

Abstract
Internalization of ligand-activated type 1 IGF receptor (IGF-1R) is followed by recycling to the plasma membrane, degradation or nuclear translocation. Nuclear IGF-1R reportedly associates with clinical response to IGF-1R inhibitory drugs, yet its role in the nucleus is poorly characterized. Here we investigated the significance of nuclear IGF-1R in clinical cancers and cell line models. In prostate cancers, IGF-1R was predominantly membrane-localized in benign glands, while malignant epithelium contained prominent internalized (nuclear/cytoplasmic) IGF-1R, and nuclear IGF-1R associated significantly with advanced tumor stage. Using ChIP-seq to assess global chromatin occupancy, we identified IGF-1R binding sites at or near transcription start sites of genes including JUN and FAM21, most sites coinciding with occupancy by RNA polymerase II (RNAPol2) and histone marks of active enhancers/promoters. IGF-1R was inducibly recruited to chromatin, directly binding DNA and interacting with RNAPol2 to upregulate expression of JUN and FAM21, shown to mediate tumor cell survival and IGF-induced migration. IGF-1 also enriched RNAPol2 on promoters containing IGF-1R binding sites. These functions were inhibited by IGF-1/2 neutralizing antibody xentuzumab (BI 836845), or by blocking receptor internalization. We detected IGF-1R on JUN and FAM21 promoters in fresh prostate cancers that contained abundant nuclear IGF-1R, with evidence of correlation between nuclear IGF-1R content and JUN expression in malignant prostatic epithelium. Taken together, these data reveal previously unrecognized molecular mechanisms through which IGFs promote tumorigenesis, with implications for therapeutic evaluation of anti-IGF drugs.
Keywords: Nuclear IGF-1R, ChIP-seq, prostate cancer, RNAPol2, JUN
Introduction
Growing evidence implicates the insulin-like growth factor (IGF) axis in promoting risk of cancer and propensity for metastasis, therapy resistance and cancer-related death (1–4). IGFs signal via cell surface type 1 IGF receptors (IGF-1Rs), activating multiple effectors including AKT and ERKs (5). Until recently, the ability of IGF-1R to regulate transcription was thought to be explained solely by these canonical signalling networks downstream of cell surface IGF-1Rs (5). This view was challenged when our group and Larsson and colleagues showed that following clathrin-dependent endocytosis, activated internalized IGF-1Rs traffic to the nucleus (6, 7). Furthermore, Larsson’s group found that IGF-1R import through the nuclear pore complex requires IGF-1R-β SUMOylation and activities of p150 Glued and importin-β/RanBP2 (6, 8). We previously reported that nuclear IGF-1R is a feature of pre-invasive lesions and invasive cancers including prostate, renal and breast cancers, and identified association between nuclear IGF-1R and adverse prognosis in renal cancer (7). Subsequent data associate nuclear IGF-1R with proliferation, tumorigenicity, resistance to EGFR inhibition and clinical response to therapeutic anti-IGF-1R antibodies (7, 9–15), suggesting that IGF-1R nuclear import requires strong IGF axis activation amounting to IGF-dependence.
While nuclear IGF-1R is known to interact with chromatin (6, 7), genomic binding sites previously-identified by chromatin-immunoprecipitation sequencing (ChIP-seq) in melanoma cells were predominantly intergenic (6), hence of uncertain significance. The aims here were to investigate whether nuclear IGF-1R is recruited to transcriptionally active regions of genomic DNA, and probe the significance of this phenomenon in clinical cancers. We now report that nuclear IGF-1R associates with advanced tumor stage, and is recruited selectively to regulatory regions of chromatin including JUN and FAM21A promoters. We identify JUN and FAM21A as mediators of cell survival and IGF-induced migration, properties that tumors require to attain advanced stage. Finally, we detect IGF-1R on JUN and FAM21A promoters in tumors that contain nuclear IGF-1R, and identify association between tumor nuclear IGF-1R content and JUN expression.
