

Abstract
A major obstacle to the success rate of chimeric antigen receptor (CAR-) T cell therapy against solid tumors is the microenvironment antagonistic to T cells that solid tumors create. Conventional checkpoint blockade can silence lymphocyte anti-survival pathways activated by tumors, but because they are systemic, these treatments disrupt immune homeostasis and induce autoimmune side effects. Thus, new technologies are required to remodel the tumor milieu without causing systemic toxicities. Here we demonstrate that targeted nanocarriers that deliver a combination of immune-modulatory agents can remove pro-tumor cell populations and simultaneously stimulate anti-tumor effector cells. We administered repeated infusions of lipid nanoparticles coated with the tumor-targeting peptide iRGD and loaded with a combination of a PI3K inhibitor to inhibit immune-suppressive tumor cells and an alpha-GalCer agonist of therapeutic T cells to synergistically sway the tumor microenvironment of solid tumors from suppressive to stimulatory. This treatment created a therapeutic window of two weeks, enabling tumor-specific CAR-T cells to home to the lesion, undergo robust expansion, and trigger tumor regression. CAR-T cells administered outside this therapeutic window had no curative effect. The lipid nanoparticles we used are easy to manufacture in substantial amounts, and we demonstrate that repeated infusions of them are safe. Our technology may therefore provide a practical and low-cost strategy to potentiate many cancer immunotherapies used to treat solid tumors, including T cell therapy, vaccines, and BITE platforms.
INTRODUCTION
The potential of immunotherapy as a cancer treatment option is evident from the positive outcomes many leukemia patients show in response to adoptive cell transfer using autologous T cells genetically modified to express disease-specific chimeric antigen receptors (CARs)(1–3). However, the vast majority of cancers, in particular the more common solid malignancies (such as those occurring in the breast, colon, and lung), fail to respond significantly to CAR-T cell infusions(4–7). This is because solid cancers present formidable barriers to adoptive cell transfer, especially by suppressing T cell functions via the inhibitory milieu they surround themselves with(8, 9).
To combat immunosuppression of T cell therapy, many clinical trials are focused on disabling checkpoint blockades(10, 11). This is not surprising, as several antibodies targeting checkpoint molecules (such as PD-1, PD-L1, and CTLA-4) have already been approved by the FDA for the treatment of certain types of cancer, and preclinical studies have demonstrated increased CAR-T cell potency when these are co-administered with them(10, 12). However, the tumor microenvironment comprises a complex network of heterogeneous cell types that express a variety of different immune inhibitory receptors, and it has become clear that blocking one pathway simply promotes the others, along with compensatory cellular mechanisms that ultimately enable tumors to develop resistance(13, 14). Moreover, the systemic autoimmune toxicity produced by these broad-acting treatments, as well as their high costs, limits widespread use of this therapy(15).
Biotechnology could solve this problem by making available inexpensive nanoparticle reagents that deliver rationally chosen combinations of immunomodulatory drugs into the tumor microenvironment without inducing adverse systemic side effects (illustrated in Fig. 1). In the research described here, we designed lipid nanoparticles containing a potent drug cocktail that can block suppressor cells within the tumor microenvironment and simultaneously stimulate key anti-tumor immune cells. Using the in vivo mouse 4T1 syngeneic breast cancer model(16, 17), we found that when administered at the optimal time and frequency, these drug nanocarriers effectively reverse the immune-hostile cancer environment and thereby create a therapeutic window of vulnerability to T cell-mediated cancer suppression. We establish that infusing tumor-specific CAR-T cells during this time frame results in disease clearance in half of the treated animals and more than doubled the survival of the others, as (in contrast to conventional CAR-T cell therapy) infused T cells were able to effectively infiltrate tumor lesions, undergo robust expansion, and ultimately clear malignant cells. These findings were confirmed in a genetically engineered mouse model of human glioma(18), which is a tumor type notoriously resistant to many currently available immunotherapies(19, 20). We found that nanoparticle-preconditioning doubled the overall survival compared to conventional CAR-T cell therapy only.
Fig. 1. Schematic depicting how targeted liposomes can improve T cell therapy by remodeling the microenvironment created by solid tumors.

We engineered lipid nanocarriers to deliver two therapeutics into tumors: one of them removes pro-tumor cell populations (“releasing the brakes”), while the other stimulates key anti-tumor effector cells (“stepping on the gas”). After immune suppression at the tumor site has been minimized and functional support has been maximized, tumor-specific CAR-T cells are administered; they can then home to the lesion, undergo robust expansion, and effect tumor regression.
MATERIALS AND METHODS
Cell lines
The murine 4T1 breast cancer cell line (American Type Culture Collection, Cat# CRL-2539) was cultured in complete RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 1.5 g/l sodium bicarbonate, 4.5 g/l glucose, 1.0 mM sodium pyruvate, 0.05 mM 2-mercaptoethanol, and 10 mM HEPES buffer (pH 7.2–7.5). To generate a 4T1 tumor cell line that expresses murine ROR1 (4T1-ROR1), we transfected cells with the ROR1 lentiviral vector (pLenti-GIII-CMV-GFP-2A-Puro) marketed by abmgood.com (Cat# LV536399). Tumor cells were cultured in complete medium containing 10 µg/ml puromycin (Invivogen) for three weeks. The Platinum-E (Plat-E) retroviral packaging cell line (Cell Biolabs, Cat# RV-101) was cultured in DMEM containing 10% FBS, 2 mM glutamate, 100 U/ml penicillin, and 100 µg/ml streptomycin. To ensure high retroviral titer, only Plat-E cells passaged no more than 5 times were used for gene transfer. Cell line authentication was not performed. All cell lines tested negative for mycoplasma using a DNA-based PCR test (DDC Medical).
Retroviral vectors and virus production
The anti-ROR1–28z CAR and the anti-EGFRvIII-28z CAR were custom-designed by Creative Biolabs (Shirley, NY). Each consists of a single-chain antibody targeting mouse ROR1 or EGFRvIII linked via a c-myc tag to a synthetic receptor skeleton comprised of the CD8 hinge, the CD28 transmembrane and signaling domains, and the signaling domain from CD3ζ. This construct was then cloned by VectorBuilder (Santa Clara, CA) into their MMLV Retrovirus Gene Expression Vector. SFG-CBR-luc vectors (which express click beetle luciferase) were kindly provided by Dr. Michel Sadelain (Memorial Sloan-Kettering Cancer Center, New York). To generate retroviral particles encoding the ROR1–28z CAR or CBR, we transfected the Plat-E retroviral packaging cell line according to the manufacturer’s instructions and harvested retroviral supernatant 48 h later.
Preparation of tumor-targeting lymphocytes
To generate ROR1-specific (CAR-transduced) T cells, spleens of BALB/cJ mice were macerated over a filter, and re-suspended in ACK lysing buffer (Biosource). To generate EGFRvIII-specific CAR-T cells, we used spleens from Ntv-a; Ink4a-Arf−/−; floxed PTEN EGFRvIII transgenic mice. CD8+ T cells were immunomagnetically selected using the EasySep™ mouse CD8+ T cell isolation kit (Stemcell Technologies). We then activated the murine T cells with anti-mouse CD3 (10 µg/ml), anti-mouse CD28 (0.1 µg/ml), and mIL-2 (19 IU/ml), and transduced them 24 h later with the Plat-E retroviral supernatant on Retronectin-coated plates. Following spinoculation (3,000×g, 2 h, 32 °C), T cells were harvested and re-suspended in complete RPMI with IL-2 (10 IU/ml) and mouse T Activator Beads (Thermofisher; 0.3×106 beads per 1×106 T cells). Following a second spinoculation in retroviral supernatant the next day, the cells were cultured for 24 h in the presence of mouse IL-15 (50 ng/ml) before using them as a tumor therapeutic.
