

Abstract
Reactive metabolites of environmental chemicals and drugs can cause site-specific damage to p53 tumor suppressor gene in a major pathway for genotoxicity. We report here a high throughput, cell-free, 96-well plate magnetic bead-enzyme system interfaced with LCMS/MS sequencing to bioactivate test chemicals and identify resulting adduction sites on genes. Bioactivated aflatoxin B1 was reacted with a 32 bp exon 7 fragment of the p53 gene using 8 microsomal cyt P450 enzymes from different organs coated on magnetic beads. All cyt P450s converted aflatoxin B1 to aflatoxin B1-8,9-epoxide that adducts guanine (G) in codon 249, with subsequent depurination to give abasic sites, then strand breaks. This is the first demonstration in a cell-free medium that aflatoxin B1 metabolite selectively causes abasic site formation and strand breaks at codon 249 of the p53 probe, corresponding to the chemical pathway and mutations of p53 in human liver cells and tumors. Molecular modeling supports the view that binding of aflatoxin B1-8,9-epoxide to G in codon 249 precedes the SN2 adduction reaction. Among a range of metabolic enzymes characteristic of different organs, human liver microsomes and cyt P450 3A5 supersomes showed the highest bioactivation rate for p53 exon 7 damage. This method to identify metabolite-related gene damage sites may facilitate predictions of organ-specific cancers for test chemicals via correlations with mutation sites.
Graphical abstract
INTRODUCTION
P53 tumor suppressor gene is known as the “the guardian of the genome” and is integral to cell cycle control, apoptosis and DNA repair.1 P53 gene codes for p53 protein that controls cellular responses by inducing cell-cycle arrest, apoptosis and senescence, and promotes impaired-function mutations in developing cancers.2,3 About half of human cancers have been found to contain mutated P53 gene 4-6 Specific mutated codons on the P53 gene are correlated to tissue specific cancers. 4,7,8 For example, characteristic p53 mutations appear for lung cancer tumors in codons 157, 158, 248, 249, for brain and prostate cancer in codon 273, for breast cancer in codons 175, 248 and 273, and for liver cancer codons 175, 282, 248, 249.9 Extensive p53 databases are available that organize p53 mutation data for human cancers.10 Mutation database (UMD) sources for p5311 include data for germline mutations and cell lines obtained from sequencing projects,12,13 cosmic database,14 cancer cell line encyclopedia,15 and the National Cancer Institute.16
Genotoxicity refers to damage to genetic material by chemical agents. Functions of the p53 gene are influenced by damage by reactive metabolites of drugs and environmental pollutants leading to toxicity resulting from gene-metabolite adducts, strand breaks, and generation of reactive oxygen species.17,18 Genotoxicity is most often associated with metabolic bioactivation of drugs and chemicals.19,20 Toxic effects may appear specific to the organ in which they are formed, or may translocate to a secondary organ.19,21 Toxicity issues drive up drug costs and engender human costs to society,34,22 so that new prediction methods are essential. Despite numerous bioassays and animal studies, and roughly 1/3 of drug candidates fail clinical testing due to previously unpredicted toxicity.23,24 Our hypothesis is that gene mutations can be predicted from damage reaction sites. Using a 32 base pair oligonucleotide that represent the structure of exon 7 p53 gene fragment, we have so far shown that this holds for a benzopyrene metabolite adduction site25 and sites from several oxidation modes.26
Cytochrome P450 (cyt P450) are major enzymes involved in oxidative biotransformation of lipophilic molecules to reactive metabolites that can react with genes and lead to genotoxicity.27,28 Common bioassays for genotoxicity include the Ames test, comet assay, chromosomal aberration analysis and the micronucleus assay.24,29, These tests are reliable, but do not provide accurate representations of human metabolism, or predict chemical pathways of gene damage that lead to mutagenicity.31,32 Bioassays that feature metabolic bioactivation often consider liver metabolism only and ignore enzymes from other organs.33 Thus, new rapid, reliable screening assays that includes bioactivation with a broad range of human metabolic enzymes are important to facilitate more realistic future predictions of drug and chemical safety.
Reactive metabolites are often electrophiles that react with nucleophilic DNA bases in SN2 addition reactions.20,24,34 They may be unstable with short half-lives, and toxicity may appear only specific to the organ in which they are formed. More stable reactive metabolites may translocate to a secondary organ and cause broader forms of toxicity. 35
Microfluidic devices utilizing multiple in-vitro 3D multiple-organ tissue cultures are under development to mimic human metabolism and predict the toxicity of drugs.36-38 Our team recently reported microfluidic microwell arrays that assess metabolite-related genotoxic pathways using metabolic enzymes from different organs simultaneously with electrochemiluminescence detection of DNA damage. We found significant differences between relative DNA damage rates of chemicals with different organ enzymes.39 However, this approach measures relative DNA damage rates but does not provide specific information about reaction sites on the DNA or chemical pathways.
We recently developed restriction enzyme-assisted LC-MS/MS sequencing to determine sequence specific gene damage sites and pathways using an 32 bp oligonucleotide, and used it to probe the kinetics of the SN2 reaction of benzo[a]pyrene metabolite benzo[a]pyrenediolepoxide with this p53 fragment.40 However, this approached lacked metabolic activation, and the pure metabolite itself was used in the reactions.
Here we describe a high throughput reaction system containing metabolic enzymes to form metabolites that can react with p53 or other genes in a cell free environment. This approach adapts magnetic bioreactor beads coated with metabolic enzymes to test compounds that can form reactive metabolites (Scheme 1).41,42 We interfaced the enzyme-coated magnetic beads in 96 well plates with restriction enzyme-assisted LC-MS/MS sequencing for the first time to convert a test chemical, aflatoxin B1, to reactive metabolites that cause sequence specific damage to the 32 bp oligonucleotide. After reaction, restriction enzymes cut the reacted 32 bp oligonucleotide into smaller strands appropriate for LC-MS/MS sequence analysis.25,40 Results provide determination of reaction sites, shed light on pathways of covalent reactions between metabolites and the 32 bp oligonucleotide, and may facilitate prediction of tissue specific cancers.
Scheme 1.
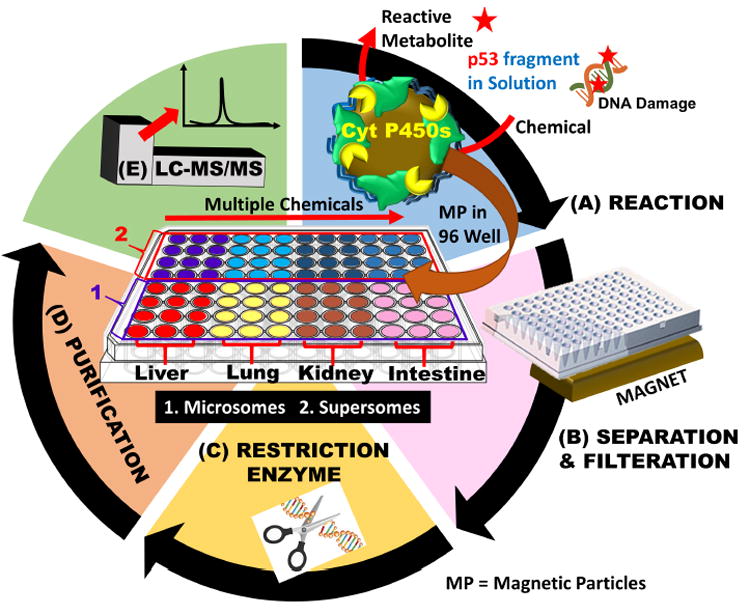
Magnetic bioreactor beads in an 96-well plate followed by LC-MS/MS to determine sequence specific on 32 bp oligonucleotide that represents p53 gene fragment, damage sites and product amounts: (A) magnetic beads coated with microsomal cyt P450 enzymes coupled to NADPH regeneration in 96 well filter plate to convert test chemicals to metabolites that can react with 32 bp oligonucleotide; B) Solutions of reacted 32 bp oligonucleotide were separated from magnetic beads, and extracted to isolate the oligonucleotides; C) restriction enzymes cut 32 bp oligonucleotide into smaller fragments; (D) purification of 32 bp oligonucleotide to remove enzymes and salts to prepare the cut 32 bp oligonucleotide for, (E) LC-MS/MS sequence analysis. Center panel shows 96-well plate with a possible experimental reaction plan.
