

Abstract
Pancreatic acinar cells synthesize, package, and secrete digestive enzymes into the duodenum to aid in nutrient absorption and meet metabolic demands. When exposed to cellular stresses and insults, acinar cells undergo a dedifferentiation process termed acinar–ductal metaplasia (ADM). ADM lesions with oncogenic mutations eventually give rise to pancreatic ductal adenocarcinoma (PDAC). In healthy pancreata, the basic helix‐loop‐helix (bHLH) factors MIST1 and PTF1a coordinate an acinar‐specific transcription network that maintains the highly developed differentiation status of the cells, protecting the pancreas from undergoing a transformative process. However, when MIST1 and PTF1a gene expression is silenced, cells are more prone to progress to PDAC. In this study, we tested whether induced MIST1 or PTF1a expression in PDAC cells could (i) re‐establish the transcriptional program of differentiated acinar cells and (ii) simultaneously reduce tumor cell properties. As predicted, PTF1a induced gene expression of digestive enzymes and acinar‐specific transcription factors, while MIST1 induced gene expression of vesicle trafficking molecules as well as activation of unfolded protein response components, all of which are essential to handle the high protein production load that is characteristic of acinar cells. Importantly, induction of PTF1a in PDAC also influenced cancer‐associated properties, leading to a decrease in cell proliferation, cancer stem cell numbers, and repression of key ATP‐binding cassette efflux transporters resulting in heightened sensitivity to gemcitabine. Thus, activation of pancreatic bHLH transcription factors rescues the acinar gene program and decreases tumorigenic properties in pancreatic cancer cells, offering unique opportunities to develop novel therapeutic intervention strategies for this deadly disease.
Keywords: ABC transporters, bHLH, exocrine pancreas, MIST1, PDAC, transcription
Abbreviations
- ABC
ATP‐binding cassette
- ADM
acinar–ductal metaplasia
- bHLH
basic helix‐loop‐helix
- CSC
cancer stem cell
- DEG
differentially expressed gene
- Dox
doxycycline
- GSEA
Gene Set Enrichment Analysis
- IC50
half‐maximal inhibitory concentration
- IHC
immunohistochemistry
- IPA
ingenuity pathway analysis
- KEGG
Kyoto encyclopedia of genes and genomes
- KPC
Cre recombinase‐driven LSL‐KrasG12D; LSL‐Trp53R172H mouse model
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
- UPR
unfolded protein response
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a dismal disease with a 5‐year survival rate of 8% (American Cancer Society, 2017). Even more alarming is the minimal progress that has been made to improve patient outcome over the past 30 years. Current efforts to treat this disease revolve around the use of gemcitabine – a nucleoside analog; FOLFIRINOX – a combinatorial drug regimen; and erlotinib – a tyrosine kinase inhibitor of the epidermal growth factor receptor (Conroy et al., 2011; Moore et al., 2007; Réjiba et al., 2009). Unfortunately, these and other agents have produced only marginal results in increasing life expectancy (American Cancer Society, 2017; Heinemann et al., 2000). Combined with issues regarding the availability of early diagnostic tools, it is clear that alternative strategies that look toward comprehensive networks are needed to more efficiently treat patients.
The formation of pancreatic tumors requires a sequence of oncogenic and tumor suppressor insults to drive healthy acinar cells toward a proliferative, dysregulated tumor cell phenotype (Bryant et al., 2014; Hruban et al., 2000; Pasca et al., 2006; Witkiewicz et al., 2015). Oncogenic KRAS is thought to be the primary driver of PDAC and readily transforms cells that have undergone acinar–ductal metaplasia (ADM), resulting in a dedifferentiated state where the proacinar basic helix‐loop‐helix (bHLH) transcription factor genes MIST1 and PTF1a are transcriptionally silenced (Adell et al., 2000; Day et al., 1996; Krah et al., 2015; Rodolosse et al., 2004; Guanglu Shi et al., 2009; Zhu et al., 2007). In healthy pancreata, MIST1 facilitates cell‐to‐cell communication, promotes acinar cell polarity, controls Ca2+ flux, promotes unfolded protein response (UPR) homeostasis, aids in vesicle assembly, and is required for regulated exocytosis (Direnzo et al., 2012; Garside et al., 2010; Hess et al., 2016; Jia et al., 2008; Rukstalis et al., 2003). PTF1a is necessary for pancreatic organogenesis and maintenance of gene expression networks associated with the production of a vast array of digestive hydrolases (Beres et al., 2006; Direnzo et al., 2012; Garside et al., 2010; Hess et al., 2016; Jia, 2008; Jiang et al., 2016; Masui et al., 2007). Importantly, both MIST1 and PTF1a negatively regulate cell cycle progression by inducing p21 CIP1/WAF1 expression (Jia et al., 2008; Rodolosse et al., 2004).
Silencing of the MIST1 or PTF1a genes results in significant changes to acinar cells, leading to widespread failure to appropriately synthesize and secrete digestive enzymes, maintain proper apical–basal polarity, and retain essential gap junctions that permit intercellular communication (Direnzo et al., 2012; Jiang et al., 2016; Krah et al., 2015; Murtaugh and Leach, 2007). In normal pancreata, the silencing of MIST1 and PTF1a during injury permits transient acinar cell regeneration, allowing the exocrine organ to recover from damage (Karki et al., 2015; Krah et al., 2015). However, in conditions where oncogenic insults have accrued, the process promotes tumorigenesis. Importantly, loss of these transcription factors is instrumental in the progression of pancreatic disease states. MIST1‐null (MIST1 KO) acinar cells with activated Kras mutations greatly accelerate the formation of precancerous pancreatic intraepithelial neoplasia (PanIN) lesions (Shi et al., 2009, 2013). Conversely, forced expression of MIST1 prevents this increased rate of KRAS‐induced transformation (Shi et al., 2013). Additionally, recent work has shown that disruption of the bHLH:HLH axis in PDAC cells through forced expression of E47 leads to accumulation of MIST1, decreased cellular proliferation, and reduction in tumor formation (Kim et al., 2015). Similar results have been observed with PTF1a where loss of PTF1a potentiates the development of ADM and PanIN lesions as the cells no longer maintain expression of the acinar gene program (Campos et al., 2013; Kondratyeva et al., 2017; Krah et al., 2015). Thus, MIST1 and PTF1a protect acinar cells against the development of the earliest stages of pancreatic cancer.
Given the importance of bHLH factors to normal cellular function, we hypothesized that induced expression of the acinar‐specific bHLH differentiation factors MIST1 and PTF1a in PDAC cells would result in stimulation of the acinar gene network and decreased tumorigenic potential. Indeed, doxycycline‐inducible MIST1 and PTF1a retained transcriptional activity in PDAC cells. MIST1 induced expression of the vesicle trafficking genes RAB26 and RAB3D as well as genes associated with the UPR, whereas PTF1a induced key acinar transcription factors and an array of digestive enzyme genes. Forced expression of PTF1a also resulted in decreased tumor‐associated gene expression profiles which led to decreased cell proliferation, decreased pancreatic cancer stem cells (CSCs), and a significant increase in sensitivity toward gemcitabine treatment. Together, these studies promote the concept that strategies to induce an acinar differentiation program in PDAC tumor cells may have high efficacy in reversing the aggressive nature of this disease.
2. Materials and methods
2.1. Plasmid constructs
The open reading frames of mouse PTF1amyc and rat MIST1myc were cloned into the Tet‐One™ plasmid (Clontech Laboratories, Inc., Mountain View, CA, USA) by standard procedures. Pgl3 RBPJ‐L (gift from Raymond McDonald) and TA‐E‐Box‐Luc reporters have been previously described (Masui et al., 2007; Tran et al., 2007).
2.2. Cell culture
The KC mouse line was generated from elastase‐CreER, LSL‐Kras G12D/+ PDAC tumors, while KPC1 and KPC2 lines were generated from elastase‐CreER, LSL‐Kras G12D/+ , Tpr53 R172H/+ PDAC tumors (Y. Yang & S. F. Konieczny, unpublished data). KC, KPC1, KPC2, and Panc‐1 cells (ATCC) were cultured in high‐glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 1% penicillin/streptomycin at 5% CO2, 37 °C. Cells were transfected with the empty Tet‐One, Tet‐PTF1amyc, and Tet‐MIST1myc plasmids using X‐tremeGENE 9 (Cat. 06365787001, Roche, Indianapolis, IN, USA), and stable transformants were selected for growth in 3.0 μg·mL−1 puromycin for a period of two weeks. Individual Panc‐1 Tet‐One, Panc‐1 Tet‐PTF1a, and Panc‐1 Tet‐MIST1 clones were screened for appropriate doxycycline induction of PTF1a and MIST1 expression, respectively, using 1 μg·mL−1 doxycycline hyclate (Cat. D3447, Sigma, St. Louis, MO, USA) for a period of 72 h unless otherwise stated. Doxycycline was replaced every 48 h along with fresh media. All cell lines were genetically authenticated by the American Type Culture Collection and pathogen‐tested by IDEXX Laboratories.
2.3. RNA‐Seq analysis
Four biological replicates of Panc‐1 Tet‐MIST1, Tet‐PTF1a, and control Tet‐One cells were incubated with or without 1 μg·mL−1 doxycycline for a period of 72 h, followed by RNA isolation using the Qiagen miRNeasy extraction kit (Cat. 217004, Qiagen, Hilden, Germany). Illumina HiSeq 4000 sequencing was used to generate 50M paired‐end reads per sample, and reads were aligned to human reference genome hg19 using TopHat. A filter of > 0.5 counts per million reads (roughly equivalent to 10 reads) in at least four samples was implemented prior to determining gene expression using edgeR (Robinson et al., 2010). Genes with an FDR < 0.05 were considered differentially expressed with no additional filter for fold change. Differentially expressed genes (DEGs) were analyzed using functional enrichment analysis with ingenuity pathway analysis software (Ingenuity Systems, Inc., Redwood City, CA, USA) and Gene Set Enrichment Analysis (GSEA, Broad Institute, Cambridge, MA, USA). The GSEA pancreatic CSC marker list was curated using published pancreatic stem cell markers (Dalla Pozza et al., 2015; Kure et al., 2012; Li et al., 2007). Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis (Kanehisa and Goto, 2000) was also performed independently on the induced and repressed DEGs.