Cytotoxicity assays
We measured in vitro cytotoxic activity of T cells using standard 51Cr release assays as described elsewhere (21). Briefly, 4T1 or 4T1-ROR1 cells were labeled with 51Cr for 1 h at 37°C, washed with RPMI containing 10% FBS, and resuspended in the same medium at a concentration of 1×105 tumor cells/ml. T cells were added to the suspensions at varying effector-to-target cell ratios in 96-well plates (final volume, 200 µl) and incubated for 4 h at 37°C, then 30 µl of supernatant from each well was transferred into Lumaplate-96 microplates for analysis with a Top Count NXT microplate scintillation counter (Packard Bioscience).
Liposome synthesis and characterization
Solutions of egg phosphatidylcholine, cholesterol, 18:0 PEG(2000)-PE and DSPE-PEG-maleimide (65:30:4:1 molar ratio, 10 mg total lipid; Avanti Polar Lipids, Alabaster, AL) in chloroform were combined in a 20-ml glass vial, then 50 µg 7DW8-5 (Funakoshi, Tokyo, Japan) and 1.0 mg PI-3065 (ChemShuttle, Hayward, CA) were dissolved in the lipid solution. The chloroform was removed via a rotary evaporator, then residual solvent was eliminated under high vacuum overnight. The resulting lipid film was hydrated in 5 ml PBS using a vortex device for 1 min, then sonicated for 5 min to produce a liposome suspension.
A solution of iRGD-SH (3 molar equivalents with respect to maleimide, GL Biochem, Shanghai, China) and TCEP (30 equivalents, ThermoFisher, Waltham, MA) was prepared in PBS to yield 5 mM iRGD-SH. This solution was incubated at room temperature for 5 min then added to the liposome suspension. After rotary mixing for 1 h, the suspension was dialyzed (Slide-A-Lyzer Dialysis Cassette, 20K MWCO, ThermoFisher) against water for 2 h. Sucrose was added to the dialyzed liposome suspension to a final concentration of 2.9% before it was lyophilized for storage at −20 °C. For application, the liposomes were re-suspended to their original concentration in sterile PBS. The hydrodynamic radius of the particles was measured with a Nanosite (Malvern), and their zeta potential was determined using dynamic light scattering detected with a Zetapals instrument (Brookhaven Instrument Corporation).
Measurements of drug encapsulation efficiencies and drug release kinetics
Encapsulation efficiency of PI-3065. Rehydrated liposomes were passed through a desalting column (Thermo Scientific Zeba spin column, 7K MWCO) to remove free PI-3065, followed by disruption with 1% Triton X-100. Encapsulated PI-3065 concentration was measured by HPLC. The total concentration of PI-3065 was obtained by repeating the procedure using unfiltered liposomes.
In vitro release of PI-3065 from liposomes. 1.8 ml of rehydrated liposomes was placed in a dialysis device (Thermo Scientific Slide-A-Lyzer MINI, 3.5K MWCO, 2 ml) then immersed in 45 ml water at 37 °C for 70 h. 200 µl of dialyzed material was removed at predetermined timepoints and treated with 1% Triton X-100. PI-3065 concentration was measured by HPLC.
HPLC analysis of PI-3065. The HPLC system consisted of an Agilent 1260 Infinity Quaternary Pump equipped with a UV detector and a Poroshell 120 C18 column, 4.6 × 50 mm, 2.7 µm (Agilent Technologies, Santa Clara, CA). 20 µl of sample was applied to the column, which was equilibrated with 90% buffer A/10% buffer B (buffer A: water + 0.1% phosphoric acid, buffer B: acetonitrile + 0.1% phosphoric acid). Elution was performed at 1 ml/min using a linear gradient to 5% buffer A/95% buffer B over 8 min, followed by a 2 min equilibration back to the starting mobile phase ratio. PI-3065 was detected at 254 nm.
Mice and tumor models
Female BALB/cJ mice (4–6 weeks old) were obtained from Jackson Laboratories (Stock # 000651) and housed in the animal facility of Fred Hutchinson Cancer Research Center. They were used in the context of an animal protocol approved by our Institutional Animal Care and Use Committee. After 1×106 4T1-ROR1 tumor cells were transplanted into the mammary gland, the disease was allowed to establish for ten days before starting treatments. As indicated, drug-loaded liposomes, non-encapsulated drugs, and anti-ROR1 CAR-T cells were administered intravenously with the exception of experiments described in Fig. 2, where CAR-T cells were injected directly into tumor lesions. Progression of tumor growth following therapy was determined by caliper measurements.
Fig. 2. Targeting solid tumors with CAR-T cells decreases immunosuppression in the tumor microenvironment.

(A) Schematic of the anti-ROR1 CAR. (B) Flow cytometry measuring surface expression of the anti-ROR1 CAR 48 h after retroviral transduction (C) 51Cr release assay. (D) Graphs comparing absolute numbers of immune suppressor cells (top panel) and anti-tumor effector cells (lower panel) in 4T1 breast tumors of untreated animals (black/white symbols) with those treated with CAR-T cells (orange symbols). To bypass the variable of T cell trafficking to tumors, 1×106 ROR1–28z CAR+ T cells were injected directly into established 4T1-ROR1 lesions. At the indicated time points for each treatment, 4T1 tumors were recovered from eight mice; these were disaggregated into single-cell suspension and labeled with the following panel of antibodies: For tumor associated macrophages (TAMs): CD45+, CD11b+, F4/80+, Ly6C−, Ly6G−, CD3−, CD19−, and NK1.1−. For myeloid-derived suppressor cells (MDSCs) monocytic: CD11b+, Ly6Chigh, Ly6G−. For MDSCs granulocytic: CD11b+, Ly6Clow, Ly6G+. For regulatory T cells (Tregs): CD4+, CD25high, FoxP3+. For CD8 T cells (host): CD3+, CD8+. For CD8 T cells (CAR-T+): Myc-tag+, CD3+, CD8+. For natural killer (NK) cells: NK1.1+, CD355+. For invariant natural killer T (iNKT) cells: CD3+, alpha GalCer:CD1d complex+. The figure is representative of two experiments which analyzed a total of eight tumors with similar results.
Nestin-specific, Cre-recombinase-responsive EGFRvIII mice were generated by crossing Nestin-Tv-a Ink4a/Arf−/− Pten−/− mice with Cre-responsive Stopfl/fl EGFRvIII mice (previously described)(22). Following nine generations of crosses, we genotyped mice for Nestin-Tv-a Ink4a/arf−/− Pten−/− Stopfl/fl EGFRvIII. These mice were then used for generating EGFRvIII-overexpressing gliomas. The initiation of PDGF-driven gliomas with RCAS has been previously described(23).
In vivo bioluminescence T cell imaging
We visualized T cells using D-luciferin (Xenogen) in PBS (15 mg/ml) as a substrate for CBR-luc. Bioluminescence images were collected with a Xenogen IVIS Spectrum Imaging System (Xenogen, Alameda, CA). Living Image software version 4.3.1 (Xenogen) was used to acquire (and subsequently quantitate) the data 10 min after intraperitoneal injection of the probe into animals anesthetized with 150 mg/kg of 2% isoflurane (Forane, Baxter Healthcare). Acquisition times ranged from 10 sec to 5 min.