Cancer is the second leading cause of death in the United States. Hepatocellular carcinoma is reported in approximately 42,220 new cases of which 30,200 will have mortality due to this cancer.43 Fungal mycotoxin, Aflatoxin B1 and viral infections are major cause for hepatocellular carcinoma. Aflatoxin B1, a mycotoxin produced by Aspergillus flavus and Aspergillus parasiticus, was chosen as the test reactant because it is a known carcinogen that forms nucleobase adducts, abasic sites and strand breaks on ds-oligonucleotides. Aflatoxin B1 caused gene damage pathways when bioactivated in liver cells by microsomal cyt P450 bioactivation into reactive metabolite aflatoxin B1-8,9-epoxide that covalently binds to N-7 of guanines.47,48 The N7-guanine adduct can also convert to a ring-opened formamidopyrimidine, which is more stable.45 Aflatoxin B1 was found to cause G-T transversion mutations at the p53 gene’s codon 249 in hepatocytes, associated with liver cancer, these mutation have been reported in cell cultures, animal models and also human specimens.49,50,51
Methodology described in this paper accomplished cell-free bioactivation of aflatoxin B1 to aflatoxin B1-8,9-epoxide, which adducted N-7 guanine of codon 249 in p53 exon 7 gene fragment. The product oligonucleotide subsequently depurinated to an abasic site resulting in strand breaks identified by LC-MS/MS sequencing. This method enabled investigation of the pathway of this Aflatoxin metabolite damage to the 32 bp oligonucleotide representing p53 gene exon 7 fragment in a cell-free chemical assay for a range of metabolic enzymes for the first time.
MATERIALS AND EXPERIMENTAL DETAILS
Chemicals & Reagents
Aflatoxin B1 (AFB1, MW=312.23), poly(diallyldimethylammonium chloride) (PDDA, average Mw= 100,000-200,000), poly(sodium-4-styrenesulfonate) (PSS, average MW= 70000), triethyl ammonium bicarbonate and other chemicals were from Sigma. 1 μm diam. carboxylated magnetic beads were from Bangs Laboratories. Custom made 32 bp, exon 7 fragment from codons 242 to 253 was from Integrated DNA technologies (IDT). Restriction enzyme NIaIII was from New England Biolabs. Enzyme containing pooled male human liver microsomes (HLM), human lung microsomes (HLuM), human intestine microsomes (HIM), human kidney microsomes (HKM), supersomes, 2A6, 1B1, 3A5 and 1A2 were from BD Gentest.
Caution
Aflatoxin B1 is a known chemical carcinogen. Protective measures including wearing protective eyewear and gloves were taken. All experiments were performed in a closed hood.
Magnetic Bead Methodology
Layer-by-layer assembly of PDDA-enzymes films on magnetic beads is described in detail in previous work.52 The magnetic bed coating was slightly modified to suit the current application. Briefly, 80 μL of 1 μm diam. carboxylated magnetic beads (128 mg) were added to 120 μL of 10 mM Tris buffer pH 7.4. To this, 200 μL of 2 mg mL-1 PDDA in water was added, and the mixture was incubated on a shaker at 25°C for 20 min. at 800 rpm. After 20 min, beads were washed 3x with 10 mM Tris buffer + 50 mM NaCl, pH 7.4 using magnetic separation. Then, 340 μL of 10 mM Tris + 50 mM NaCl buffer, pH 7.4 and 60 μL of enzyme (microsomes or supersomes) were added to these magnetic beads and incubated at 4°C for 30 min. on a shaker at 800 rpm. After incubation. the beads were washed 3x with 10 mM Tris buffer + 50 mM NaCl, pH 7.4.
For 32 bp oligonucleotide, damage reactions, these enzyme-coated beads were reacted at 37°C with stirring in the wells of a 96 well plate for 4 hr in pH 7.4, 10 mM PBS solution containing 150 μM aflatoxin B1, 150 μg of ds-32 bp, exon 7 p53 fragment, and an enzyme-based NADPH regeneration system53 at final volume 200 μL. After reaction, magnetic beads were filtered using a magnet into another 96 well plate to collect supernatant solution containing the reacted exon 7 fragment and excess AFB1 were collected. Then, 3000 da molecular weight cut off filters (EMD Millipore, UFC500396) were used to remove excess AFB 1 from exon 7 solutions, which were place into new 96 well plate for the restriction enzyme treatment.
Restriction Enzyme treatment and sample workup
In each well of the 96-well plate, 150 μg of the reacted 32 bp oligonucleotide was combined with 15 μL (150 units) of NIaIII enzyme, 20 μL of 10 X NE buffer (New England Biolabs) and the volume made up to 200 μL using water, then incubated for 8 hours at 37 °C. Unreacted 32 bp oligonucleotide, was also processed in the same way as a control. Then, the resulting oligonucleotide samples were recovered from the mixture using a previously described extraction employing phenol/chloroform/isoamylalcohol, 25/24/1 and chloroform/isoamylalcohol, 24/1.25 Briefly the mixture of oligonucleotides and NIaIII was vortexed with equal volume of phenol/chloroform/isoamyl alcohol for 15 min followed by centrifugation for 10 min, organic phase discarded and aqueous phase was collected (3x). Then, the same process was repeated with chloroform/isoamylalcohol (2x) and the aqueous phase was collected. Before LCMS/MS analysis, these samples were desalted using solid phase extraction (SPE) cartridges (Waters Oasis HLB cartridges, WAT094226). The cartridges were first equilibrated with methanol, followed by water then samples were loaded on them and washed with 5% methanol to remove salts. These samples were then eluted from the cartridges using 100% methanol, then dried using a roto-vap and dissolved in 100 μL HPLC grade water. These samples were then heated and cooled rapidly to convert ds-oligonucleotides to single strands and stored at -20 °C until analysis.
LC-MS/MS Parameters
Two different UPLC-MS/MS methods were employed to (i) identify the specific site of the modification on the oligonucleotides and (ii) quantitation of the oligonucleotide reaction products, and all samples were run in triplicate. To measure the site of reactions of oligonucleotides, ion pair reverse phase chromatography was used to separate the ss-oligonucleotides using 25 mM triethylammonium bicarbonate as ion pairing agent. A binary solvent with 25 mM triethylammoniun bicarbonate as mobile phase A and 100 % methanol as mobile phase B was used. Gemini C-18 column (0.5 mm ID, 3 μ particle size and 150 mm length) was used along with Thermo Scientific Ultimate 3000 UPLC. Separation protocol was 17% B (in phase A) for 2 min, then increasing gradient from 17 to 33 % B for 16 min, held gradient at 33% B for one min and then back to 17% B for 1 min. Between each run, the column was washed with gradient from 0%-60 % B for 7 min, held at 60% B for 8 min then back to 0% B for 5 min at 10 μL/min. Post column m-Nitrobenzylalcohol was added via syringe pump through a three way connector to increase the detection sensitivity and charge states of oligonucleotide fragments.54 The choice of ion pairing agent and LC-MS/MS parameters were slightly modified based on previous studies to suit the current application.25,55,56,57 An AB Sciex QSTAR MS was used in negative mode. Product ion scanning was used to identify the reactive site of modification of the ss-oligonucleotides. Ionization parameters were -4500 ion spray voltage, ion source gas 1 at 30 units and ion source gas 2 at 25 units, -80 V declustering potential, -280 V focusing potential at 300°C was used with collision energy from -42 to -50 eV for MS/MS analysis.