AME from meme suite v4.12.0 was used to determine the enrichment of PTF1a and MIST1 motifs within the promoter regions of DEGs (McLeay and Bailey, 2010). Promoter regions were defined as −1000 to +100 base pairs from the TSS. Promoter sequences were retrieved from genome build hg19 using BEDTools v.2.27.1 (Quinlan and Hall, 2010). PTF1a motifs containing an E box and TC box separated by either one or two helical turns midpoint to midpoint, as described previously, were used as input motifs (Cockell et al., 1989; Rose and MacDonald, 1997; Thompson et al., 2012) for analysis of the Tet‐PTF1a dataset. GC and TC E box motifs were used as motif inputs for analysis of the Tet‐MIST1 dataset. Additionally, de novo motif discovery was performed using homer v4.8 (Salk Institute, San Diego, CA, USA) (Heinz et al., 2010). For all motif analyses, induced and repressed genes were analyzed independently.
For comparison of the RNA‐Seq results to published ChIP‐Seqs of PTF1a (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE86262) and MIST1 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE86289), previously called peaks were first downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo/). great v3.0.0 software (McLean et al., 2010) was then used, with a limitation on distal associations of up to 20 kb, to determine what genes were associated with ChIP‐Seq peaks. Because the ChIP‐Seq experiments were performed on mouse samples while the RNA‐Seqs were generated from human cells, genes with ChIP‐Seq peaks were first converted to human orthologs using Ensembl BioMart and then examined to determine overlaps between datasets.
2.4. Immunofluorescence microscopy
Pancreatic tissue sections were processed as previously described (Karki et al., 2015). Panc‐1 cells were fixed in 10% neutral buffered formalin, washed with PBS, and permeabilized with 0.3% Triton X‐100 in PBS. Cells were incubated for 1 h at RT with the following primary antibodies: CPA1 (1 : 100, cat. 131901; BD Biosciences, San Jose, CA, USA), PRSS2 (1:100, cat. sab1400226; Sigma), MIST1 (1 : 500, in‐house affinity‐purified antibody), MYC (1 : 500, cat. 9E10; Developmental Hybridoma Bank, Iowa City, IA, USA), and PTF1a (1 : 2000, gift from Chris Wright). Samples were incubated for 1 h with a 1 : 200 dilution of goat anti‐mouse or goat anti‐rabbit secondary antibody conjugated to biotin followed by a 5‐min incubation with Avidin–Alexa Fluor 488 (Cat. S11223, Invitrogen, Carlsbad, CA, USA) or Avidin–Alexa Fluor 594 (Cat. S11227, Invitrogen). Nuclei were counterstained using Vectashield hard mount with DAPI (Cat. H‐1500, Vector Labs, Burlingame, CA, USA). A full listing of antibodies used in this study is provided in Table S1.
2.5. Real‐time qPCR analysis
Total RNA was extracted using the RNeasy Mini Kit (Cat. 217004, Qiagen). cDNA was synthesized (1 μg per sample) using the iScript cDNA synthesis kit (Cat. 1708891, Bio‐Rad, Hercules, CA, USA). Quantitative RT‐PCR was performed on a Roche LightCycler 96 System with SYBR Green (Cat. 04913850001, Roche). Human pancreas RNA used for RT‐qPCR analysis was obtained from Life Technologies (Cat. AM7954, lot 1908572, Carlsbad, CA, USA). All amplicons were sequenced at the Purdue University Sequencing Core to verify correct targets. Gene expression was normalized to 18S rRNA. Primer sequences are provided in Table S2. Three to six biological replicates were performed with two technical replicates for each assay. Data were analyzed using the delta‐delta CT method to determine fold change differences.
2.6. Flow cytometry
Panc‐1 Tet‐One, Tet‐MIST1, and Tet‐PTF1a cells were dosed with 1 μg·mL−1 doxycycline and incubated at 5% CO2, 37 °C. The medium was changed after 48 h, and cells were harvested at 72 h. Cells were pelleted, washed, and incubated with blocking antibody CD16/32 (Cat. 2.4G2, BD Pharmingen, Franklin Lakes, NJ, USA), washed twice with PBS, and then incubated with FITC‐conjugated mouse anti‐human CD24 (1 : 100, Cat. 560992; BD Biosciences) and Cy5‐conjugated CD44 (1 : 100, Cat. 553135; BD Biosciences) for 30 min at 4 °C. Cells were analyzed for coexpression of CD24 and CD44 antigens using the BD Fortessa Cell Analyzer in the Purdue University Bindley Biosciences Flow Cytometry Core. Gates were set using a no‐antibody control. Assays were performed with a minimum of three biological replicates.
2.7. Immunoblotting
Cells were treated for 72 h with 1 μg·mL−1 doxycycline unless otherwise stated. Protein lysates were harvested using standard RIPA buffer containing sodium vanadate (Cat. 450253; Sigma) as well as protease inhibitors (Cat. COEDTAF‐RO; Roche) and phosphatase inhibitors (Cat. P5726 and P0044; Roche) and sonicated using a Vibra Cell Sonicator. Wild‐type pancreata and tumor tissues were harvested in RIPA as above, homogenized using a Tissue Tearor (model 985370) and sonicated using a Vibra Cell Sonicator. Membranes were incubated for 1 h at room temperature with primary antibodies to Myc (1 : 500, Cat. 9E10; Developmental Hybridoma Bank), α/β HSP90 (1 : 5000, Cat. SC‐7947; Santa Cruz, Santa Cruz, CA, USA), MIST1 (1 : 1000, in‐house affinity‐purified antibody), PTF1a (1 : 4000, gift from Chris Wright), and SOX9 (1 : 3000, Cat. ab5535; Millipore, Burlington, MA, USA) for 1 h. Immunoblots were washed and blotted with HRP‐conjugated anti‐mouse and anti‐rabbit secondary antibodies (1 : 5000, Cat. BA‐1000; Vector Labs). Peroxidase reactions were performed using ECL (Cat. 32106; Thermo Scientific, Waltham, MA, USA).
2.8. Soft agar assays
Soft agar colony formation assays were performed using 5000 cells in 0.4% agar on a base layer of 0.5% agar in DMEM. Cells were dosed immediately with doxycycline and were allowed to grow for 21 days. Colonies were stained using 0.005% crystal violet. The number of colonies was counted using imagej software (NIH, Bethesda, MD, USA) where clusters of cells 60 μm2 or greater were considered an individual colony. Three to six biological replicates were performed for each cell line. Protein was harvested from colonies as stated above, where the agar was first minced and softened via heat, and then, cells were filtered through a cell strainer to remove agar. Cells were lysed using standard RIPA buffer.
2.9. Orthotopic tumor assays
NSG (NOD.Cg‐Prkdc scid Il2rg tm1Wjl/SzJ) or NRG (NOD.Cg‐Rag1 tm1Mom Il2rg tm1Wjl/SzJ) mice 8–20 weeks old were injected with 5.0 × 105 cells in 20 μL PBS directly into the pancreas. Incisions were healed using sutures on the intraperitoneal cavity and staples to mend the skin wound. Mice were given acidified water and experimental mice were provided with gamma‐irradiated doxycycline chow replaced every 6 days (Cat. TD 08541, Envigo Laboratories, Madison, WI, USA). In some instances, mice were treated with gemcitabine (Cat. G6423; Sigma) where animals were IP injected with 25 mg·kg−1 every 3 days for 14 days. Each experimental group/time point consisted of 12–14 mice. All animal studies were conducted in strict compliance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the Purdue University IACUC guidelines (Protocol Approval 1110000037).
2.10. Cell viability assays
Cell viability was assessed using the MTT assay (Cat. 11465007001, Roche). Panc‐1 Tet‐One, Tet‐MIST1, and Tet‐PTF1a cells were seeded at 5000 cells per well in 100 μL hgDMEM in a 96‐well plate. Cells were allowed to adhere for 4 h and then dosed with 2 μg·mL−1 doxycycline in 100 μL to bring the final concentration to 1 μg·mL−1 doxycycline. Twenty‐four hours postseeding, cells were dosed with increasing concentrations of gemcitabine from 1.0 × 10−10 to 1.0 × 10−3 m. Seventy‐two hours post‐gemcitabine treatment, 10 μL MTT was added to each well for a 4‐h period. Formazan was then solubilized using 100 μL solubilization reagent. Formazan product was measured at 560 nm in a 96‐well plate reader. GraphPad Prism was used to plot the data and calculate the log‐dose vs. response and the IC50 of each group. Four biological replicates were used for each data point.
2.11. Statistical analysis
Statistical significance was determined using unpaired one‐tailed t‐tests. In instances where samples displayed unequal variance (based on an F‐test), Welch's correction was applied. All P‐values ≤ 0.05 were considered statistically significant. Error bars represent the standard error of the mean. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
2.12. Data accessibility
RNA‐Seq data have been deposited in the NCBI GEO database under accession number https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106290.
3. Results
3.1. MIST1 and PTF1a loci are silenced early in the development of pancreatic cancer
A hallmark of PDAC is a dramatic shift in pancreas tissue architecture and gene expression. During tumorigenesis in mouse models of PDAC, quiescent acinar cells that synthesize and secrete large quantities of digestive hydrolases convert into tumor epithelial cells that acquire a duct‐like phenotype and activate genes normally associated with the ductal epithelium (Murtaugh and Leach, 2007; Storz, 2017; Wang et al., 2009). For example, in the KPC mouse model of PDAC (induced by acinar‐specific Kras G12D and Trp53 R172H mutations), acinar cells transition into pancreatic intraepithelial neoplasia lesions (PanIN) that progress to PDAC (Habbe et al., 2008; Hruban et al., 2000; Murtaugh and Leach, 2007). During this transition, acinar‐specific genes (e.g., AMYLASE) become silenced, while ductal‐specific genes (e.g., KERATIN19) are rapidly induced (Fig. 1A) (Jonckheere et al., 2010; Karki et al., 2015). Driving these changes are key duct‐restricted transcription factors such as SOX9 (Fig. 1A) (Jacquemin et al., 2000; Kopp et al., 2012). Importantly, the silencing of two critical acinar‐restricted transcription factor genes – MIST1 and PTF1a – is a common feature of PDAC tumors (Adell et al., 2000; Zhu et al., 2007) (Fig. 1B). Indeed, silencing MIST1 and PTF1a loci is an early event as mouse ADM and PanIN lesions no longer accumulate MIST1 and PTF1a proteins when acinar cells transition toward a transformed phenotype (Fig. 1C). A similar loss of MIST1 and PTF1a proteins is observed in murine PDAC cell lines derived from KC and KPC mice (KC, KPC1, KPC2) as well as from human PDAC tumors (BxPC‐3, Panc‐1, MiaPaCa‐2) (Fig. 1D). Together, these results highlight a significant feature of acinar‐derived PDAC – silencing of the acinar transcription factor genes MIST1 and PTF1a.
Figure 1.

