

Abstract
Periodontal diseases are among the most prevalent worldwide, but largely silent, chronic diseases. They affect the tooth-supporting tissues with multiple ramifications on life quality. Their early diagnosis is still challenging, due to lack of appropriate molecular diagnostic methods. Saliva offers a non-invasively collectable reservoir of clinically relevant biomarkers, which, if utilized efficiently, could facilitate early diagnosis and monitoring of ongoing disease. Despite several novel protein markers being recently enlisted by discovery proteomics, their routine diagnostic application is hampered by the lack of validation platforms that allow for rapid, accurate and simultaneous quantification of multiple proteins in large cohorts. Here we carried out a pipeline of two proteomic platforms; firstly, we applied open ended label-free quantitative (LFQ) proteomics for discovery in saliva (n = 67, including individuals with health, gingivitis, and periodontitis), followed by selected-reaction monitoring (SRM)-targeted proteomics for validation in an independent cohort (n = 82). The LFQ platform led to the discovery of 119 proteins with at least 2-fold significant difference between health and disease. The 65 proteins chosen for the subsequent SRM platform included 50 functionally related proteins derived from the significantly enriched processes of the LFQ data, 11 from literature-mining, and four house-keeping ones. Among those, 60 were reproducibly quantifiable proteins (92% success rate), represented by a total of 143 peptides. Machine-learning modeling led to a narrowed-down panel of five proteins of high predictive value for periodontal diseases with maximum area under the receiver operating curve >0.97 (higher in disease: Matrix metalloproteinase-9, Ras-related protein-1, Actin-related protein 2/3 complex subunit 5; lower in disease: Clusterin, Deleted in Malignant Brain Tumors 1). This panel enriches the pool of credible clinical biomarker candidates for diagnostic assay development. Yet, the quantum leap brought into the field of periodontal diagnostics by this study is the application of the biomarker discovery-through-verification pipeline, which can be used for validation in further cohorts.
Keywords: Biofluids*, Biomarker: Diagnostic, Biomarker: Prognostic, Selected reaction monitoring, Label-free quantification, Inflammation, Infectious disease, Diagnostic, Inflammatory response, Host-Pathogen Interaction, agressive periodontitis, chronic periodontitis, oral biofluids, periodontal disease, saliva
Periodontal diseases are oral biofilm induced chronic inflammatory diseases of the tooth-supporting (periodontal) tissues. Despite major improvements in oral hygiene practices in industrialized countries, severe periodontitis remains the sixth-most prevalent chronic disease worldwide, affecting almost 11.5% of many populations (1, 2). This cluster of oral diseases do not only affect the tooth-supporting tissues but also the other body parts by contributing to the development of life threating conditions, namely, cardiovascular disease or stroke (3, 4). Therefore, identifying early and abolishing the onset of these diseases is highly desirable.
Similar to the other chronic diseases in humans, there are still considerable challenges in diagnosis and classification for the cases of different forms of periodontal diseases and current diagnosis is based on subjective indices, mainly evaluating the past disease (5). The poor performance of clinical tools and unpredictability in the progression of the disease has led to a search for new, more accurate biomarkers in oral biofluids for periodontal disease screening, classification monitoring, and management since 1960s (6–9). The use of quantitative proteomics for characterizing periodontal diseases offers significant potential for providing “periodontal disease related finger prints” (10, 11). In this respect, saliva is a useful reservoir of clinically relevant biomarkers for reflection of periodontal diseases as well as general health (12–14). Early studied biomarkers in saliva were more etiology oriented and ranged from specific bacteria or their secreted products to host immune markers or tissue lysis products (15). A few biomarkers have been marketed for chair-side use but the most have disappeared from the market because of their low specificity (16–18). A single protein marker is less likely to reliably detect early periodontal disease or to provide a differential diagnosis between different forms of the disease (19). A better approach is to aim for a panel of related markers for conclusive prediction.
We have demonstrated earlier that label-free quantitative (LFQ) 1 mass spectrometry methods are able to facilitate characterization and concurrent quantitative analysis of the proteome in periodontal health and disease (10, 20). Although the non-targeted, shotgun proteomic workflows are considerably successful in the discovery of novel candidate markers and in generating hypotheses for periodontal diseases, no direct effect on improved diagnostic capacity has been demonstrated. This goes in line with the fact that no protein biomarker has been incorporated into the daily dental practice or in a clinical assay, despite more than 600 proteins have been linked to the disease by proteomics work (21). The main reason is the lack of validation platforms that allow for rapid, accurate and simultaneous quantification of multiple proteins in large cohorts. Targeted mass spectrometry (MS) methods are at the fore front for accurate (high specificity and sensitivity) measurement of dozens of proteins simultaneously in complex biological samples decreasing the requirements for (individual target) antibody-based assays (22, 23). The separation and detection methodology, termed selected reaction monitoring (SRM) or multiple reaction monitoring (MRM), has matured into a robust technology for reproducible and reliable quantification of protein panels in complex sample backgrounds. These advancements within the field has led to an increasing interest in using liquid chromatography (LC)-MS as a primary biomarker discovery and validation platform (24). Despite numerous reports describing the fast-growing application of SRM-based workflows for quantification of target peptides (proteins) in plasma (25–27), studies in saliva has been very limited (28, 29) and there has been no application to the field of periodontics.
We have followed two main mass-spectrometry guided strategies to identify biomarkers of periodontal diseases in human saliva. First, the discovery study with cross-sectional case-control design (n = 67) was conducted to dissect comparative saliva proteome in (1) periodontal health (2) during inflammatory but not destructive disease stage (gingivitis) (3) during advanced disease stage in healthy young individuals (generalized aggressive periodontitis) (4) during advanced but chronic disease stage in older individuals (generalized chronic periodontitis) by LFQ. Second, we have conducted multiplex LC-SRM assays with an independent cohort (n = 82) in order to qualify or validate the identified candidate markers by the discovery approach.
EXPERIMENTAL PROCEDURES
Phase 1: Discovery Phase (Exploratory Study)
Experimental Design and Rationale
The whole saliva samples were obtained from 67 systemically healthy subjects (age range 20–64 years) consisting of patients with generalized chronic periodontitis (CP), generalized aggressive periodontitis (AP), gingivitis (G) and individuals with periodontal health (H). The use of humans for study satisfied the requirements of the Ege University Institutional Review Board (Ethics number 16–12.1/16) and was conducted in accordance with the guidelines of the World Medical Association Declaration of Helsinki. It is confirmed that this cross-sectional case-control study conforms to STROBE guidelines for observational studies. Complete medical and dental histories were obtained from all participants. Systemic exclusion criteria were the presence of cardiovascular and respiratory diseases, diabetes mellitus, HIV infection, systemic inflammatory conditions or non-plaque-induced oral inflammatory conditions, immunosuppressive chemotherapy, and current pregnancy or lactation or smoking. None of the patients had taken medication such as antibiotics or anti-inflammatory drugs that could affect their periodontal status for at least 6 months before the study. Patients eligible for the study returned to the clinic for clinical measurement screening one-week after being pre-screened. Before being enrolled in the study, participants provided written and informed consent for use of their saliva samples and clinical data for scientific research purposes.
The clinical periodontal indices including probing depth (PD), clinical attachment loss (CAL), plaque index (PI) and bleeding on probing (BOP) were recorded by a manual periodontal probe by a trained and calibrated examiner (V.Ö.Ö.). The extent and severity of alveolar bone support was evaluated radiographically in each patient. The participants were classified into four groups based on their periodontal conditions according to the criteria proposed by the 1999 International Workshop for a Classification of Periodontal Diseases and Conditions (30). The AP group included 17 patients with ≥16 teeth. The patients had a non- contributory medical history and demonstrated with an early age of clinical manifestations with a generalized pattern of rapid attachment loss and bone destruction disproportionate to the magnitude of local etiological factors. Additionally, self-reported family history of periodontitis was a strong indicator of the diagnosis. These individuals had minimum of CAL greater 5 mm and PD greater 6 mm on eight or more teeth; at least three of these were other than central incisors or first molars. Radiographic bone loss was above 30% of root length affecting more than 3 permanent teeth other than first molars and incisors. The CP group (n = 17) included individuals who had a minimum four non-adjacent teeth with sites with CAL greater 5 mm and PPD greater 6 mm, and above 50% alveolar bone loss in at least two quadrants which was commensurate with the amount of plaque accumulation. They also had the mean BOP values above 63%. The G group (n = 17) had varying degrees of gingival inflammation with the mean BOP values above 50%, but no clinical attachment loss >2 mm, no sites with alveolar bone loss present in radiography (the distance between the cementoenamel junction and bone crest less 3 mm at above 95% of the proximal tooth sites). The individuals with periodontal health had no sites with PD greater 3 mm and CAL greater 2 mm, a mean BOP below 15% at the time of examination, and no detectable alveolar bone loss. The demographic and clinical details of details of the participants included in the analysis are presented in supplemental File S1.