To quantitate the oligonucleotide reaction products, a Thermo Scientific, Hypersil gold column (C18, 100 mm X 0.3mm id and 3 μ particle size) was used. Reverse phase chromatography employed a binary solvent system containing 10 mM Ammonium acetate as mobile phase A and Methanol as mobile phase B with 0.2 % Formic acid was used. Separation involved a total run time of 15 min with 5 % B (for 2 min) increased to 70 %B (2-12 min) and then back to 5 % B (12- 15min). An AB Sciex Qtrap 4000 MS was used for quantitation in positive mode and multiple reaction monitoring (MRM) was used to quantitate the oligonucleotide adducts. Conditions were 5000 ion spray voltage, curtain gas at 10 units, ion source gas 1 at 12 and ion source gas 2 at 5 units and temperature 300°C with collision energy 45 eV. The LC-MS/MS parameters were based of our previous studies.39
Modeling
Molecular modeling of metabolite aflatoxin b1-8,9-epoxide binding onto was done using Autodock 4.2.658,59. A model of the 32 bp oligonucleotide that represents the p53 exon 7 fragment was made using make-na software60 and solvated with water using chimera software.61 Lamarckian genetic algorithm in Autodock 4.2.6 was used to determine the binding energies, binding constant and distance between N7 position of the reactive guanine and epoxide carbon of aflatoxin B1-8-9-epoxide. Complete details of docking parameters for related oligonucleotide systems were reported earlier.40
RESULTS
The 32 bp oligonucleotide was reacted using aflatoxin B1 in microwells with magnetic beads coated with different microsomal enzyme sources. The enzyme reactions driven by an NADPH regeneration system dissolved in solution converts aflatoxin B1 to metabolites, which then react with 32 bp oligonucleotide in solution. This experimental plan facilitates identification of the specific sites and relative amounts of damage to 32 bp oligonucleotide that represents the p53 gene fragment caused by the metabolites formed by the different enzymes. Eight different cyt P450 enzyme sources were used, including human liver microsomes (HLM), human lung microsomes (HLuM), human kidney microsomes (HKM), and human intestine microsomes (HIM). Single cyt P450 supersomes (also containing cyt P450 reductase) were chosen that contain major enzymes in specific human organs including cyt P450s 1A2 (liver), 2A6 (lung), 1B1 (kidney) and 3A5 (liver and intestine).62,63,64
The different enzymes were attached to magnetic beads and placed into different wells of the 96-well plates along with aflatoxin B1 and the NADPH regeneration system. Cyt P450 enzymes biotransformed aflatoxin B1 to aflatoxin B1-8,9-epoxide,65 with the aid of cyt P450 reductase and NADPH. Aflatoxin B1-8,9-epoxide reacts with the N-7 position of guanine to form covalent adducts (Scheme 2). These adducts are unstable and undergo depurination resulting in the formation of an abasic site, Scheme 3.66 These abasic sites can further undergo β-elimination resulting in strand breaks. 48,67
Scheme 2.
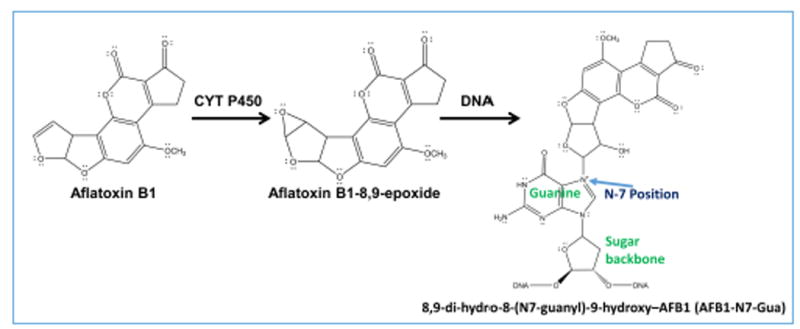
Cytochrome P450-mediated biotransformation of aflatoxin B1. Aflatoxin B1 undergoes enzyme catalyzed oxidation to form reactive metabolite, aflatoxin B1-8,9-epoxide, which undergoes an SN2 addition reaction at N-7 position of guanine to form a guanine adduct on the right.
Scheme 3.
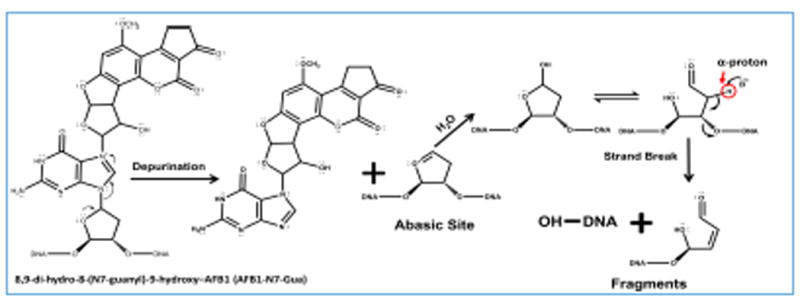
Abasic site formation and strand break after oligonucleotide reaction with aflatoxin B1-8,9-epoxide. Proposed pathway (refs. 48,66,67) for depurination (base loss) from 8,9-dihydro-8-(7-guanyl)-9-hydroxy-AFB1 (AFB1-N7-Gua) and subsequent strand break via β-elimination.
Restriction enzyme treatment of the unreacted 32 bp oligonucleotide, will result in four fragments (Scheme S1, SI file). After the reaction with bioactivated aflatoxin B1 and sample workup, 6 single-stranded fragments were found (Scheme 4). MS analysis of the reacted oligonucleotide sample identified the undamaged fragments 1 (m/z=1002.7279, z=-4), 2 (m/z=969.3836 z=-5), 3 (m/z=1421.2875 z=-5) and 4 (m/z=912.1931, z=-3), and identified the damaged fragment 2 broken into two smaller fragments, 2_1 and 2_2 (Scheme 4, Table S1, SI). Figure 1 shows the extracted ion chromatogram of the undamaged fragments 1-4 along with isotopic distribution of the multiply charged fragment ions obtained that confirms charge state of the fragment ion. The total ion chromatogram (TIC) obtained for the reaction between Aflatoxin B1 and 32 base pair exon 7 probe is as shown in Figure S1, SI. These results suggest covalent adduction of aflatoxin B1-8,9-epoxide onto guanine (G) on codon 249 of 32 bp oligonucleotide that represents P53 gene fragment, with subsequent depurination of G on codon 249 (Scheme 3), then strand cleavage at the depurinated site possibly assisted by the heating step used for dehybridization or insource ionization conditions. A ring-opened formamidopyrimidine adduct (Scheme S2) of fragment 2 was found in trace amounts not amenable to quantitation. Mainly a single product was found for the reaction between aflatoxin b1 and the 32 bp oligonucleotide which resulted in a strand break at codon 249. These results are consistent with published data in cell lines, animal models and human specimens.49,51
Scheme 4.
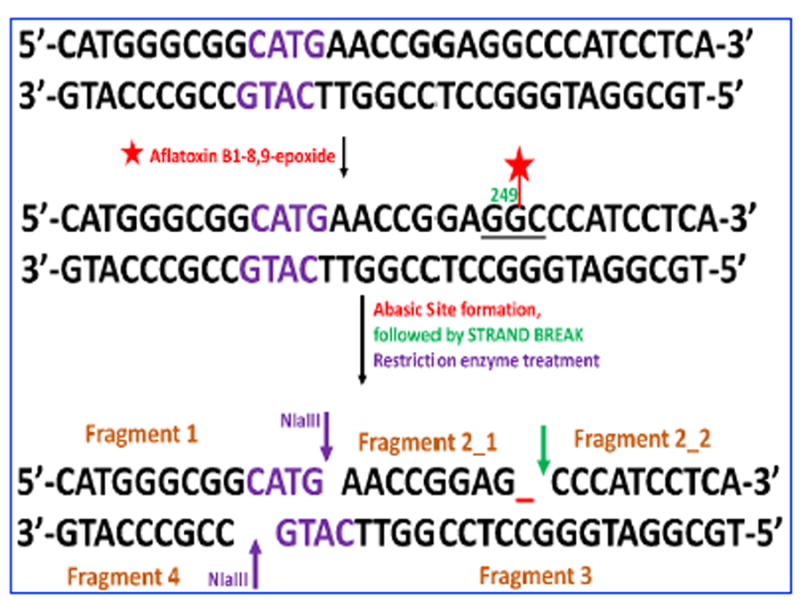
Representation of ds-exon 7 p53 fragment after reaction with aflatoxin metabolite B1-8,9 epoxide formed by cyt P450 enzymes catalysis. The reactive metabolite reacts with guanine in codon 249, which subsequently undergoes abasic site formation and strand break resulting in two damaged fragments of fragment 2 and four undamaged fragments 1-4 after restriction enzyme treatment.