MIST1 and PTF1a expression in pancreatic cancer. (A) RT‐qPCR analysis of healthy adult murine pancreata (N) compared to murine KPC PDAC tumors (T). Amylase, a pancreatic acinar marker, is not expressed in PDAC tissue. Conversely, PDAC tissue has elevated transcript levels of the ductal genes K19 and SOX9 (n = 3 mice per data point). (B) MIST1 and PTF1a transcripts are lost in KPC PDAC tumors (n = 3 mice per data point). (C) Tissue from KPC tumors stained with AMYLASE and MIST1 or AMYLASE and PTF1a. MIST1 and PTF1a are localized in the nuclei of normal acinar cells, but there is no protein detected in acinar‐derived PanIN (white arrows) and PDAC tissues (scale bar = 50 μm). Normal pancreas tissue is from 5‐month‐old control mice, PanIN tissue is from 11‐week‐old mice (21 days post‐tamoxifen), and PDAC tumors are from 5‐month‐old mice (4.1 months post‐tamoxifen). All histology is representative from 3 to 5 mice per disease state. (D) Immunoblot analysis of healthy adult murine pancreata compared to KPC PDAC tissue and a panel of mouse and human pancreatic cancer cell lines. MIST1 and PTF1a proteins are detected in normal pancreas tissue but lost in PDAC and all pancreatic cancer cell lines. *P ≤ 0.05.
3.2. Generation of MIST1‐ and PTF1a‐induced Panc‐1 cell lines
Given the dramatic repression of MIST1 and PTF1a expression in PDAC, we asked whether these key acinar bHLH factors could retain functional transcriptional activity in the context of a transformed PDAC cell. If transcriptional activity could be restored, we speculated that induced MIST1 or PTF1a might drive PDAC cells toward their original acinar‐like state. To test this hypothesis, we took advantage of a bidirectional inducible vector (Tet‐One) in which MIST1 myc or PTF1a myc cDNA were inserted downstream of a tetracycline‐regulated promoter (Fig. S1A). Tet‐MIST1 and Tet‐PTF1a transgenes were introduced into human PDAC Panc‐1 cells, and transgene expression was activated by incubation of stable cell lines with doxycycline (Dox). As expected, MIST1 and PTF1a proteins were not detected in untreated cells (Fig. S1B,C). However, by 4 h post‐Dox addition, prominent MIST1 and PTF1a proteins were readily observed in the corresponding Tet‐MIST1 and Tet‐PTF1a lines, with maximal expression occurring by 24 h post‐Dox treatment (Fig. S1B,C). This level of expression remained stable in the presence of Dox for at least 21 days, the longest time examined (data not shown). Immunofluorescence analysis confirmed that greater than 85% of the Panc‐1 Tet‐MIST1 and Tet‐PTF1a cells expressed their respective proteins (Fig. S1B,C). Additionally, both factors exhibited transcriptional activity as each line activated specific target reporter genes (Fig. S1B,C).
3.3. MIST1 and PTF1a activate their respective acinar gene programs in PDAC cells
To verify that the Panc‐1 Tet‐MIST1 and Tet‐PTF1a lines respond to activation of these key transcription factors, we examined the response of known MIST1 and PTF1a gene targets in the Panc‐1 cells treated with Dox, where the cells failed to exhibit any overt morphological changes upon Dox treatment (Fig. 2A). MIST1 is responsible for organizing acinar cell structure and regulated exocytosis, and part of this function is to activate expression of individual RAB genes to facilitate organelle and zymogen granule movement (Hou et al., 2015; Tian et al., 2010). As shown in Fig. 2B, MIST1 readily induced expression of RAB3D and RAB26, whereas induced PTF1a had no effect on these genes. Likewise, MIST1 promoted expression of the MIST1 target genes associated with the UPR pathway, including SEC61b, SEC62, SEC63, PERK, and DNAJc1 (Fig. 2B) (Direnzo et al., 2012; Hess et al., 2011, 2016). Importantly, these data suggest that MIST1 can access target gene promoters in pancreatic cancer cells despite the inherent mutations, genomic instability, and altered epigenetic marks that are associated with these transformed cells (Deer et al., 2010; Fazio et al., 2017; Mehmood et al., 2014; Pin et al., 2015; Tan et al., 2009).
Figure 2.
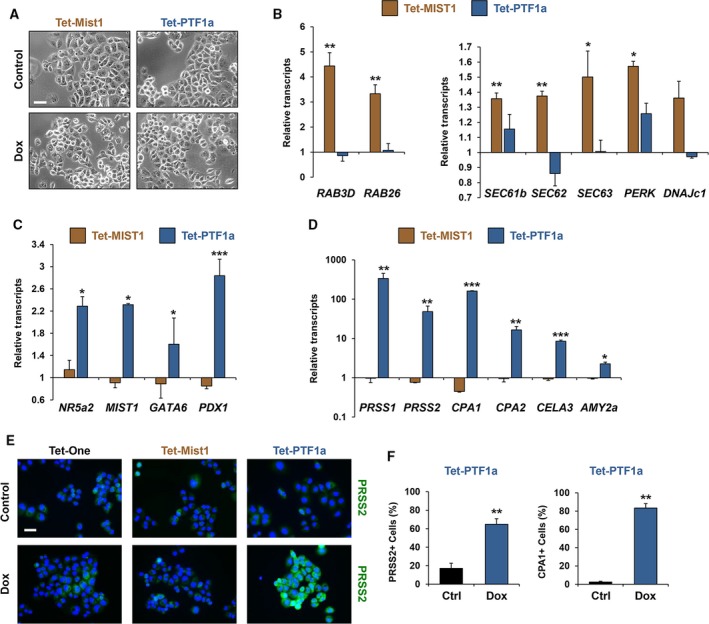
MIST1 and PTF1a activate known target genes in PDAC cells. (A) Phase contrast images of Panc‐1 Tet‐MIST1 and Tet‐PTF1a cells ± Dox (scale bar = 50 μm). (B) RT‐qPCR analysis of Panc‐1 Tet‐MIST1 and Tet‐PTF1a cells. Data are normalized to the same cell lines without doxycycline treatment and displayed as fold induction. MIST1 vesicle trafficking target genes RAB3D and RAB26 and ER chaperones SEC61b, SEC62, SEC63, PERK, and DNAJc1 are induced upon MIST1 expression. (C) Expression of acinar transcription factors NR5a2, MIST1, GATA6, and PDX1 is induced in PDAC cells upon PTF1a induction. (D) Pancreas digestive enzymes PRSS1, PRSS2, CPA1, CPA2, CELA3b, and AMY2a transcripts accumulate upon PTF1a expression. (E) Trypsinogen (PRSS2) staining of Panc‐1 Tet‐One, Tet‐MIST1, and Tet‐PTF1a cells 72 h post‐doxycycline treatment (scale bar = 50 μm). (F) Percentage of Tet‐PTF1a cells expressing PRSS2 or carboxypeptidase (CPA1) upon doxycycline treatment. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
A similar response was observed in the Panc‐1 Tet‐PTF1a cells. PTF1a is a master regulator of acinar cell development and as such orchestrates expression of key acinar transcription factors including NR5a2, RBPJL, GATA6, and PDX1 (Jiang et al., 2016; Krah et al., 2015; Thompson et al., 2012; Topno et al., 2017). As predicted, induced PTF1a in Panc‐1 cells produced significant increases in the downstream transcription factors NR5a2, MIST1, GATA6, and PDX1 (Fig. 2C). In contrast, induced MIST1 expression failed to influence expression of this gene set. We next examined downstream hydrolase genes to determine whether they were similarly influenced by induced PTF1a expression. Although MIST1 failed to induce expression of this gene set, PTF1a readily activated expression of a large number of digestive enzyme genes including trypsinogens (PRSS1, PRSS2), carboxypeptidases (CPA1, CPA2), elastase (CELA3), and amylase (AMY2a) (Fig. 2D). Importantly, the response to induced PTF1a was also observed when cells were stained for PRSS2 and CPA1 protein, where up to 83% of the cells accumulated digestive enzymes (Fig. 2E,F). In contrast, control Tet‐One and Tet‐MIST1 cells failed to show evidence of PRSS2 and CPA1 protein output (Fig. 2E). In all cases, Tet‐MIST1 and Tet‐PTF1a and their respective target genes responded to Dox in a dose‐dependent fashion with maximal levels of products obtained with 1 μg·mL−1 Dox treatment (Fig. S1D,E).
Given the induced expression of MIST1 and PTF1a gene targets in human Panc‐1 cells, we next asked how Panc‐1 expression levels compared to gene transcript levels in human pancreas samples. Several RT‐qPCR analyses were performed, comparing Panc‐1 Tet‐MIST1 ± Dox, Panc‐1 Tet‐PTF1a ± Dox, and human pancreas RNA. As shown in Fig. S2A,B, most of the induced genes in Panc‐1 Tet‐MIST1 and Tet‐PTF1a cells were expressed to 20–40% levels of human pancreatic acinar cells, a remarkably robust response considering that the Panc‐1 line is a well‐established PDAC model. The only exception was that Tet‐PTF1a cells expressed significantly lower levels of digestive hydrolase genes (PRSS1, CPA1) when compared to intact pancreata. This is an interesting finding which suggests that although PTF1a can activate aspects of the PTF1a transcriptome in human PDAC cells, there remain limitations to the robustness of the responses. Indeed, it is possible that PTF1a primarily activates pancreas progenitor genes (e.g., NR5a2, PDX1) but not classic differentiation genes (e.g., PRSS1) to the maximal levels found in patient pancreata.
We next examined whether the ability of PDAC cells to respond to Tet‐MIST1 and Tet‐PTF1a was unique and/or restricted to only Panc‐1 cells. For these studies, we generated new Tet‐MIST1 and Tet‐PTF1a lines using human BxPC‐3 PDAC cells and mouse KPC PDAC cells that were derived by activating mutations in adult acinar cells. As predicted, BxPC‐3 and KPC cells activated expression of the corresponding MIST1 and PTF1a target genes only in the presence of Dox (Fig. S3A,B) in an analogous fashion as shown for Panc‐1 cells. Thus, expression of MIST1 and PTF1a leads to universal cell responses. Together, these data reveal that PTF1a and MIST1 retain functional capacity in pancreatic cancer cells and can activate their respective transcriptional networks, providing evidence that genes involved in acinar development can be reactivated in human and mouse PDAC cells.