Saliva Sampling and Processing
The whole saliva samples were obtained in the morning between 8.00 am–10.00 am, as this is the least variable time point during the day for saliva composition (31). The unstimulated saliva samples were collected by expectorating into sterile 50 ml tubes for 5 min as described earlier (32). Briefly, the participants were asked to avoid oral hygiene practices including flossing, brushing, and mouth-rinses as well as eating, and drinking for at least 2 h before collection. Before clinical periodontal measurements, each participant was asked first to rinse the mouth completely with water for 2 min, wait for 10 min, and then expectorate into sterile tubes for 5 min. On the day of analysis, the samples were thawed on ice and centrifuged at 10,000 × g for 15 min at 4 °C. The obtained supernatants were supplemented with the EDTA-free Protease Inhibitor Mixture (Sigma-Aldrich, Dorset, UK).
Label-free Quantitative Proteomic Analysis
Protein Digestion and C18 Clean Up
Total protein content of the collected supernatants were measured with Qubit® Protein Assay Kit (Thermo Scientific, Wohlen, Switzerland). Despite that the saliva collection was standardized by time, there were considerable inter-individual variations in the total protein concentrations. The total protein concentrations for H, G, CP, and AP (μg/ml, median (min-max)) were 764.3 (522–1290), 1110 (637–1970), 1140 (693–1850), 1118 (694–1760), respectively. The median total protein levels were found to be significantly different among the groups (p < 0.01). Therefore, total protein amount per sample was controlled. Solutions of 80 μg of total protein per sample were subjected to in-solution trypsin digestion according to the RapiGest protocol. Briefly, the supernatants were diluted with ammonium bicarbonate buffer to reach a neutral pH, then RapiGest was added to the samples at the final concentration of 0.1%. Afterward, the samples were reduced with dithiothreitol by incubation at 60 °C for 30 min and carbamidomethylated using iodoacetamide at a final concentration of 15 mm for 30 min in dark. The samples were digested with trypsin in 0.05 m triethylammonium bicarbonate (1:100 w:w) overnight at 37 °C. Trifluoroacetic acid (TFA) was added to a final concentration of 0.5% and the samples were incubated for 30 min at 37 °C. Peptide mixtures were desalted using reverse phase cartridges Finisterre SPE C18 (Wicom International AG, Maienfeld, Switzerland) according to the manufacturer's specifications. Each sample was evaporated using a Speedvac (Thermo Scientific) and subsequentially reconstituted in 3% acetonitrile (ACN) and 0.1% formic acid (FA).
Shotgun-MS Proteomics
Tryptic digests were analyzed on a LTQ Orbitrap Velos equipped with a nanospray ion source. Chromatographic separations of peptides on a Eksigent nanoLC-1D device (ABSciex, Concord, Ontario) coupled to an in-house pulled and packed tip column, 75 μm diameter, packed with Magic C18 AQ beads (3 μm bead size, 200 Å pore size) (Bishoff Chromatography, Leonberg, Germany). Peptides were loaded on the column from a cooled (4 °C) Eksigent autosampler and separated with a linear gradient of acetonitrile/water, containing 0.1% formic acid, at a flow rate of 200 nl/min. A gradient from 2 to 30% acetonitrile in 60 min was used. Mass spectra were acquired in a data-dependent manner, with an automatic switch between MS and MS/MS using a top 10 method. MS spectra were acquired in the Orbitrap analyzer with a mass range of 300–2000 m/z, with a resolution of 30,000 in the Orbitrap. Collision-induced dissociation (CID) peptide fragments were acquired in the ion trap with a collision energy of 35, activation energy of 0.25 and 30 ms activation time, excluding singly charged ions for fragmentation. Fragmented peptides were put on a dynamic exclusion list with a list size of 500 and an expiration time fo 90 s.
Protein Identification and Quantification
The raw files from the mass spectrometer were uploaded onto the Progenesis LC-MS (version 4.1, Nonlinear Dynamics, Newcastle upon Tyne, UK). The LC-MS data were normalized and aligned according to the manufacturer's specifications. The Mascot generic file (.mgf file format) generated with Progenesis LC-MS (using up to five tandem mass spectra for each feature with the top 200 fragment ion peaks, charge deconvolution and deisotoping option applied) was searched against an in-house built database, constructed using human, bacterial and fungal species, including combination of common contaminants and reversed sequences (a total of 249,061 sequences) using the Mascot 2.4.1 search engine (Matrix Science, London, UK) in order to evaluate the false discovery rate (FDR) using the target-decoy strategy (http://fgcz-ms/FASTA/p963_db1_d_20111201.fasta). All sequences were downloaded from NCBI on May 27, 2016 and concatenated to 261-sequences known as MS contaminants and reversed (decoyed) to generate the search database. The selected parameters included precursor tolerance (15 ppm) and [ss2] fragment ion tolerance (0.6 Da). Trypsin was used as the protein-cleaving enzyme, and three missed cleavages were allowed. Variable modifications included oxidation of methionine, deamidation from glutamine and asparagine and N-terminal acetylation of proteins whereas carbamidomethylation of cysteine was selected as a fixed modification. The mascot result was loaded into Scaffold v4.1.1 using 95% PeptideProphet and ProteinProphet thresholds and protein cluster analysis. The spectrum report was exported and loaded into Progenesis LC-MS. The experimental design consisted of the following groups: H Versus G or CP or AP. For quantification, all proteins identified with at least 2 peptide ions were assessed. For normalization, the default function was used.
Visualization by Heat Map and Cluster Analysis
The expression trends of salivary human proteins were visualized by use of heat-maps generated using the R software (R: A Language and Environment for Statistical Computing, R Development Core Team) and in particular the packages SRMService (https://github.com/protViz/SRMService) which implements the precursor and peptide summaries (See supplemental material) and quantable (https://cran.r-project.org/web/packages/quantable/index.html).
Statistical Analysis
Normality of the data was assessed by D'Agostino & Pearson normality test. Chi-squared and one-way ANOVA tests were used for demographic and clinical data analysis (GraphPad Software, La Jolla, CA). Differences were considered as statistically significant at p value <0.05. For quantitative protein expression analysis, significant differences between the pair-wise group comparisons were done in the Progenesis LC-MS using normalized protein abundances in arcsinh transformation. Differences were considered as statistically significant at fold-change ≥2, p value <0.05.
Phase 2: SRM Based Candidate Validation in an Independent Cohort
Experimental Design and Rationale
We aimed to develop a SRM-based workflow for detecting and quantitating the relative abundance of the candidate proteins in an independent saliva cohort. Eighty-two subjects (age range 24–59 years) were recruited for the study at the Department of Periodontology, School of Dentistry, Adnan Menderes University, Aydın, Turkey. Ethical clearance was obtained from the Ethics Committee of the School of Medicine, Ege University with the protocol number (Ethics number 70198063–050.06.04). Each participant gave written and verbal informed consent after the purpose and procedures of the study were explained. It is confirmed that this cross-sectional case-control study conforms to STROBE guidelines for observational studies. A dental and medical history was compiled for all subjects as detailed in phase 1 and the participants were clustered into four groups according to the criteria as detailed at Phase 1. The demographic and clinical details of details of the Phase 2 participants are presented in supplemental File S1.
SRM Assay Development
Selection of Target Proteins by Prioritization
The discovery LFQ experiments yielded 119 differentially expressed candidates between health versus periodontal disease. The next step involved SRM to establish sensitive, accurate assays for candidate marker measurements. However, reagent costs limited the practicality of developing assays for all identified candidates, necessitating a further prioritization step. To functionally overview the differentially expressed proteins between health and disease (fold change ≥ 2, p value <0.05), the proteins from the LFQ experiment were processed with MetaCore to build an analysis of functional ontologies including canonical pathway maps, Gene Ontology (GO) processes, diseases by biomarkers, and process networks as described previously (33).
Peptide Selection
Targeted SRM assays for the 65 proteins were then developed and optimized in saliva samples and can be found in supplemental Files S8–S9. For each protein, a set of three proteotypic peptides preferentially observed in previous LFQ experiments was selected for SRM analysis. For proteins with no or less than three proteotypic peptides, additional peptides were selected from the SRM Atlas (http://www.srmatlas.org). All peptides were ranging between 6 and 20 amino acids in length and were containing tryptic ends with no missed cleavages. Stable isotope-labeled standard peptides corresponding to the proteotypic peptides and containing either a C-terminal (13C(6) 15N(4)) arginine or a (13C(6) 15N(2)) lysine residue were chemically synthesized via SPOT synthesis (JPT Peptide Technologies, Germany) and used in unpurified form for the SRM analysis.
Sample Preparation–Protein Digestion
The samples were prepared in the same way as described in Phase 1. The total protein concentrations for H, G, CP and AP ((μg/ml, median (min-max)) were 664 (422–982), 794 (502–972), 816 (134.5–2830), 830 (345.5–3910), respectively. The median total protein levels were found to be significantly different among the groups (p < 0.05). Therefore, total protein amount per sample was controlled. The samples were blinded and alternated in random order in each analysis during both the sample preparation and the measurements. Solutions of 80 μg of proteins per sample were subjected to in-solution trypsin digestion according to the RapiGest protocol as described above in the discovery phase. Peptide mixtures were desalted using reverse phase cartridges Finisterre SPE C18 (Wicom International AG, Maienfeld, Switzerland) according to the manufacturer's specifications. Peptides were dried using a vacuum centrifuge, resolubilized with 30 μl 3% Acetonitrile (ACN) in 0.1% formic acid, and frozen at −20 °C, until further use.