Figure 1.
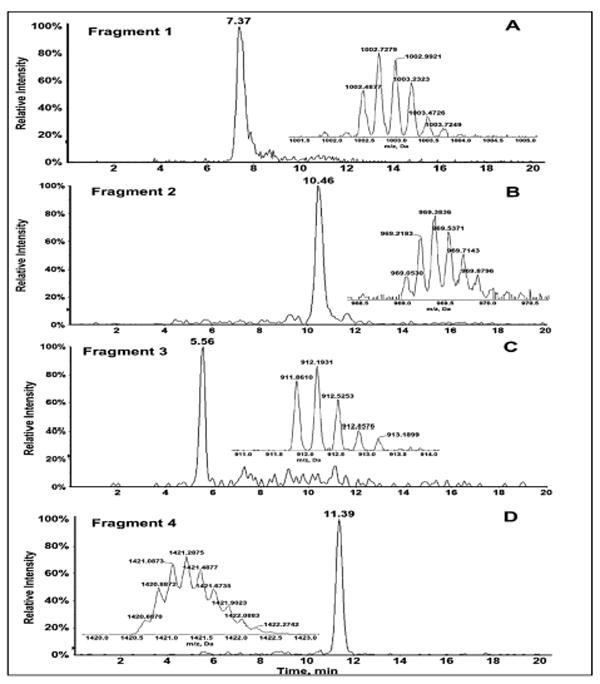
LC-MS of Fragment 1-4 of ds-32 bp oligonucleotide: (A) Extracted ion chromatogram of unadducted fragment 1, m/z 1002.7279, z=-4. (B) Extracted ion chromatogram of unadducted fragment 2, m/z 969/3836, z=-6. (C) Extracted ion chromatogram of unadducted fragment 4, m/z 912.1931, z=-3. (D) Extracted ion chromatogram of unadducted fragment 3, m/z 1421.2875, z=-5. Insets showing isotopic distribution of the multiply charged fragment ions.
Fragment 2_1 m/z=674.4, z=-4 corresponds to the starting fragment AACCGGAGG with loss of guanine to give AACCGGAG, consistent with the difference in mass between AACCGGAGG and AACCGGAG of ~151 daltons corresponding to guanine. The position of guanine loss was determined by MS/MS analysis (Figure 2). Collision-induced (CID) mass spectra of damaged oligonucleotides give characteristic an-bn and wn ions, when compared with the spectra of unreacted fragments in this case give the exact position of guanine loss. For fragment 2_1 all wn ions have m/z less than the unreacted control standard (standard without base loss), hence the base lost is the terminal G. Presence of w1 ion with m/z 195 was an indication that last guanine has been depurinated along with other w ions with decreased m/z. Most of the fragment ions confirmed for AACCGGAG_have a charge state of -1 or -2. There is a possibility of AACCG_AGG, based on ions with m/z 303 (Z=-3), 328 (Z=-4), but the charge state of these ions do not match and absence of confirmatory ion a7-b7 with decreased m/z corresponding to ~151 da adduction/depurination at suggested position could not be confirmed. Fragment 2_2 formed due to strand break at codon 249, which is very significant also reflects adduction at terminal guanine of Fragment 2_1. Fragment 2_2 has m/z of 994.9, z=-3. This fragment is intact without any base loss, as confirmed by MS/MS (Figure 3). Presence of all an-bn and wn ions with m/z similar that of the standard CCCATCCTCA sequence confirm the depurination at codon 249 leading to a strand break between guanine and cytosine. We estimated approximate conversion at 44% by comparing the peak area between undamaged Fragment 2 in a control sample (reaction in the absence of aflatoxin B1) and Aflatoxin B1 reacted sample for most reactive cytochrome P450 enzyme source (3A5 supersomes), to be approximately 44%.
Figure 2.
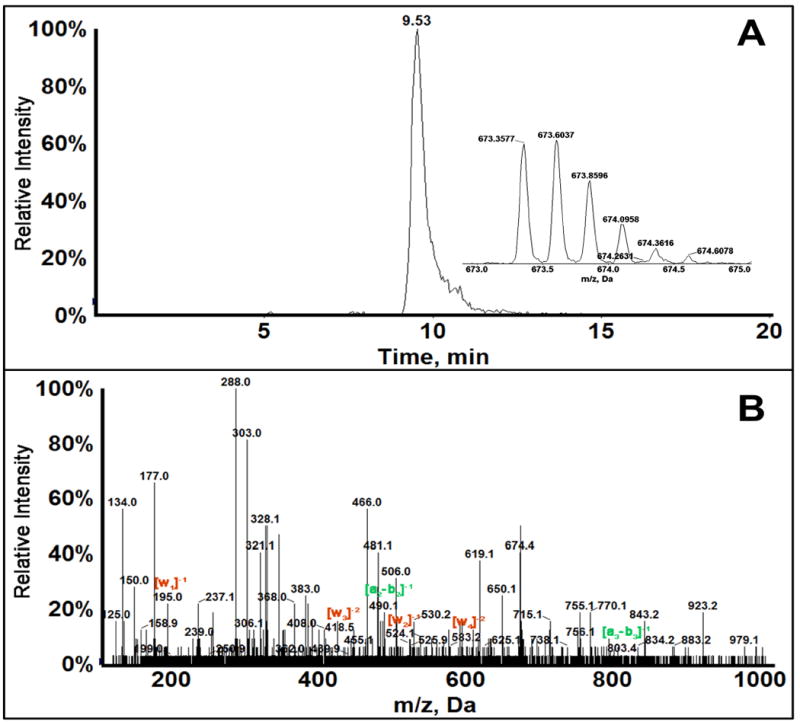
LC-MS/MS of Fragment 2_1 of 32 bp oligonucleotide: (A) Extracted ion chromatogram of singly adducted fragment 2_1, m/z 673.6037, z=-4. (B) MS/MS spectrum of Fragment 2_1 showing the obtained an-bn and wn ions. Ions with m/z same as the unreacted control are shown in green and ions with m/z decreased below control shown in red.
Figure 3.
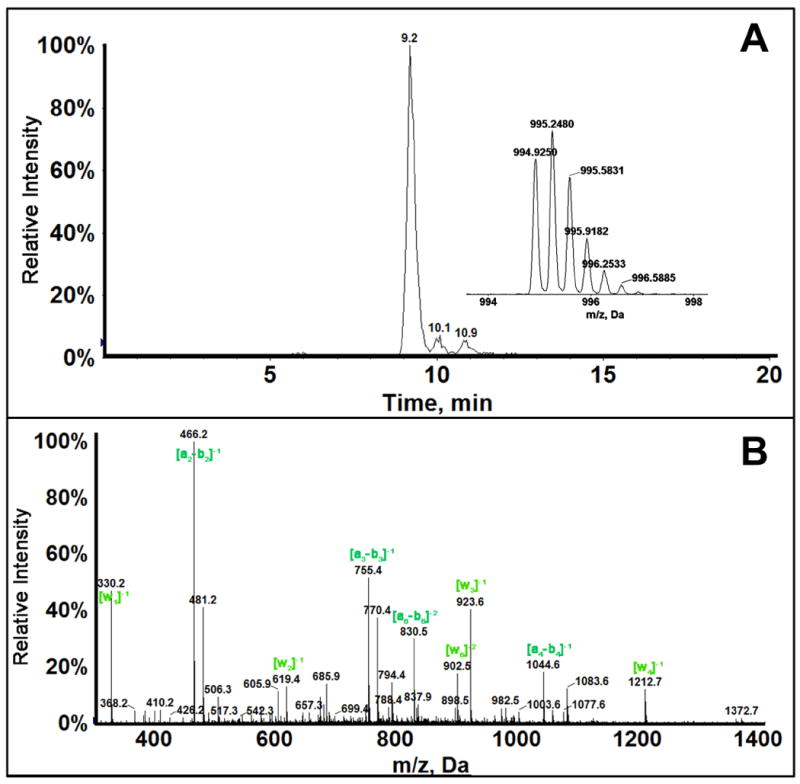
LC-MS/MS of fragment 2_2 of 32 bp oligonucleotide: (A) Extracted ion chromatogram of (CCCATCCTCA) fragment 2_2, m/z 995.2, z=-3. (B) MS/MS spectra of Fragment 2_2 showing the obtained an-bn and wn ions. Ions with m/z the same as that of standard CCCATCCTCA are shown in green.
