

Abstract
Arsenic exposure is known to disrupt innate immune functions in humans and in experimental animals. In this study, we provide a mechanism by which arsenic trioxide (ATO) disrupts macrophage functions. ATO treatment of murine macrophage cells diminished internalization of FITC-labeled latex beads, impaired clearance of phagocytosed fluorescent bacteria and reduced secretion of pro-inflammatory cytokines. These impairments in macrophage functions are associated with ATO-induced unfolded protein response (UPR) signaling pathway characterized by the enhancement in proteins such as GRP78, p-PERK, p-eIF2α, ATF4 and CHOP. The expression of these proteins is altered both at transcriptional and translational levels. Pretreatment with chemical chaperon, 4-phenylbutyric acid (PBA) attenuated the ATO-induced activation in UPR signaling and afforded protection against ATO-induced disruption of macrophage functions. This treatment also reduced ATO-mediated reactive oxygen species (ROS) generation. Interestingly, treatment with antioxidant N-acetylcysteine (NAC) prior to ATO exposure, not only reduced ROS production and UPR signaling but also improved macrophage functions. These data demonstrate that UPR signaling and ROS generation are interdependent and are involved in the arsenic-induced pathobiology of macrophage. These data also provide a novel strategy to block the ATO-dependent impairment in innate immune responses.
Keywords: Inorganic arsenic, Innate immune function, Macrophage, ROS, UPR, PBA
Introduction
Chronic exposure to arsenic-contaminated drinking water is a significant global environmental and public health problem (Abhyankar et al., 2012; Brown et al., 2002; Hughes et al., 2011). Arsenic exposure has been associated with increased risk of cancer affecting skin, bladder, kidney, lung and prostate (Benbrahim-Tallaa and Waalkes, 2008; Gibb et al., 2011; Mink et al., 2008). At the same time, this metalloid as arsenic trioxide (ATO) is also known for medication in the treatment of refractory and relapsed acute promyeloid leukemia (APL) (Beauchamp and Uren, 2012; Shen et al., 1997). ATO is also an industrial chemical and recently be classified as a chemical war threat agent (http://www.newenv.com/resources/chemical_and_biological_agents).
In addition to its carcinogenic effects, arsenic is also known to possess immunosuppressive properties. Environmentally relevant levels of arsenic inhibit the ability of innate immune system to eliminate bacterial and viral infection. Employing various murine models, exposure to arsenic has been shown to inhibit the cutaneous contact hypersensitivity response (Patterson et al., 2004; Zhou et al., 2006). Its administration to animals results in impaired T cell functions (Burchiel et al., 2009; Martin-Chouly et al., 2011). Inhalation of ATO has been shown to decrease pulmonary antibacterial defense in mice (Aranyi et al., 1985; Burchiel et al., 2009). In an in vitro study, ATO was found to enhance the HIV-1 infection (Pion et al., 2007). Another study showed that arsenic exposure is associated with an exacerbated risk of influenza A (H1N1) infection and-associated lung function impairment (Kozul et al., 2009a, 2009b). Similar immunosuppressive effects of arsenic also occur in fish. Exposure to arsenic (2–10 ppb) resulted in a greater than 50-fold increase in viral load and at least 17-fold increase in bacterial load in zebra fish embryos (Nayak et al., 2007). Parallel to the observations in experimental animal models, human arsenic poisoning is also associated with an increased risk of infections. For example, in Bangladesh lower respiratory tract infections and diarrhea are more common among arsenic-exposed human populations, particularly in children (Mazumder et al., 2000; Rahman et al., 2011; Raqib et al., 2009; Smith et al., 2011). Patients’ receiving arsenic trioxide (ATO) treatment also showed impaired immune functions. In this regard, Nouri et al. observed recurrent herpes simplex and herpes zoster in patients with multiple myeloma and colon cancer treated with ATO (Nouri et al., 2006).
The exact molecular mechanism by which arsenic impairs immune functions is not known. However, arsenic affects the functions of various immune regulatory cell populations. For example, it alters neutrophil functions and also induces apoptotic cell death in these cells (Binet et al., 2010). In patient population receiving ATO treatment for APL, myelocyte-like cells showing partial differentiation have been noticed in bone marrow and peripheral blood. ATO treatment also affects survival of human monocytes during macrophage differentiation (Lemarie et al., 2006a, 2006b). Recently, arsenic was found to decrease cystic fibrosis transmembrane conductance regulator (CFTR)-mediated chloride secretion which results in a diminished clearance of respiratory pathogens (Bomberger et al., 2012). Macrophages which are important regulator of innate immunity have been shown as an important cellular target population of arsenic toxicity (Burchiel et al., 2009; Lemarie et al., 2006a; Sakurai et al., 2005). Macrophages derived from chronically arsenic-exposed human populations show changes in their phenotype. Significant loss of cell adhesion capacity, impaired phagocytic activity and decreased expression of CD54/F-actin were noticed with these immune regulatory cells (Lemarie et al., 2006a).
In response to disturbed endoplasmic reticulum (ER) homeostasis, signaling pathways known as unfolded protein response (UPR) signaling are induced to restore the protein folding capacity of ER. These signaling pathways are transduced by three sensor proteins PERK, IRE1α, and ATF6α to restore protein folding in a translational-and transcriptional-dependent manner (Wiseman et al., 2010). In addition to this fundamental chaperon function, UPR is found to be associated with the pathogenesis of various diseases/disorders such as inflammation, obesity, diabetes, atherosclerosis and neurodegeneration (Cnop et al., 2012; Wang and Kaufman, 2012; Zhang, 2010; Zhang and Kaufman, 2008). Earlier, we demonstrated a role of all three UPR signaling pathways in arsenic-induced cutaneous inflammatory responses in SKH-1 mice (Li et al., 2011). Based on published reports and our own observations, we tested whether arsenic-modulated innate functions in macrophages are regulated through this signaling pathway.
In this study, we unraveled the molecular mechanism by which ATO disrupts macrophage functions. We demonstrate that ATO alters UPR signaling pathway which mediates impairment in the ability of macrophage to engulf and clear phagocytosed bacteria and produce cytokines. Treatment with chemical chaperone, 4-phenylbutyric acid (PBA) or antioxidant, N-acetylcysteine (NAC) blocks ATO-mediated impairment in macrophage functions. Both of these agents are FDA approved for arsenic-unrelated conditions and therefore, provide an opportunity to repurpose them for attenuating arsenic toxicity.