SRM Measurements
SRM-triggered MS2 and SRM measurements were performed on a QTRAP 5500 instrument (ABSciex, Concord, Ontario) equipped with a nanoelectrospray ion source. Chromatographic separations of peptides were performed on a NanoLC-2D HPLC system (Eksigent, Dublin, CA) coupled to a 15 cm fused silica emitter, 75 μm diameter, packed with a ReproSil-Pur C18-AQ 120 A and 1.9 μm resin (Dr. Maisch HPLC GmbH). Peptides were loaded on the column from a cooled (4 °C) Eksigent autosampler and separated with a linear gradient of acetonitrile/water, containing 0.1% formic acid, at a flow rate of 300 nl/min. A gradient from 1 to 40% acetonitrile in 29 min was used. For the SRM triggered MS2 measurements, MS2 spectra were recorded upon detection of an SRM trace above a threshold of 1000 counts per second. An average of 100 transitions (scan time 10 ms/transition) per run was used and in SRM mode Q1 and Q3 were set to 0.7-unit mass resolution. MS2 spectra were recorded in enhanced product ion (EPI) mode for the highest SRM transitions with the following measurement parameters; dynamic fill time, Q1 at unit resolution, scan speed 10000 Da/s and m/z range to 300–1000. Collision energies parameters for each transition were calculated according to the following equations: CE = 0.036 * (m/z) + 8.857 and CE = 0.0544 * (m/z) − 2.4099 (CE, collision energy and m/z, mass to charge ratio) for doubly and triply charged precursor ions, respectively.
Spectra Library Building
SRM-triggered MS2 spectra measurements of the stable isotopically labeled standard peptides were searched with the Mascot search engine (v. 2.4.1, MatrixScience) against a database containing all the target proteins, concatenated to a forward and reversed Escherischia coli database as well as common protein contaminants. Data were searched with full tryptic cleavage using 2.0 Da for the parent-ion mass tolerance and 0.8 Da for the fragment-ion mass tolerance. Carboxyamidomethylation was used as a fixed modification on cysteine and oxidation as variable modification on methionine residues. Additionally, lysine and arginine were searched with 13(C6)15(N2)(K) and 13(C6)15(N4)(R) as fixed labels, because the C-terminal K or R residues of the synthetic peptides were substituted with the corresponding heavy version. The output mascot search results (i.e. dat files) from the measurements of stable isotopically labeled standard peptides were exported and a spectral library (i.e. blib file) was built using the skyline open source software. For each peptide (i.e. light and heavy form), three transitions with optimal SRM properties (34, 35) were selected. To ensure high sensitivity for the low abundant proteins, two different time-scheduled SRM acquisition methods were generated (supplemental File S2 and supplemental File S3).
SRM Data Processing
Acquired SRM raw files were imported into skyline daily. Automatic peak picking was performed based on the MProphet algorithm for SRM data processing and statistical error estimation (36). The second-best peaks were used as controls in training the model. The skyline files (supplemental File S4a) and skyline files quantification results generated with both time-scheduled SRM acquisition methods were exported without filtering and merged in a single table (supplemental File S4b). The file contains the results from 75 samples, with measurements from 65 Proteins (190 Peptide Sequences, both heavy and light) with in total 92024 quantitative values. To ensure high data quality, the peptide assays were filtered according to their Q Value. All assays with a Q Value greater 0.05 were removed. Also, all assays with intensities reported as 0 were removed. Subsequentially, all transitions with more than 30 NA's (out of 75 measurements) were removed from the dataset and the log2 foldchange of light to heavy ration were computed. The correlation between transitions of a peptide was also assessed and applied for data quality filtering (see. 3A). Transitions and peptides with a Pearson correlation below 0.5 were removed (supplemental File S5 and supplemental File S6). To obtain peptide quantification the median of the log2(l/h) ratio of the transitions was taken. The median was also used to obtain the protein log2(l/h) ratios for a protein. After filtering, 60 proteins remained for analysis (supplemental File S7).
Statistical Analysis
Based on the log2(l/h) fold changes, p values using a two-sided, not paired t test were computed (r-base). The p values were adjusted by the Benjamini and Hochberg method (p.adjust) to obtain the False Discovery Rate (FDR). Receiver Operating Charecteristic (ROC) curves and their Area Under Curve (AUC) were computed using the package pROC for single proteins to show their specificity and sensitivity to differentiate the two selected conditions. To further improve the classification accuracy, the generalized linear models (glm) were used. A binomial family generalized linear model with logit link using 3 proteins as explanatory variable was fitted. To examine the performance of all 60 proteins, all combinations of 3 proteins out of 60 was generated, which resulted in 34220 set. The models were fitted with only 3 variables (proteins) to prevent model overfitting. For all the fitted models, the response was predicted, and the AUC was computed using the predictions. Based on the AUC, the models were ranked. The top 10% of the models (3422 models with an AUC larger than 0.916), were examined as to how frequently a protein was utilized as well as the magnitude of its average coefficient. Both these statistics, frequency of use and average value of the coefficient were utilized to asses the utility of the proteins to differentiate the healthy controls from the diseased groups (Fig. 5A).
Fig. 5.


A, Histogram of AUC for all 34220 fitted models. Red line - AUC 90% quantile. Variables frequency (y axis) and average absolute coefficient weights (x axis) in top 10% of models (3422) with AUC range 0.90–0.97. B, STRING visualization of the proteins quantified by SRM. The nodes are the proteins and the connecting lines represent STRING interaction. Highlighted green are the proteins with a significant p value and log2 fold change larger or smaller than 1. In blue are marked the proteins which have passed the p value and fold change threshold as well as were most frequently used by the logistic regression models (CLUS and DMBT1). In red are the proteins which were used by the logistic regression models but did not have a fold change larger than 2 (MMP9, ARPC5 and RAP1A).
String Data Analysis
The STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) was used for critical assessment and integration of protein-protein interactions (http://string-db.org/). The interactions are drawn from experimental evidence as well as predictions based on knowledge gained from the other organisms (37) By using STRING, the 60 proteins that were prioritized earlier were mapped and a network image was created (Fig. 5B).
RESULTS
Phase 1. Discovery Label-free Quantitative Proteomics
General Overview of Discovery Stage, Non-targeted Quantitative Proteomic Findings—
In analyzing the collected saliva samples, the first approach was to use a non-targeted shotgun proteomics combined with a label-free quantitative approach whereby comparative precursor-ion pattern is used, based on the direct comparison of MS peptide signals (20, 38) (Fig. 1). Four hundred eighty-six proteins were quantified including 24 bacterial (4.9%) and 8 fungal (1.6%) proteins (FDR 5.76% at protein level) (supplemental File S10). After exclusion of reverse sequences, the identified bacterial proteins originated from Aggregatibacter actinomycetemcomitans (n = 3), Campylobacter rectus (n = 1), Fusobacterium nucleatum (n = 2), Porphyromonas gingivalis (n = 2), Prevotella oralis (n = 1), Streptococcus anginosus (n = 1), Treponema denticola (n = 1), Veillonella dispar (n = 2), Streptococcus oralis (n = 2). The identified fungal proteins were derived from Candida albicans (n = 2) and Saccharomyces cerevisiae (n = 1). Among those, only 7 were represented by more than one peptide, therefore, they were included in further comparative quantification.
Fig. 1.

Schematic representation of the protein discovery and validation workflow by use of label-free quantitative (LFQ) proteomics and Selected Reaction Monitoring (SRM) analysis. The shotgun proteomics study with case-control design (n = 67) was conducted to dissect comparative saliva proteome along the different disease stages. An independent cohort (n = 82) was used to verify sixty-five candidate markers by SRM. More details can be found under Experimental Procedures.
Among 486 identified proteins, 126 was represented by a single peptide whereas 360 proteins with more than 2 peptides (10 of them were microbial origin) were used for further comparative quantification (FDR 0.83% at protein level) (supplemental File S11).
Discovery of Potential Candidate Proteins as Markers of Periodontal Diseases
Further comparative analysis aimed to determine whether significant quantitative differences could be found between healthy controls and the diseased groups (G, CP, AP). Only nine proteins were different between the healthy and gingivitis and groups (Fig. 2A, supplemental File S11). Clustering of the periodontitis cohort into “chronic (CP)” and “aggressive (AP)” form subgroups, resulted into 67 and 100 proteins being significantly changed compared with health, respectively (Fig. 2A, supplemental File S11). Among those 62 and 63 were found at lower levels, whereas 5 and 37 were found at higher levels, in the CP and AP groups, respectively. In the case of the 7 shared regulated proteins among the studied groups, the expression levels of all were significantly reduced in disease compared with the health (Fig. 2). These included calmodulin-like protein 5 (CALML5), cystatin-B (CSTB), extracellular matrix protein 1 (ECM1), carboxylesterase 2 (EST2), Ig heavy chain V-III region, antileukoproteinase (SLPI) and cornifin-B (SPRR1B). All these proteins were downregulated in disease compared with health, but the magnitude of downregulation was much higher in periodontitis compared with gingivitis.
Fig. 2.