Molecular modeling using Autodock 4.2.6 of aflatoxin B1-8-9-epoxide binding to the 32 bp oligonucleotide that represents the p53 exon 7 fragment was done to gain a better understanding of binding selectivity and efficiency. Aflatoxin B1-8,9-epoxide has two isomers, exo and endo, and previous work showed that exo isomer of aflatoxin B1-8,9-epoxide is the major isomer reacting with DNA.65 Docking studies that sample all binding configurations of exo-aflatoxin B1-8,9-epoxide with 32 bp oligonucleotide that represents the p53 exon 7 fragment revealed a best binding energy (ΔGb) of -4.44 kcal/mol for binding of the metabolite closest to G in codon 249. The distance between the epoxide carbon of aflatoxin B1-8,9-epoxide and the N7 position of the reactive G in codon 249 is 3.81 Å (Figure 4). This result is consistent with a model involving initial non-covalent binding of the epoxide at the exon 7 reaction site, followed by the SN2 chemical reaction with N7-G.
Figure 4.
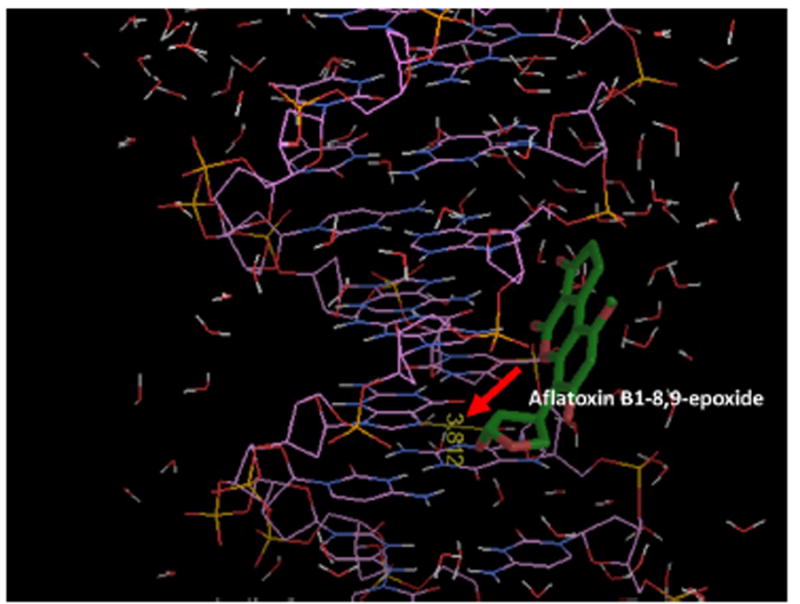
Model of energy minimum docking of aflatoxin B1-8,9-epoxide near G in codon 249 of the 32 bp oligonucleotide. Red arrow shows distance of 3.812 Å between the epoxide carbon of aflatoxin B1-8,9-epoxide and the N7 position of G codon 249. Inclusion of water molecules in the model are necessary to obtain this result.
Relative amounts of oligonucleotide damage found with different cyt P540 (CYP) sources were estimated by determining the amount of AFB1-N7-Gua depurinated into solution after the metabolite-oligonucleotide reaction. A product ion MS/MS of the adduct AFB1-N7-Gua (Figure 4) spectra show two major peaks at m/z 152 and 329 corresponding to protonated guanine (152) and aflatoxin B1 alcohol (329). Multiple reaction monitoring (MRM) was used to quantify the amounts of this adduct using 7-methylguanosine as an internal standard. MRM transitions 480→ 152 for 8,9-dihydro-8-(7-guanyl)-9-hydroxy-AFB1 (AFB1-N7-Gua) adduct and 298→166 for 7-methylguanosine were selected. Figure 6 shows the relative amounts of AFB1-N7-Gua obtained from ratios of area under the curve from extracted ion chromatograms to that of internal standard transition for different microsomes enzyme sources. Results were normalized for the amount of protein on the magnetic beads, concentration of af1atoxin B1 used, and time of reaction. Similarly, Figure 7 shows relative normalized amounts of AFB1-N7-Gua adducts for different single cyt P450 supersomes. The order of bioactivation enzyme activity found from these data was HLM > HLuM > HIM > HKM. Order of bioactivation activity of the supersomes was CYP 3A5 >> 2A6 > 1A2 > 1B1. The overall order of bioactivation activity was 3A5 >> HLM > HLuM > 2A6 > HIM > 1A2 > 1B1 > HKM.
Figure 6.
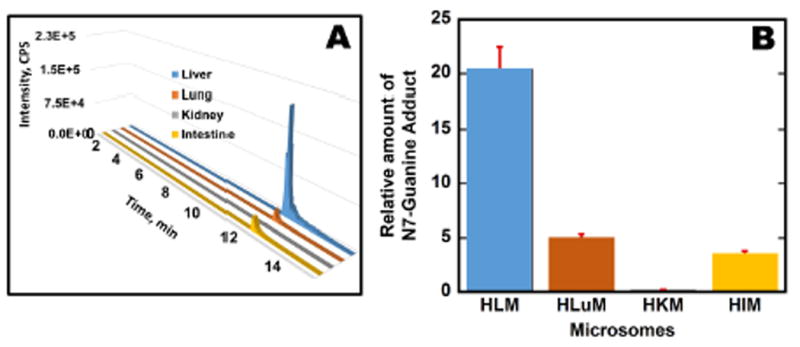
AFB1-N7-Gua production using microsomes: (A) Selected reaction monitoring (SRM) transition chromatogram for AFB1-N7-Gua (m/z 480 → 152) for different organ microsomes; (B) Relative amounts of AFB1-N7-Gua {μg of protein}-1, {mM aflatoxin B1}-1 s-1 formed as ratios of area under SRM peak transitions to that of internal standard 7-methylguanosine after reaction for 4 hr at 37°C.
Figure 7.
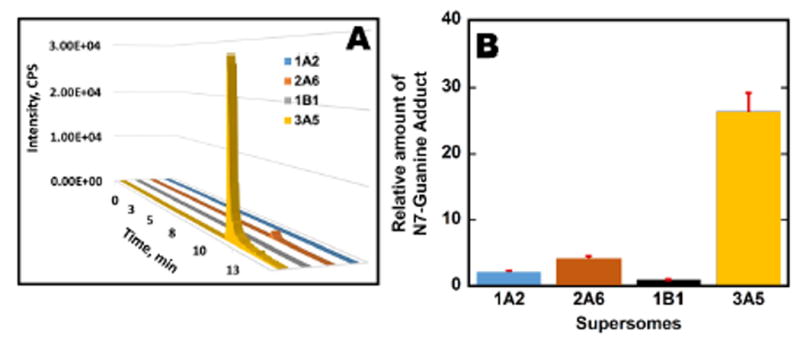
AFB1-N7-Gua production using supersomes: (A) SRM transition chromatogram for AFB1-N7-Gua (m/z 480 → 152) from metabolic reactions involving supersomal enzyme sources (cyt P450 1A2, 2A6, 1B1, 3A5). (B) the relative amount of AFB1-N7-Gua {μg of protein}-1, {mM aflatoxin B1}-1 s-1 as ratios of area under peak from SRM transitions to that internal standard 7-Methylguanosine after reaction for 4 hr at 37°C.