Toxicity studies
To measure potential in vivo toxicities of repeated liposome infusions, we injected mice intravenously with a total of nine doses of 7×1013 liposomes carrying PI-3065 and 7DW8-5 (3 doses × 3 weeks), or with phosphate-buffered saline as a control. To measure effects of the nanocarriers per se, a third cohort of mice was injected with empty liposomes. Each experimental group consisted of 8 mice. Twenty-four hours after the final liposome infusion, the animals were anesthetized and blood was collected by retro-orbital bleed using a heparinized microcapillary tube. Blood was collected into microcontainers containing ethylenediaminetetraacetic acid for basic analysis, which included a white blood cell count with differential, a red blood cell count, hemoglobin, hematocrit, and a platelet count. Blood was also collected into serum separator tubes for serum chemistry and cytokine profiles performed by Ani Lytics, Inc. (Gaithersburg, MD). Animals were then euthanized with carbon dioxide to retrieve organs, which were washed with deionized water and fixed in 4% paraformaldehyde. The tissues were processed routinely, and tissue sections were stained with hematoxylin and eosin before evaluation by Dr. Smitha Pillai MVSc, PhD, DACVP, a board-certified staff pathologist, in a blinded fashion.
Flow cytometry
All flow cytometry antibodies were purchased from Ebioscience, including their anti-mouse FoxP3 staining set to detect regulatory T cells. We used CD1d tetramer from Proimmune to stain mouse iNKT cells. We used a mouse tumor dissociation kit from Miltenyi Biotec (Bergisch Gladbach, Germany) to generate single-cell suspensions from 4T1-ROR1 tumors. Subsequently, erythrocytes and dead cells were removed by Percoll density gradient. Signals were acquired using a FACSCanto Flow Cytometer (BD Biosciences, San Diego, CA).
Statistical methods
Pairwise differences in bioluminescent signals, or cell numbers per tumor weights, were analyzed at selected time points using the Wilcoxon rank-sum test, and we characterized survival data using the Log-rank test. A P value less than 0.05 was considered significant. We treated 8 animals per group, which provided 80% power to detect an effect size of 1 standard deviation between groups, based on a t-test with a 2-sided significance level of 0.05. With the exception of toxicity studies, investigators conducting the experiments were not blinded. All statistical analyses were performed using GraphPad Prism software version 6.0.
Study approval
Experiments and handling of mice were conducted following federal, state, and local guidelines under an IACUC protocol and with approval from the Fred Hutchinson Cancer Research Center IACUC.
RESULTS
Therapeutic limitations of conventional CAR-T cell therapy
In our first set of experiments, we examined why injections of cancer-specific CAR-T cells fail to eradicate solid tumors, using an orthotopic 4T1 mouse mammary tumor model that recapitulates the immunosuppressive microenvironment and metastatic pattern typified by human breast cancer(16, 17). To create therapeutic lymphocytes against these tumors, we transduced mouse T cells with a retrovirus encoding an ROR1–28z CAR specific for the tyrosine kinase-like orphan receptor ROR1(24), which is strongly expressed at the cell surface of breast cancers (particularly triple-negative breast cancer; (25, 26)). This CAR is comprised of an anti-mouse ROR1 scFv fused to the murine CD8 transmembrane region, murine CD28 signal transduction domain, and murine CD3 cytoplasmic domains (Fig. 2A). We confirmed successful transduction of mouse T lymphocytes with ROR1–28z transgenes (Fig. 2B), which enables them to destroy 4T1 tumor cells existing in culture (Fig. 2C). To determine whether these CAR-T cells can persist and proliferate in the tumor microenvironment in vivo and identify how tumors adapt to these attacks, we locally injected 1×106 ROR1–28z CAR-T cells into established 4T1 tumors (thereby bypassing the problem of tumor targeting) and compared cellular compositions of the tumor microenvironment to that of untreated 4T1 tumors over time. We found that ROR-28z CAR-T cells did not thrive in 4T1 tumors, and only expanded 1.65-fold over a 12-day period (Fig. 2D, lower panel) before tumors reached endpoint criteria based on their size. Tumor growth was associated with an increased accumulation of cells known to inhibit tumor-specific T cell responses in cancer patients, including tumor associated macrophages (TAMs)(27), myeloid derived suppressor cells (MDSCs)(28), and regulatory T cells (Tregs)(29). Notably, CAR-T cell treatment was associated with higher numbers of these suppressor cells (in particular populations of monocytic- and granulocytic- MDSCs; Fig. 2D, upper panel) compared to controls, indicating that tumors can amplify their populations as a strategy to evade T cell attacks.
We next examined the therapeutic benefit provided by a clinically representative dose of ROR1–28z CAR-T cells (10×106) administered systemically rather than locally. In order to track and quantify the in vivo migration and accumulation of the transferred T cells in relation to 4T1 tumors, we included vectors for click beetle luciferase (CBR-luc)(30) in the plasmid. The results establish that, although intravenously infused T cells accumulate at high levels in the spleen and the liver, they inefficiently traffic to tumor sites (Fig. 3A–C), yielding a modest 6-day survival advantage compared to untreated control animals (Fig. 3D, E).
Fig. 3. Systemic infusions of tumor-specific CAR-T cells produce only modest therapeutic benefits.

Ten days after 4T1-ROR1 tumor cells were transplanted into the mammary glands of BALB/cJ mice, the animals were intravenously injected with 107 ROR1–28z CAR-transduced T cells or tumor-irrelevant (anti-GP75) control lymphocytes. The CAR-T cells co-expressed click beetle red luciferase (CBR-luc) reporter so we could measure the dynamics of programmed lymphocyte targeting. (A) Sequential bioluminescence imaging of the adoptively transferred T cells in three representative mice from each cohort (n = 8). (B) CBR-luc signal intensities from T cells at the tumor site examined via sequential bioluminescence imaging every two days after cell transfer. At the indicated time points, pairwise differences in photon counts between treatment groups were analyzed with the Wilcoxon rank-sum test. (C) Multicolor flow cytometry of cells recovered from tumors six days after CAR-T cell infusion. Adoptively transferred ROR1–28z CAR-transduced T cells were identified by positive labeling for CD3 and the c-myc tag incorporated in the CAR (see Fig. 2A). (D) Progression of 4T1-ROR1 tumor growth following therapy. Every line represents one animal and the dots indicate tumor sizes, as determined by caliper measurements. Shown are results from eight animals pooled from three independent experiments. (E) Kaplan-Meier survival curves for treated versus control mice. ms, median survival. Statistical analysis was performed using the log-rank test and P < 0.05 was considered significant.
Rational design of nanocarriers to reshape the tumor milieu
In the case of conventional nanocarrier-mediated chemotherapy, it is desirable for the tumor cells to internalize the drug-loaded particle(31, 32). By contrast, immune modulators are more effective if they are slowly released from nanocarriers situated in the stroma, so they must be engineered in a way different from those designed to deliver chemotherapy or siRNA.