Materials and Methods
Immunohistochemistry (IHC)
Formalin-fixed paraffin-embedded (FFPE) radical prostatectomy (RP) sections were used for IHC using IGF-1R antibody #9750 (Cell Signaling Technology) as described (16, 17) (see Supplementary Methods). IGF-1R was scored blinded by Uro-Pathologist CV for intensity and percentage of tumor stained, generating immunoreactive scores (range 0-12) for membrane, cytoplasmic and nuclear IGF-1R, and also internalized (cytoplasmic/nuclear, 0-24), and total (membrane/cytoplasmic/nuclear, 0-36) IGF-1R. We utilized the same method and scoring system for JUN IHC on adjacent sections using antibody ab32137 (Abcam). The study was approved by National Research Ethics Service Committee Oxfordshire Committee C (study 07/H0606/120). All patients provided written informed consent to use of tissue in research.
Cell lines, reagents
DU145 prostate cancer (from Cancer Research UK Clare Hall Laboratories, UK), and SK-N-MC Ewing Sarcoma Family Tumor (ESFT) cells (from Professor Nicholas Athanasou, University of Oxford UK) were cultured in RPMI 1640 medium with 10% fetal calf serum (FCS). Both were mycoplasma-free when tested with MycoAlert (Lonza Rockland Inc.). Cultures were used within 20 passages of authentication by STR genotyping (Eurofins Medigenomix Forensik GmbH). Xentuzumab (BI 836845) was provided by Boehringer Ingelheim, Bafilomycin A1 (BafA1) and long R3-IGF-1 purchased from Sigma-Aldrich.
Chromatin-immunoprecipitation (ChIP) and ChIP-sequencing (ChIP-seq)
Serum-starved DU145 and SK-N-MC cultures (50 x106 cells per condition) were treated with 50nM IGF-1 for 30 min, fixed, lysed and subjected to ChIP using antibodies to IGF-1R (#3027, Cell Signalling), H3K4me1 (ab8895, Abcam), H3K4me3 (ab8580, Abcam), RNAPol2 (ab5095, Abcam), or IgG (Santa Cruz, negative control) and the ChIP Assay Kit (17-295, Millipore) according to the manufacturer’s instructions (see Supplementary Methods). Independent replicate ChIP-DNAs underwent paired-end sequencing (HiSeq, Illumina). ChIP-Seq reads were mapped using Bowtie2 (18) aligned to the human reference genome (hg19) from UCSC. Aligned reads were filtered against IgG DNA and analysed with MACS2 for peak calling (19). These softwares reported peaks with assigned FDR values and p-values that identify DNA regions with statistically significant binding enrichment. ChIP-seq identified peaks were validated on triplicate independent samples by ChIP-quantitative Polymerase Chain Reaction (qPCR).
Reverse transcription, qPCR
RNAs were extracted and reverse transcribed using Pure Link RNA Mini RNA extraction kits (Ambion) and SuperScript III First-Strand Synthesis SuperMix (Invitrogen). ChIP DNAs and cDNAs were amplified using primers shown in Supplementary Table S1 and Sybr Green PCR Mix (Applied Biosystems) on a 7500 Fast RT-PCR System (Applied Biosystems).
Electrophoretic Mobility Shift Assay (EMSA)
ChIP-seq data were used to design ˜80 bp oligonucleotides, each 5’ biotin end-labelled on the sense strand (Supplementary Table S1). After annealing (95°C for 5min, cooling to 23°C over 2hr), biotinylated double-stranded (ds) oligonucleotides were used in EMSA with recombinant human IGF-1R residues 960-1397 (rhIGF-1R, ThermoFisher Scientific) using the EMSA assay kit (Active Motif), according to (20) and the manufacturer’s protocol with minor modifications. Each reaction used 100pmol bioinylated oligonucleotide probe with 0.2µg rhIGF-1R in the absence or presence of 500-fold excess unlabelled probe.
Western blotting, Immunoprecipitation and Immunofluorescence were performed as previously with minor modifications (7); see Supplementary Methods.