DISCUSSION
Results described above demonstrate a high throughput, cell-free reaction system that metabolizes test chemicals using microsomal enzyme sources to produce potentially reactive metabolites that can react with gene fragments and prepares products for analysis by LC-MS/MS sequencing. The aim is to identify sites and pathways of gene damage by potentially genotoxic test compounds, as well as to evaluate a range of metabolic enzymes that initiate the reactions. Magnetic beads coated with metabolic enzymes facilitate investigating a wide range of cyt P450s and bioconjugation enzymes, and includes a downstream restriction enzyme step to cut the reacted oligonucleotides into <20 base strands in preparation for MS/MS sequencing.25
The new method was demonstrated by results above documenting the chemical pathway for reaction of bioactivated Aflotoxin B1 with 32 bp oligonucleotide that represents exon 7 p53 gene fragment containing several known mutatable codons. The microsomal cyt P450 oxidation product of aflotoxin B1, aflatoxin B1-8,9-epoxide, was shown to undergo an SN2 reaction with guanine (G) in codon 249, with subsequent depurination to form an abasic site leading to a strand break. This reaction pathway was verified here for the first time in a cell-free chemical system, and is the same as that found in hepatic and other cells upon bioactivation of aflatoxin B1 with cyt P450 microsomes.44-45,46,47 The abasic site on fragment 2 (Scheme 4) of p53 exon 7 is from loss of G from codon 249. These reactions presumably result in the downstream G→T transversion of p53 codon 249 found in liver cells challenged with bioactivated aflatoxin B1,49 and the same codon mutated in liver cancer.9
Results above are chemically reasonable, since N-7 guanine is the most nucleophilic site on oligonucleotides and the preferred site of SN2 reactions with electrophiles like epoxides.68,69 N7-guanine adducts are inherently unstable due to the positive formal charge developed on the guanine ring (Scheme S2). In particular, large electrophiles like aflatoxin B1-8,9-epoxide linked with N7-G makes the adducted oligonucleotide prone to depurination.70 Abasic sites due to aflatoxin B1 metabolite reactions undergo strand breaks catalyzed by strong nucleophiles and heat (Schemes 3 and Scheme S1,71 so in our method heating to unwind the ds-oligonucleotides may drive the strand break.
All the cyt P450 enzyme sources in this work led to the same products. The largest amount of depurinated adduct AFB1-N7-Gua was formed when human liver microsomes were used as the cyt P450 source (Figure 5). Human lung and kidney microsomes provided intermediate amounts of AFB1-N7-Gua, but kidney microsomes had low bioactivation activity for aflatoxin B1. Among the supersomes used, CYP (cyt P450) 3A5 produced the largest amount of AFB1-N7-Gua (Figure 6). CYP 3A5 are highly expressed in human liver and intestine,72,73 and our results are consistent with a high rate of aflatoxin B1 biotransformation in these organs.74 CYP 3A6 provided moderate bioactivation, but the activities of CYP 1A2 and CYP 1B1 supersomes were quite low.
Figure 5.
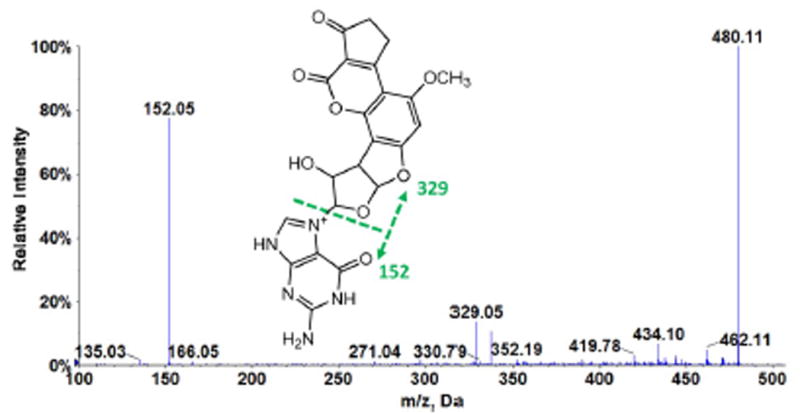
Product ion scan MS/MS of AFB1-N7-Gua, m/z 480.1, which undergoes fragmentation at collision energy 45 eV to give base peak m/z 152 corresponding to protonated guanine.
Molecular modeling results support high selectivity for AFB1-N7-Gua docking near the G of codon 249, thereby targeting the highly mutated p53 codon in liver cancer (Figure 3) for noncovalent binding. A thermodynamically favorable negative binding energy was predicted, and binding distance of 3.812 Å found between the epoxide carbon of aflatoxin b1-8,9-epoxide and N7 of G in codon 249. These two reactive groups form the eventual covalent linkage in the adduct reaction, suggesting a reaction pathway whereby a preceding reactant binding step facilitates subsequent covalent adduct formation at the binding site. This is consistent with our results on the kinetics of the direct reaction of benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE) with p53 exon 7, in which a G in codon 248 was the preferred binding site and subsequent reaction site.40
Reactive site information on 32 bp oligonucleotide uncovered with our methodology may be useful to predict cancer assuming that the damaged base site correlates with codon mutations found in organ specific cancers (see P53 database).9 We previously found site specific covalent adduction at p53 codon 248 for BPDE corresponding to p53 mutations in lung and oral cancers.40 In the present study, the reaction site of codon 249 is correlated with the liver cancer mutation at the same codon related to aflatoxin B1 exposure. Our results are consistent with previous studies in which aflatoxin B1 exposure was correlated with mutations at codon 249 of 32 bp oligonucleotide that represents the P53 gene fragments that leads to G to T transversions in hepatocellular carcinoma patients.75 Similar studies have shown that bioactivation of aflatoxin B1 form reactive metabolites to induce DNA damage leading to mutations in p53 gene.45,76 While a broader range of examples are needed, there is good probability that this cell-free in-vitro method may be able to identify reactive sites on the p53 gene fragments that can be used as predictors of organ specific cancers for test drugs, environmental pollutants and other potentially toxic chemicals.
In summary, the above high-throughput enzyme-bead reaction system coupled to restriction enzyme-assisted LC-MS/MS sequencing facilitates bioactivation of potentially genotoxic chemicals for reactions with genes to profile metabolite-related damage reactions. The experimental plan can be configured to investigate a wide range of metabolic enzymes from virtually any source. The most reactive enzymes for bioactivation related to gene damage can be readily identified, and reaction pathway analysis is facilitated. The method has the potential to identify codon-specific gene damage sites, and possibly predict organ-specific cancers for test compounds via correlation with mutation sites. Further work will focus on adapting this approach for both moderately and strongly bioactivated compounds, and validating the cancer prediction capability of gene adduction sites.
Supplementary Material
Acknowledgments
The authors thank the National Institute of Environmental Health Sciences (NIEHS), NIH, USA, Grant No. ES03154 for financial support. They also thank Prof. Emeritus John Schenkman for helpful discussions and suggestions during the inception of this project.
Footnotes
Including M/z data from mass spectrometry analyses, and figures showing the sites of restriction enzyme cutting, and possible pathway of depurination.
References
- 1.Efeyan A, Serrano M. p53: guardian of the genome and policeman of the oncogenes. Cell cycle. 2007;6:1006–1010. doi: 10.4161/cc.6.9.4211. [DOI] [PubMed] [Google Scholar]
- 2.Haber D, Harlow E. Tumor Suppressor Genes: Evolving Definitions in the Genetic Age. Nature Gen. 1997;16:320–322. doi: 10.1038/ng0897-320. [DOI] [PubMed] [Google Scholar]
- 3.Zilfou JT, Lowe SW. Tumor Suppressive Functions of p53. Cold Spring Harb Perspect Biol. 2009;00:a001883. doi: 10.1101/cshperspect.a001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soussi T. p53 alterations in human cancer: more questions than answers. Oncogene. 2007;26:2145–2156. doi: 10.1038/sj.onc.1210280. [DOI] [PubMed] [Google Scholar]
- 5.Pfeifer GP, Besaratinia A. Mutational spectra of human cancer. Hum Genet. 2009;125:493–506. doi: 10.1007/s00439-009-0657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogelstein B, Sur S, Prives C. p53: the most frequently altered gene in human cancers. Nature Education. 2010;3:6. [Google Scholar]
- 7.Leroy B, Anderson M, Soussi T. TP53 Mutations in Human Cancer: Database Reassessment and Prospects for the Next Decade. Hum Mutat. 2014;35:672–688. doi: 10.1002/humu.22552. [DOI] [PubMed] [Google Scholar]
- 8.Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Adv Cancer Res. 2011;110:107–139. doi: 10.1016/B978-0-12-386469-7.00005-0. [DOI] [PubMed] [Google Scholar]
- 9. [May 11, 2018]; http://p53.fr/the-database.