We decided upon a liposomal drug carrier composed of egg phosphatidylcholine, cholesterol, and DSPE–PEG–maleimide (Fig. 4A) rather than polymeric nanoparticles because liposomes tend to be non-toxic, biodegradable, and non-immunogenic(33, 34). Furthermore, their flexible physicochemical properties facilitate manipulation in order to address various delivery considerations. To propel homing of the particles to tumors, we decorated them with iRGD peptide so they can bind to αv integrins(35). Proteolytic processing of iRGD then unmasks a second sequence motif, the cryptic C-terminal CendR motif (R/KXXR/K), which binds to neuropilin-1 and activates an endocytic bulk transport pathway through tumor tissue(36). Once activated, this pathway can take iRGD-bound and -unbound payloads deep into solid malignancies (as demonstrated in human tumor explants as well as mouse models) ((35, 37–39)). We confirmed the tumor-targeting effect of iRGD in a mouse model of glioma, which we use as one of our in vivo test systems (see Therapeutic studies below). Decorating liposomes with iRGD peptide improved tumor localization of systemically infused liposomes by an average of 3.2-fold, when compared to non-targeted liposomes (P = 0.0074; Supplementary Fig. S1A, B). To attenuate immunosuppression in the tumor microenvironment and simultaneously increase the number and activity of tumor-specific effector immune cells, we chose to combine two immunomodulator agents as nanoparticle cargo: PI-3065, a selective inhibitor of the p110δ PI3K kinase that has activity against immune-suppressive regulatory T cell subsets and tumor-associated myeloid cells(40), and 7DW8-5, an immunostimulant-invariant natural killer T cell (iNKT) agonist with 100-times higher potency than its parental compound, α-GalCer(41, 42). We decided to stimulate iNKT rather than T cells, as activated iNKT cells themselves orchestrate broad anti-tumor attacks that include several other cell types, including natural killer (NK-) cells, T cells, macrophages, neutrophils, and dendritic cells(43). Both PI-3065 and 7DW8-5 are highly hydrophobic and can be efficiently placed inside the bilayer of the liposome (Fig. 4A, Supplementary Fig. S2A–C).
Fig. 4. Nanoparticle-based delivery systems carrying synergistic immunomodulatory drug combinations can sway the tumor microenvironment of 4T1 breast tumors from suppressive to permissive.

(A) Schematic description of the targeted drug nanocarrier used in our experiments. Also depicted are the chemical structures of the two compounds we encapsulated into the nanoparticles and the iRGD targeting ligand that we coupled to their surface. (B) Mice with established (d 10) 4T1 breast tumors were treated with 7×1013 liposomes (2 mg lipid) carrying 150 µg PI-3065 and 10 µg 7DW8-5, either alone or in combination, or with equivalent non-encapsulated drugs every three days. These graphs display absolute numbers of immune suppressor cells (top panel) and anti-tumor effector cells (lower panel) in 4T1 breast tumors after six treatments (day 26). The flow cytometry antibody panel used to phenotype these cells is the same as that described in Fig. 2D. (C) Progression of 4T1-ROR1 tumor growth following therapy. Every line represents one animal and each dot reflects tumor size, as determined by caliper measurements. Shown are eight animals per treatment group pooled from three independent experiments. (D) Overview graph displaying the average tumor volumes ± S.E.M. for each treatment regimen over time. (E) Graphs displaying absolute numbers of immune suppressor cells (top panel) and anti-tumor effector cells (lower panel) in 4T1 breast tumors that ultimately developed resistance and relapsed despite repeated PI-3065 + 7DW8-5 liposome infusions.
To evaluate the advantage targeted carriers provide in comparison to freely circulating therapeutics, we treated mice bearing 4T1 tumors with triweekly intravenous infusions of iRGD-liposomes loaded with PI-3065 and 7DW8-5 (either alone or in combination), or for comparison systemic injections of the carrier-free drugs in equal amounts. Afterwards, differences in the composition of immune suppressor cells and anti-tumor cells were measured, and tumor progression rates were compared. We observed that only the nanoparticle-formulated combination of PI-3065 and 7DW8-5 potently reduced the concentration of immune suppressors at the tumor site, in particular TAMs (9.4-fold), monocytic MDSCs (4.6-fold) and Tregs (4.8-fold). At the same time, this combination increased the number of anti-tumor immune effector cells, such as CD8+ T cells (6.2-fold) and iNKT cells (29.8-fold; Fig. 4B). As a result, the disease remained stable in such treated animals for as long as 30 days following the first nanoparticle injection, whereas tumors progressed in all other treatment cohorts. However, despite continued triweekly nanoparticle infusions, tumors eventually developed resistance and relapsed (Fig. 4C, D), displaying high numbers of monocytic MDSCs and TAMs (Fig. 4E). These data indicate that rationally designed nanocarriers can significantly improve the effect of immunomodulatory agents compared to non-targeted drugs and achieve substantial tumor stasis even in relatively non-immunogenic cancers, such as our 4T1 model.
Safety studies
Guided by the therapeutic benefits achieved with PI-3065/7DW8-5 liposome injections, we next assessed whether these nanoreagents are biocompatible and safe for repeated dosing. Mice were injected with a total of nine doses of 7×1013 liposomes (3 doses/week × 3 weeks), or phosphate-buffered saline as a control. To measure effects of the nanocarriers per se, a third cohort of mice was injected with empty control liposomes. Mice were euthanized 24 h after the final dose, body weight was recorded, blood was collected by retro-orbital bleed for serum chemistry, and a complete gross necropsy was performed. There was no difference in body weights between groups. The following tissues were evaluated by a board-certified staff pathologist: liver, kidney, heart, spleen, and lungs. Histopathological evaluation revealed moderate mononuclear infiltrates in the liver and heart of PI-3065/7DW8-5 liposome-treated mice (Fig. 5), which is a mild version of the severe hepatitis reported upon conventional α-GalCer treatment(44). Cellular infiltrates in the parenchyma were occasionally associated with hepatocellular loss and hepatocytes showing high mitotic activity (up to 5 mitotic figures were present in a single 400× field, Fig. 5). Importantly, overall liver function was normal in all PI-3065/7DW8-5 liposome-treated mice, with only minimally elevated blood levels of the liver enzyme alanine transaminase (ALT; Fig. 5). Also, kidneys of PI-3065/7DW8-5 liposome-treated mice showed slight multifocal interstitial, perivascular, and periglomerular infiltrates of mononuclear cells (Fig. 5); these lesions were subtle, and there was no associated proteinuria or necrosis. Serum chemistry revealed mild leukocytosis in the PI-3065/7DW8-5 liposome treatment group, accounted for by neutrophilia, monocytosis, and lymphocytosis, which reflects the proinflammatory condition caused by treatment with α-GalCer. We also detected a mild anemia in this group, which is likely associated with the inflammatory phenotype (Fig. 5). In “anemia of inflammation”, erythropoiesis is iron-restricted by hepcidin-mediated hypoferremia and erythrocyte production is suppressed by cytokines acting on erythroid progenitors(45). Levels of glucose were within normal limits for all mice tested (106–278 mg/dl). Hyperglycemia, a documented side effect of PI3K inhibitors(46), was not present in treated mice. All serum chemistry, blood counts, and histopathology of animals injected with empty control liposomes were normal. Taken together, these results indicate PI-3065/7DW8-5 liposomes are biocompatible and safe for repeated dosing. The most prominent histological lesions, mononuclear infiltrates around portal tracts, central veins, and within the parenchyma of the liver, were minor reactions that usually resolve with minimal or no clinical intervention(47).
Fig. 5. PI-3065/7DW8-5 liposomes are safe for repeated dosing.