Material and methods
Cell culture, reagents and exposure
Mouse macrophage cell line RAW 264.7 (TIB-71™) was obtained from the ATCC and maintained in DMEM medium containing 10% fetal bovine serum (FBS) (Gibco, NY, USA), 1% antibiotic–antimycotic solution at 37 °C in 5% CO2 incubator. Arsenic trioxide (202673), N-acetylcysteine (NAC, A7250), lipopolysaccharides from Escherichia coli (LPS, L6529), thapsigargin (TG, T9033) and 4-phenylbutyric acid (PBA, P21005) were obtained from Sigma Aldrich (MO, USA). 2′,7′-Dichlorofluorescin diacetate (DCFH-DA, D-399), Hoechst 33342 (H21492) and E. coli (K-12 strain) bioParticles® fluorescents conjugated bacteria (E-2861) were obtained from Life Technology (NY, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (M92050-1.0) was purchased from Research Product International Corp. (IL, USA). NE-PER nuclear and cytoplasmic extraction kit (78833) was procured from Thermo Scientific (IL, USA). Phagocytosis assay kit (500290) containing latex beads-rabbit IgG-FITC complex was purchased from Cayman Chemicals Company (MI, USA). Primers used in the study were obtained from Invitrogen (Supplementary Table S1). Primary antibodies and horseradish peroxidase (HRP) conjugated secondary antibodies were purchased as described in Supplementary Table S2. Prior to conducting experiments, cells were checked for their viability using trypan blue dye exclusion assay as described earlier (Srivastava et al., 2013). Cells showing viability more than 95% were used in the experiments.
The experimental design includes exposure to ATO at concentrations 0.2–2 μM for a period ranging between 1.5 and 48 h as described for individual experiments. NAC (10 mM, 2 and 24 h) and PBA (1 mM, 24 h) were used as a pre-treatment.
MTT assay
The non-cytotoxic concentration range of ATO was determined by MTT assay. ATO-exposed cells were washed with PBS and incubated with MTT (0.5 mg/ml) for 3 h at 37 °C. At the end of incubation period, the reaction mixture was replaced with 200 μl of DMSO to dissolve formazan crystals and plates were read at 490 nm using 1420 multilable counter VICTOR3™-V (Perkin Elmer, Shelton, CT, USA). Saline-treated sets of cells were also run under identical conditions and served as basal control.
Morphological changes
Morphological changes were assessed in 4% paraformaldehyde fixed cultured cells and observed for any phenotypic alteration (cell roundness, cell blebbing) from the normal morphology under the Phase contrast microscopy (Olympus1X-S8F2, Japan).
RNA isolation and reverse transcriptase PCR
Total RNA was extracted from cells using TRIzol method (Invitrogen). mRNA (1 μg) was reverse-transcribed into cDNA by iScript cDNA synthesis kit (Bio-Rad, CA, USA). PCR products were run on 1.5% agarose gels and the relative quantification was done using Gel Documentation System (Bio-Rad Laboratories, Segrate, Italy). 18sRNA was used as endogenous control. Further details regarding primers and condition used in this study are described in Supplementary Table-S1.
Quantitative real time PCR (qRT-PCR)
qRT-PCR analysis was carried out using either SYBR Green (Bio-Rad, CA, USA) fluorescent dye or TaqMan (Applied Biosystems, NY, USA) PCR master mix. Total cDNA (250 ng) was used in a 20 μl reaction mixture with sequence specific primers. qRT-PCR were carried out in triplicate using 7500 fast Real-Time PCR system (Applied Biosystems, NY, USA). Cycling conditions were 20 s at 95 °C followed by 40 cycles at 95 °C for 3 s and 60 °C for 30 s. Relative quantification of the steady state target mRNA levels was calculated after normalization of total amount of cDNA to β-actin endogenous reference. TaqMan Primers employed in this study are listed in Supplementary Table S1.
Fractional studies
Cytoplasmic and nuclear extracts from ATO-treated and non-treated Raw 264.7 cells were prepared and fractionated as per manufacturer’s protocol using the NE-PER nuclear and cytoplasmic extraction reagent kit (Thermo Scientific, USA) and were analyzed by western blotting.
Protein quantification and western blot (WB) analysis
Lysate of cells receiving various treatments were prepared using an ice cold lysis buffer (50 mmol/l Tris, pH 7.5 Triton X-100, 0.25% NaF, 10 mmol/l β-glycerolphosphate, 2 mmol/l EDTA, 5 mmol/l sodium pyrophosphate, 1 mmol/l Na3VO4, 10 mmol/l DTT and protease inhibitor) followed by centrifugation at 5000 rpm for 10 min to obtain a clear supernatant. Protein assay was done using a DC kit (Bio-Rad). Lysate were mixed with 5× sample buffer (312.5 mM Tris–HCl, pH-6.8, 5% β-mercaptoethanol, 10% SDS, 0.5% bromophenol blue, 50% glycerol), boiled for 5 min at 95 °C and subjected to SDS-PAGE. Proteins were electrophoretically transferred to polyvinylidene difluoride membrane and then nonspecific site was blocked with 5% nonfat dry milk in Tris buffer saline Tween-20 (TBST) (25 mmol/l Tris–HCl, pH-7.5, 150 mmol/l NaCl, 0.05% Tween-20) for 1 h at room temperature followed by probing with primary antibody overnight at 4 °C or 2 h at room temperature. After washing, the membranes were incubated for 1.5 h with respective (anti-mouse, anti-rabbit or anti-goat immunoglobulin) HRP conjugated secondary antibody. The blots were developed with enhanced chemiluminescence (ECL) according to manufacturer’s instructions (Amersham Bioscience, NJ, USA). The integrated density of bands was measured with Image J. software.
Immunofluorescence (IF) staining
Cells were cultured in four wells chamber slide (Bio-Tek, VT, USA). After completion of ATO treatment cells were washed with PBS two times, fixed in methanol for 30 min, permeabilized with 0.1% Triton X-100 for 15 min followed by PBS wash, and blocked for 1 h with 2% bovine serum albumin (BSA). Cells were then incubated with primary antibodies overnight at 4 °C. After three times of PBS wash, cells were incubated with fluorescein-conjugated secondary antibodies for 1 h at room temperature. After a second rinsing step with PBS, the cells were incubated with Hoechst 33342 for 5 min at room temperature to counterstain cell nuclei. Labeled cells were examined with upright fluorescence microscopy (Olympus 1X-S8F2, Japan).
Reactive oxygen species (ROS) generation
ROS generation was assessed using fluorescent dichlorodihydrofluorescein di-acetate (DCFH-DA) dye. In brief, (1 × 104 cells per well) were seeded in 96-wells black bottom culture plates and allowed to adhere them for 24 h in CO2 incubator at 37 °C. Following various treatments for the desired time interval cells were incubated at a final concentration of 2.5 μM of DCFH-DA in serum-free medium for 20 min at 37 °C. The reaction mixture was then aspirated, washed and replaced by 200 μl of PBS in each well. Fluorescence intensity was measured at excitation wavelength at 485 nm and emission wavelength at 528 nm. Saline-treated sets were also run under identical conditions and served as basal control.
In a parallel experiment, 2.5 × 105 cells in 6 well tissue culture plates were analyzed for intracellular fluorescence imaging using upright fluorescence microscope (Olympus 1X-S8F2, Japan).