3.4. MIST1 and PTF1a promote similar but distinct acinar‐associated pathways in PDAC cells
MIST1 and PTF1a retain functional activity in the context of PDAC cells as they each promote expression of their known pancreatic acinar gene networks. To obtain a broader profile of the effects of induced expression of MIST1 and PTF1a in PDAC, RNA‐Seq studies were conducted on Panc‐1 Tet‐One, Tet‐MIST1, and Tet‐PTF1a cells ±Dox treatment (Fig. 3A). As predicted, control Tet‐One cells exhibited minor responses, displaying only nine DEGs when transcript profiles were compared in ±Dox‐treated groups (Fig. 3C). In contrast, 874 DEGs were identified in the Tet‐MIST1 line (Fig. 3B,C). A similar but more robust gene expression response was observed with Tet‐PTF1a cells where 6688 DEGs were identified (Fig. 3B,C). The sevenfold difference in DEGs between MIST1 and PTF1a cells is likely due to the functional nature of each transcription factor, where PTF1a is a master regulator for pancreatic acinar development while MIST1 is considered a scaling factor, augmenting specific transcription programs (Hess et al., 2016; Jiang et al., 2016; Kondratyeva et al., 2017; Mills and Taghert, 2012; Tian et al., 2010). Nonetheless, there was considerable overlap in DEGs regulated by both transcription factors, with 167 target genes induced by PTF1a and MIST1, representing 29% of the upregulated MIST1 targets (Fig. 3D). Likewise, 33% of MIST1 repressed targets were shared with PTF1a (Fig. 3D).
Figure 3.
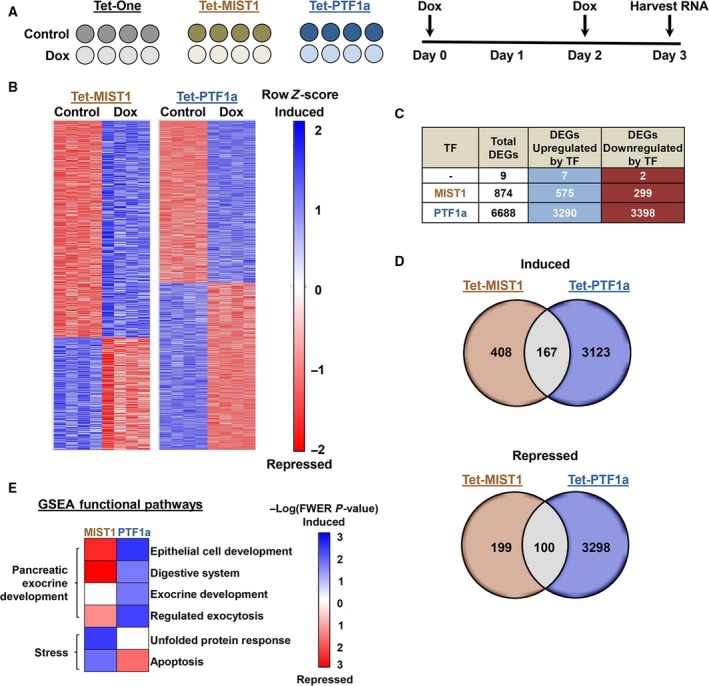
RNA sequencing of Tet‐MIST1 and Tet‐PTF1a. (A) Experimental strategy and timeline for cells subjected to RNA‐Seq. (B) Representative heatmaps of four control samples compared to four doxycycline‐treated samples for the Tet‐MIST1 and Tet‐PTF1a cells. Heatmaps depict the comparison of log2‐transformed read counts for each gene. (C) A total of 874 genes are differentially expressed in the Tet‐MIST1 cell line, while 6688 genes were differentially expressed in Tet‐PTF1a cells. (D) Venn diagrams comparing induced and repressed genes in Tet‐MIST1 vs. Tet‐PTF1a samples. (E) Summary heatmap displaying −Log (FWER P‐value) for biological and molecular pathways identified by GSEA.
As transcriptional activators, PTF1a and MIST1 likely act directly on a subset of the induced DEGs which in turn produce additional downstream changes in gene expression. Interestingly, PTF1a and MIST1 motifs were more significantly enriched in the promoter regions of induced DEGs compared to repressed genes (Fig. S4A) although the biological relevance of these sites, as well as motifs discovered de novo (Doc. S1), will require further analysis. Nonetheless, to gain additional insight into possible direct PTF1a‐ and MIST1‐regulated genes within our RNA‐Seq data, DEGs were compared to previously published PTF1a ChIP‐Seq (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE86262) and MIST1 ChIP‐Seq (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE86289) mouse studies (Hoang et al., 2016; Jiang et al., 2016a). As predicted, a similar proportion of induced and repressed DEGs were found to have ChIP‐Seq peaks in near proximity with several well‐characterized PTF1a‐ and MIST1‐regulated genes highlighted in the overlap between the ChIP‐Seq and induced DEGs (Fig. S4B) (Ahnfelt‐Rønne et al., 2012; Direnzo et al., 2012; Hoang et al., 2016; Jiang et al., 2016a; Lo et al., 2017; Wiebe et al., 2007). This discovery further shows that PTF1a and MIST1 maintain regulation of their transcriptional networks, even in the context of PDAC. Indeed, KEGG pathway analyses showed enrichment for ER protein processing and protein export in the PTF1a‐ and MIST1‐induced DEGs, respectively, including several known direct target genes in both categories (Fig. S5A–D).
Gene Set Enrichment Analysis, which is capable of analyzing each dataset as a whole, was also performed taking into account the directionality of DEG expression. As expected, a wide range of cellular processes involved in pancreatic development and function were identified. For example, MIST1 promoted expression of genes involved in stress responses, including key genes of the UPR and apoptosis pathways (Fig. 3E), which agrees with previous studies showing that MIST1 functions to alleviate UPR stress by increasing expression of chaperones and other UPR genes (Hess et al., 2011, 2016; Jiang et al., 2016). Surprisingly, MIST1 expression in Panc‐1 cells negatively influenced genes involved in exocrine development (Fig. 3E), suggesting that MIST1 may function differently in PDAC cells compared to embryonic and adult acinar cells (Direnzo et al., 2012; Jiang et al., 2016; Karki et al., 2015; Pin et al., 2001). In contrast, PTF1a exhibited a more potent transcriptional response with a number of enriched pathways being induced that are involved in pancreatic exocrine development, including ‘Digestive System’, ‘Epithelial Cell Development’, and ‘Exocrine Development’ (Fig. 3E).
3.5. PDAC‐associated pathways are negatively influenced by MIST1 and PTF1a expression
Ingenuity pathway analysis identified ‘cancer’ as the top disease pathway altered in both Tet‐MIST1 and Tet‐PTF1a cell lines, followed by ‘organismal injury and abnormalities’ and ‘gastrointestinal disease’ (Fig. 4A, Doc. s2). In all three cases, PTF1a exhibited a greater footprint within the pathways than MIST1, again demonstrating the wider transcriptional activity and gene expression responses of this bHLH factor. Identification of ‘cancer’ pathways suggested that tumor‐associated properties may be decreased when MIST1 and PTF1a are induced in PDAC cells. GSEA revealed that genes which are classically repressed in PDAC were significantly induced upon bHLH induction (Fig. 4B). Similarly, PTF1a expression negatively influenced genes associated with a ‘PDAC signature’ (Fig. 4B). Interestingly, there was little overlap in DEGs in the ‘repressed in PDAC’ signature when comparing MIST1 and PTF1a, suggesting that different MIST1 and PTF1a target genes are involved in affecting PDAC properties (Fig. 4C). Consistent with this assumption, we observed several antitumorigenic trends in PTF1a cells which were not seen with MIST1, including increased ‘stem cell differentiation’ and decreased ‘PDAC stem cell markers’ and ‘multiple drug resistance’ pathways (Fig. 4D).
Figure 4.
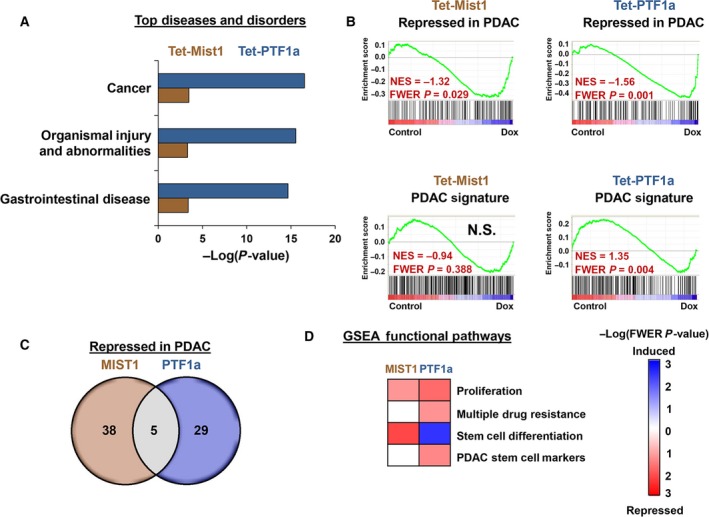
Tet‐MIST1 and Tet‐PTF1a gene expression analysis. (A) Top three disease pathways identified by IPA. (B) GSEA of genes associated with pancreatic cancer using the ‘Gruetzman Pancreatic Cancer Up’ list (PDAC Signature) and genes classically repressed in PDAC (‘Gruetzman Pancreatic Cancer Down’ list). (C) Venn diagram comparison of genes differentially regulated in ‘repressed in PDAC’. (D) Heatmaps of disease‐associated pathways from GSEA altered upon MIST1 or PTF1a induction. N.S., not significant.
3.6. PTF1a induction reduces cancer stem cell characteristics
GSEA suggested that induced PTF1a activity promotes decreased CSC properties. To examine this further, we curated a list of eight (CD24, CD44, EPCAM, ABCg2, ALDH1a3, MET, DCLK1, ALDH2) pancreatic stem cell markers and examined their overall expression in our RNA‐Seq dataset. Interestingly, PTF1a induction significantly reduced seven of the eight CSC markers, with the only exception being DCLK1 (Fig. 5A). Consistent with these findings, RT‐qPCR analysis confirmed that CD24, CD44, CD44v6, ALDH1a3, and EPCAM transcripts were all significantly decreased upon PTF1a induction (Fig. 5B). In contrast, MIST1 induction had little to no effect on CSC marker expression (data not shown). Flow cytometry corroborated our initial findings, revealing ~ 70% reduction in CD24+/CD44+ cells upon PTF1a expression (Fig. 5C,D). The decrease in CD24+/CD44+ cells also translated into a substantial reduction in soft agar colony formation when Tet‐PTF1a cells were induced to express PTF1a (Fig. 6A,B). As predicted, the reduced colony formation was reflected in decreased CD44 and EPCAM protein levels (Fig. 6C). Importantly, Dox treatment had no effect on colony formation in the Panc‐1 Tet‐One cells (Fig. 6A,B) or with parental Panc‐1 cells (data not shown). Finally, to assess the ability of CSC properties to support tumor initiation and growth in vivo, we performed pancreatic orthotopic tumor assays. As shown in Fig. 6D, a 30% reduction in tumor growth was consistently observed when mice were maintained on a Dox diet. Collectively, these data provide evidence that induced expression of PTF1a reduces pancreatic CSC properties and overall PDAC tumor growth in orthotopic assays.