- 10.Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37:865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- 11.Leroy B, Ballinger ML, Baran-Marszak F, Bond GL, Braithwaite A, Concin N, Donehower LA, El-Deiry WS, Fenaux P, Gaidano G, Langerod A, Hellstrom Lindberg E, Iggo R, Lehmann-Che J, Mai PL, Malkin D, Moll UM, Myers JN, Nichols KE, Pospisilova S, Ashton-Prolla P, Rossi D, Savage SA, Strong LC, Tonin PN, Zeillinger R, Zenz T, Fraumeni JF, Jr, Taschner PE, Hainaut P, Soussi T. Recommended Guidelines for Validation, Quality Control, and Reporting of TP53 Variants in Clinical Practice. Cancer Res. 2017;77:1250–1260. doi: 10.1158/0008-5472.CAN-16-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. [15 February 2018]; https://icgc.org/icgc.
- 13. [15 February 2018]; http://www.cbioportal.org/index.do.
- 14. [15 February 2018]; http://cancer.sanger.ac.uk/cell_lines.
- 15. [15 February 2018]; https://portals.broadinstitute.org/ccle.
- 16. [15 February 2018]; https://discover.nci.nih.gov/cellminer/
- 17.Amundson SA, Myers TG, Fornace AJ., Jr Roles for p53 in growth arrest and apoptosis: putting on the brakes after genotoxic stress. Oncogene. 1998;17:3287–3300. doi: 10.1038/sj.onc.1202576. [DOI] [PubMed] [Google Scholar]
- 18.Bagchi D, Balmoori J, Bagchi M, Ye X, Williams CB, Stohs SJ. Role of p53 tumor suppressor gene in the toxicity of TCDD, endrin, naphthalene, and chromium (VI) in liver and brain tissues of mice. Free Rad Biol Med. 2000;28:895–903. doi: 10.1016/s0891-5849(00)00173-8. [DOI] [PubMed] [Google Scholar]
- 19.Thompson RA, Isin EM, Ogese MO, Mettetal JT, Williams DP. Reactive metabolites: current and emerging risk and hazard assessments. Chem Res Toxicol. 2016;29:505–533. doi: 10.1021/acs.chemrestox.5b00410. [DOI] [PubMed] [Google Scholar]
- 20.Liebler DC, Guengerich FP. Elucidating mechanisms of drug-induced toxicity. Nature reviews Drug Discov. 2005;4:410. doi: 10.1038/nrd1720. [DOI] [PubMed] [Google Scholar]
- 21.Irving RM, Elfarra AA. Role of reactive metabolites in the circulation in extrahepatic toxicity. Exp Opin Drug Metab & Toxicol. 2012;8:1157–1172. doi: 10.1517/17425255.2012.695347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. [15 February 2018]; http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.html.
- 23.Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Disc. 2012;11:191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 24.Hvastkovs EG, Rusling JF. State-of-the-Art Metabolic Toxicity Screening and Pathway Evaluation. Anal Chem. 2016;88:4584–4599. doi: 10.1021/acs.analchem.5b04772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malla S, Kadimisetty K, Fu Y, Choudhary D, Jansson I, Schenkman JB, Rusling JF. Chemical selectivity of nucleobase adduction relative to in vivo mutation sites on exon 7 fragment of p53 tumor suppressor gene. Chem Sci. 2015;6:5554–5563. doi: 10.1039/c5sc01403d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang D, Malla S, Fu Y-J, Choudhary D, Rusling JF. Direct LC-MS/MS detection of oxidations in Exon 7 of the p53 tumor suppressor gene. Anal Chem. 2017;89:12872–12879. doi: 10.1021/acs.analchem.7b03487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Isin EM, Guengerich FP. Substrate binding to cytochromes P450. Anal Bioanal Chem. 2008;392:1019–1030. doi: 10.1007/s00216-008-2244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brendler-Schwaab S, Hartmann A, Pfuhler S, Speit G. The in vivo comet assay: use and status in genotoxicity testing. Mutagenesis. 2005;20:245–254. doi: 10.1093/mutage/gei033. [DOI] [PubMed] [Google Scholar]
- 30.Matsumoto ST, Mantovani MS, Malaguttii MIA, Dias AL, Fonseca IC, Marin-Morales MA. Genotoxicity and mutagenicity of water contaminated with tannery effluents, as evaluated by the micronucleus test and comet assay using the fish Oreochromis niloticus and chromosome aberrations in onion root-tips. Gen & Molec Biol. 2006;29:148–158. [Google Scholar]
- 31.Russell C, Rahman A, Mohammed AR. Application of genomics, proteomics and metabolomics in Drug discovery, development and clinic. Therapeutic Deliv. 2013;4:395–413. doi: 10.4155/tde.13.4. [DOI] [PubMed] [Google Scholar]
- 32.Kang SH, Kwon JY, Lee JK, Seo YR. Recent advances in in vivo genotoxicity testing: prediction of carcinogenic potential using comet and micronucleus assay in animal models. J Can Prev. 2013;18:277–288. doi: 10.15430/JCP.2013.18.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: Function in Xenobiotic Metabolism and Tissue-Selective Chemical Toxicity in the Respiratory and Gastrointestinal Tracts. Annu Rev Pharmacol Toxicol. 2003;43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- 34.Guengerich FP. Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug metab & Pharmacokin. 2011;26:3–14. doi: 10.2133/dmpk.dmpk-10-rv-062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irving RM, Elfarra A. A Role of reactive metabolites in the circulation in extrahepatic toxicity. Exp Opin Drug Metab & Toxicol. 2012;8:1157–1172. doi: 10.1517/17425255.2012.695347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esch MB, Smith AS, Prot J, Oleaga C, Hickman JJ, Shuler ML. How multi-organ microdevices can help foster drug development. Adv Drug Deliv Rev. 2014;69:158–169. doi: 10.1016/j.addr.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagner I, Materne E, Brincker S, Süßbier U, Frädrich C, Busek M, Sonntag F, Sakharov DA, Trushkin EV, Tonevitsky AG. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab on Chip. 2013;13:3538–3547. doi: 10.1039/c3lc50234a. [DOI] [PubMed] [Google Scholar]
- 38.Materne E, Tonevitsky AG, Marx U. Chip-based liver equivalents for toxicity testing–organotypicalness versus cost-efficient high throughput. Lab on chip. 2013;13:3481–3495. doi: 10.1039/c3lc50240f. [DOI] [PubMed] [Google Scholar]
- 39.Wasalathanthri DP, Li D, Song D, Zheng Z, Choudhary D, Jansson I, Lu X, Schenkman JB, Rusling JF. Elucidating organ-specific metabolic toxicity chemistry from electrochemiluminescent enzyme/DNA arrays and bioreactor bead-LC-MS/MS. Chem Sci. 2015;6:2457–2468. doi: 10.1039/c4sc03401e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malla S, Kadimisetty K, Fu Y, Choudhary D, Schenkman JB, Rusling JF. Methyl-Cytosine-Driven Structural Changes Enhance Adduction Kinetics of an Exon 7 fragment of the p53 Gene. Sci Rep. 2017;7:40890. doi: 10.1038/srep40890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rusling JF, Wasalathanthri DP, Schenkman JB. Thin multicomponent films for functional enzyme devices and bioreactor particles. Soft Matter. 2014;10:8145–8156. doi: 10.1039/c4sm01679c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao L, Schenkman JB, Rusling JF. High-throughput metabolic toxicity screening using magnetic biocolloid reactors and LC– MS/MS. Anal Chem. 2010;82:10172–10178. doi: 10.1021/ac102317a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: a cancer journal for clinicians. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 44.Wild CP, Turner PC. The toxicology of aflatoxins as a basis for public health decisions. Mutagenesis. 2002;17:471–481. doi: 10.1093/mutage/17.6.471. [DOI] [PubMed] [Google Scholar]