Latex bead phagocytosis assay
Cayman’s Phagocytosis assay kit (IgG FITC) that employs latex beads coated with fluorescently labeled rabbit-IgG was used as a probe for the identification of phagocytosis process in saline-treated or ATO-treated (alone as well as in combination with PBA, NAC and siRNA transfection) Raw 264.7 cells as per instructions provided by the manufacturer. In brief, following completion of exposure period, cells were incubated with 30 μl of latex beads coated with fluorescently labeled rabbit-IgG for 1 h at 37 °C. Cells were then washed three times with assay buffer followed by trypan blue quenching solution. The engulfed fluorescent-beads were observed using a fluorescent microscope at excitation at 485 and emission at 535 nm. Bead engulfment was also confirmed by flow cytometry at 488 nm. A negative control was performed in parallel by incubating cells with latex beads at 4 °C instead of 37 °C.
Clearance of phagocytized bacteria
For studying the kinetics of clearance of engulfed bacteria by Raw 264.7 cells, we used opsonized E. coli (K-12 strain) BioParticles® fluorescents conjugated bacteria. Briefly, 2 × 105 cells were plated in 6 well tissue culture plates and the next day cells were pretreated with PBA and NAC followed by ATO treatment. At the completion of respective treatments cells were incubated with 6.66 μl (Bacteria:cells 50:1 ratio) of fluorescent tagged E. coli bacteria at 37 °C for 2 h. Cells were then washed three times with warm PBS and imaged by fluorescent microscopy to capture the baseline. Cells were further incubated at 37 °C to complete the process of bacterial clearance. After 24 h, cells were recaptured for fluorescent imaging.
Inflammatory cytokine levels
Inflammatory cytokine levels were determined by real time PCR using SYBR Green methodology as described under quantitative real time PCR subheading. Primers for cytokines used have been listed in Supplementary Table S1.
Statistical analysis
Results were expressed as mean ± standard error of mean (SEM). Statistical analysis between two groups was performed using Student’s t test. In all cases, #/*P < 0.05, ##/**P < 0.01 and ###/***P < 0.001 were considered to be statistically significant.
Results
ATO alters cell phenotype and viability in a dose- and time-dependent manner in murine macrophage Raw 264.7 cells
To determine the suitable concentrations and time-point for this study, we first conducted MTT assay (Supplementary Fig. 1A) at various concentrations (0.0–2 μM) and time-points (6–48 h). From this data, we selected 0.2 and 2 μM concentrations which had no detectable effects on cell viability up to 6 h. However, at 24 and 48 h time-points a dose- and time-dependent decrease in cell viability occurred. Morphological changes began at 12 h (data not shown) but significant changes could be seen at 16 h (Supplementary Fig. 1B). These alterations include cell rounding, loss of cell adhesion and blebbing. Based on these data, we concluded that the molecular changes occurring at early time intervals are involved in the pathophysiological derangements observed in these cells at later time-points, ultimately leading to cell death. We therefore, selected ATO concentrations 0.2 and 2 μM for majority of experiments in this study.
ATO activates PERK/ATF4 and XBP-1 branch of UPR signaling in murine macrophage Raw 264.7
First we tested whether UPR signaling plays a key role in the functional disruption of macrophage. Since, UPR signaling pathways are transduced by the three sensor proteins PERK, IRE1α and ATF6α, we examined the effects of ATO on these three UPR signaling pathways. Reverse transcriptase PCR analysis of ATO-treated cells showed that GRP78, GRP94, ATF4, CHOP, HRD-1 and spliced XBP-1 were upregulated in a dose- and time-dependent manner (Fig. 1A). In this study, the graphs represent densitometry analysis of band intensity of UPR genes (Fig. 1A). Consistently, western blot analysis confirmed the result of PCR analyses. A dose-dependent increase in the expression of p-PERK, p-eIF2α, ATF4, CHOP and spliced XBP-1 was observed (Fig. 1B and Supplementary Fig. 1D–I) which peaked at 3 h though at other early time-points gradual increases in these proteins were noted (Supplementary Fig. 1C). Immunofluorescence staining of ATF4 and spliced XBP-1 showed that ATO-treatment dose-dependently induced nuclear translocation of both ATF4 and spliced XBP-1 (Fig. 1C and Supplementary Fig. 1E–I). Western blot analysis of cytoplasmic and nuclear fractions showed that treatment of Raw 264.7 cells with ATO-induced nuclear translocation of ATF4 and also enhanced the protein expression of its downstream molecule CHOP (Fig. 1D). However, there were no discernible changes seen in ATF6α expression either in the western blot analysis or by immunofluorescence staining of ATO-treated cells (Supplementary Fig. 1D-II & E-II). These results indicate that ATO-induced UPR signaling in murine macrophage cells occurs mainly via PERK/ATF4 and XBP-1.
Fig. 1.
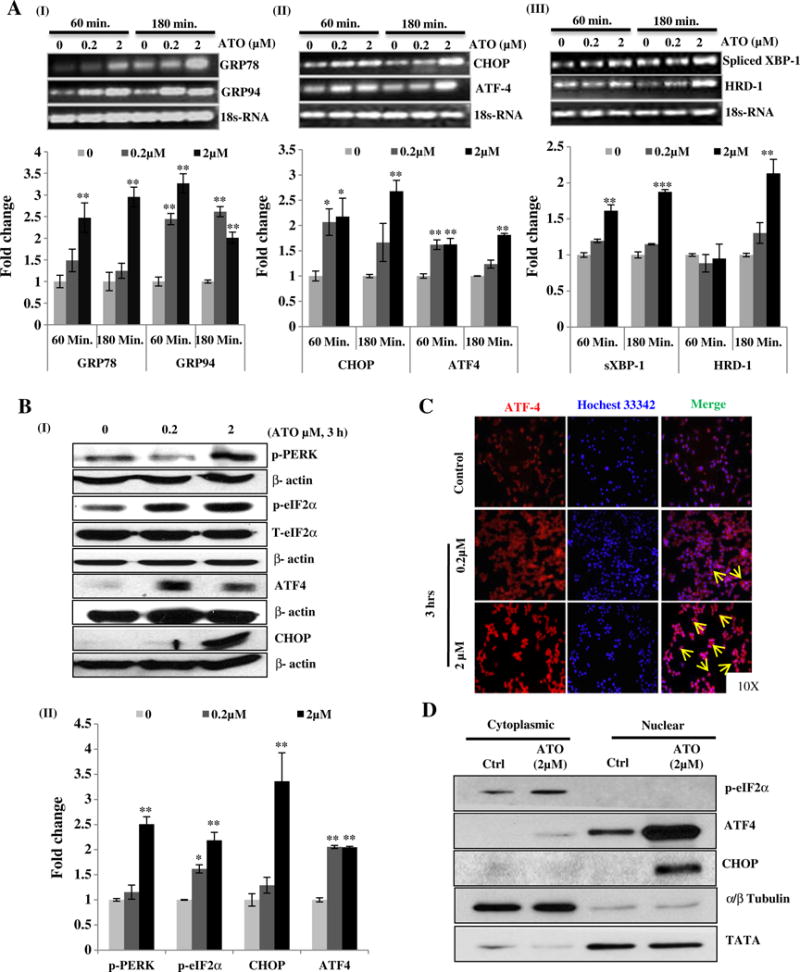
Arsenic trioxide activates UPR signaling in murine macrophage Raw 264.7 cells. Reverse transcriptase PCR analysis of UPR responsive genes in ATO-treated Raw 264.7 cells was performed. For this, cells were treated with ATO at two different concentrations (0.2 and 2 μM) for 60 and 180 min. Band intensity showing transcriptional expression levels of GRP78 and GRP94 (A-I), CHOP and ATF4 (A-II) and spliced XBP-1 and HRD1 (A-III) is depicted. Graphs indicate the densitometry analysis of band intensity in terms of fold change. 18sRNA was used as endogenous control. (B-I) Western blots analysis of UPR signaling proteins (p-PERK, p-eIF2α, ATF4 and CHOP) in ATO (0.2 and 2 μM)-treated Raw 264.7 cells for 3 h. (B-II) Relative densitometry analysis of band intensity expressed in terms of fold change. β-Actin was used as an endogenous control. (C) Immunofluorescence staining showing nuclear localization of ATF4 in ATO (0.2 and 2 μM for 3 h)-treated Raw 264.7 cells. Nuclei were stained with Hoechst 33342 dye. Arrows indicate the migration and localization of ATF4 from cytosol to nucleus in ATO-treated cells as compared to saline-treated control cells. (D) Western blot analysis of ATO-induced translocation and activation of ATF4 and its downstream molecule CHOP. Fraction purity was confirmed by α/β-tubulin and TATA binding protein for cytoplasmic and nuclear fractions respectively. Data are expressed as Mean ± SE of at least three independent samples. Statistical significance was determined using Student’s t test. *P > 0.05, **P > 0.01 and ***P > 0.001 show significance levels.