JUN promoter reporter
DU145 genomic DNA was used as a template to amplify nucleotides –982 to +394 of the JUN promoter (21) (see Supplementary Methods). The ˜1.4 kb PCR product was digested with XhoI and HindIII-HF (New England Biolabs), cloned into similarly-digested pNLCol2 vector (Promega) and the sequence confirmed by DNA sequencing (Source Bioscience). DU145 cells were transfected with pNLCol2-JUN or pNLCol2 empty vector (EV) using Lipofectamine 3000 (InVitrogen), selected with 500µg/mL hygromycin, and stable clones screened for promoter activity in ONE-Glo EX Luciferase assays (Promega) on a POLARstar Omega platereader (BMG Labtech). DU145 clones incorporating EV or JUN promoter plasmid were serum-starved overnight, treated with 50nM IGF-1 for 24hr and luciferase assays performed as above.
Assays for proliferation, cell survival, motility and migration were performed as described in (2) and Supplementary Methods.
Statistics
T-tests were used to analyze two groups, one-way or two-way ANOVA for >2 groups, and Wilcoxon matched pairs signed rank test for non-parametric data. We assessed the significance of variation in IGF-1R with clinical parameters with Chi-square, Mann-Whitney U tests and correlation analyses using Prism v6 (GraphPad Software) and Stata package release 11.2 (Stata Corporation, Texas, USA). All tests were 2-sided and p value <0.05 was considered significant.
Results
Nuclear IGF-1R associates with advanced stage in clinical prostate cancers
As a first approach to investigate the significance of IGF-1R subcellular localization, we used IGF-1R IHC to score IGF-1R in the membrane, cytoplasm and nucleus of 137 RPs from British men with prostate cancer recruited to the Prostate Cancer Mechanisms of Progression and Treatment (ProMPT) study (Supplementary Table S2). IGF-1R was detected in benign and malignant epithelium of all RPs, with a luminal-basal IGF-1R gradient in benign epithelia that was lost in the cancers (Figure 1A). Total IGF-1R in the cancers was greater than in benign areas of the same RPs (Figure 1B, Supplementary Figure S1A), supporting our previous report of IGF-1R over-expression in primary prostate cancers (22). Malignant epithelium contained significantly more internalized (nuclear/cytoplasmic) IGF-1R, while IGF-1R was predominantly in the plasma membranes of benign glands (Figure 1C, Supplementary Figure S1B). This difference in subcellular localization is novel, and may reflect increased IGF-1R activation in malignant vs benign epithelium. Importantly, nuclear IGF-1R associated with higher pathological tumor stage (pT1-2 vs 3, p=0.011, Figure 1D, Table 1). We also identified borderline association between internalized (nuclear plus cytoplasmic) IGF-1R and higher pathological grade (primary Gleason grade 3 vs 4-5, p=0.057; Table 1, Supplementary Figure S1C). There were no significant associations between clinical parameters and total IGF-1R or IGF-1R in the plasma membrane or cytoplasm (Supplementary Table S3).
Figure 1. Nuclear IGF1R is associated with advanced tumor stage.

A. IGF-1R IHC in radical prostatectomy: a) Benign epithelium showing membrane IGF-1R, with cytoplasmic IGF-1R in basal cells; b) Mixed Gleason 3 (grey arrow) and 4 (black arrow) cancer containing more IGF-1R than benign epithelium, prominent cytoplasmic and nuclear IGF-1R, and perineural invasion (white arrow). Scale bar 20μm. B.IGF-1R IHC scored for total IGF-1R (n= 137 RPs). Graph: total IGF-1R score (bars, mean ± SEM, in red) in benign and malignant epithelia. The cancers contained significantly more IGF-1R than benign prostatic epithelium from the same RP (***p=0.001, Wilcoxon matched pairs signed rank test).C. IGF-1R quantification in plasma membrane, cytoplasm, nucleus (n=137 RPs, ***p<0.001, Wilcoxon test). D. Stage pT3 prostate cancers contain more nuclear IGF-1R than stage pT1-2 cancers (p=0.011).