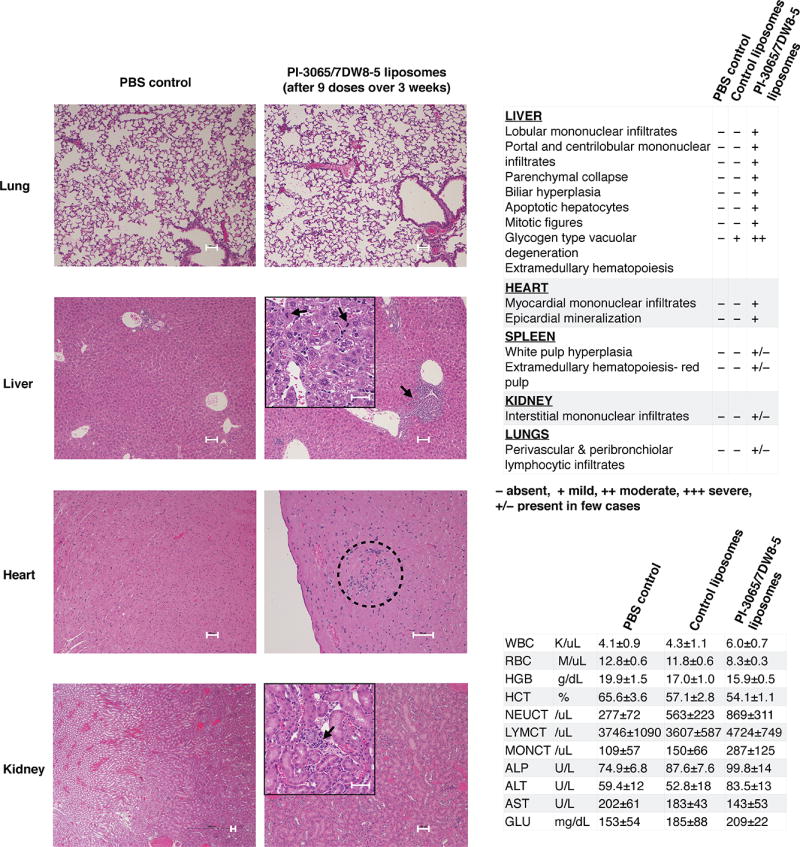
Representative hematoxylin and eosin-stained sections of several organs from controls or PI-3065/7DW8-5 liposome-treated animals (nine doses of 7×1013 empty control liposomes: 2 mg lipid; 3 doses × 3 weeks, or liposomes carrying 150 g PI-3065 and 10 µg 7DW8-5). Scale bars, 100 µm. Serum chemistry and blood counts for animals treated with PI-3065/7DW8-5 liposomes, or with phosphate-buffered saline as a control are shown on the right.
Therapeutic studies
The results described above indicate that repeated infusions of targeted PI-3065/7DW8-5 liposomes are a safe and effective method to create a tumor microenvironment where suppression of immune effector cells is minimal and functional support for them is maximal. Accordingly, we next sought to synergistically combine nanoparticle conditioning and adoptive T cell therapy to test whether liposomes can create a therapeutic window for externally programmed tumor-specific T cells to be effective. To accomplish this, we repeated our in vivo studies in the 4T1 breast cancer model (which did not respond well to conventional infusions of CAR-T cells; Fig. 2), but this time administered liposome infusions according to the treatment regimens illustrated in Fig. 6A: (i) 9 doses of PI-3065/7DW8-5 liposomes before CAR-T cell infusion, or (ii) CAR-T cell administration first, followed by 9 doses of PI-3065/7DW8-5 liposomes. As in our previous experiments, we retrovirally transduced adoptively transferred CAR-T cells with luciferase to track their migration and accumulation in relation to 4T1 tumors. Serial imaging of mice revealed that ROR1–28z CAR-T cells concurrently injected with the starting dose of PI-3065/7DW8-5 liposomes showed only a modest (yet significant) improvement in their ability to expand in established 4T1 tumors (3.4-fold by day 6 after infusion; Fig. 6B, C, middle panel). By contrast, T cells infused after 3-weeks of PI-3065/7DW8-5 liposome pre-conditioning accumulated in breast tumors at 22-fold higher levels by day 6 post-infusion relative to T cells injected into mice that received no preconditioning (Fig. 6B, C, right panel). Quantitatively similar results were obtained by dissecting tumors 6 days after T cell transfer and assessing the frequency of infiltrating ROR1–28z CAR-T cells by flow cytometry (Fig. 6D, E). Liposome preconditioning followed by CAR-T cell therapy eradicated 4T1 tumors in four of eight treated mice (Fig. 6F–H), and the other four showed substantial tumor regression with a doubling (average 42-day improvement) in survival compared to conventional CAR-T cell therapy. This regimen was safe and well-tolerated (Supplementary Fig. S3A, B). Although PI-3065/7DW8-5 liposomes administered during and after T cell therapy never produced complete clearance, they increased survival by 17 days compared to injecting T cells only (Fig. 6F–H). We already established in Fig. 4 that unformulated PI-3065 as monotherapy has no significant therapeutic effect in our 4T1 breast tumor model. To test whether repeated PI-3065 infusions can boost the anti-tumor activity of adoptively transferred CAR-T cells in this model, we administered CAR-T cells followed by 9 doses (150 µg each) of PI-3065 (Supplementary Fig. S4A). We measured a modest increase in bioluminescent CAR-T cell signal at the tumor site (1.4-fold higher by day 6 after T cell infusion, P = 0.14, ns) in animals that received PI-3065 (Supplementary Fig. S4B, C), which translated into an average 8-day survival benefit (Supplementary Fig. S4D, E).
Fig. 6. Nanocarrier-mediated remodeling of the tumor microenvironment improves function and persistence of adoptively transferred CAR-T cells.
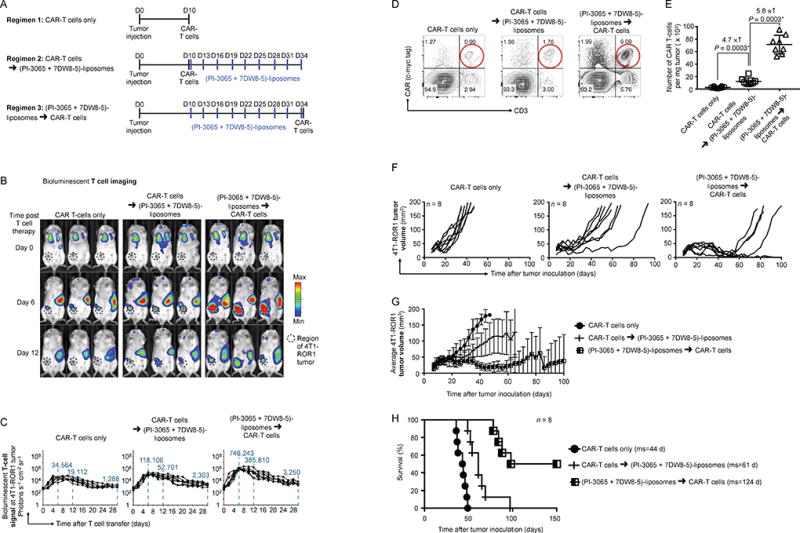
(A) Time lines and dosing regimens. (B) Sequential bioluminescence imaging of the adoptively transferred T cells in three representative mice from each cohort (n = 8/cohort). (C) CBR-luc T cell signal intensities at the tumor site obtained by sequential bioluminescence imaging every two days after cell transfer. (D) Multicolor flow cytometry of cells recovered from tumors six days after CAR-T cell infusion. Adoptively transferred ROR1–28z CAR-transduced T cells were identified by positive labeling for CD3 and c-myc tag (see Fig. 2A). (E) Graphs displaying absolute numbers of ROR1–28z CAR-T cells in 4T1-ROR tumors. Pairwise differences in cell counts between treatment groups were analyzed with the Wilcoxon rank-sum test. (F) Progression of 4T1-ROR1 tumor growth following therapy. Every line represents one animal and each dot reflects tumor size. Shown are eight animals pooled from three independent experiments. (G) Overview graph displaying the average tumor volumes ± S.E.M for each treatment regimen over time. (H) Kaplan-Meier survival curves for treated versus control mice. ms, median survival. Statistical analysis was performed using the log-rank test and P < 0.05 was considered significant.