ATO-mediated ROS production and UPR signaling activation are interdependent
Fluorescent microscopy and plate reader based analysis using DCFH-DA fluorescent probe revealed that ATO enhances ROS generation dose-and time-dependently (Fig. 2A-I & II). This was further confirmed by studies showing that antioxidant, NAC abrogated ATO-dependent ROS generation in these cells (Fig. 2A-I & II). Since ROS generation is known to induce UPR signaling, we also determined whether ATO-dependent alterations in this signaling are ROS-mediated. Pretreatment with NAC (2 and 24 h) significantly attenuated the ATO-induced expression of p-PERK, GRP78, GRP94, p-eIF2α, and CHOP (Fig. 2B and Supplementary Fig. 2A). A similar protective effect of NAC on CHOP expression was also observed at the transcriptional level (Supplementary Fig. 2B). We found that NAC protects against the ATO-mediated decreases in cell viability as determined by both MTT assay and cell morphology assessment (data not shown). Next, we investigated whether ATO-mediated ROS production and ATO-dependent decreases in cell viability are regulated by UPR signaling. For this, we employed a chemical chaperon PBA (1 mM for 24 h). PBA has been demonstrated to attenuate UPR signaling in other biological systems (Li et al., 2012, 2013; Wang et al., 2012). As confirmed by western blot analysis, PBA acts in similar manner in macrophage as shown in Fig. 2C, while thapsigargin (Tg), an ER stress inducer, augmented these effects. Then, we determined whether blocking UPR signaling activation by PBA can afford protection against ATO-induced decrease in cell viability. Pretreatment with PBA resulted in the recovery of the phenotypic alterations observed in ATO-exposed cells (Supplementary Fig. 1B). This was accompanied by a significant reduction in the ATO-mediated ROS production in PBA-treated cells (Fig. 2A–I). These results suggest that ATO-induced ROS generation and UPR signaling activation are interdependently regulated via a feedback loop.
Fig. 2.
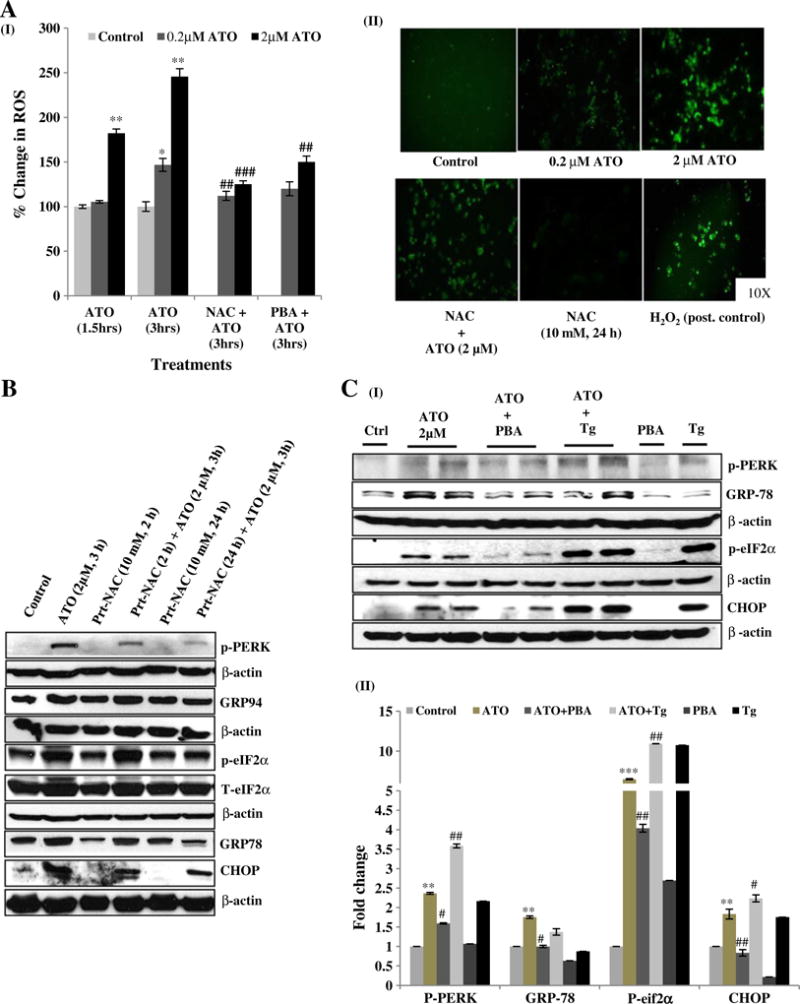
Arsenic trioxide-mediated ROS generation and UPR signaling activation are inter-dependent. ROS generation was assessed using fluorescent DCFH-DA dye. (A-I) Raw 264.7 cells were exposed to ATO (0.2 and 2 μM) treatment for 1.5 and 3 h alone as well as in the presence of NAC (10 mM for 24 h) and PBA (1 mM for 24 h). Fluorescence intensity was measured at excitation wavelength at 485 nm and emission wavelength at 528 nm by micro-plate reader and changes were calculated as percent of control. (A-II) Representative microphotographs showing ATO-induced ROS generation in Raw 264.7 cells. Images were snapped using upright fluorescence microscope. The positive control (Post. control) group consisted of Raw 264.7 cells pretreated (1 h) with 500 μM of H2O2. (B) Cells were pre-incubated with NAC (10 mM for 2 h and 10 mM for 24 h) followed by ATO (2 μM for 3 h) treatment. UPR regulating proteins p-PERK, GRP78, GRP94, p-eIF2α and CHOP were assessed by western blot analysis. (C-I) Raw 264.7 cells were pretreated with PBA and co-treated with thapsigargin at concentrations 1 mM and 2.5 μM for 24 h and 3 h respectively. Following various pretreatments/co-treatments, cells were incubated with ATO (2 μM for 3 h) and levels of proteins including p-PERK, GRP78, p-eIF2α and CHOP were examined by western blot analysis. (C-II) Relative densitometry analysis of band intensity expressed in fold change. β-Actin was used as an endogenous control. Saline-treated cells served as control in all experiments. Data are expressed as Mean ± SE of at least three independent samples. Statistical significance was determined using Student’s t test. #,*P > 0.05, ##,**P > 0.01 and ###,***P > 0.001 show significance levels. * indicates significant level when compared to saline-treated control cells. # indicates significant level when compared to ATO-treated cells.