Table 1. Nuclear IGF-1R associates with advanced stage prostate cancer.
IGF-1R IHC was performed on 137 radical prostatectomies. Internalized (nuclear plus cytoplasmic) IGF-1R showed borderline association with higher Gleason grade tumors, and nuclear IGF-1R was significantly associated with tumours of higher pathological stage (Chi-square test). There were no significant associations between clinical parameters and total IGF-1R or IGF-1R in the plasma membrane or cytoplasm (Supplementary Table S3).
| Internalized IGF-1R | |||
| IGF-1R ≤ 6 | IGF-1R > 6 | p-value | |
| Stage | |||
| Stage pT1-2 | 40 | 27 | 0.293 |
| Stage pT3 | 35 | 34 | |
| Grade: | |||
| Gleason grade 6 + 7(3+4) | 59 | 39 | 0.057 |
| Gleason grade 7(4+3) + 8-9 | 16 | 22 | |
| PSA | |||
| 0-10 | 60 | 43 | 0.422 |
| > 10 | 15 | 15 | |
| Nuclear IGF-1R | |||
| IGF-1R = 0 | IGF-1R > 0 | p-value | |
| Stage | |||
| Stage pT1-2 | 50 | 17 | 0.011 |
| Stage pT3 | 37 | 32 | |
| Grade: | |||
| Gleason grade 6 + 7(3+4) | 64 | 34 | 0.602 |
| Gleason grade 7(4+3) + 8-9 | 23 | 15 | |
| PSA | |||
| 0-10 | 67 | 36 | 0.612 |
| > 10 | 18 | 12 | |
Nuclear IGF-1R undergoes IGF-induced recruitment to transcriptionally active regions of DNA
Having identified association between nuclear IGF-1R and adverse clinical factors in men with prostate cancer, we next investigated nuclear IGF-1R function by ChIP-seq in human DU145 prostate cancer cells. We also performed ChIP-seq for RNAPol2, and H3K4me1 and H3K4me3 that mark active enhancers and promoters respectively (23). Of ˜7-14x106 reads per sample, ≥85% were mapped to the human genome (Supplementary Table S4). Peak calling identified 16,239, 19,759 and 21,782 peaks of RNAPol2, H3K4me1 and H3K4me3 enrichment respectively, consistent with findings in other cell lines, with a pattern of sharp peaks of RNAPol2 and H3K4me1 recruitment, and broader peaks of H3K4me3 (Supplementary Figure S2A), reportedly associated with increased transcriptional consistency (23, 24). In contrast, we identified 62 regions with a clear increase in IGF-1R ChIP fragment depth compared with control (IgG) ChIP (Supplementary Figure S2B-C). To test the robustness of our data we repeated IGF-1R ChIP-seq in a second model, SK-N-MC Ewing sarcoma cells, which like DU145 showed nuclear IGF-1R positivity and inhibitory response to IGF neutralizing antibody xentuzumab (25) (Supplementary Figure S2D-F). The genome of SK-N-MC cells contained 66 IGF-1R binding peaks, of which 25 were shared with DU145 (Supplementary Figure S2C).
By comparison with peaks called in RNAPol2 and H3K4me1/3 ChIP-seq, we explored the genomic locations of sites of IGF-1R recruitment. Predictably, most RNAPol2 and H3K4me1/3 peaks were within 300kb of the transcription start site (TSS). Unexpectedly, given the intergenic location of the majority of IGF-1R binding sites reported by (6), our analysis showed that IGF-1R peaks also clustered near a TSS (Figure 2A). Supplementary Table S5 lists the coordinates of IGF-1R peaks, the distance from the nearest TSS and the identity of the nearest gene. Of the 62 unique regions of IGF-1R binding, 59 (95%) were coincident with RNAPol2 peaks, 54 (87%) with H3K4me1 peaks and 31 (50%) with H3K4me3. We detected only two peaks in common with IGF-1R peaks identified by ChIP-seq in melanoma cells, on chromosome 8 (6) (Supplementary Figure S2G).