Figure 5.
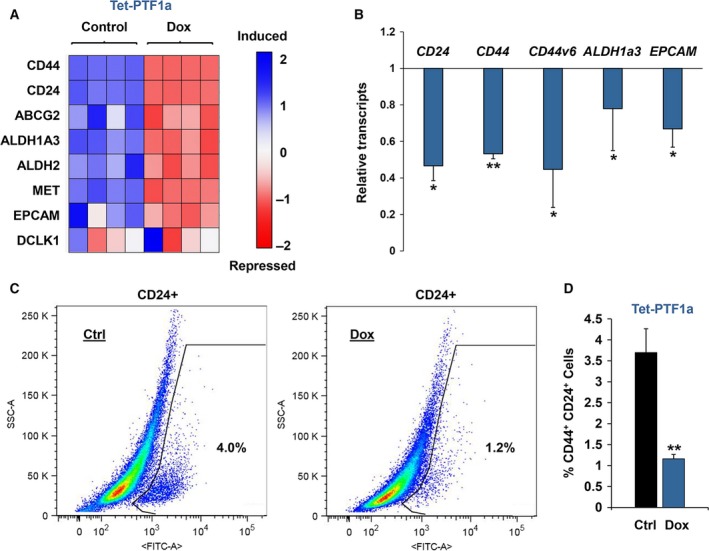
PTF1a expression decreases CSC properties. (A) Heatmap of a curated list of pancreatic CSC markers in Tet‐PTF1a cells ± Dox. (B) RT‐qPCR of selected pancreatic CSC markers. (C) Flow cytometry of CD24+ Tet‐PTF1a cells ± Dox. (D) CD44+/CD24+ cells quantified from Tet‐PTF1a cells ± Dox. *P ≤ 0.05, **P ≤ 0.01.
Figure 6.
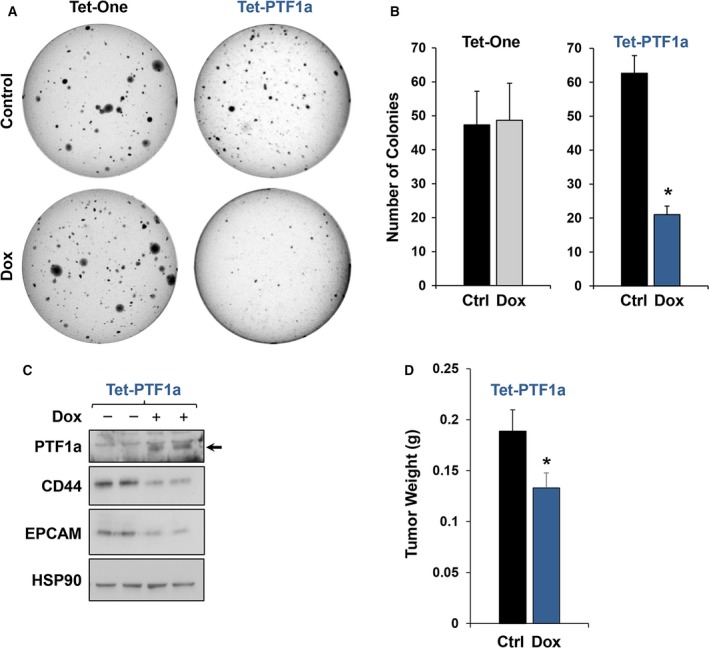
PTF1a expression decreases soft agar colony and orthotopic pancreatic cancer tumor growth. (A,B) Soft agar colony formation assay of Tet‐One and Tet‐PTF1a cells ± Dox. (C) Immunoblot analysis of CD44 and EPCAM from soft agar colonies of Tet‐PTF1a cells ± Dox. CD44 and EPCAM show marked reductions in the PTF1a expressing colonies. (D) Orthotopic xenograft assays testing Panc‐1 Tet‐PTF1a tumor growth in mice subjected to ± Dox chow. *P ≤ 0.05.
3.7. PTF1a sensitizes PDAC cells to gemcitabine treatment
A key feature of CSCs is the ability to sustain multidrug resistance. Given that PTF1a expression leads to a decreased stem cell population, we asked whether PTF1a activity also altered CSC characteristics. As a first step, we screened expression profiles of ATP‐binding cassette (ABC) efflux transporters known to be critical to clinical PDAC drug resistance because they pump gemcitabine out of cells (Dean, 2009; Ding et al., 2010; Moitra, 2015; Tanaka et al., 2011). Notably, five transporter genes showed decreased transcript levels following PTF1a activity, while expression of the remaining three genes was either undetected (ABCB1) or showed a similar (but statistically insignificant) decrease in expression upon PTF1a induction (Fig. 7A). As predicted, Tet‐MIST1 and Tet‐One control cells did not alter expression of any of the ABC efflux transporters (data not shown). Interestingly, the gemcitabine importer ENT1 also exhibited a significant change in gene expression upon PTF1a induction, but in this case expression was elevated (Fig. 7B). Taken together, these results suggest that the steady‐state level of gemcitabine in the cytosol may be altered in the presence of PTF1a. Indeed, direct testing of gemcitabine on the Panc‐1 Tet‐PTF1a cells revealed an increased cell death efficacy in cells expressing PTF1a when compared to control cells (Fig. 7C), with an order of magnitude increase in sensitivity to gemcitabine upon PTF1a induction (Fig. 7D) and a threefold lower IC50 value for these cells when treated with Dox (Fig. 7E). As predicted, cleaved caspase‐3 was also elevated in the PTF1a‐expressing cells when treated with gemcitabine compared to control cells treated with gemcitabine alone (Fig. 7F).
Figure 7.

PTF1a sensitizes Panc‐1 cells to gemcitabine treatment. (A) RT‐qPCR of gemcitabine efflux transporters for Tet‐PTF1a cells ± Dox. (B) RT‐qPCR of gemcitabine influx transporters ENT1 and CNT1. (C) IC 50 kill curve of Tet‐inducible cell lines using a gemcitabine gradient for 72 h. (D,E) Log10 fold change of IC 50 when cells are treated with doxycycline to induce PTF1a. (F) Immunoblot analysis of Tet‐inducible cells using 10 μm gemcitabine for 72 h. Cleaved caspase‐3 is elevated upon PTF1a induction, while Tet‐One cells display no change. *P ≤ 0.05, **P ≤ 0.01. ND, not detected.
As PTF1a expression resulted in increased sensitivity to gemcitabine, we next tested the ability of induced PTF1a to increase tumor sensitivity to gemcitabine in vivo. Panc‐1 Tet‐PTF1a cells were orthotopically injected into immunocompromised mice and allowed to grow for 27 days, at which time the mice were randomly assigned to groups receiving either no treatment, Dox only, gemcitabine only, or Dox and gemcitabine together. Gemcitabine was administered every 3 days for the following 14‐day time span (Fig. 8A). This strategy permitted an assessment of PTF1a's ability to sensitize an already established PDAC tumor. As expected, PTF1a expression was observed solely in the +Dox groups (Fig. 8B,C). Similarly, PTF1a target genes (NR5a2, PRSS1, CPA1) exhibited the predicted induction in the +Dox samples (Fig. 8C). Despite H&E sections failing to reveal any distinct morphological differences between groups, PRSS2 protein accumulation was readily detected and restricted to samples expressing PTF1a (Fig. 8D). Analysis of control vs. +Dox groups revealed that inducing PTF1a expression in established tumors produced only minor and statistically insignificant decreases in tumor burden (Fig. 8D,E). Treatment with gemcitabine alone resulted in a 38% decrease in tumor burden in cells not expressing PTF1a. However, when established tumors were provided both Dox (to activate PTF1a) and gemcitabine, total tumor burden decreased by 55%, a 1.4‐fold enhancement in gemcitabine sensitivity (Fig. 8D,E). These results support the concept that induced PTF1a sensitizes PDAC cells to gemcitabine treatment.
Figure 8.
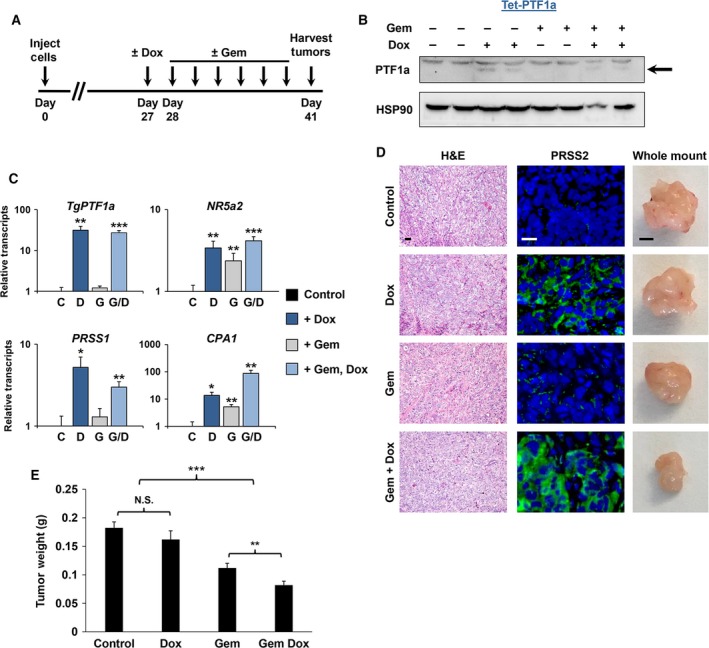
PTF1a sensitizes PDAC cells to gemcitabine in vivo. (A) Experimental timeline. (B) Immunoblot of PTF1a expression from excised tumors in mice treated ± Dox and ± Gem. (C) RT‐qPCR of tumor samples from the indicated groups for the Tet‐induced PTF1a transgene (TgPTF1a) as well as for endogenous NR5a2,PRSS1, and CPA1 genes. (D) Hematoxylin and eosin (H&E) staining, PRSS2 immunofluorescence (green), and whole‐mount images of tumors isolated from mice treated ± Dox and ± Gem (scale bars = 50 μm for sections; 2.5 mm for whole‐mount images). (E) Tumor weights obtained from Panc‐1 Tet‐PTF1a cells ± Dox and ± Gem (n = 12–14 mice per experimental group). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, N.S., not significant.