UPR regulates ATO-inhibited bacterial engulfment and clearance of engulfed bacteria by murine macrophage
Next, we ascertained whether ATO could alter the functions of macrophage. In this regard, first we assessed the pathogen-engulfment activity of macrophage. ATO treatment dose-dependently reduced the engulfment of fluorescently labeled (IgG FITC) latex beads as compared to PBS-treated control cells (Fig. 3A). Similar to its effects on UPR signaling and ROS production, pretreatment with PBA significantly improved the engulfment activity of ATO-treated murine macrophage (Fig. 3A). Essentially, identical results were obtained when assessed by plate reader based assay or flow cytometry analyses (Fig. 3B and Supplementary Fig. 3). Similar to PBA, the antioxidant NAC also afforded protection against ATO-induced impairment in the pathogen engulfment activity of the macrophage (Fig. 3B and Supplementary Fig. 3). After demonstrating its effect on bacterial engulfment, we further investigated the effects of ATO on the ability of macrophage to regulate clearance of engulfed bacteria using Phagotest, which is a functional assay for demonstrating phagocytosis of bacteria (Lemarie et al., 2006a). We observed that PBS-treated control cells effectively cleared the fluorescence tagged E. coli bacteria within 24 h (Fig. 3C–I). However, ATO-treated macrophage were not efficient to clear the engulfed bacterial load and continued to show the presence of fluorescent tagged opsonized bacteria over a period of 24 h observation (Fig. 3C-II). Interestingly, pretreatment of macrophage with PBA and NAC restored ATO-impaired clearance/phagocytosis of engulfed bacteria (Fig. 3C-III and IV).
Fig. 3.
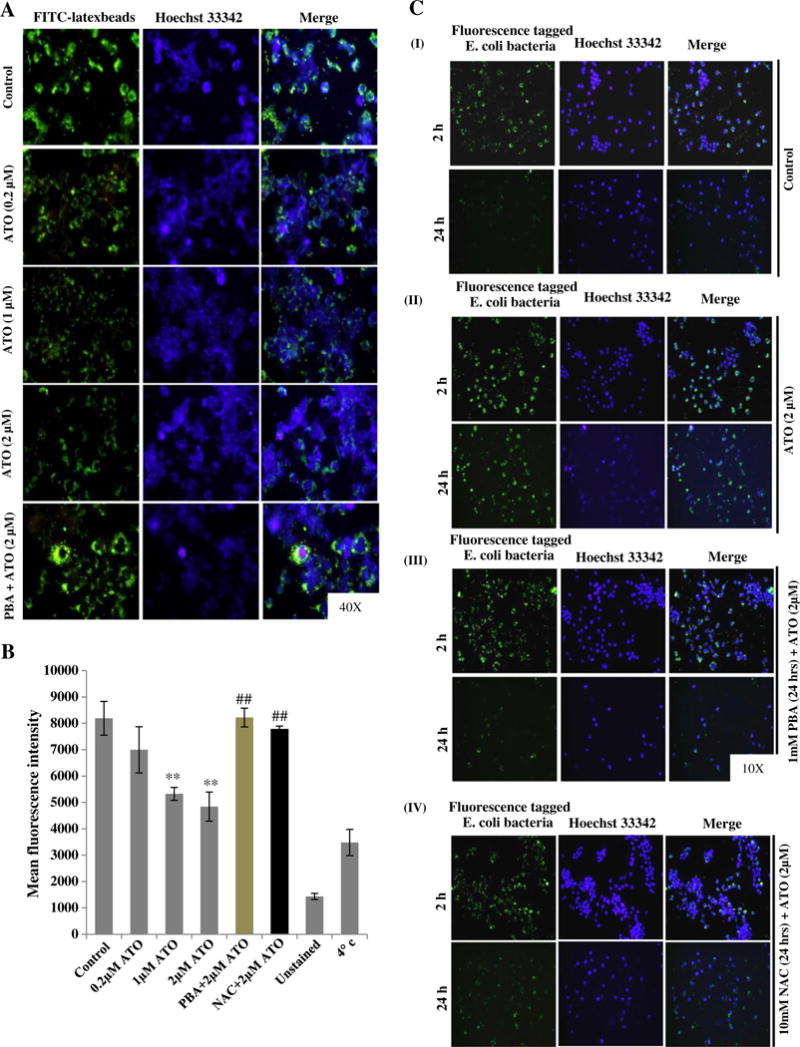
UPR signaling regulates arsenic trioxide-inhibited bacterial engulfment and clearance of E. coli (K-12 strain) BioParticles® fluorescents conjugated bacteria by murine macrophage. (A) Raw 264.7 cells were treated with ATO (2 μM for 16 h) alone as well as in combination of PBA (1 mM for 24 h) followed by incubation with latex beads coated with fluorescently labeled rabbit-IgG for 45 min at 37 °C. Engulfed fluorescent-beads were observed using a fluorescent microscope. Intensity of green fluorescence indicates the engulfed beads. (B) Mean fluorescence intensity (MFI) of engulfed fluorescence beads were recorded in 96 wells black bottom plate at excitation 485 nm and emission at 535 nm by microplate reader. Saline-treated cells receiving an identical treatment with similar procedure as described above except for the incubation temperature which was 4 °C instead of 37 °C served as negative control. (C) Infection load of opsonized E. coli (K-12 strain) BioParticles® fluorescents conjugated bacteria in Raw 264.7 cells-treated with ATO (2 μM for 24 h) in the presence and absence of PBA and NAC were observed under fluorescent microscopy. At the end of treatment, cells were incubated with fluorescent tagged E. coli bacteria (Bacteria:cells 50:1 ratio) at 37 °C for 2 h and baseline was imaged under the fluorescent microscope. Cells were further incubated at 37 °C to complete the process of bacterial clearance. After 24 h, cells were recaptured for fluorescent imaging. (C-I) Saline-treated control cells effectively cleared the fluorescence tagged E. coli bacteria within 24 h. (C-II) ATO-treated macrophage could not clear this bacterial load and continued to show the presence of fluorescent tagged opsonized bacteria. (C-III & IV) PBA and NAC pretreatment of Raw 264.7 cells restored the ATO-impaired clearance/phagocytosis of engulfed bacteria.