Figure 2. IGF-1R is recruited to regulatory regions of DNA.

A. Distance from TSS for ChIP-seq-identified peaks. B. UCSC browser images: IGF-1R binding sites (red bars) within JUN and FAM21A/C promoters in DU145 cells treated with or without IGF-1 (dark/light grey, duplicate ChIPs), and for IgG, RNAPol2 and H3K4me1/3. H3K27Ac mark, often found near active regulatory elements, from ENCODE (https://genome.ucsc.edu/ENCODE/). C. EMSA: rhIGF-1R retards mobility of dsDNA probes corresponding to IGF-1R binding peaks in promoters of JUN (lanes 1-3) and FAM21A (lanes 4-6). White arrow: mobility of free probes; black, biotinylated probes bound to rhIGF-1R. Mobility shift abolished by excess unlabelled probes (lanes 3, 6), supporting specificity. No signal in absence of biotinylated probe (lane 7). D. Serum-starved DU145 cells treated with 50nM IGF-1 for 5-30 min, subjected to IGF-1R ChIP-qPCR to amplify IGF-1R peaks in JUN and FAM21A/C promoters. Graphs: mean ± SEM fold enrichment over serum-starved controls. IGF-1 increased IGF-1R recruitment, peaking at 10min (***p<0.001). E. IGF-1R ChIP performed as D) on serum-starved cells treated with 50nM IGF-1 for 10min alone or with 1hr 100nM xentuzumab pre-treatment. Graphs: mean ± SEM fold enrichment of IGF-1R binding to promoters of: left, JUN; center, FAM21A; right, FAM21C (*p<0.05, **p<0.01).
We focused on IGF-1R binding sites within the JUN and FAM21A/C genes that coincided with RNAPol2 and H3K4me1 peaks in both DU145 and SK-N-MC cells (Figure 2B, Supplementary Figure S3A), suggesting conserved binding to active regulatory regions. In EMSA, IGF-1R bound directly to dsDNA probes representing IGF-1R binding regions of JUN and FAM21A promoters (lanes 2, 5, Figure 2C). The specificity of this interaction is supported by its abolition in reactions containing excess unlabelled probe (lanes 3, 6, Figure 2C). We then performed ChIP-qPCR to validate ChIP-seq-detected IGF-1R binding, first confirming that IGF-1 activated IGF-1R over 30 minutes (Supplementary Figure S3B). IGF-1R recruitment to JUN and FAM21A/C promoters was enhanced by IGF-1 and suppressed by xentuzumab (Figure 2D-E), supporting requirement for IGF-1R activation. Similar ChIP-qPCR would be required to validate the additional IGF-1R peaks we identified in ChIp-seq. Contrasting with data from breast cancer and melanoma cells (26, 27), we did not detect IGF-1R on CCND1 or IGF1R promoters (Supplementary Figure S3C-D).
Nuclear IGF-1R promotes RNAPol2 recruitment and expression of pro-tumorigenic genes
Identification of nuclear IGF-1R at the TSS of the JUN and FAM21A/C promoters (Figure 3A) suggests regulatory function. As an initial step to explore this hypothesis, we cloned the proximal JUN promoter, representing the peak of IGF-1R recruitment (nucleotides –982 to +394), into a luciferase reporter. In DU145 cells stably-transfected with JUN promoter reporter, we detected luciferase activity significantly greater than that in empty-vector transfectants, and in serum-starved cells, reporter activity was enhanced by IGF-1 (Figure 3B). We noted that IGF-1R binding regions of the JUN and FAM21 promoters contained GATA-2 binding motifs, and the JUN promoter peak also contained a KU80-binding motif and AP-1 (FOS/JUN)-like site (Figure 3A, Supplementary Figure S4A). This prompted us to question whether nuclear IGF-1R interacts with these transcriptional effectors. Therefore, after confirming IGF-1R detection in DU145 nuclear extract (Figure 3C), we performed reciprocal co-immunoprecipitation (co-IP) experiments, revealing evidence of interaction between IGF-1R and RNAPol2. This appeared to be IGF-dependent when detected by IGF-1R IP but constitutive (ie present in serum-starved cells) in RNAPol2 IPs. We also detected ligand-independent interaction of IGF-1R with KU80 and GATA-2 (Figure 3D-E, Supplementary Figure S4B). Noting the abundance of IGF-1R in cytoplasmic extract (Figure 3C), we also tested for interaction between RNAPol2 and cytoplasmic IGF-1R. However, RNAPol2 was almost undetectable in the cytoplasm, with no evidence of IGF-1R co-IP (Supplementary Figure S4C).