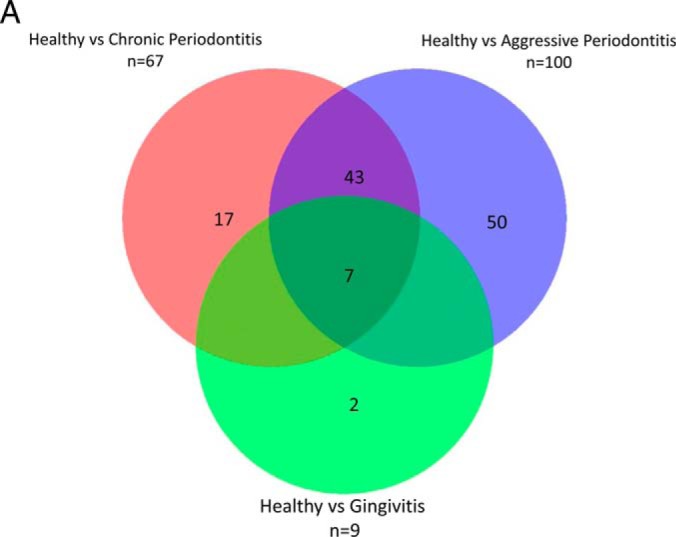

A, Venn diagram summarizing the number of differentially expressed proteins (p value <0.05 and ≥2 fold) and overlaps between health (H) and the diseased groups (G, AP, CP) by use of label-free quantitative proteomics (LFQ). Gingivitis (G), Aggressive Periodontitis (AP), Chronic Periodontitis (CP). B, Heat map of proteins significantly regulated between H and the diseased groups (G, AP, CP). Only significant and regulated proteins are represented (≥2 fold and p value <0.05). Each column represents an individual saliva sample, and each row represents an individual protein. Clustering separating samples into sub-groups as Healthy (H): Yellow, Gingivitis (G): Green, Aggressive Periodontitis (AP): Red, Chronic Periodontitis (CP): Orange.
Although the abundance of the most microbial proteins was not significantly altered between health and disease, the levels of metallo-beta-lactamase of Treponema denticola and glucose-6-phosphate isomerase of Prevotella oralis were significantly higher in AP compared with H (by 1.80-fold, 1.88-fold, respectively, p < 0.05). In contrast, the level of glyceraldehyde-3-phosphate dehydrogenase of Streptococcus oralis was significantly lower by 3.7-fold (p < 0.05) in AP compared with health.
A visual representation of protein abundances are provided in the heat-maps of Fig. 2B where proteins are clustered in rows and samples in columns. The scale represents arcsinh-normalized protein abundance levels and ranges from low (red) to high (blue) protein abundance. The colors at the top of the heat map show the studied groups. The divisions between the samples were not only based on the specified clinical diagnosis, despite the individuals from both periodontitis groups tend to form clusters which excluded the H and G groups.
These significantly regulated human derived proteins between healthy controls and the diseased groups were further categorized using the MetaCore software (https://portal.genego.com, Thomson Reuters), as described earlier whereby relevant pathways were then prioritized according to statistical significance (log2 ratio and p value) (39). The GO pathway mapping revealed that the three most significantly regulated processes in CP compared with H were “complement (C5a) 5a-induced chemotaxis”, “immunological synapse formation” and “IC3b-induced phagocytosis” (supplemental File S12). The specific proteins mapped on in these pathways were Heat shock 27 kDa protein (HSP27), Cell division control protein 42 homolog (CDC42), RAP-1A, Ras-related C3 botulinum toxin substrate 2 (RAC-2). The proteins found to be differentially expressed in AP compared with H showed significant enrichment of cellular and molecular processes involved in “cytoskeleton rearrangement,” “ECM and connective tissue proteolysis,” “immune response,” “response to hypoxia and oxidative stress.” The top three scored GO pathway maps were “ACM3 signaling,” “Interleukin (IL)-13 signaling,” and “IL-17 signaling” (supplemental File S13), and the specific proteins involved in these pathways were mucin 5B, mucin 7, lactoferrin, Solute carrier family 4 member 1 (SLC4A1), CD14, lacritin, MMP-8, MMP-9, Interleukin-1 receptor antagonist protein (IL-1RN), UGRP2, NGAL.
Phase 2: Verification of Candidate Proteins in Saliva for Periodontal Disease by LC-SRM-MS
To further verify the findings of our discovery label-free quantitative proteomics experiments, we conducted a similar case-control study with an independent large series of saliva samples (n = 82) using a SRM-based targeted proteomics approach (Fig. 1). The characteristics of the saliva samples belonging to the different groups of patients can be found in supplemental File S1. The proteins chosen for this targeted study included fifty proteins selected from the top ten most significantly enriched pathways, GO processes and process networks (supplemental File S12, S13) of our LFQ data using GeneGo′s MetaCore Pathway tool. Additionally, we selected eleven proteins from our LFQ data that were reported to be involved in disease process or differentially expressed according to earlier literature. Although, for SRM quantification, normalization of different samples can be done based on total protein amounts, a more accurate approach is the use of invariantly expressed housekeeping proteins (35). Therefore, four invariant proteins (Alpha-2-macroglobulin (A2M), Enolase (ENOA), Hemopexin (HEMO), Fibrinogen beta chain (FIBB)) were included in our target protein list as housekeeping proteins. Hence, this led to a list of 65 target proteins that were monitored across our cohort of saliva samples by SRM. Proteotypic peptides sequences for each protein were chosen based on the selection criteria described in the experimental procedures, which resulted in a total of 193 proteotypic peptides with up to 3 peptides per protein. These SRM assays fall in Tier 2 category as described earlier (26).
Our SRM assays successfully detected and quantified 143 peptides and 60 proteins across 75 saliva samples, which correspond to a success rate of 74 and 92% at the peptide and protein level, respectively. A comprehensive list of peptide/protein identification and quantification can be found in supplemental File S14. We observed that the use of ENOA for normalization decreases within group variance and improves prediction. This may be expected as enolases are highly conserved proteins found in almost all human tissues (40). An example of quality check of SRM measurements on precursor and protein level can be found in Fig. 3A and the expression trends of these proteins were visualized by heat-maps in Fig. 3B.
Fig. 3.


A, Quality check (QC) of SRM measurements on precursor and protein level. Top panel shows a line plot of the light to heavy ratios of the tree fragments y4, y5, y8 for peptide LFDQAFGLPR in all samples, whereas the bottom panel shows the light to heavy ratios of the two peptides quantified for protein HSPB1. The heatmap on the bottom envisages the correlation between the petides, to the right one can see the color scale used to encode the correlation [−1,1]. B, Heat map of all quantified proteins in the studied groups (H, G, AP, CP). Each column represents an individual saliva sample, and each row represents an individual protein. Clustering separating samples into sub-groups as Healthy (H): Yellow, Gingivitis (G): Green, Aggressive Periodontitis (AP): Red, Chronic Periodontitis (CP): Orange.
SRM Verification of Candidate Proteins as Biomarkers for Health Versus Periodontal Disease
Further comparative analysis aimed to determine whether significant quantitative differences could be found between “healthy” (H) and “diseased” saliva (G, CP, AP). We used different algorithms to distinguish protein signatures between health and disease, to provide the diagnostic performance of each protein. Out of 60 proteins, 22 were differentially expressed between health and disease with a fold change greater than 2 and a p value of less than 0.05 (Fig. 4A). Only two of those were significantly higher in disease, whereas 20 were significantly lower in disease. Predictive power of these proteins are further reported as ROC area under the curve (AUC). In Figs. 4B and 4C, twelve proteins with the highest AUC (AUC 0.83–0.91) between health and disease are presented.
Fig. 4.



A, Volcano plot of proteins significantly regulated between periodontal health and disease. The vertical dashed lines indicate a 2 fold change whereas the horizontal lines indicate the p value of 0.05. In blue triangles are marked the proteins which were the most frequently used by the logistic regression models. B, Receiver-operating characteristic (ROC) curve and corresponding area under the curve (AUC) statistics for the selected markers. The true positive rate (sensitivity) is plotted as a function of the false positive rate (1-specificity). The area under the ROC curve is a measure of how well the model distinguishes health from disease. C, The regulation pattern of the selected markers between health and disease.
Although it was observed that already a single protein can have a good discriminating power between health and disease state (see Fig. 4B), we further assessed if the combination of three proteins does increase the sensitivity and specificity. Using the expression levels of all possible protein triplets, we build 34220 logistic regression models as described under “Experimental Procedures” and used them to predict health and disease states. The models were ranked by AUC and then the top 10% of the models 3422 was selected. It was examined as to how frequently a protein was utilized and its average coefficient was computed. Both these statistics, frequency of use and average value of the coefficient are show in Fig. 5A. These statistics are a measure of the utility of the proteins to differentiate the healthy controls and diseased groups (Fig. 5A).
The top three proteins associated with positive coefficients in the logistic regression model are MMP9, RAP1A, ARPC5 which all have higher expression levels in disease than in health. These proteins can be considered as counterindicative of health. The top 2 proteins associated with positive coefficients in the logistic regression model are CLUS and DBMT1, which have higher levels in health than in disease. We furthermore examined which pairs of protein give the best predictions (Table I). The protein pair with the highest predictive power is ARPC5 and CLUS. All the models which include the pair (58 in total) have an AUC of between 0.96–0.98 with an average AUC of 0.97. The second best and third best pairs of proteins which can be used for prediction are: DMTB1 and RAP1A with an average AUC of 0.95 and DMTB1 and MMP9 with an average AUC of 0.94.