ATO-mediated dampening in LPS-induced cytokine production by macrophage is under the control of UPR
Another most important function of macrophage is to produce cytokines in response to pathogens. Therefore, employing real time qRT-PCR, we determined whether ATO affects the ability of macrophage to induce inflammatory cytokine production. This was done in both unstimulated and LPS-stimulated cells. ATO-treatment diminished the baseline production of IL-1β (0.30 fold), TNFα (0.46 fold), TGFβ (0.69 fold), and IL-10 (0.27 fold) in comparison to PBS-treated control cells (Fig. 4A). Pretreatment with ATO also significantly reduced the LPS-induced cytokine production (Fig. 4A). However, interestingly LPS-stimulated cells when subsequently treated with ATO further induced cytokine production (Supplementary Fig. 4). These results were similar to those published earlier (Liu et al., 2012). Pretreatment of cells with PBA significantly attenuated the ATO-impaired production of cytokines (Fig. 4A).
Fig. 4.
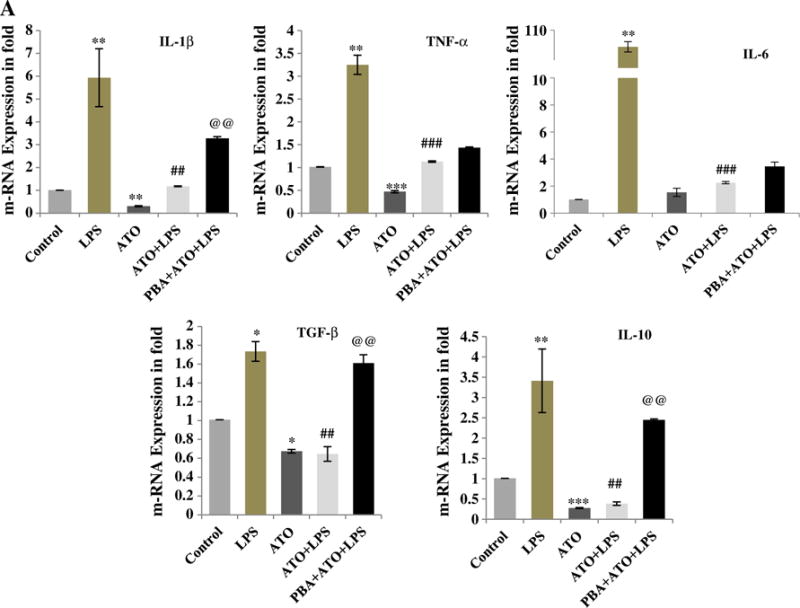
Diminished cytokine production in ATO-treated Raw 264.7 cells was dependent on UPR signaling: Cytokine production was quantified by real time quantitative PCR (qRT-PCR) using SYBR Green methodology. Transcription levels of IL-1β, TNF-α, IL-6, TGF-β, and IL-10 were determined and presented in terms of fold change. (A) Cells received five different treatments. (1) Control-treated with normal saline, (2) LPS-treated with 250 ng/ml for 3 h, (3) ATO-treated with 2 μM for 3 h, (4) pre-treated with ATO followed by LPS, and (5) pretreated with PBA (1 mM for 24 h) followed by ATO and LPS. ATO-treatment to murine macrophage reduced the cytokine production alone as well as in combination of LPS stimulation. PBA pretreatment to the macrophage restored the cytokine production of ATO and LPS-induced cytokine production. Data are expressed as Mean ± SE of at least three independent samples. Statistical significance was determined using Student’s t test. #,*P > 0.05, ##,**P > 0.01 and ###,***P > 0.001 show significance levels. * indicates significant level when compared to saline-treated control cells. # indicates significant level when compared to LPS-treated cells. @ indicates significant level when compared to ATO + LPS-treated cells.
Discussion
Phagocytes such as neutrophils and macrophages effectively engulf and eliminate invading microorganisms (Sarantis and Grinstein, 2012). These cells provide an important component of the innate immune system’s first line of defense against infections (Akira et al., 2006). Since it is known that various inorganic arsenicals such as sodium arsenite and arsenic trioxide impair innate immune responses both in exposed human populations (Parvez et al., 2010; Raqib et al., 2009; Smith et al., 2011) and in experimental animal models (Aranyi et al., 1985; Burchiel et al., 2009; Nayak et al., 2007), we sought to determine the molecular mechanism by which inorganic arsenic deranges these functions. We found that arsenic impairs both bacterial engulfment ability of macrophages as well as their ability to digest the engulfed bacteria. These changes were accompanied by the impaired production of cytokines. It is known that in response to infection these cells often produce copious amounts of pro-inflammatory cytokines which under certain conditions lead to a life threatening condition called septic shock (Qi et al., 2012). Our data suggest that arsenic-mediated alterations in innate immune functions of macrophage are related to the ability of arsenic to suppress multiple biological functions of macrophages.
We also demonstrate here that the mechanism of these alterations is related to the activation of UPR signaling pathway. Although arsenic is known to induce UPR signaling in multiple cell-types (Binet et al., 2010; Lu et al., 2011; Oh et al., 2012; Yen et al., 2012), it was not shown whether arsenic can induce UPR signaling in macrophages and whether activated UPR signaling plays key role(s) in arsenic-induced impairment of macrophage functions. The demonstration in the present study that chemical chaperone PBA protects against arsenic-induced alterations in macrophage functions and survival by attenuating UPR signaling provides a novel mechanism by which arsenic mediates its toxicity to disrupt innate immune functions. Earlier, PBA was shown to act by functionally rescuing misfolded proteins (Li et al., 2013; Wang et al., 2012) and to afford protection against ER stress-induced alterations in neuronal functions (Wiley et al., 2011). We and others have shown that arsenic acts by elaborating ROS production (Li et al., 2011; Yen et al., 2012). However, the exact mechanism by which arsenic-mediated ROS production is regulated remains unclear. Studies as described here also provide first evidence that arsenic-induced ROS and UPR signaling are co-regulated in macrophage as treatment with the known antioxidant NAC reduces arsenic-induced UPR signaling and PBA treatment impedes ROS production (Fig. 2). In this regard, it is known that ROS augments ER stress (Li et al., 2011; Liu et al., 2013; Yen et al., 2012). However, co-regulation of UPR signaling and ROS during the manifestation of immune-toxicity of arsenic provides a novel approach to block multiple disruptive biological effects of arsenic by attenuating UPR signaling.
In summary, these data provide evidence that arsenic trioxide treatment disrupts macrophage functions by activating UPR signaling pathways particularly by PERK/ATF4 regulated signaling. Attenuation of arsenic-mediated activation of UPR signaling by administering chemical chaperon, PBA or by antioxidant, NAC protects against arsenic-mediated disruption of macrophage functions as depicted in Fig. 5.
Fig. 5.