To evaluate the therapeutic efficacy of our technology in a second clinically relevant system, we used a genetically engineered mouse model of human glioblastoma (GBM)(18). Adoptive cell therapy using T cells engineered with chimeric antigen receptor (CAR) targeting an ideal molecular marker in GBM, e.g., epidermal growth factor receptor type III (EGFRvIII), has demonstrated satisfactory efficacy in treating malignant brain tumors(48). To model this clinical scenario, we induced EGFRvIII+ glioma driven by RCAS-PDGF and RCAS-Cre in Ntv-a; Ink4a/Arf−/−; floxed PTEN transgenic mice(18). Following tumor initiation, these mice establish 4–6-mm diameter tumors, with nearly complete tumor penetrance within 17–21 days (Fig. 7A). To treat these lesions, we created a CAR directed against EGFRvIII by linking a single-chain antibody to a synthetic receptor skeleton comprised of the CD8 hinge, the CD28 transmembrane and signaling domains, and CD3ζ signaling domains (Fig. 7B). We transduced murine T cells using recombinant retroviruses expressing these EGFRvIII-specific CARs, which enabled them to recognize and lyse brain tumor cells isolated from PDGF-EGFRvIII tumors (Fig. 7C). We then treated mice with these CAR-expressing T cells, either as monotherapy or followed by four doses of PI-3065/7DW8-5 liposomes (Fig. 7D). In order to track and quantify the in vivo migration of these cells in relation to brain tumors, we included vectors encoding click beetle luciferase in the plasmid. Results based on this PDGF-EGFRvIII tumor model were similar to those from 4T1 breast tumor (Fig. 6): although EGFRvIII-specific CAR-T cells traffic to PDGF-EGFRvIII tumors, they do so very inefficiently and do not persist or expand (Fig. 7E, F), producing only a 4-day survival advantage compared to untreated controls (Fig. 7G). By contrast, CAR-T cells infused after nanoparticle preconditioning underwent significant proliferation at the tumor site (12.1-fold higher peak photon count relative to conventional T cell therapy on day 6, P < 0.0001, Fig. 7F) and substantially reduced tumor growth (they more than doubled the survival of treated mice, Fig. 7G).
Fig. 7. Nanoparticle preconditioning improves CAR-T cell therapy of glioblastoma.
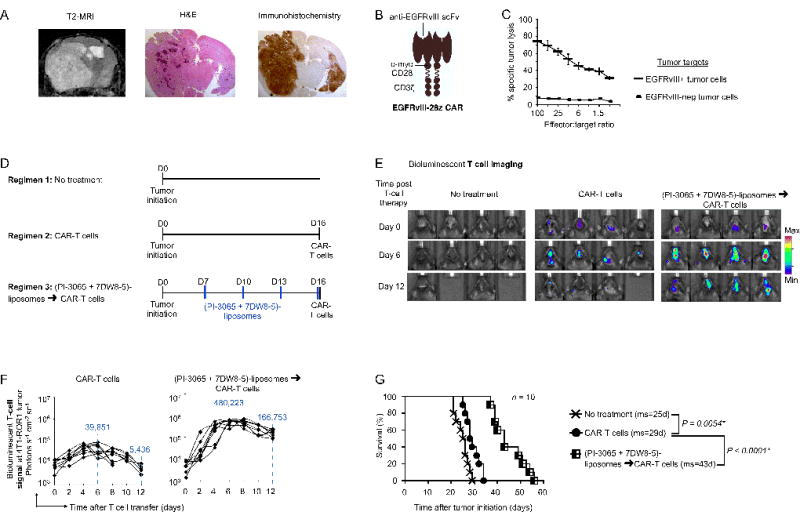
(A) T2 MRI scan, H&E and immunohistochemical analysis following initiation of a PDGFB-EGFRvIII tumor in Tg(NES-TVA);Cdkn2a (Ink4a-Arf)−/−;Ptenfl/fl; LSL EGFRvIII mice on post-induction day 21. Images are shown at 1.5× magnification. (B) Schematic of the chimeric receptor we used to recognize EGFRvIII. (C) 51Cr release cytotoxicity assay of anti-EGFRvIII CAR-transduced T cells reacting with tumor cells isolated from PDGFB-EGFRvIII tumor lesions. Data are representative of two independent experiments. (D) Time lines and dosing regimens. (E) Sequential bioluminescence imaging of the adoptively transferred T cells in four representative mice from each cohort. (F) CBR-luc T cell signal intensities at the tumor site obtained by sequential bioluminescence imaging every two days after cell transfer. (G) Kaplan-Meier survival curves for treated versus control mice. ms, median survival. Statistical analysis was performed using the log-rank test and P < 0.05 was considered significant.
In conclusion, our results demonstrate that nanoparticle-induced remodeling of the tumor microenvironment is safe and can substantially increase the therapeutic activity of CAR-T cells. This adjuvant nanotherapy is most effective when administered repeatedly several weeks prior to T cell infusion, thereby permitting the delivered immune modulating drugs to first complete their effects on components of the tumor microenvironment.
DISCUSSION
Solid cancers present formidable barriers against the use of adoptive cell therapies, including functional suppression of T cells and inhibition of their localization. In this project, we developed a nanoreagent that can – as a singular therapeutic – safely decrease the numbers of immunosuppressive cells in the tumor microenvironment and, at the same time, boost key mediators of anti-tumor immune responses. We demonstrate that repeated administration of these functionalized nanocarriers favorably preconditions the tumor milieu for a limited but defined time period, during which adoptively-transferred T cells can have curative effects.
The dosing and frequency required to achieve this transient state will likely require optimization in a clinical setting, and may prove to be different for various tumor types and disease stages. In our preclinical test system, we intravenously infused PI-3065/7DW8-5 liposomes every three days and induced tumor regression followed by stasis between 10 and 15 days after the first liposome injection (Fig. 4C, D). In a clinical scenario, the same amount of time is required for apheresis, manufacturing, and re-infusion of engineered CAR-T cells into the patient (which typically involves from 2.5 to 3 weeks); our evidence indicates this provides a period long enough to pre-condition patients with PI-3065/7DW8-5 liposomes. An advantage of the engineered nanoparticles described here is, after they are configured for specificity and functionality, they can safely be lyophilized for long-term storage. This enables them to be available for “off the shelf” use as early as the day of diagnosis.
We selected 7DW8-5 to improve the immune response to tumors because it has the strongest adjuvant effect measured for a CD1d-binding NKT cell ligand in humans(41). Originally identified by Li. et al. out of a library of 25 synthetic analogues of α-GalCer, 7DW8-5 has been shown in multiple vaccine trials to induce a significantly higher Th1 response compared to α-GalCer(42, 49, 50). The selective p110δ inhibitor PI-3065 was chosen as a desirable co-modulator to accompany 7DW8-5, based on previous work establishing it can delay 4T1 tumor growth and reduce Treg suppression in mice(40). Also, other PI3K- δ inhibitors such as CAL-101 have been reported to decrease the numbers of suppressive Treg and increase the numbers of antitumor T cells (51, 52). In our 4T1 test system, even repeated administration of unencapsulated PI-3065 did not significantly reduce immune suppressor cells (1.2-fold ↓ Tregs, P = 0.38), but required delivery by targeted liposomal nanocarriers (2.7-fold ↓ Tregs, P = 0.0051; Fig. 4B) to be effective.