- 45.Bedard LL, Massey TE. Aflatoxin B 1-induced DNA damage and its repair. Cancer Lett. 2006;241:174–183. doi: 10.1016/j.canlet.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 46.Besaratinia A, Kim SI, Hainaut P, Pfeifer GP. In vitro recapitulating of TP53 mutagenesis in hepatocellular carcinoma associated with dietary aflatoxin B1 exposure. Gastroenterol. 2009;137:1127–1137. doi: 10.1053/j.gastro.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Denissenko MF, Cahill J, Koudriakova TB, Gerber N, Pfeifer GP. Quantitation and mapping of aflatoxin B1-induced DNA damage in genomic DNA using aflatoxin B1-8, 9-epoxide and microsomal activation systems. Mutation Res. 1999;425:205–211. doi: 10.1016/s0027-5107(99)00038-x. [DOI] [PubMed] [Google Scholar]
- 48.Xia Q, Huang X, Xue F, Zhang J, Zhai B, Kong D, Wang C, Huang Z, Long X. Genetic polymorphisms of DNA repair genes and DNA repair capacity related to aflatoxin b1 (AFB1)-induced DNA damages. New Res Dir DNA Repair. 2013;1:377–412. [Google Scholar]
- 49.Puisar F, Hussain SP, Cerutti P. Aflatoxin B1 induces the transversion of G-->T in codon 249 of the p53 tumor suppressor gene in human hepatocytes. Proc Natl Acad Sci U S A. 1993;90:8586–8590. doi: 10.1073/pnas.90.18.8586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chawanthayatham S, Valentine CC, 3rd, Fedeles BI, Fox EJ, Loeb LA, Levine SS, Slocum SL, Wogan GN, Croy RG, Essigmann JM. Mutational spectra of aflatoxin B1 in vivo establish biomarkers of exposure for human hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2017;114:E3101–E3109. doi: 10.1073/pnas.1700759114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smela ME, Currier SS, Bailey EA, Essigmann JM. The chemistry and biology of aflatoxin B1: from mutational spectrometry to carcinogenesis. Carcinogenesis. 2001;22:535–545. doi: 10.1093/carcin/22.4.535. [DOI] [PubMed] [Google Scholar]
- 52.Zhao L, Schenkman JB, Rusling JF. High-throughput metabolic toxicity screening using magnetic biocolloid reactors and LC– MS/MS. Anal Chem. 2010;82:10172–10178. doi: 10.1021/ac102317a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meunier B, De Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 54.Brahim B, Alves S, Cole RB, Tabet J. Charge enhancement of single-stranded dna in negative electrospray ionization using the supercharging reagent meta-nitrobenzyl alcohol. J Am Soc Mass Spectrom. 2013;24:1988–1996. doi: 10.1007/s13361-013-0732-8. [DOI] [PubMed] [Google Scholar]
- 55.Sharma VK, Glick J, Vouros P. Reversed-phase ion-pair liquid chromatography electrospray ionization tandem mass spectrometry for separation, sequencing and mapping of sites of base modification of isomeric oligonucleotide adducts using monolithic column. Journal of Chromatography A. 2012;1245:65–74. doi: 10.1016/j.chroma.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma VK, Xiong W, Glick J, Vouros P. Determination of site selectivity of different carcinogens for preferential mutational hot spots in oligonucleotide fragments by ion-pair reversed-phase nano liquid chromatography tandem mass spectrometry. European Journal of Mass Spectrometry. 2014;20:63–72. doi: 10.1255/ejms.1268. [DOI] [PubMed] [Google Scholar]
- 57.Gong L. Comparing ion-pairing reagents and counter anions for ion-pair reverse phase liquid chromatography/electrospray ionization mass spectrometry analysis of synthetic oligonucleotides. Rapid Communications in Mass Spectrometry. 2015;29:2402–2410. doi: 10.1002/rcm.7409. [DOI] [PubMed] [Google Scholar]
- 58.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comp Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comp Chem. 1998;19:1639–1662. [Google Scholar]
- 60. [January 25, 2018]; http://structure.usc.edu/make-na/server.html.
- 61.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comp Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 62.Ding X, Kaminsky LS. Human extrahepatic cytochromes p450: Function in Xenobiotic Metabolism and Tissue-Selective Chemical Toxicity in the Respiratory and Gastrointestinal Tracts*. Annu Rev Pharmacol Toxicol. 2003;43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- 63.Bieche I, Narjoz C, Asselah T, Vacher S, Marcellin P, Lidereau R, Beaune P, de Waziers I. Reverse transcriptase-PCR quantification of mRNA levels from cytochrome (CYP)1, CYP2 and CYP3 families in 22 different human tissues. Pharmacogen Genomics. 2007;17:731–742. doi: 10.1097/FPC.0b013e32810f2e58. [DOI] [PubMed] [Google Scholar]
- 64.Anzenbacher P, Zanger UM. Metabolism of drugs and other xenobiotics. John Wiley & Sons; 2012. pp. 27–66. [Google Scholar]
- 65.Johnson WW, Guengerich FP. Reaction of aflatoxin B1 exo-8,9-epoxide with DNA: kinetic analysis of covalent binding and DNA-induced hydrolysis. Proc Natl Acad Sci U S A. 1997;94:6121–6125. doi: 10.1073/pnas.94.12.6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gates KS. An overview of chemical processes that damage cellular DNA: spontaneous hydrolysis, alkylation, and reactions with radicals. Chem Res Toxicol. 2009;22:1747–1760. doi: 10.1021/tx900242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fortini P, Dogliotti E. Base damage and single-strand break repair: mechanisms and functional significance of short-and long-patch repair subpathways. DNA repair. 2007;6:398–409. doi: 10.1016/j.dnarep.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 68.Reiner B, Zamenhof S. Studies on the chemically reactive groups of deoxyribonucleic acids. J Biol Chem. 1957;228:475–486. [PubMed] [Google Scholar]
- 69.Gates KS, Nooner T, Dutta S. Biologically relevant chemical reactions of N7-alkylguanine residues in DNA. Chem Res Toxicol. 2004;17:839–856. doi: 10.1021/tx049965c. [DOI] [PubMed] [Google Scholar]
- 70.Boysen G, Pachkowski BF, Nakamura J, Swenberg JA. The formation and biological significance of N7-guanine adducts. Mutation Research/Gen Toxicol Environ Mutagen s. 2009;678:76–94. doi: 10.1016/j.mrgentox.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Benasutti M, Ejadi S, Whitlow MD, Loechler EL. Mapping the binding site of aflatoxin B1 in DNA: systematic analysis of the reactivity of aflatoxin B1 with guanines in different DNA sequences. Biochemistry. 1988;27:472–481. doi: 10.1021/bi00401a068. [DOI] [PubMed] [Google Scholar]
- 72.McCune JS, Risler LJ, Phillips BR, Thummel KE, Blough D, Shen DD. Contribution of CYP3A5 to hepatic and renal ifosfamide N-dechloroethylation. Drug Metab Dispos. 2005;33:1074–1081. doi: 10.1124/dmd.104.002279. [DOI] [PubMed] [Google Scholar]
- 73.Finta C, Zaphiropoulos PG. Intergenic mRNA molecules resulting from trans-splicing. J Biol Chem. 2002;277:5882–5890. doi: 10.1074/jbc.M109175200. [DOI] [PubMed] [Google Scholar]
- 74.Wang H, Dick R, Yin H, Licad-Coles E, Kroetz DL, Szklarz G, Harlow G, Halpert JR, Correia MA. Structure-function relationships of human liver cytochromes P450 3A: aflatoxin B1 metabolism as a probe. Biochemistry. 1998;37:12536–12545. doi: 10.1021/bi980895g. [DOI] [PubMed] [Google Scholar]
- 75.Stern MC, Umbach DM, Yu MC, London SJ, Zhang ZQ, Taylor JA, Hepatitis B. aflatoxin B(1), and p53 codon 249 mutation in hepatocellular carcinomas from Guangxi, People’s Republic of China, and a meta-analysis of existing studies. Cancer Epidemiol Biomarkers Prev. 2001;10:617–625. [PubMed] [Google Scholar]
- 76.Weng MW, Lee HW, Choi B, Wang HT, Hu Y, Mehta M, Desai D, Amin S, Zheng Y, Tang MS. AFB1 hepatocarcinogenesis is via lipid peroxidation that inhibits DNA repair, sensitizes mutation susceptibility and induces aldehyde-DNA adducts at p53 mutational hotspot codon 249. Oncotarget. 2017;8:18213–18226. doi: 10.18632/oncotarget.15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.