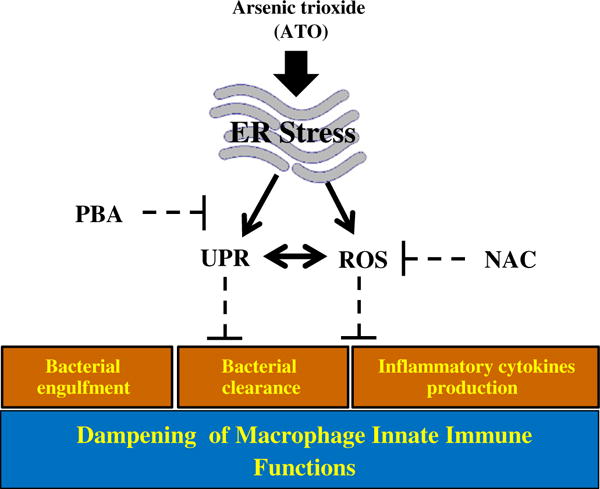
Arsenic treatment impaired macrophage functions are UPR regulated: ATO which causes endoplasmic reticulum stress, induces UPR signaling and ROS concomitantly in macrophage. In addition, ATO-treated macrophage show impairment in innate immune functions including diminished phagocytosis of FITC-labeled latex beads, impaired clearance of phagocytosed fluorescent bacteria and reduced secretion of pro-inflammatory cytokines. However, treatment of macrophage with a chemical chaperon, 4-phenylbutyric acid attenuated both UPR signaling and ROS generation in these cells. Furthermore, this treatment afforded significant protection against ATO-mediated impairment of macrophage functions. Similar protective effects against ATO-mediated impairment in innate immune functions were noted following treatment of these cells with antioxidant NAC. Interestingly the mechanism by which NAC affords protection against immunotoxicity of ATO also involves diminution in UPR signaling pathway.
Supplementary Material
Acknowledgments
This work has been partially supported by funding provided by NIH/NIEHS R21ES017494 and NIH/NIAMS R21AR064595 to MA.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.taap.2013.08.004.
Footnotes
Conflict of interest
The authors disclose that there is no conflict of interest.
References
- Abhyankar LN, Jones MR, Guallar E, Navas-Acien A. Arsenic exposure and hypertension: a systematic review. Environ Health Perspect. 2012;120:494–500. doi: 10.1289/ehp.1103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Aranyi C, Bradof JN, O’Shea WJ, Graham JA, Miller FJ. Effects of arsenic trioxide inhalation exposure on pulmonary antibacterial defenses in mice. J Toxicol Environ Health. 1985;15:163–172. doi: 10.1080/15287398509530643. [DOI] [PubMed] [Google Scholar]
- Beauchamp EM, Uren A. A new era for an ancient drug: arsenic trioxide and Hedgehog signaling. Vitam Horm. 2012;88:333–354. doi: 10.1016/B978-0-12-394622-5.00015-8. [DOI] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Waalkes MP. Inorganic arsenic and human prostate cancer. Environ Health Perspect. 2008;116:158–164. doi: 10.1289/ehp.10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binet F, Chiasson S, Girard D. Arsenic trioxide induces endoplasmic reticulum stress-related events in neutrophils. Int Immunopharmacol. 2010;10:508–512. doi: 10.1016/j.intimp.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Bomberger JM, Coutermarsh BA, Barnaby RL, Stanton BA. Arsenic promotes ubiquitination and lysosomal degradation of cystic fibrosis transmembrane conductance regulator (CFTR) chloride channels in human airway epithelial cells. J Biol Chem. 2012;287:17130–17139. doi: 10.1074/jbc.M111.338855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KG, Ross GL, American Council on Science and Health Arsenic, drinking water, and health: a position paper of the American Council on Science and Health. Regul Toxicol Pharmacol. 2002;36:162–174. doi: 10.1006/rtph.2002.1573. [DOI] [PubMed] [Google Scholar]
- Burchiel SW, Mitchell LA, Lauer FT, Sun X, McDonald JD, Hudson LG, Liu KJ. Immunotoxicity and biodistribution analysis of arsenic trioxide in C57Bl/6 mice following a 2-week inhalation exposure. Toxicol Appl Pharmacol. 2009;241:253–259. doi: 10.1016/j.taap.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18:59–68. doi: 10.1016/j.molmed.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Gibb H, Haver C, Gaylor D, Ramasamy S, Lee JS, Lobdell D, Wade T, Chen C, White P, Sams R. Utility of recent studies to assess the National Research Council 2001 estimates of cancer risk from ingested arsenic. Environ Health Perspect. 2011;119:284–290. doi: 10.1289/ehp.1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MF, Beck BD, Chen Y, Lewis AS, Thomas DJ. Arsenic exposure and toxicology: a historical perspective. Toxicol Sci. 2011;123:305–332. doi: 10.1093/toxsci/kfr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozul CD, Ely KH, Enelow RI, Hamilton JW. Low-dose arsenic compromises the immune response to influenza A infection in vivo. Environ Health Perspect. 2009a;117:1441–1447. doi: 10.1289/ehp.0900911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozul CD, Hampton TH, Davey JC, Gosse JA, Nomikos AP, Eisenhauer PL, Weiss DJ, Thorpe JE, Ihnat MA, Hamilton JW. Chronic exposure to arsenic in the drinking water alters the expression of immune response genes in mouse lung. Environ Health Perspect. 2009b;117:1108–1115. doi: 10.1289/ehp.0800199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemarie A, Morzadec C, Bourdonnay E, Fardel O, Vernhet L. Human macrophages constitute targets for immunotoxic inorganic arsenic. J Immunol. 2006a;177:3019–3027. doi: 10.4049/jimmunol.177.5.3019. [DOI] [PubMed] [Google Scholar]
- Lemarie A, Morzadec C, Merino D, Micheau O, Fardel O, Vernhet L. Arsenic trioxide induces apoptosis of human monocytes during macrophagic differentiation through nuclear factor-kappaB-related survival pathway down-regulation. J Pharmacol Exp Ther. 2006b;316:304–314. doi: 10.1124/jpet.105.092874. [DOI] [PubMed] [Google Scholar]
- Li C, Xu J, Li F, Chaudhary SC, Weng Z, Wen J, Elmets CA, Ahsan H, Athar M. Unfolded protein response signaling and MAP kinase pathways underlie pathogenesis of arsenic-induced cutaneous inflammation. Cancer Prev Res. 2011;4:2101–2109. doi: 10.1158/1940-6207.CAPR-11-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LZ, Deng HX, Lou WZ, Sun XY, Song MW, Tao J, Xiao BX, Guo JM. Growth inhibitory effect of 4-phenyl butyric acid on human gastric cancer cells is associated with cell cycle arrest. World J Gastroenterol. 2012;18:79–83. doi: 10.3748/wjg.v18.i1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Xu C, Yang P. c-Jun NH2-terminal kinase 1/2 and endoplasmic reticulum stress as interdependent and reciprocal causation in diabetic embryopathy. Diabetes. 2013;62:599–608. doi: 10.2337/db12-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ren F, Cheng Q, Bai L, Shen X, Gao F, Busuttil RW, Kupiec-Weglinski JW, Zhai Y. Endoplasmic reticulum stress modulates liver inflammatory immune response in the pathogenesis of liver ischemia and reperfusion injury. Transplantation. 2012;94:211–217. doi: 10.1097/TP.0b013e318259d38e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CM, Zheng GH, Ming QL, Sun JM, Cheng C. Protective effect of quercetin on lead-induced oxidative stress and endoplasmic reticulum stress in rat liver via the IRE1/JNK and PI3K/Akt pathway. Free Radic Res. 2013;47:192–201. doi: 10.3109/10715762.2012.760198. [DOI] [PubMed] [Google Scholar]