Table I. Prediction accuracy of protein pairs.
| a | b | mean (AUC) | min (AUC) | max (AUC) |
|---|---|---|---|---|
| ARPC5 | CLUS | 0.966061904 | 0.961873638 | 0.976034858 |
| DMBT1 | RAP1A | 0.95404177 | 0.946623094 | 0.970588235 |
| DMBT1 | MMP9 | 0.944200285 | 0.935729847 | 0.959694989 |
| ARPC5 | DMBT1 | 0.941026219 | 0.924836601 | 0.969498911 |
| CLUS | RAP1A | 0.941026219 | 0.927015251 | 0.968409586 |
| ARPC5 | HSPB1 | 0.937852152 | 0.927015251 | 0.970588235 |
Protein Network Analysis
The nature of differentially expressed proteins was further charecterized by generating protein-protein interaction maps by use of STRING (Fig. 5B). We used 60 proteins as “seeds,” and checked the interconnectivity between them. The results indicated that several of highly expressed proteins in health were host-derived proteolytic enzymes involved in anti-microbial activities such as SERPIN family proteins whereas many of the low abundant proteins were directly involved in matrix degradation and hyperinflammatory responses such as MMP-9 and TREM-1.
DISCUSSION
To compile a robust list of potential biomarkers for periodontal disease we carried out a comprehensive, non-targeted and targeted quantitative proteomic analysis in two large independent cohorts, using saliva as a diagnostic biological fluid. To the best of our knowledge, this is the first report combining discovery and targeted proteomics workflows for the study of periodontal diseases. The distilled proteomic profiling resulted in a reasonable number of proteins that can be followed practically, but also successfully identify a novel panel of candidate disease markers, thus marking a substantial improvement over the current state of biomarker evaluation. In a first non-targeted label-free LC-MS step, all detectable candidate markers were surveyed for a differential expression between health and disease, in an unbiased manner. Then, together with the already known markers from the literature, the newly identified candidates from LFQ were subjected to a more rigorous quantitative analysis using targeted LC-SRM in an independent cohort, for qualifying and validating the candidate biomarker panel.
The advantage conferred by utilizing a label-free platform was to simultaneously identify and quantify more than 360 proteins with more than two peptides. This lead to the discovery of many less known regulated proteins, both of host and microbial origin, beyond those previously established in the literature. In the case of the 10 identified microbial proteins, the levels of most of them were not significantly altered between health and disease. Yet, select poteins of T. denticola and P. oralis were significantly higher, and one of S. oralis was significantly lower in disease, corroborating an upregulatory trend of pathogen proteins and a downregulatory trend of commensal proteins in disease. The numbers of quantified proteins in this study were considerably higher than those reported earlier by qualitative workflows such as two-dimensional gel electrophoresis, which identified only few proteins with altered abundance and pin-pointed already known highly abundant proteins, such as ones belonging to the S100 family (41). Such differences may be attributable to different study designs, including clinical classification criteria, source of saliva, collection time and stimulation status as well as differences in instrumentation. A recent in-depth analysis of the saliva proteome in individuals with a good oral health status demonstrated that there are considareble changes in the saliva proteome throughout the day (42). During the morning, the salivary proteome is possibly more reflective of an undisturbed host-bacterial interaction status. We have also identified considerable inter-individual variations in total protein concentrations despite that the saliva collection was standardized by time (31). As the sample to sample variability is inevitable in saliva, for meaningful quantification of proteins in saliva, an appropriate normalization strategy including uniform loading with equal amounts of total protein is necessary.
In the present data set, 67 of the proteins were more than 2-fold regulated in chronic periodontitis compared with health. In a more recent small-scale study using label-free proteomic workflow in saliva, a total of 152 human proteins with more than one unique peptide were reported, only 3 of which (S100P, defensin 3, and Plastin-2) showed a more than 2-fold difference in abundance between health and chronic periodontitis (43). In the case of Plastin-2, the present findings corroborate our earlier work showing lack of regulation in chronic periodontitis (44). Considering the interindividual variability among humans (45, 46), large sample sizes would be expected to lead to more precise estimations of the analyzed data. The next analysis applied to the regulated set of proteins was the enrichment of functional terms as defined in the Metacore database, instead of individual proteins, which is a more innovative approach toward biomarker discovery. When considering the GO classifications for the pathway maps affected in chronic periodontitis, ten pathways passed the significance level defined by an FDR of 0.01. The analysis confirmed the C5a-induced chemotaxis pathway at rank 1, consistent with current knowledge of the biological pathways implicated in the disease, as its pharmacological inhibition in experimental models can abrogate periodontitis (47). The present study also reports that there are at least 100 proteins which are diffentially expressed in the saliva of healthy individuals and aggressive periodontitis patients. The ontology enrichment of proteins with altered expression led to identification of the most represented processes including “cytoskeleton rearrangement,” “extracellular matrix and connective tissue proteolysis,” “immune response,” and “response to hypoxia and oxidative stress.” These findings are well in line with our earlier work employing label-free LC/MSe in local gingival tissue exudates collected from a smaller cohort of aggressive periodontitis patients (10). Although the pathogenesis of aggressive periodontitis remains still elusive, the role of a hyperactive local immune response and oxidative stress could be important in disease initiation and progression (48). A significant overpresentation of certain unexpected signaling pathways was also identified, such as “Transport_Muscarinic acetylcholine receptor M3 (ACM3) signaling” and “Immune response_IL-13 signaling via Janus kinase-signal transducer and activator of transcription (JAK-STAT)”, along with well known ones such as the “IL-17 signaling” pathway. The top-scored pathway “ACM3 signaling” is a mediator of both electrolyte and water secretion in salivary and lacrimal glands (49). Signaling dysfunction of this pathway can lead to decrease in salivary secretion, which could favor microbial biofilm accumulation on the tooth surfaces. Agonists of ACM3 are often used clinically to stimulate salivary secretion in patients with Sjögren's syndrome (50), which is associated with higher incidence and severity of periodontitis (51). The regulated proteins in this pathway included lactoferrin, lacritin, sCD14, Mucin 5B and Mucin 7, which all were down regulated in disease, and SLC4A1, which was upregulated. The reduced levels of lactoferrin and lacritin suggest that anti-microbial properties of saliva are diminished in these patients with aggressive periodontitis. Moreover, the reduced levels of mucins suggest a decline in their defensive role in saliva, and consequently a higher susceptibility for local infection (52). The IL-17 family is known to play a critical role in periodontal inflammation, with a strinking influence in young individuals with immune deficiencies (53). It is therefore not surprising that the IL-17 signaling pathway was deregulated in this cohort of aggressive periodontitis patients. Although IL-17 protein itself was not detected or differentially expressed in the present data set, it may still be present and involved in the associated disease processes. This is supported by the fact that, despite its reasonable accuracy, precision at predicting protein abundance and its good coverage (54), LFQ it still lacks absolute accuracy and achievable sensitivity at lower protein concentrations such as pg/ml (55), where IL-17 is expected to range. Collectively, these proteomic changes corroborate the nature of aggressive periodontitis, in that a local impairment of the immune defense in otherwise systemically healthy patients.
As a next step, we applied the SRM-based targeted proteomics approach, which confers greater sensitivity toward low abundance proteins, high-level of reproducibility and sample throughput capabilities (23, 56). Accurate quantification is crucial, as periodontal diseases reflect modifications of the abundance of the proteins, rather than a binary state. Although we have not used all the identified proteins from the LFQ step and selected approximately half of them from the top ten most significantly enriched pathways, this approach does not disqualify the non-selected ones. In fact, these constitute a database for future validations. The developed SRM assays detected simultaneously 60 of the 65 LFQ-driven candidate protein biomarkers in an independent cohort. Choosing the proteotypic peptides with high ionization properties was essential for the successful detection of the proteins in saliva by SRM. Most of the peptides that were selected from our LFQ-driven part of the study resulted into a high detection rate (92%) in saliva by SRM. Additionally, we used the publicly available SRM database such as SRM atlas to increase the number of detectable proteotypic peptides per protein (57). However, a well-known trade-off between the number of peptides/transitions and the sensitivity of the SRM assay narrowed down our assay list to only three peptides/protein during the assay development phase. Novel emerging targeted technologies such as Parallel Reaction Monitoring (PRM) (58), Sequential Window Acquisition of all Theoretical Spectra (SWATH) (59), or data independent analysis (DIA)-based workflows (60) may well improve the degree of multiplexing in terms of analytes, for which our study could serve as a benchmark for other saliva-related studies. Although SRM have several advantages for detection and accurate quantification of a predetermined proteins in saliva, selection of signature peptides that uniquely represent the target protein may be a major challenge in the case of some class of proteins. Out of 193 peptides, 26 peptides were not unique for a given protein (supplemental File S15). However, these peptides were mainly matching proteins belonging to the same families. Though this approach may be effective for most protein targets, it was not so for serpins, histones and cystatins. Earlier studies have shown that top-down approaches may be advantageous for detection of detection of post-translational modifications of cystatins, as well as their sequence polymorphisms (61–63).