But beyond the PI-3065/7DW8-5 combination described here, our treatment concept of “releasing immune brakes” while at the same time “stepping on the gas” using a targeted nanocarrier to co-deliver synergistic immunomodulatory agents could be applied to other potent combinations of effectors. Promising candidates to remove suppressor cells are, for example: (1) BLZ945: an inhibitor of colony stimulating factor 1 receptor (CSF1R) that selectively binds to it on tumor-associated macrophages, thereby inhibiting signal transduction pathways mediated by this receptor in TAMs(23, 53); and (2) PLX3397: a small-molecule receptor tyrosine kinase inhibitor which binds to and inhibits phosphorylation of stem cell factor receptor (c-KIT), CSF1R, and FMS-like tyrosine kinase 3 (FLT3), thereby causing the down-modulation of TAMs(54). Drug candidates that can stimulate an adaptive immune response similar to that resulting from the α-GalCer agonist 7DW8-5 include: (1) TLR-based immune adjuvants, such as monophosphoryl lipid A (MPLA), which could easily be incorporated into the liposome envelope(55), or (2) STING agonists, which have shown dramatic effects against solid tumors in rodent models when administered locally and repeatedly(56, 57).
Clinical implementation of liposome-mediated tumor preconditioning for adoptive T cell therapy of cancer will rely heavily on the safety of the procedure. Many liposome products are already available for human use (e.g., Doxil®, Ambisome®, and DepoDur™), and various others are undergoing clinical testing, based on their favorable safety profile (along with the robust, rapid, and affordable manufacturing processes that are available)(58). As indicated above, even repeated infusions of empty control nanocarriers did not result in significant macroscopic or microscopic lesions in our test system. Furthermore, liposomes that co-delivered a potent α-GalCer agonist induced only mild inflammation (Fig. 5). Notably, these treatments did not significantly affect organ function, animal weight, or their overall well-being, which highlights the safety and clinical applicability of the approach. This is in sharp contrast to systemic administration of α-GalCer (which fails to promote anti-tumor immune responses; Fig. 4C, D), a treatment known to trigger liver injury and auto-immune hepatitis(44).
In summary, our work describes a new nanotechnology approach that can promote T cell therapy for solid tumors. Implemented in the clinic as a preconditioning regimen, targeted nanocarriers that transiently reset the tumor microenvironment may improve the success of such treatments. More broadly, we believe that this platform could be explored clinically to maximize the outcomes of other immunotherapy approaches that rely on robust T cell responses to treat solid tumors (such as personalized cancer vaccines, neoantigen-targeted methods, and bispecific antibody therapies).
Supplementary Material
SIGNIFICANCE.
A new nanotechnology approach can promote T cell therapy for solid tumors.
Acknowledgments
This work was supported by the Fred Hutchinson Cancer Research Center’s Immunotherapy Initiative with funds provided by the Bezos Family Foundation (M.T. Stephan), the NCI (RO1CA195718; M.T. Stephan and E.C. Holland), and the Immunotherapy Integrated Research Center (IIRC) at Fred Hutchinson Cancer Research Center (Collaborative Grant Accelerator; M.T. Stephan and E.C. Holland).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J Clin Oncol. 2017;35(26):3010–20. doi: 10.1200/JCO.2017.72.8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. 2016;59(5):468–79. doi: 10.1007/s11427-016-5023-8. [DOI] [PubMed] [Google Scholar]
- 5.Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. 2017;66(11):1425–36. doi: 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamers CH, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans. 2016;44(3):951–9. doi: 10.1042/BST20160037. [DOI] [PubMed] [Google Scholar]
- 7.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–20. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostrand-Rosenberg S, Fenselau C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol. 2018;200(2):422–31. doi: 10.4049/jimmunol.1701019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davoodzadeh Gholami M, Kardar GA, Saeedi Y, Heydari S, Garssen J, Falak R. Exhaustion of T lymphocytes in the tumor microenvironment: Significance and effective mechanisms. Cell Immunol. 2017;322:1–14. doi: 10.1016/j.cellimm.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 10.John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19(20):5636–46. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 11.Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther. 2017;25(9):2214–24. doi: 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20(16):4262–73. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology. 2017;6(1):e1249561. doi: 10.1080/2162402X.2016.1249561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramos RN, Piaggio E, Romano E. Mechanisms of Resistance to Immune Checkpoint Antibodies. Handb Exp Pharmacol. 2017 doi: 10.1007/164_2017_11. [DOI] [PubMed] [Google Scholar]
- 15.Kourie HR, Klastersky JA. Side-effects of checkpoint inhibitor-based combination therapy. Curr Opin Oncol. 2016;28(4):306–13. doi: 10.1097/CCO.0000000000000295. [DOI] [PubMed] [Google Scholar]
- 16.Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol. 2001 doi: 10.1002/0471142735.im2002s39. Chapter 20:Unit 20 2. [DOI] [PubMed] [Google Scholar]
- 17.Bailey-Downs LC, Thorpe JE, Disch BC, Bastian A, Hauser PJ, Farasyn T, et al. Development and characterization of a preclinical model of breast cancer lung micrometastatic to macrometastatic progression. PLoS One. 2014;9(5):e98624. doi: 10.1371/journal.pone.0098624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling Adult Gliomas Using RCAS/t-va Technology. Transl Oncol. 2009;2(2):89–95. doi: 10.1593/tlo.09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18(10):1373–85. doi: 10.1016/S1470-2045(17)30517-X. [DOI] [PubMed] [Google Scholar]
- 20.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017;3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erskine CL, Henle AM, Knutson KL. Determining optimal cytotoxic activity of human Her2neu specific CD8 T cells by comparing the Cr51 release assay to the xCELLigence system. J Vis Exp. 2012;(66):e3683. doi: 10.3791/3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu H, Acquaviva J, Ramachandran P, Boskovitz A, Woolfenden S, Pfannl R, et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci U S A. 2009;106(8):2712–6. doi: 10.1073/pnas.0813314106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–72. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang S, Chen L, Wang-Rodriguez J, Zhang L, Cui B, Frankel W, et al. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. Am J Pathol. 2012;181(6):1903–10. doi: 10.1016/j.ajpath.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Chen L, Cui B, Chuang HY, Yu J, Wang-Rodriguez J, et al. ROR1 is expressed in human breast cancer and associated with enhanced tumor-cell growth. PLoS One. 2012;7(3):e31127. doi: 10.1371/journal.pone.0031127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chien HP, Ueng SH, Chen SC, Chang YS, Lin YC, Lo YF, et al. Expression of ROR1 has prognostic significance in triple negative breast cancer. Virchows Arch. 2016;468(5):589–95. doi: 10.1007/s00428-016-1911-3. [DOI] [PubMed] [Google Scholar]