- Lu TH, Su CC, Chen YW, Yang CY, Wu CC, Hung DZ, Chen CH, Cheng PW, Liu SH, Huang CF. Arsenic induces pancreatic beta-cell apoptosis via the oxidative stress-regulated mitochondria-dependent and endoplasmic reticulum stress-triggered signaling pathways. Toxicol Lett. 2011;201:15–26. doi: 10.1016/j.toxlet.2010.11.019. [DOI] [PubMed] [Google Scholar]
- Martin-Chouly C, Morzadec C, Bonvalet M, Galibert MD, Fardel O, Vernhet L. Inorganic arsenic alters expression of immune and stress response genes in activated primary human T lymphocytes. Mol Immunol. 2011;48:956–965. doi: 10.1016/j.molimm.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Mazumder DN, Haque R, Ghosh N, De BK, Santra A, Chakraborti D, Smith AH. Arsenic in drinking water and the prevalence of respiratory effects in West Bengal, India. Int J Epidemiol. 2000;29:1047–1052. doi: 10.1093/ije/29.6.1047. [DOI] [PubMed] [Google Scholar]
- Mink PJ, Alexander DD, Barraj LM, Kelsh MA, Tsuji JS. Low-level arsenic exposure in drinking water and bladder cancer: a review and meta-analysis. Regul Toxicol Pharmacol. 2008;52:299–310. doi: 10.1016/j.yrtph.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Nayak AS, Lage CR, Kim CH. Effects of low concentrations of arsenic on the innate immune system of the zebrafish (Danio rerio) Toxicol Sci. 2007;98:118–124. doi: 10.1093/toxsci/kfm072. [DOI] [PubMed] [Google Scholar]
- Nouri K, Ricotti CA, Jr, Bouzari N, Chen H, Ahn E, Bach A. The incidence of recurrent herpes simplex and herpes zoster infection during treatment with arsenic trioxide. J Drugs Dermatol. 2006;5:182–185. [PubMed] [Google Scholar]
- Oh RS, Pan WC, Yalcin A, Zhang H, Guilarte TR, Hotamisligil GS, Christiani DC, Lu Q. Functional RNA interference (RNAi) screen identifies system A neutral amino acid transporter 2 (SNAT2) as a mediator of arsenic-induced endoplasmic reticulum stress. J Biol Chem. 2012;287:6025–6034. doi: 10.1074/jbc.M111.311217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez F, Chen Y, Brandt-Rauf PW, Slavkovich V, Islam T, Ahmed A, Argos M, Hassan R, Yunus M, Haque SE, Balac O, Graziano JH, Ahsan H. A prospective study of respiratory symptoms associated with chronic arsenic exposure in Bangladesh: findings from the Health Effects of Arsenic Longitudinal Study (HEALS) Thorax. 2010;65:528–533. doi: 10.1136/thx.2009.119347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson R, Vega L, Trouba K, Bortner C, Germolec D. Arsenic-induced alterations in the contact hypersensitivity response in Balb/c mice. Toxicol Appl Pharmacol. 2004;198:434–443. doi: 10.1016/j.taap.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Pion M, Stalder R, Correa R, Mangeat B, Towers GJ, Piguet V. Identification of an arsenic-sensitive block to primate lentiviral infection of human dendritic cells. J Virol. 2007;81:12086–12090. doi: 10.1128/JVI.00800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Qiao Y, Wang P, Li S, Zhao W, Gao C. microRNA-210 negatively regulates LPS-induced production of proinflammatory cytokines by targeting NF-kappaB1 in murine macrophages. FEBS Lett. 2012;586:1201–1207. doi: 10.1016/j.febslet.2012.03.011. [DOI] [PubMed] [Google Scholar]
- Rahman A, Vahter M, Ekstrom EC, Persson LA. Arsenic exposure in pregnancy increases the risk of lower respiratory tract infection and diarrhea during infancy in Bangladesh. Environ Health Perspect. 2011;119:719–724. doi: 10.1289/ehp.1002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raqib R, Ahmed S, Sultana R, Wagatsuma Y, Mondal D, Hoque AM, Nermell B, Yunus M, Roy S, Persson LA, Arifeen SE, Moore S, Vahter M. Effects of in utero arsenic exposure on child immunity and morbidity in rural Bangladesh. Toxicol Lett. 2009;185:197–202. doi: 10.1016/j.toxlet.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Ohta T, Fujiwara K. Inorganic arsenite alters macrophage generation from human peripheral blood monocytes. Toxicol Appl Parmacol. 2005;203:145–153. doi: 10.1016/j.taap.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Sarantis H, Grinstein S. Subversion of phagocytosis for pathogen survival. Cell Host Microbe. 2012;12:419–431. doi: 10.1016/j.chom.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW, Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z, Wang ZY. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89:3354–3360. [PubMed] [Google Scholar]
- Smith AH, Marshall G, Yuan Y, Liaw J, Ferreccio C, Steinmaus C. Evidence from Chile that arsenic in drinking water may increase mortality from pulmonary tuberculosis. Am J Epidemiol. 2011;173:414–420. doi: 10.1093/aje/kwq383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava R, Rahman Q, Kashyap M, Singh A, Jain G, Jahan S, Lohani M, Lantow M, Pant A. Nano-titanium dioxide induces genotoxicity and apoptosis in human lung cancer cell line, A549. Hum Exp Txicol. 2013;32:153–156. doi: 10.1177/0960327112462725. [DOI] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JQ, Chen X, Zhang C, Tao L, Zhang ZH, Liu XQ, Xu YB, Wang H, Li J, Xu DX. Phenylbutyric acid protects against carbon tetrachloride-induced hepatic fibrogenesis in mice. Toxicol Appl Pharmacol. 2012;266:307–316. doi: 10.1016/j.taap.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Wiley JC, Pettan-Brewer C, Ladiges WC. Phenylbutyric acid reduces amyloid plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell. 2011;10:418–428. doi: 10.1111/j.1474-9726.2011.00680.x. [DOI] [PubMed] [Google Scholar]
- Wiseman RL, Haynes CM, Ron D. SnapShot: the unfolded protein response. Cell. 2010;140:590–590. doi: 10.1016/j.cell.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, Liu SH. Arsenic induces apoptosis in myoblasts through a reactive oxygen species-induced endoplasmic reticulum stress and mitochondrial dysfunction pathway. Arch Toxicol. 2012;86:923–933. doi: 10.1007/s00204-012-0864-9. [DOI] [PubMed] [Google Scholar]
- Zhang K. Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Int J Clin Exp Med. 2010;3:33–40. [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LF, Zhu Y, Cui XF, Xie WP, Hu AH, Yin KS. Arsenic trioxide, a potent inhibitor of NF-kappaB, abrogates allergen-induced airway hyper responsiveness and inflammation. Respir Res. 2006;7:146. doi: 10.1186/1465-9921-7-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.