Performance of a diagnostic test is often measured by paired comparisons, or sensitivity and specificity, rather than one single statistics summary. Selection of a small, highly predictive set of markers is a general problem in any study of classification nature. Common approaches used to evaluate the quality of biomarker signatures include the determination of ROC and the measurement of AUC (64). In our set of data, sensitivity and specificity analysis by ROC narrowed-down to 12 out of the 60 SRM-measured proteins giving good predictive power between periodontal diseases and health. Yet, only Cystatin SN exhibited predictive power with AUC > 0.90. Cystatin SN is a major secreted cysteine proteinase inhibitor and its levels are reportedly higher in the saliva of periodontally healthy individuals, compared with diseased ones (65, 66). Logistic regression is another common method for selecting the best set of markers, for improving sensitivity and specificity simultaneously (64). In the present study, this approach has led to improvement of the classification accuracy by increasing ROC values from 0.83–0.90, to up to 0.97. The analysis has shown that building a good predictive model requires the inclusion of proteins which are both down-, as well as upregulated in the disease. Among the most frequently utilized proteins in the best performing logistic regression models were MMP9, RAP1A, ARPC5, CLUS, and DBMT1 (Table I). CLUS and DBMT1 are significantly upregulated with a fold change greater than two, whereas MMP9, ARPC5, and RAP1A are significantly downregulated (Fig. 4A). Although the available literature does not directly implicate CLUS and DMBT1 in periodontal diseases, DMBT1 (deleted in malignant brain tumors 1), or namely glycoprotein-340, is an abundant protein in multiple body fluids including saliva (67, 68). DMBT1 is an antimicrobial and inflammation-regulating molecule involved mucosal innate immunity, and its saliva-purified form has been shown to suppress a variety of bacteria and viruses (69). Hence, reduced DMBT1 levels in saliva may impair innate immunity, and it is therefore reasonable to postulate that it could serve as a risk indicator for the disease. Clusterin (CLUS) is a secreted multifunctional glycoprotein with important roles in protein homeostasis/proteostasis. Although it has been functionally implicated in age-related diseases, including tumorigenesis, cardiovascular and metabolic syndrome (70) its role in periodontal disease is under-investigated. CLUS is reportedly detected more often in gingival crevicular fluid in health than periodontal disease, as identified by LCMS (71). As the main role of CLUS is to counter-balance the deleterious effects of oxidative stress, it is reasonable to hypothesize that its reduced levels in the periodontal milieu may lead impaired resistance against oxidative stress, one of the driving forces of periodontal tissue damage (72).
To investigate whether these five statistically significant associations described above have a proven biological functional association, we further characterized protein-protein interactions between the selected biomarkers. Understanding protein-protein interactions and their functional implications is imperative for dissecting mechanisms of disease at the molecular level (73). The analysis by STRING reveled that CLUS was one of the center nodes strongly over-connected with RAP1A, MMP-9, and several SERPIN family proteins. MMP-9 has a well-established role in disease, demonstrated also by the higher salivary levels of MMP-9 in periodontitis compared with health (74, 75). RAP1A, a unique member of the Ras G protein family, is known to regulate several proteins controling diverse biological functions (76), including inflammation (77) and regulation of osteoclast function and bone resorption (78), consistent with the histopathological traits of periodontitis.
The present study approached salivary biomarker discovery and validation for periodontal diseases by coupling label-free untargeted and SRM-based targeted proteomics methods. The initial open-ended LFQ-proteomics platform led to the discovery of more than a hundred protein candidates which is becoming community available resource. These were subsequently utilized for SRM-guided validation of 60 reproducibly quantifiable proteins, which finally yielded a panel of five biomarkers with high predictive value for periodontal diseases. This narrowed-down protein panel displayed high specificity and sensitivity, enriching the pool of credible clinical biomarkers for the development of more sensitive antibody-based diagnostic assays. Yet, the quantum leap brought by this study lies in the possibility to apply in the field of periodontal diagnostics an earlier proposed innovative top-to-bottom operational pipeline (79), confirming that this platform can be pursued further for periodontal biomarker discovery and clinical validation in periodontal patient cohorts.
DATA AVAILABILITY
The mass spectrometry proteomics data have been depositedto the ProteomeXchange Consortium via the PRIDE (80) partner repository with the dataset identifier “PXD007535”.
Supplementary Material
Footnotes
* The study was supported the author′s institutional funds. The authors declare no conflict of interest.
1 The abbreviations used are:
- LFQ
- label-free quantitative
- ACN
- acetonitrile
- ACM3
- muscarinic acetylcholine receptor M3
- AP
- aggressive periodontitis
- AUC
- area under curve
- A2M
- alpha-2-macroglobulin
- ARPC5
- actin-related protein 2/3 complex subunit 5
- BOP
- bleeding on probing
- CAL
- clinical attachment loss
- CDC42
- cell division control protein 42 homolog
- CID
- collision-induced dissociation
- CLUS
- clusterin
- CP
- chronic periodontitis
- DIA
- data independent analysis
- DMBT1
- deleted in malignant brain tumors 1
- EDTA
- ethylenediaminetetraacetic acid
- ENOA
- enolase
- FDR
- false discovery rate
- FIBB
- fibrinogen beta chain
- H
- periodontal health
- HSP27
- heat shock 27 kDa protein
- G
- gingivitis
- GO
- Gene Ontology
- GCF
- gingival crevicular fluid
- JAK-STAT
- Janus kinase-signal transducer and activator of transcription
- IL
- Interleukin
- IL-1RN
- Interleukin-1 receptor antagonist protein
- LC
- liquid chromatography
- MMPs
- matrix metalloproteinass
- MRM
- multiple reaction monitoring
- MS/M
- tandem mass spectrometry
- PD
- probing depth
- PI
- plaque index
- PMNs
- polymorphonuclear cells
- PRM
- parallel reaction monitoring
- RAP1A
- Ras-related protein Rap-1
- ROC
- receiver operating characteristic
- SLC4A1
- solute carrier family 4 member 1
- SRM
- selected reaction monitoring
- SWATH
- sequential window acquisition of all theoretical spectra.
REFERENCES
- 1. Kassebaum N. J., Bernabe E., Dahiya M., Bhandari B., Murray C. J., and Marcenes W. (2014) Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J. Dental Res. 93, 1045–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Albandar J. M., Brunelle J. A., and Kingman A. (1999) Destructive periodontal disease in adults 30 years of age and older in the United States, 1988–1994. J. Periodontol. 70, 13–29 [DOI] [PubMed] [Google Scholar]
- 3. Papapanou P. N. (2015) Systemic effects of periodontitis: lessons learned from research on atherosclerotic vascular disease and adverse pregnancy outcomes. Int. Dent. J. 65, 283–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ryden L., Buhlin K., Ekstrand E., de Faire U., Gustafsson A., Holmer J., Kjellstrom B., Lindahl B., Norhammar A., Nygren A., Nasman P., Rathnayake N., Svenungsson E., and Klinge B. (2016) Periodontitis increases the risk of a first myocardial infarction: a report from the PAROKRANK Study. Circulation 133, 576–583 [DOI] [PubMed] [Google Scholar]
- 5. Chapple I. L. (2009) Periodontal diagnosis and treatment–where does the future lie? Periodontol. 2000 51, 9–24 [DOI] [PubMed] [Google Scholar]
- 6. Bao K., Bostanci N., Selevsek N., Thurnheer T., and Belibasakis G. N. (2015) Quantitative proteomics reveal distinct protein regulations caused by Aggregatibacter actinomycetemcomitans within subgingival biofilms. PloS one 10, e0119222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belibasakis G. N., and Bostanci N. (2012) The RANKL-OPG system in clinical periodontology. J. Clin. Periodontol. 39, 239–248 [DOI] [PubMed] [Google Scholar]
- 8. Bostanci N., Emingil G., Afacan B., Han B., Ilgenli T., Atilla G., Hughes F. J., and Belibasakis G. N. (2008) Tumor necrosis factor-alpha-converting enzyme (TACE) levels in periodontal diseases. J. Dental Res. 87, 273–277 [DOI] [PubMed] [Google Scholar]
- 9. Bostanci N., Ozturk V. O., Emingil G., and Belibasakis G. N. (2013) Elevated oral and systemic levels of soluble triggering receptor expressed on myeloid cells-1 (sTREM-1) in periodontitis. J. Dental Res. 92, 161–165 [DOI] [PubMed] [Google Scholar]
