11 protein. Virology 376:31–41. doi: 10.1016/j.virol.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 34.Lussignol M, Queval C, Bernet-Camard MF, Cotte-Laffitte J, Beau I, Codogno P, Esclatine A. 2013. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol 87:859–871. doi: 10.1128/JVI.01158-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang K, Shi H, Qi R, Sun S, Tang Y, Zhang B, Wang C. 2006. Hsp90 regulates activation of interferon regulatory factor 3 and TBK-1 stabilization in Sendai virus-infected cells. Mol Biol Cell 17:1461–1471. doi: 10.1091/mbc.e05-09-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verpooten D, Ma Y, Hou S, Yan Z, He B. 2009. Control of TANK-binding kinase 1-mediated signaling by the γ134.5 protein of herpes simplex virus 1. J Biol Chem 284:1097–1105. doi: 10.1074/jbc.M805905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. 2012. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson PA, MacLean C, Marsden HS, Dalziel RG, Everett RD. 1986. The product of gene US11 of herpes simplex virus type 1 is expressed as a true late gene. J Gen Virol 67(Pt 5):871–883. [DOI] [PubMed] [Google Scholar]
- 39.Mulvey M, Poppers J, Sternberg D, Mohr I. 2003. Regulation of eIF2alpha phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J Virol 77:10917–10928. doi: 10.1128/JVI.77.20.10917-10928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma Y, Jin H, Valyi-Nagy T, Cao Y, Yan Z, He B. 2012. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J Virol 86:2188–2196. doi: 10.1128/JVI.05376-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schopf FH, Biebl MM, Buchner J. 2017. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol 18:345–360. doi: 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 42.Lee MN, Roy M, Ong SE, Mertins P, Villani AC, Li W, Dotiwala F, Sen J, Doench JG, Orzalli MH, Kramnik I, Knipe DM, Lieberman J, Carr SA, Hacohen N. 2013. Identification of regulators of the innate immune response to cytosolic DNA and retroviral infection by an integrative approach. Nat Immunol 14:179–185. doi: 10.1038/ni.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z, Zhou L, Prodromou C, Savic V, Pearl LH. 2017. HECTD3 mediates an HSP90-dependent degradation pathway for protein kinase clients. Cell Rep 19:2515–2528. doi: 10.1016/j.celrep.2017.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat Immunol 7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 45.Deschamps T, Kalamvoki M. 27 July 2017. Evasion of the STING DNA-sensing pathway by VP11/12 of herpes simplex virus 1. J Virol doi: 10.1128/JVI.00535-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rasmussen SB, Sorensen LN, Malmgaard L, Ank N, Baines JD, Chen ZJ, Paludan SR. 2007. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J Virol 81:13315–13324. doi: 10.1128/JVI.01167-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 48.Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to γ134.5, a gene nonessential for growth in culture. Science 250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 49.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]