4. Discussion
Pancreatic cancer is a lethal disease that has failed to be therapeutically controlled, despite enormous research and clinical efforts. Recent studies utilizing mouse models have revealed that adult duct cells expressing Hnf1b or Sox9 can serve as the cell of origin to PDAC if the cells are subjected to activating Kras mutations and inactivating mutations in TP53 (deletion or Tp53 R172H) (Bailey et al., 2016a; Lee et al., 2018). Another PDAC progression model suggests that the disease can originate from acinar cells harboring an oncogenic Kras mutation (Collins et al., 2012, 2014; Habbe et al., 2008; Kopp et al., 2012; Krah et al., 2015; Shi et al., 2009, 2013). Interestingly, despite both duct and acinar cells involved in disease initiation, the cell of origin can differentially affect tumor development schemes (Bailey et al., 2016a; Lee et al., 2018). Nonetheless, the well‐described acinar → PDAC progression model provides opportunities to develop new strategies to interrupt key processes along the entire disease spectrum and reverse the malignant properties of PDAC cells. Indeed, work from the Pasca di Magliano Lab (Collins et al., 2012, 2014) has shown that precancerous PDAC lesions exhibit plasticity and can be reverted back to healthy pancreatic acinar tissue upon loss of oncogenic KRAS signaling. These studies have provided a key insight into possibilities for reprogramming PDAC cells.
A complementary approach to reversing KRAS signaling in PDAC cells is to manipulate transcriptional networks to drive cells toward a nontumorigenic phenotype. Acinar cells have been particularly amenable to reprogramming as these cells convert to duct‐like cells during ADM and PDAC development through a SOX9‐dependent network (Kopp et al., 2012). This phenomenon is driven in part by upregulation of ERBB2 expression and elevated ERBB signaling (Grimont et al., 2014). Additionally, acinar cells convert to insulin‐expressing β‐cells when the transcription factors NGN3, PDX1, and MAFA are exogenously expressed (Zhou et al., 2008). A similar reprogramming phenomenon has been observed in PDAC cells engineered to express elevated levels of the bHLH factor E47 (Kim et al., 2015). Increased E47 shifts the bHLH:ID axis in these cells, ultimately activating acinar cell properties and decreasing tumorigenic characteristics, providing one of the first reports that PDAC cells are susceptible to alterations in their transcription networks. In this study, we tested whether other bHLH transcription factors could be instrumental in driving PDAC cells toward their acinar origin and subsequently decrease cancer‐associated properties. We chose to focus on two key pancreas bHLH factors (MIST1 and PTF1a) because (i) MIST1 and PTF1a are silenced in KRAS‐expressing acinar cells, (ii) their silencing promotes the progression of precancerous lesions (Krah et al., 2015; Shi et al., 2009), and (iii) forced expression of MIST1 in the context of KRAS signaling mitigates the effects of silencing the endogenous alleles on PDAC formation (Shi et al., 2013), suggesting that these bHLH factors function as tumor suppressors. Notably, both PTF1a and MIST1 are essential for the acinar gene network where PTF1a drives an acinar transcription system and expression of digestive enzyme genes while MIST1 regulates genes essential to acinar cell–cell communication, calcium flux, and acinar cell secretion (Beres et al., 2006; Direnzo et al., 2012; Garside et al., 2010; Hess et al., 2016; Jiang et al., 2016; Krah et al., 2015; Pin et al., 2001)
An important outcome from our study was that KRASG12D, p53R273H, CDKN2A/p16KO PDAC cells (Panc‐1) retain the capacity to activate the acinar gene program following forced expression of PTF1a or MIST1. Indeed, PDAC cells induced to express PTF1a or MIST1 promoted acinar‐specific gene expression. For example, induced expression of PTF1a activated a previously silent acinar transcription network that included triggering expression of the key transcription factor genes NR5a2, GATA6, PDX1, and MIST1, which subsequently led to elevated expression of acinar‐specific digestive hydrolase genes PRSS1, PRSS2, CPA1, CPA2, CELA3b, and AMY2a. Interestingly, although PTF1a activity in human PDAC cells induced expression of acinar transcription factors to 20–40% the levels found in normal, intact human pancreata, PTF1a was inefficient in inducing comparable levels of digestive enzyme genes (e.g., PRSS1). Many of the PTF1a‐induced transcription factors (NR5a2, GATA6, PDX1) are known to mark and be critical to pancreatic progenitor cells (Zhou et al., 2008). Thus, it remains possible that PTF1a primarily activates an early developmental program in PDAC cells which may help the cells achieve a more mature acinar phenotype. Similar results were obtained with induced MIST1 where a large number of MIST1 target genes, including RAB3D, RAB26, SEC61b, SEC62, and SEC63, were expressed in PDAC cells. Consistent with inducing a differentiated phenotype, MIST1 and PTF1a also activated genes that are normally repressed in cancer, including NR5a2, SGK1, XBP1, SEC61b, DMPK, RBP1, and CLDN10 (Grützmann et al., 2005). However, in all cases PTF1a was more potent in decreasing cancer‐associated gene expression when compared to MIST1‐induced profiles, suggesting that PTF1a has a greater global impact in driving cells toward a quiescent, acinar‐specific phenotype. These findings are consistent with a number of studies showing that PTF1a functions as a transcriptional master regulator while MIST1 serves as a scaling factor, primarily augmenting gene expression patterns (Capoccia et al., 2013; Hess et al., 2016; Jiang et al., 2016; Krapp et al., 1996, 1998; Mills and Taghert, 2012). Thus, progenitor cell and acinar genes within PDAC cells remained accessible to activation by PTF1a and MIST1, revealing that PDAC cells are not irreversibly locked into an undifferentiated cancer cell state.
The phenomenon of MIST1 and PTF1a to reprogram the PDAC transcriptome was not restricted to Panc‐1 cells as similar results were obtained when human BxPC‐3 PDAC cells and mouse KPC PDAC cells were engineered to express induced MIST1 and PTF1a. This was an intriguing finding given that each cell line harbors unique oncogene and tumor suppressor gene mutations. Additionally, although Panc‐1 and BxPC‐3 cells are thought to be derived from duct cells, the KPC line was derived from acinar cells. Our results are particularly interesting given the recent discovery that human PDAC tumors can be classified into four different molecular subtypes (squamous; pancreatic progenitor; immunogenic; and aberrantly differentiated endocrine exocrine tumors) (Bailey et al., 2016b). Thus, despite potential differences in the molecular evolution and signature of PDAC subtypes, strategies to alter transcription networks may still be effective in most PDAC examples.
One of the more striking phenotypes associated with PTF1a expression in PDAC cells was the consistent decrease in CSCs and CSC properties. It is well established that pancreatic CSCs initiate and propagate PDAC tumor formation and are a major contributor to drug and radiotherapy resistance (Li et al., 2007; Vaz et al., 2014; Westphalen et al., 2016). Interestingly, PTF1a expression produced a marked reduction in expression of a number of CSC genes including transcript decreases in CD24, CD44, CD44v6, ALDH1a3, and EPCAM. Indeed, PTF1a induction generated a threefold reduction in CD44+/CD24+ CSCs as well as a threefold reduction in soft agar colony formation. The decrease in CSC numbers was also associated with 30% reductions in orthotopic tumor burden when mice injected with Tet‐PTF1a Panc‐1 cells were provided Dox. Thus, activating/inducing PTF1a in PDAC tumor cells may provide a new therapeutic venue to decrease PDAC progression by lowering CSC properties.
Given that CSCs are resistant to therapeutics, we also examined whether PTF1a expression sensitized cells to gemcitabine treatment, a standard therapy for patients with pancreatic cancer (Heinemann et al., 2000; Kumar Jena et al., 2012). PTF1a expression produced marked reductions in expression of a large family of ABC efflux transporters, suggesting that gemcitabine cannot be actively pumped out of PDAC cells that express PTF1a. Indeed, gemcitabine flux was significantly altered upon activation of PTF1a, resulting in a 17‐fold enhancement in gemcitabine efficacy when tested in vitro. A similar increase in efficacy was observed in pancreatic orthotopic tumor assays where PDAC cells expressing PTF1a exhibited significantly greater sensitivity to gemcitabine compared to controls. To our knowledge, these are the first studies to show that rescuing PTF1a expression in the context in PDAC cells provides a reduction in CSCs and increased sensitivity to gemcitabine treatment.
The ability to reprogram cells with key transcription factors has evolved rapidly since the pioneering studies of Takahashi and Yamanaka in generating iPS cells from adult somatic cells (Takahashi and Yamanaka, 2006). And although strategies to convert iPS cells into many different differentiated cell types are now commonplace (Shi et al., 2017), there has been limited progress in shifting transcription networks to restore normal cell properties to cancer cells. Many of the remaining obstacles concern reversing multiple mutations that exist in a heterogeneous tumor cell population (Bailey et al., 2016b). Nonetheless, we now show that (i) loci normally silent in PDAC cells can be reactivated when key transcription factors are present, (ii) activation of acinar gene expression profiles can have a profound effect on CSC properties, and (iii) induction of PTF1a leads to decreases in ABC efflux transporters, ultimately producing higher efficacy potential of antitumor therapeutics. Future challenges will be to develop strategies to activate expression of master regulator genes directly in tumor cells. Because PTF1a and MIST1 loci are hypermethylated in PDAC cells (B. Marshall, B. Jakubison & S. F. Konieczny, unpublished data), our next direction will be to activate endogenous MIST1/PTF1a gene expression by reversing the methylated status of these loci. Technologies such as modified CRISPR targeting (e.g., dCas9‐SunTag; CRISPR/Cas9 TGA) to demethylate CpG islands at the MIST1 and PTF1a regulatory regions could have a significant impact on PDAC and progenitor and acinar cell transcription networks, possibly leading to reduced aggressiveness of the disease (Liao et al., 2017; Morita et al., 2016). Additional studies investigating expression of other acinar transcription factors such as GATA6 and NR5a2 will also be of importance as each is essential to the acinar differentiation program and each suppresses KRAS‐mediated ADM/preneoplastic transformation (von Figura et al., 2014; Flandez et al., 2014; Martinelli et al., 2016). These and other efforts will provide additional insights regarding the early stages of PDAC development and facilitate the advancement of novel strategies to successfully treat the disease.