- 10. Bostanci N., Heywood W., Mills K., Parkar M., Nibali L., and Donos N. (2010) Application of label-free absolute quantitative proteomics in human gingival crevicular fluid by LC/MS E (gingival exudatome). J. Proteome Res. 9, 2191–2199 [DOI] [PubMed] [Google Scholar]
- 11. Guzman Y. A., Sakellari D., Arsenakis M., and Floudas C. A. (2014) Proteomics for the discovery of biomarkers and diagnosis of periodontitis: a critical review. Expert Rev. Proteomics 11, 31–41 [DOI] [PubMed] [Google Scholar]
- 12. Nylund K. M., Ruokonen H., Sorsa T., Heikkinen A. M., Meurman J. H., Ortiz F., Tervahartiala T., Furuholm J., and Bostanci N. (2017) Association of the Salivary Triggering Receptor Expressed on Myeloid Cells/ its Ligand Peptidoglycan Recognition Protein 1 Axis With Oral Inflammation in Kidney Disease. J. Periodontol. 28, 1–17 [DOI] [PubMed] [Google Scholar]
- 13. Rathnayake N., Gustafsson A., Norhammar A., Kjellstrom B., Klinge B., Ryden L., Tervahartiala T., Sorsa T., and Group P. S. (2015) Salivary matrix metalloproteinase-8 and -9 and myeloperoxidase in relation to coronary heart and periodontal diseases: a subgroup report from the PAROKRANK Study (Periodontitis and Its Relation to Coronary Artery Disease). PloS one 10, e0126370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu S., Loo J. A., and Wong D. T. (2006) Human body fluid proteome analysis. Proteomics 6, 6326–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaufman E., and Lamster I. B. (2000) Analysis of saliva for periodontal diagnosis–a review. J. Clin. Periodontol. 27, 453–465 [DOI] [PubMed] [Google Scholar]
- 16. Nomura Y., Tamaki Y., Tanaka T., Arakawa H., Tsurumoto A., Kirimura K., Sato T., Hanada N., and Kamoi K. (2006) Screening of periodontitis with salivary enzyme tests. J. Oral Sci. 48, 177–183 [DOI] [PubMed] [Google Scholar]
- 17. Hemmings K. W., Griffiths G. S., and Bulman J. S. (1997) Detection of neutral protease (Periocheck) and BANA hydrolase (Perioscan) compared with traditional clinical methods of diagnosis and monitoring of chronic inflammatory periodontal disease. J. Clin. Periodontol. 24, 110–114 [DOI] [PubMed] [Google Scholar]
- 18. Bretz W. A., Eklund S. A., Radicchi R., Schork M. A., Schork N., Schottenfeld D., Lopatin D. E., and Loesche W. J. (1993) The use of a rapid enzymatic assay in the field for the detection of infections associated with adult periodontitis. J. Public Health Dent. 53, 235–240 [DOI] [PubMed] [Google Scholar]
- 19. Kinney J. S., Morelli T., Braun T., Ramseier C. A., Herr A. E., Sugai J. V., Shelburne C. E., Rayburn L. A., Singh A. K., and Giannobile W. V. (2011) Saliva/pathogen biomarker signatures and periodontal disease progression. J. Dental Res. 90, 752–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bostanci N., Ramberg P., Wahlander A., Grossman J., Jonsson D., Barnes V. M., and Papapanou P. N. (2013) Label-free quantitative proteomics reveals differentially regulated proteins in experimental gingivitis. J. Proteome Res. 12, 657–678 [DOI] [PubMed] [Google Scholar]
- 21. Bostanci N., and Bao K. (2017) Contribution of proteomics to our understanding of periodontal inflammation. Proteomics 17 [DOI] [PubMed] [Google Scholar]
- 22. Aebersold R., Bensimon A., Collins B. C., Ludwig C., and Sabido E. (2016) Applications and developments in targeted proteomics: From SRM to DIA/SWATH. Proteomics 16, 2065–2067 [DOI] [PubMed] [Google Scholar]
- 23. Aebersold R., Burlingame A. L., and Bradshaw R. A. (2013) Western blots versus selected reaction monitoring assays: time to turn the tables? Mol. Cell. Proteomics 12, 2381–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ebhardt H. A., Root A., Sander C., and Aebersold R. (2015) Applications of targeted proteomics in systems biology and translational medicine. Proteomics 15, 3193–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., and Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carr S. A., Abbatiello S. E., Ackermann B. L., Borchers C., Domon B., Deutsch E. W., Grant R. P., Hoofnagle A. N., Huttenhain R., Koomen J. M., Liebler D. C., Liu T., MacLean B., Mani D. R., Mansfield E., Neubert H., Paulovich A. G., Reiter L., Vitek O., Aebersold R., Anderson L., Bethem R., Blonder J., Boja E., Botelho J., Boyne M., Bradshaw R. A., Burlingame A. L., Chan D., Keshishian H., Kuhn E., Kinsinger C., Lee J. S., Lee S. W., Moritz R., Oses-Prieto J., Rifai N., Ritchie J., Rodriguez H., Srinivas P. R., Townsend R. R., Van Eyk J., Whiteley G., Wiita A., and Weintraub S. (2014) Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics 13, 907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huttenhain R., Soste M., Selevsek N., Rost H., Sethi A., Carapito C., Farrah T., Deutsch E. W., Kusebauch U., Moritz R. L., Nimeus-Malmstrom E., Rinner O., and Aebersold R. (2012) Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci. Transl. Med. 4, 142ra194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kawahara R., Bollinger J. G., Rivera C., Ribeiro A. C., Brandao T. B., Paes Leme A. F., and MacCoss M. J. (2016) A targeted proteomic strategy for the measurement of oral cancer candidate biomarkers in human saliva. Proteomics 16, 159–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Percy A. J., Hardie D. B., Jardim A., Yang J., Elliott M. H., Zhang S., Mohammed Y., and Borchers C. H. (2017) Multiplexed panel of precisely quantified salivary proteins for biomarker assessment. Proteomics 17 [DOI] [PubMed] [Google Scholar]
- 30. Armitage G. C. (1999) Development of a classification system for periodontal diseases and conditions. Ann. Periodontol. 4, 1–6 [DOI] [PubMed] [Google Scholar]
- 31. Dawes C. (1972) Circadian rhythms in human salivary flow rate and composition. J. Physiol. 220, 529–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gumus P., Emingil G., Ozturk V. O., Belibasakis G. N., and Bostanci N. (2015) Oxidative stress markers in saliva and periodontal disease status: modulation during pregnancy and postpartum. BMC Infect. Dis. 15, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bessarabova M., Ishkin A., JeBailey L., Nikolskaya T., and Nikolsky Y. (2012) Knowledge-based analysis of proteomics data. BMC Bioinformatics 13, S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Picotti P., Rinner O., Stallmach R., Dautel F., Farrah T., Domon B., Wenschuh H., and Aebersold R. (2010) High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods 7, 43–46 [DOI] [PubMed] [Google Scholar]
- 35. Lange V., Picotti P., Domon B., and Aebersold R. (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reiter L., Rinner O., Picotti P., Huttenhain R., Beck M., Brusniak M. Y., Hengartner M. O., and Aebersold R. (2011) mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods 8, 430–435 [DOI] [PubMed] [Google Scholar]
- 37. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., and von Mering C. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grossmann J., Roschitzki B., Panse C., Fortes C., Barkow-Oesterreicher S., Rutishauser D., and Schlapbach R. (2010) Implementation and evaluation of relative and absolute quantification in shotgun proteomics with label-free methods. J. Proteomics 73, 1740–1746 [DOI] [PubMed] [Google Scholar]
- 39. Bostanci N., Bao K., Wahlander A., Grossmann J., Thurnheer T., and Belibasakis G. N. (2015) Secretome of gingival epithelium in response to subgingival biofilms. Mol. Oral Microbiol. 30, 323–335 [DOI] [PubMed] [Google Scholar]
- 40. Piast M., Kustrzeba-Wojcicka I., Matusiewicz M., and Banas T. (2005) Molecular evolution of enolase. Acta Biochim. Pol. 52, 507–513 [PubMed] [Google Scholar]
- 41. Haigh B. J., Stewart K. W., Whelan J. R., Barnett M. P., Smolenski G. A., and Wheeler T. T. (2010) Alterations in the salivary proteome associated with periodontitis. J. Clin. Periodontol. 37, 241–247 [DOI] [PubMed] [Google Scholar]
- 42. Grassl N., Kulak N. A., Pichler G., Geyer P. E., Jung J., Schubert S., Sinitcyn P., Cox J., and Mann M. (2016) Ultra-deep and quantitative saliva proteome reveals dynamics of the oral microbiome. Genome Med. 8, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Salazar M. G., Jehmlich N., Murr A., Dhople V. M., Holtfreter B., Hammer E., Volker U., and Kocher T. (2013) Identification of periodontitis associated changes in the proteome of whole human saliva by mass spectrometric analysis. J. Clin. Periodontol. 40, 825–832 [DOI] [PubMed] [Google Scholar]
- 44. Ozturk V. O., Emingil G., Osterwalder V., and Bostanci N. (2014) The actin-bundling protein L-plastin: a novel local inflammatory marker associated with periodontitis. J. Periodontal Res. 50, 337–346 [DOI] [PubMed] [Google Scholar]
- 45. Millea K. M., Krull I. S., Chakraborty A. B., Gebler J. C., and Berger S. J. (2007) Comparative profiling of human saliva by intact protein LC/ESI-TOF mass spectrometry. Biochim. Biophys. Acta 1774, 897–906 [DOI] [PubMed] [Google Scholar]