- 27.Zhao X, Qu J, Sun Y, Wang J, Liu X, Wang F, et al. Prognostic significance of tumor-associated macrophages in breast cancer: a meta-analysis of the literature. Oncotarget. 2017;8(18):30576–86. doi: 10.18632/oncotarget.15736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang Z, Wen C, Chen X, Yin R, Zhang C, Wang X, et al. Myeloid-derived suppressor cell and macrophage exert distinct angiogenic and immunosuppressive effects in breast cancer. Oncotarget. 2017;8(33):54173–86. doi: 10.18632/oncotarget.17013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banin-Hirata BK, de Oliveira CEC, Losi-Guembarovski R, Ozawa PMM, Vitiello GAF, de Almeida FC, et al. The prognostic value of regulatory T cells infiltration in HER2-enriched breast cancer microenvironment. Int Rev Immunol. 2017:1–7. doi: 10.1080/08830185.2017.1401620. [DOI] [PubMed] [Google Scholar]
- 30.Dobrenkov K, Olszewska M, Likar Y, Shenker L, Gunset G, Cai S, et al. Monitoring the efficacy of adoptively transferred prostate cancer-targeted human T lymphocytes with PET and bioluminescence imaging. J Nucl Med. 2008;49(7):1162–70. doi: 10.2967/jnumed.107.047324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4(128):128ra39. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 32.Taghavi S, Ramezani M, Alibolandi M, Abnous K, Taghdisi SM. Chitosan-modified PLGA nanoparticles tagged with 5TR1 aptamer for in vivo tumor-targeted drug delivery. Cancer Lett. 2017;400:1–8. doi: 10.1016/j.canlet.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Hua S, Wu SY. The use of lipid-based nanocarriers for targeted pain therapies. Front Pharmacol. 2013;4:143. doi: 10.3389/fphar.2013.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmad A, Sheikh S, Taran R, Srivastav SP, Prasad K, Rajappa SJ, et al. Therapeutic efficacy of a novel nanosomal docetaxel lipid suspension compared with taxotere in locally advanced or metastatic breast cancer patients. Clin Breast Cancer. 2014;14(3):177–81. doi: 10.1016/j.clbc.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 35.Yin H, Yang J, Zhang Q, Yang J, Wang H, Xu J, et al. iRGD as a tumorpenetrating peptide for cancer therapy (Review) Mol Med Rep. 2017;15(5):2925–30. doi: 10.3892/mmr.2017.6419. [DOI] [PubMed] [Google Scholar]
- 36.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328(5981):1031–5. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamilton AM, Aidoudi-Ahmed S, Sharma S, Kotamraju VR, Foster PJ, Sugahara KN, et al. Nanoparticles coated with the tumor-penetrating peptide iRGD reduce experimental breast cancer metastasis in the brain. J Mol Med (Berl) 2015;93(9):991–1001. doi: 10.1007/s00109-015-1279-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Wang L, Fai Chan H, Xie W, Chen S, He C, et al. Co-delivery of paclitaxel and tetrandrine via iRGD peptide conjugated lipid-polymer hybrid nanoparticles overcome multidrug resistance in cancer cells. Sci Rep. 2017;7:46057. doi: 10.1038/srep46057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Lin P, Perrett I, Lin J, Liao YP, Chang CH, et al. Tumor-penetrating peptide enhances transcytosis of silicasome-based chemotherapy for pancreatic cancer. J Clin Invest. 2017;127(5):2007–18. doi: 10.1172/JCI92284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510(7505):407–11. doi: 10.1038/nature13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, Fujio M, Imamura M, Wu D, Vasan S, Wong CH, et al. Design of a potent CD1d-binding NKT cell ligand as a vaccine adjuvant. Proc Natl Acad Sci U S A. 2010;107(29):13010–5. doi: 10.1073/pnas.1006662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Padte NN, Boente-Carrera M, Andrews CD, McManus J, Grasperge BF, Gettie A, et al. A glycolipid adjuvant, 7DW8-5, enhances CD8+ T cell responses induced by an adenovirus-vectored malaria vaccine in non-human primates. PLoS One. 2013;8(10):e78407. doi: 10.1371/journal.pone.0078407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bedard M, Salio M, Cerundolo V. Harnessing the Power of Invariant Natural Killer T Cells in Cancer Immunotherapy. Front Immunol. 2017;8:1829. doi: 10.3389/fimmu.2017.01829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biburger M, Tiegs G. Alpha-galactosylceramide-induced liver injury in mice is mediated by TNF-alpha but independent of Kupffer cells. J Immunol. 2005;175(3):1540–50. doi: 10.4049/jimmunol.175.3.1540. [DOI] [PubMed] [Google Scholar]
- 45.Andrews NC. Anemia of inflammation: the cytokine-hepcidin link. J Clin Invest. 2004;113(9):1251–3. doi: 10.1172/JCI21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geuna E, Roda D, Rafii S, Jimenez B, Capelan M, Rihawi K, et al. Complications of hyperglycaemia with PI3K-AKT-mTOR inhibitors in patients with advanced solid tumours on Phase I clinical trials. Br J Cancer. 2015;113(11):1541–7. doi: 10.1038/bjc.2015.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol. 2016;13(3):316–27. doi: 10.1038/cmi.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399) doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghnewa YG, O'Reilly VP, Vandenberghe E, Browne PV, McElligott AM, Doherty DG. Retinoic acid induction of CD1d expression primes chronic lymphocytic leukemia B cells for killing by CD8(+) invariant natural killer T cells. Clin Immunol. 2017;183:91–8. doi: 10.1016/j.clim.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 50.Li X, Huang J, Kaneko I, Zhang M, Iwanaga S, Yuda M, et al. A potent adjuvant effect of a CD1d-binding NKT cell ligand in human immune system mice. Expert Rev Vaccines. 2017;16(1):73–80. doi: 10.1080/14760584.2017.1256208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahmad S, Abu-Eid R, Shrimali R, Webb M, Verma V, Doroodchi A, et al. Differential PI3Kdelta Signaling in CD4(+) T-cell Subsets Enables Selective Targeting of T Regulatory Cells to Enhance Cancer Immunotherapy. Cancer Res. 2017;77(8):1892–904. doi: 10.1158/0008-5472.CAN-16-1839. [DOI] [PubMed] [Google Scholar]
- 52.Abu Eid R, Ahmad S, Lin Y, Webb M, Berrong Z, Shrimali R, et al. Enhanced Therapeutic Efficacy and Memory of Tumor-Specific CD8 T Cells by Ex Vivo PI3K-delta Inhibition. Cancer Res. 2017;77(15):4135–45. doi: 10.1158/0008-5472.CAN-16-1925. [DOI] [PubMed] [Google Scholar]
- 53.Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352(6288):aad3018. doi: 10.1126/science.aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dammeijer F, Lievense LA, Kaijen-Lambers ME, van Nimwegen M, Bezemer K, Hegmans JP, et al. Depletion of Tumor-Associated Macrophages with a CSF-1R Kinase Inhibitor Enhances Antitumor Immunity and Survival Induced by DC Immunotherapy. Cancer Immunol Res. 2017;5(7):535–46. doi: 10.1158/2326-6066.CIR-16-0309. [DOI] [PubMed] [Google Scholar]
- 55.Beck Z, Matyas GR, Jalah R, Rao M, Polonis VR, Alving CR. Differential immune responses to HIV-1 envelope protein induced by liposomal adjuvant formulations containing monophosphoryl lipid A with or without QS21. Vaccine. 2015;33(42):5578–87. doi: 10.1016/j.vaccine.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 56.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11(7):1018–30. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohkuri T, Kosaka A, Ishibashi K, Kumai T, Hirata Y, Ohara K, et al. Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunol Immunother. 2017;66(6):705–16. doi: 10.1007/s00262-017-1975-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics. 2017;9(2) doi: 10.3390/pharmaceutics9020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.