5. Conclusions
In this study, we asked whether induced expression of acinar transcription factors in PDAC tumor cells could partially activate the acinar gene program and at the same time reduce tumor cell properties. Both MIST and PTF1a induced target genes that are normally associated with differentiated acinar cells. Additionally, induction of PTF1a in PDAC influenced cancer‐associated properties, leading to decreases in cell proliferation, CSC numbers, and repression of ABC efflux transporters resulting in heightened sensitivity to gemcitabine. These results suggest that manipulation of the PDAC transcriptome offers promising opportunities to develop novel therapeutic intervention strategies for this deadly disease.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
BLJ, PGS, SM, and YY designed, performed, and analyzed experiments; HG and YL assisted with bioinformatics; KS and PI‐A provided cellular reagents and advice; and BLJ, PGS, and SFK wrote the manuscript.
Supporting information
Doc. S1. Differentially expressed gene promoter de novo motif analysis.
doc . S2. IPA cancer‐associated molecules.
Fig. S1. Generation of the Tet‐inducible cell lines.
Fig. S2. Comparison of Tet‐MIST1‐ and Tet‐PTF1a‐induced targets with human pancreas samples.
Fig. S3. BxPC‐3 and KPC cells engineered to express Tet‐MIST1 and Tet‐PTF1a.
Fig. S4. PTF1a and MIST1 differentially expressed promoter motif enrichment and ChIP‐Seq intersect.
Fig. S5. KEGG pathway analysis of the Tet‐PTF1a and Tet‐MIST1 RNA‐Seq.
Table S1. Antibodies.
Table S2. Mouse and human RT‐qPCR primers.
Acknowledgments
We thank Christopher Wright for providing the PTF1a antibody, Raymond MacDonald for providing the pGL3 RBPJ‐L reporter plasmid, and Yan Sun for generating the TA‐E box MIST1 reporter plasmid. We also thank David Hess for his consultation, technical expertise, and development of the project. This work was supported by grants to SFK (NIH DK55489, NIH CA124586) and the Indiana Clinical and Translational Sciences Institute (ICTSI 22‐808‐25). The authors also gratefully acknowledge support from the Purdue University Center for Cancer Research, NIH Grant P30 CA023168.
References
- Adell T, Gómez‐Cuadrado A, Skoudy A, Pettengill OS, Longnecker DS, Real FX (2000) Role of the basic helix‐loop‐helix transcription factor p48 in the differentiation phenotype of exocrine pancreas cancer cells. Cell Growth Differ 11, 137–147. [PubMed] [Google Scholar]
- Ahnfelt‐Rønne J, Jørgensen MC, Klinck R, Jensen JN, Füchtbauer E‐M, Deering T, MacDonald RJ, Wright CVE, Madsen OD, Serup P (2012) Ptf1a‐mediated control of Dll1 reveals an alternative to the lateral inhibition mechanism. Development 139, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Cancer Society (2017). Key Statistics for Pancreatic Cancer. American Cancer Society, Atlanta, GA. [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC et al (2016b) Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Hendley AM, Lafaro KJ, Pruski MA, Jones NC, Alsina J, Younes M, Maitra A, McAllister F, Iacobuzio‐Donahue CA et al (2016a) p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene 35, 4282–4288. [DOI] [PubMed] [Google Scholar]
- Beres TM, Masui T, Swift GH, Shi L, Henke RM and MacDonald RJ (2006) PTF1 is an organ‐specific and Notch‐independent basic helix‐loop‐helix complex containing the mammalian Suppressor of Hairless (RBP‐J) or its paralogue, RBP‐L. Mol Cell Biol 26, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant KL, Mancias JD, Kimmelman AC, Der CJ (2014) KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci 39, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos ML, Sánchez‐Arévalo Lobo VJ, Rodolosse A, Gottardi CJ, Mafficini A, Beghelli S, Scardoni M, Bassi C, Scarpa A and Real FX (2013) ICAT is a novel Ptf1a interactor that regulates pancreatic acinar differentiation and displays altered expression in tumours. Biochem J 451, 395–405. [DOI] [PubMed] [Google Scholar]
- Capoccia BJ, Jin RU, Kong YY, Peek RMJ, Fassan M, Rugge M and Mills JC (2013) The ubiquitin ligase mindbomb 1 coordinates gastrointestinal secretory cell maturation. J Clin Invest 123, 1475–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockell M, Stevenson BJ, Strubin M, Hagenbüchle O and Wellauer PK (1989) Identification of a cell‐specific DNA‐binding activity that interacts with a transcriptional activator of genes expressed in the acinar pancreas. Mol Cell Biol 9, 2464–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Bednar F, Zhang Y, Brisset J, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca M (2012) Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 122, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Yan W, Sebolt‐Leopold JS, Pasca di Magliano M (2014) MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology 146, 822–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul J‐L, Gourgou‐Bourgade S, de la Fouchardière C et al (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364, 1817–1825. [DOI] [PubMed] [Google Scholar]
- Dalla Pozza E, Dando I, Biondani G, Brandi J, Costanzo C, Zoratti E, Fassan M, Boschi F, Melisi D, Cecconi D et al (2015) Pancreatic ductal adenocarcinoma cell lines display a plastic ability to bi‐directionally convert into cancer stem cells. Int J Oncol 46, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Day JD, Digiuseppe JA, Yeo C, Lai‐Goldman M, Anderson SM, Goodman SN, Kern SE and Hruban RH (1996) Immunohistochemical evaluation of HER‐2/neu expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasms. Hum Pathol 27, 119–124. [DOI] [PubMed] [Google Scholar]
- Dean M (2009) ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia 14, 3–9. [DOI] [PubMed] [Google Scholar]
- Deer EL, González‐Hernández J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ (2010) Phenotype and genotype of pancreatic cancer cell lines. Pancreas 39, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Wu J and Jiang C (2010) ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci 86, 631–637. [DOI] [PubMed] [Google Scholar]
- Direnzo D, Hess DA, Damsz B, Hallett JE, Marshall B, Goswami C, Liu Y, Deering T, MacDonald RJ and Konieczny SF (2012) Induced Mist1 expression promotes remodeling of mouse pancreatic acinar cells. Gastroenterology 143, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio EN, Young CC, Toma J, Levy M, Berger KR, Johnson CL, Mehmood R, Swan P, Chu A, Cregan SP et al (2017) Activating transcription factor 3 promotes loss of the acinar cell phenotype in response to cerulein‐induced pancreatitis in mice. Mol Biol Cell 28, 2347–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Figura G, Morris JP, Wright CVE and Hebrok M (2014) Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras‐mediated pancreatic neoplastic initiation. Gut 63, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flandez M, Cendrowski J, Cañamero M, Salas A, del Pozo N, Schoonjans K and Real FX (2014) Nr5a2 heterozygosity sensitises to, and cooperates with, inflammation in KRas G12V ‐driven pancreatic tumourigenesis. Gut 63, 647–655. [DOI] [PubMed] [Google Scholar]
- Garside VC, Kowalik AS, Johnson CL, DiRenzo D, Konieczny SF and Pin CL (2010) MIST1 regulates the pancreatic acinar cell expression of Atp2c2, the gene encoding secretory pathway calcium ATPase 2. Exp Cell Res 316, 2859–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimont A, Pinho AV, Cowley MJ, Augereau C, Mawson A, Giry‐Laterrière M, Van den Steen G, Waddell N, Pajic M, Sempoux C et al (2014) SOX9 regulates ERBB signalling in pancreatic cancer development. Gut 64, 1–10. [DOI] [PubMed] [Google Scholar]
- Grützmann R, Boriss H, Ammerpohl O, Lüttges J, Kalthoff H, Schackert HK, Klöppel G, Saeger HD and Pilarsky C (2005) Meta‐analysis of microarray data on pancreatic cancer defines a set of commonly dysregulated genes. Oncogene 24, 5079–5088. [DOI] [PubMed] [Google Scholar]
- Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD et al (2008) Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A 105, 18913–18918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann V, Wilke H, Mergenthaler H‐G, Clemens M, König H, Illiger HJ, Arning M, Schalhorn A, Possinger K and Fink U (2000) Gemcitabine and cisplatin in the treatment of advanced or metastatic pancreatic cancer. Ann Oncol 11, 1399–1403. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H and Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DA, Humphrey SE, Ishibashi J, Damsz B, Lee A, Glimcher LH and Konieczny SF (2011) Extensive pancreas regeneration following acinar‐specific disruption of Xbp1 in mice. Gastroenterology 141, 1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DA, Strelau KM, Karki A, Jiang M, Azevedo‐Pouly AC, Lee A‐H, Deering TG, Hoang CQ, MacDonald RJ and Konieczny SF (2016) MIST1 links secretion and stress as both target and regulator of the unfolded protein response. Mol Cell Biol 36, 2931–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang CQ, Hale MA, Azevedo‐Pouly A, Elsässer HP, Deering TG, Willet SG, Pan FC, Magnuson MA, Wright CVE, Swift GH et al (2016) Transcriptional maintenance of pancreatic acinar identity, differentiation and homeostasis by PTF1A. Mol Cell Biol 36, MCB.00358‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Ernst SA, Stuenkel EL, Lentz SI and Williams JA (2015) Rab27A is present in mouse pancreatic acinar cells and is required for digestive enzyme secretion. PLoS One 10, e0125596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruban RH, Goggins M, Parsons J and Kern SE (2000) Progression model for pancreatic cancer. Clin Cancer Res 6, 2969–2972. [PubMed] [Google Scholar]
- Jacquemin P, Durviaux SM, Jensen J, Godfraind C, Gradwohl G, Guillemot F, Madsen OD, Carmeliet P, Dewerchin M, Collen D et al (2000) Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol Cell Biol 20, 4445–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D (2008) Deciphering the bHLH transcription factor Mist1: dual roles in pancreas development. Theses and Dissertations Available from ProQuest 3418662, 1–171. [Google Scholar]
- Jia D, Sun Y and Konieczny SF (2008) Mist1 regulates pancreatic acinar cell proliferation through p21 CIP1/WAF1. Gastroenterology 135, 1687–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Azevedo‐Pouly A, Deering TG, Hoang CQ, DiRenzo D, Hess DA, Konieczny SF, Swift GH and MacDonald RJ (2016) MIST1 and PTF1 collaborate in feed‐forward regulatory loops that maintain the pancreatic acinar phenotype in adult mice. Mol Cell Biol 36, 2945–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckheere N, Skrypek N and Van Seuningen I (2010) Mucins and pancreatic cancer. Cancers 2, 1794–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M and Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki A, Humphrey SE, Steele RE, Hess DA, Taparowsky EJ and Konieczny SF (2015) Silencing Mist1 gene expression is essential for recovery from acute pancreatitis. PLoS One 10, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lahmy R, Riha C, Yang C, Jakubison BL, van Niekerk J, Staub C, Wu Y, Gates K, Dong DS et al (2015) The basic helix‐loop‐helix transcription factor E47 reprograms human pancreatic cancer cells to a quiescent acinar state with reduced tumorigenic potential. Pancreas 44, 718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratyeva LG, Chernov IP, Zinovyeva MV, Kopantzev EP and Sverdlov ED (2017) Expression of master regulatory genes of embryonic development in pancreatic tumors. Dokl Biochem Biophys 475, 250–252. [DOI] [PubMed] [Google Scholar]