- 46. Prodan A., Brand H. S., Ligtenberg A. J., Imangaliyev S., Tsivtsivadze E., van der Weijden F., Crielaard W., Keijser B. J., and Veerman E. C. (2015) Interindividual variation, correlations, and sex-related differences in the salivary biochemistry of young healthy adults. Eur. J. Oral Sci. 123, 149–157 [DOI] [PubMed] [Google Scholar]
- 47. Abe T., Hosur K. B., Hajishengallis E., Reis E. S., Ricklin D., Lambris J. D., and Hajishengallis G. (2012) Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J. Immunol. 189, 5442–5448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kantarci A., Oyaizu K., and Van Dyke T. E. (2003) Neutrophil-mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. J. Periodontal. 74, 66–75 [DOI] [PubMed] [Google Scholar]
- 49. Matsui M., Motomura D., Karasawa H., Fujikawa T., Jiang J., Komiya Y., Takahashi S., and Taketo M. M. (2000) Multiple functional defects in peripheral autonomic organs in mice lacking muscarinic acetylcholine receptor gene for the M3 subtype. Proc. Natl. Acad. Sci. U.S.A. 97, 9579–9584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mavragani C. P., and Moutsopoulos H. M. (2007) Conventional therapy of Sjogren's syndrome. Clin. Rev. Allergy Immunol. 32, 284–291 [DOI] [PubMed] [Google Scholar]
- 51. Celenligil H., Eratalay K., Kansu E., and Ebersole J. L. (1998) Periodontal status and serum antibody responses to oral microorganisms in Sjogren's syndrome. J. Periodontal. 69, 571–577 [DOI] [PubMed] [Google Scholar]
- 52. Groenink J., Walgreen-Weterings E., Nazmi K., Bolscher J. G., Veerman E. C., van Winkelhoff A. J., and Nieuw Amerongen A. V. (1999) Salivary lactoferrin and low-Mr mucin MG2 in Actinobacillus actinomycetemcomitans-associated periodontitis. J. Clin. Periodontol. 26, 269–275 [DOI] [PubMed] [Google Scholar]
- 53. Moutsopoulos N. M., Konkel J., Sarmadi M., Eskan M. A., Wild T., Dutzan N., Abusleme L., Zenobia C., Hosur K. B., Abe T., Uzel G., Chen W., Chavakis T., Holland S. M., and Hajishengallis G. (2014) Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci. Transl. Med. 6, 229ra240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krey J. F., Wilmarth P. A., Shin J. B., Klimek J., Sherman N. E., Jeffery E. D., Choi D., David L. L., and Barr-Gillespie P. G. (2014) Accurate label-free protein quantitation with high- and low-resolution mass spectrometers. J. Proteome Res. 13, 1034–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anderson N. L., and Anderson N. G. (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 56. Domon B., and Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 [DOI] [PubMed] [Google Scholar]
- 57. Kusebauch U., Campbell D. S., Deutsch E. W., Chu C. S., Spicer D. A., Brusniak M. Y., Slagel J., Sun Z., Stevens J., Grimes B., Shteynberg D., Hoopmann M. R., Blattmann P., Ratushny A. V., Rinner O., Picotti P., Carapito C., Huang C. Y., Kapousouz M., Lam H., Tran T., Demir E., Aitchison J. D., Sander C., Hood L., Aebersold R., and Moritz R. L. (2016) Human SRMAtlas: a resource of targeted assays to quantify the complete human proteome. Cell 166, 766–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Peterson A. C., Russell J. D., Bailey D. J., Westphall M. S., and Coon J. J. (2012) Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gillet L. C., Navarro P., Tate S., Rost H., Selevsek N., Reiter L., Bonner R., and Aebersold R. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Venable J. D., Dong M. Q., Wohlschlegel J., Dillin A., and Yates J. R. (2004) Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 1, 39–45 [DOI] [PubMed] [Google Scholar]
- 61. Ryan C. M., Souda P., Halgand F., Wong D. T., Loo J. A., Faull K. F., and Whitelegge J. P. (2010) Confident assignment of intact mass tags to human salivary cystatins using top-down Fourier-transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 21, 908–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Whitelegge J. P., Zabrouskov V., Halgand F., Souda P., Bassilian S., Yan W., Wolinsky L., Loo J. A., Wong D. T., and Faull K. F. (2007) Protein-sequence polymorphisms and post-translational modifications in proteins from human saliva using top-down fourier-transform ion cyclotron resonance mass spectrometry. Int. J.. Mass Spectrom. 268, 190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Messana I., Cabras T., Pisano E., Sanna M. T., Olianas A., Manconi B., Pellegrini M., Paludetti G., Scarano E., Fiorita A., Agostino S., Contucci A. M., Calo L., Picciotti P. M., Manni A., Bennick A., Vitali A., Fanali C., Inzitari R., and Castagnola M. (2008) Trafficking and postsecretory events responsible for the formation of secreted human salivary peptides: a proteomics approach. Mol. Cell. Proteomics 7, 911–926 [DOI] [PubMed] [Google Scholar]
- 64. McDermott J. E., Wang J., Mitchell H., Webb-Robertson B. J., Hafen R., Ramey J., and Rodland K. D. (2013) Challenges in Biomarker Discovery: Combining Expert Insights with Statistical Analysis of Complex Omics Data. Expert Opin. Med. Diagn. 7, 37–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dickinson D. P. (2002) Cysteine peptidases of mammals: their biological roles and potential effects in the oral cavity and other tissues in health and disease. Crit. Rev. Oral Biol. Med. 13, 238–275 [DOI] [PubMed] [Google Scholar]
- 66. Baron A. C., Gansky S. A., Ryder M. I., and Featherstone J. D. (1999) Cysteine protease inhibitory activity and levels of salivary cystatins in whole saliva of periodontally diseased patients. J. Periodontal Res. 34, 437–444 [DOI] [PubMed] [Google Scholar]
- 67. Madsen J., Mollenhauer J., and Holmskov U. (2010) Review: Gp-340/DMBT1 in mucosal innate immunity. Innate Immun. 16, 160–167 [DOI] [PubMed] [Google Scholar]
- 68. Loimaranta V., Jakubovics N. S., Hytonen J., Finne J., Jenkinson H. F., and Stromberg N. (2005) Fluid- or surface-phase human salivary scavenger protein gp340 exposes different bacterial recognition properties. Infection Immunity 73, 2245–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li J., Metruccio M. M. E., Evans D. J., and Fleiszig S. M. J. (2017) Mucosal fluid glycoprotein DMBT1 suppresses twitching motility and virulence of the opportunistic pathogen Pseudomonas aeruginosa. PLoS Pathog. 13, e1006392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Van Dijk A., Vermond R. A., Krijnen P. A., Juffermans L. J., Hahn N. E., Makker S. P., Aarden L. A., Hack E., Spreeuwenberg M., van Rossum B. C., Meischl C., Paulus W. J., Van Milligen F. J., and Niessen H. W. (2010) Intravenous clusterin administration reduces myocardial infarct size in rats. Eur. J. Clin. Invest. 40, 893–902 [DOI] [PubMed] [Google Scholar]
- 71. Baliban R. C., Sakellari D., Li Z., Dimaggio P. A., Garcia B. A., and Floudas C. A. (2012) Novel protein identification methods for biomarker discovery via a proteomic analysis of periodontally healthy and diseased gingival crevicular fluid samples. J. Clin. Periodontol. 39, 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee Y. N., Shim Y. J., Kang B. H., Park J. J., and Min B. H. (2012) Over-expression of human clusterin increases stress resistance and extends lifespan in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 420, 851–856 [DOI] [PubMed] [Google Scholar]
- 73. Lage K. (2014) Protein-protein interactions and genetic diseases: The interactome. Biochim. Biophys. Acta 1842, 1971–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Morelli T., Stella M., Barros S. P., Marchesan J. T., Moss K. L., Kim S. J., Yu N., Aspiras M. B., Ward M., and Offenbacher S. (2014) Salivary biomarkers in a biofilm overgrowth model. J. Periodontal. 85, 1770–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ingman T., Tervahartiala T., Ding Y., Tschesche H., Haerian A., Kinane D. F., Konttinen Y. T., and Sorsa T. (1996) Matrix metalloproteinases and their inhibitors in gingival crevicular fluid and saliva of periodontitis patients. J. Clin. Periodontol. 23, 1127–1132 [DOI] [PubMed] [Google Scholar]
- 76. Stork P. J. (2003) Does Rap1 deserve a bad Rap? Trends Biochem. Sci. 28, 267–275 [DOI] [PubMed] [Google Scholar]
- 77. Dorn A., Zoellner A., Follo M., Martin S., Weber F., Marks R., Melchinger W., Zeiser R., Fisch P., and Scheele J. S. (2012) Rap1a deficiency modifies cytokine responses and MAPK-signaling in vitro and impairs the in vivo inflammatory response. Cell. Immunol. 276, 187–195 [DOI] [PubMed] [Google Scholar]
- 78. Zou W., Izawa T., Zhu T., Chappel J., Otero K., Monkley S. J., Critchley D. R., Petrich B. G., Morozov A., Ginsberg M. H., and Teitelbaum S. L. (2013) Talin1 and Rap1 are critical for osteoclast function. Mol. Cell. Biol. 33, 830–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rifai N., Gillette M. A., and Carr S. A. (2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 24, 971–983 [DOI] [PubMed] [Google Scholar]
- 80. Vizcaino J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been depositedto the ProteomeXchange Consortium via the PRIDE (80) partner repository with the dataset identifier “PXD007535”.