- Kopp JL, von Figura G, Mayes E, Liu F‐F, Dubois CL, Morris JP, Pan FC, Akiyama H, Wright CVE, Jensen K et al (2012) Identification of Sox9‐dependent acinar‐to‐ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22, 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krah NM, De La O J‐P, Swift GH, Hoang CQ, Willet SG, Chen Pan F, Cash GM, Bronner MP, Wright CV, MacDonald RJ et al (2015) The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 4, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapp A, Knöfler M, Frutiger S, Hughes GJ, Hagenbüchle O and Wellauer PK (1996) The p48 DNA‐binding subunit of transcription factor PTF1 is a new exocrine pancreas‐specific basic helix‐loop‐helix protein. EMBO J 15, 4317–4329. [PMC free article] [PubMed] [Google Scholar]
- Krapp A, Knöfler M, Ledermann B, Bürki K, Berney C, Zoerkler N, Hagenbüchle O and Wellauer PK (1998) The bHLH protein PTF1‐p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev 12, 3752–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Jena R, Kansurkar SS, Swain TR (2012) Cancer stem cell‐essence of tumorigenesis. J Carcinog Mutagen 1, 1–6. [Google Scholar]
- Kure S, Matsuda Y, Hagio M, Ueda J, Naito Z and Ishiwata T (2012) Expression of cancer stem cell markers in pancreatic intraepithelial neoplasias and pancreatic ductal adenocarcinomas. Int J Oncol 41, 1314–1324. [DOI] [PubMed] [Google Scholar]
- Lee AYL, Dubois CL, Sarai K, Zarei S, Schaeffer DF, Sander M, Kopp JL (2018) Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut 1–12. https://doi.org/10.1136/gutjnl-2017-314426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF and Simeone DM (2007) Identification of pancreatic cancer stem cells. Cancer Res 67, 1030–1037. [DOI] [PubMed] [Google Scholar]
- Liao H‐K, Hatanaka F, Araoka T, Reddy P, Wu M‐Z, Sui Y, Yamauchi T, Sakurai M, O'Keefe DD, Núñez‐Delicado E et al (2017) In vivo target gene activation via CRISPR/Cas9‐mediated trans‐epigenetic modulation. Cell 171, 1495–1507.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HG, Jin RU, Sibbel G, Liu D, Karki A, Joens MS, Madison BB, Zhang B, Blanc V, Fitzpatrick JAJ et al (2017) A single transcription factor is sufficient to induce and maintain secretory cell architecture. Genes Dev 31, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli P, Madriles F, Cañamero M, Pau ECS, del Pozo N, Guerra C and Real FX (2016) The acinar regulator Gata6 suppresses Kras G12V‐driven pancreatic tumorigenesis in mice. Gut 65, 476–486. [DOI] [PubMed] [Google Scholar]
- Masui T, Long Q, Beres TM, Magnuson MA and MacDonald RJ (2007) Early pancreatic development requires the vertebrate Suppressor of Hairless (RBPJ) in the PTF1 bHLH complex. Genes Dev 21, 2629–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM and Bejerano G (2010) GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeay RC, Bailey TL (2010) Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmood R, Varga G, Mohanty SQ, Laing SW, Lu Y, Johnson CL, Kharitonenkov A, Pin CL (2014) Epigenetic reprogramming in Mist1 2/2 mice predicts the molecular response to cerulein‐induced pancreatitis. PLoS One 9, e84182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JC and Taghert PH (2012) Scaling factors: transcription factors regulating subcellular domains. Bioessays 34, 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitra K (2015) Overcoming multidrug resistance in cancer stem cells. Biomed Res Int 2015, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA et al (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25, 1960–1966. [DOI] [PubMed] [Google Scholar]
- Morita S, Noguchi H, Horii T, Nakabayashi K, Kimura M, Okamura K, Sakai A, Nakashima H, Hata K, Nakashima K et al (2016) Targeted DNA demethylation in vivo using dCas9–peptide repeat and scFv–TET1 catalytic domain fusions. Nat Biotechnol 34, 1060–1065. [DOI] [PubMed] [Google Scholar]
- Murtaugh LC and Leach SD (2007) A case of mistaken identity? Nonductal origins of pancreatic “ductal” cancers. Cancer Cell 11, 211–213. [DOI] [PubMed] [Google Scholar]
- Pasca M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M (2006) Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev 20, 3161–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin CL, Rukstalis JM, Johnson C and Konieczny SF (2001) The bHLH transcription factor Mist1 is required to maintain exocrine pancreas cell organization and acinar cell identity. J Cell Biol 155, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin CL, Ryan JF and Mehmood R (2015) Acinar cell reprogramming: a clinically important target in pancreatic disease. Epigenomics 7, 267–281. [DOI] [PubMed] [Google Scholar]
- Quinlan AR and Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Réjiba S, Bigand C, Parmentier C, Hajri A (2009) Gemcitabine‐based chemogene therapy for pancreatic cancer using Ad‐dCK::UMK GDEPT and TS/RR siRNA strategies. Neoplasia 11, 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ and Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodolosse A, Chalaux E, Adell T, Hagège H, Skoudy A and Real FX (2004) PTF1α/p48 and cell proliferation. Gastroenterology 127, 937–949. [DOI] [PubMed] [Google Scholar]
- Rose SD and MacDonald RJ (1997) Evolutionary silencing of the human elastase I gene (ELA1). Hum Mol Genet 6, 897–903. [DOI] [PubMed] [Google Scholar]
- Rukstalis JM, Kowalik A, Zhu L, Lidington D, Pin CL and Konieczny SF (2003) Exocrine specific expression of Connexin32 is dependent on the basic helix‐loop‐helix transcription factor Mist1. J Cell Sci 116, 3315–3325. [DOI] [PubMed] [Google Scholar]
- Shi G, DiRenzo D, Qu C, Barney D, Miley D and Konieczny SF (2013) Maintenance of acinar cell organization is critical to preventing Kras‐induced acinar‐ductal metaplasia. Oncogene 32, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Inoue H, Wu JC and Yamanaka S (2017) Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov 16, 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, Zhu L, Sun Y, Bettencourt R, Damsz B, Hruban RH and Konieczny SF (2009) Loss of the acinar‐restricted transcription factor Mist1 accelerates Kras‐induced pancreatic intraepithelial neoplasia. Gastroenterology 136, 1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P (2017) Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol 14, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K and Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Tan AC, Jimeno A, Lin SH, Wheelhouse J, Chan F, Solomon A, Rajeshkumar NV, Rubio‐Viqueira B and Hidalgo M (2009) Characterizing DNA methylation patterns in pancreatic cancer genome. Mol Oncol 3, 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Okazaki T, Suzuki H, Abbruzzese JL and Li D (2011) Association of multi‐drug resistance gene polymorphisms with pancreatic cancer outcome. Cancer 117, 744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson N, Gesina E, Scheinert P, Bucher P and Grapin‐Botton A (2012) RNA profiling and chromatin immunoprecipitation‐sequencing reveal that PTF1a stabilizes pancreas progenitor identity via the control of MNX1/HLXB9 and a network of other transcription factors. Mol Cell Biol 32, 1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Jin RU, Bredemeyer AJ, Oates EJ, Błazewska KM, McKenna CE and Mills JC (2010) RAB26 and RAB3D are direct transcriptional targets of MIST1 that regulate exocrine granule maturation. Mol Cell Biol 30, 1269–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topno NS, Kannan M, Krishna R (2017) Mechanistic insights into the activity of Ptf1‐p48 (pancreas transcription factor 1a): probing the interactions levels of Ptf1‐p48 with E2A‐E47 (transcription factor E2‐alpha) and ID3 (inhibitor of DNA binding 3). J Biomol Struct Dyn 36, 1–19. [DOI] [PubMed] [Google Scholar]
- Tran T, Jia D, Sun Y and Konieczny SF (2007) The bHLH domain of Mist1 is sufficient to activate gene transcription. Gene Expr 13, 241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz AP, Ponnusamy MP, Seshacharyulu P and Batra SK (2014) A concise review on the current understanding of pancreatic cancer stem cells. J Cancer Stem Cell Res 2, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G‐J, Gao C‐F, Wei D, Wang C and Ding S‐Q (2009) Acute pancreatitis: etiology and common pathogenesis. World J Gastroenterol 15, 1427–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphalen CB, Takemoto Y, Tanaka T, Macchini M, Jiang Z, Renz BW, Chen X, Ormanns S, Nagar K, Tailor Y et al (2016) Dclk1 defines quiescent pancreatic progenitors that promote injury‐induced regeneration and tumorigenesis. Cell Stem Cell 18, 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiebe PO, Kormish JD, Roper VT, Fujitani Y, Alston NI, Zaret KS, Wright CVE, Stein RW and Gannon M (2007) Ptf1a binds to and activates area III, a highly conserved region of the Pdx1 promoter that mediates early pancreas‐wide Pdx1 expression. Mol Cell Biol 27, 4093–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin W‐C, Mansour J, Mollaee M, Wagner K‐U, Koduru P, Yopp A et al (2015) Whole‐exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6, 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J and Melton DA (2008) In vivo reprogramming of adult pancreatic exocrine cells to beta‐cells. Nature 455, 627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Shi G, Schmidt CM, Hruban RH and Konieczny SF (2007) Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol 171, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Doc. S1. Differentially expressed gene promoter de novo motif analysis.
doc . S2. IPA cancer‐associated molecules.
Fig. S1. Generation of the Tet‐inducible cell lines.
Fig. S2. Comparison of Tet‐MIST1‐ and Tet‐PTF1a‐induced targets with human pancreas samples.
Fig. S3. BxPC‐3 and KPC cells engineered to express Tet‐MIST1 and Tet‐PTF1a.
Fig. S4. PTF1a and MIST1 differentially expressed promoter motif enrichment and ChIP‐Seq intersect.
Fig. S5. KEGG pathway analysis of the Tet‐PTF1a and Tet‐MIST1 RNA‐Seq.
Table S1. Antibodies.
Table S2. Mouse and human RT‐qPCR primers.
Data Availability Statement
RNA‐Seq data have been deposited in the NCBI GEO database under accession number https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106290.