Figure 3. Nuclear IGF-1R interacts with RNAPol2.

A. JUN and FAM21A/C promoters showing regions bound by IGF-1R (dashed square bracket, coordinates of binding) that overlaps TSS (arrow) and contains binding sites for GATA-2, KU80, and AP-1-like sites. B. Luciferase activity generated by: left, stably-integrated EV or JUN promoter reporter in DU145 cells; right, JUN promoter reporter in serum-starved DU145 cells treated with solvent or 50 nM IGF-1 for 24 hr (n=3 assays in each case, ***p<0.001). C. Serum-starved DU145 cells were treated with 50nM IGF-1 for 30 min and cytoplasmic and nuclear extracts were analysed by western blot. D-E. Nuclear extracts were immunoprecipitated for D: IGF-1R, E: RNAPol2 (left), GATA2 (right). The same results were obtained in two independent experiments. F. IGF-treated DU145 cells were analysed by RNAPol2 ChIP-qPCR to amplify IGF-1R binding regions of JUN and FAM21A/C promoters (mean ± SEM of triplicate independent ChIPs). After 5 min, IGF-1 enhanced RNAPol2 recruitment (**p<0.01, ***p<0.001).
Identification of IGF-induced interaction between nuclear IGF-1R and RNAPol2 led us to speculate that IGF axis activation might influence RNAPol2 recruitment to these sites. We used three approaches to test the dependence of RNAPol2 recruitment on nuclear IGF-1R. Firstly, using RNAPol2 ChIP-qPCR, we found that IGF-1 enhanced recruitment of RNAPol2 to JUN and FAM21A/C promoters (Figure 3F). Secondly, we assessed RNAPol2 recruitment to the TSS of β2-microglobulin and FOS genes that lack IGF-1R peaks. We detected RNAPol2 on these promoters, but found no enrichment of RNAPol2 binding upon IGF-1 treatment (Supplementary Figure S4D). Thirdly, to differentiate functions of cell surface and nuclear IGF-1Rs, we used BafA1, a vacuolar H+-ATPase inhibitor that blocks vesicular trafficking (28). Both xentuzumab and BafA1 blocked IGF-1R nuclear translocation; xentuzumab did this by inhibiting IGF-1R activation, hence also suppressing downstream signaling, while BafA1 prevented IGF-1R internalization without preventing ligand-induced activation of cell surface IGF-1Rs and their ability to signal via AKT and ERKs (Figure 4A-C, Supplementary Figure S4E). We found that IGF-induced enhancement of RNAPol2 recruitment to the JUN and FAM21A promoters was suppressed by both xentuzumab and BafA1 (Figure 4D). Furthermore, IGF-1 up-regulated expression of JUN and FAM21A; as with RNAPol2 recruitment, these effects were also inhibited by xentuzumab and BafA1, although only partially in the case of IGF-induced JUN upregulation (Figure 4E). While BafA1 blocks internalization of many proteins (28), inhibition of IGF-induced recruitment and transcription does associate this effect with IGF-1R. We also detected IGF-induced RNAPol2 recruitment to the FAM21C promoter, but this transcript was not upregulated by IGF-1 (Figure 4D-E, right panels).
Figure 4. IGF axis blockade and inhibition of IGF-1R internalization induce comparable suppression of IGF-induced RNAPol2 recruitment and gene expression.
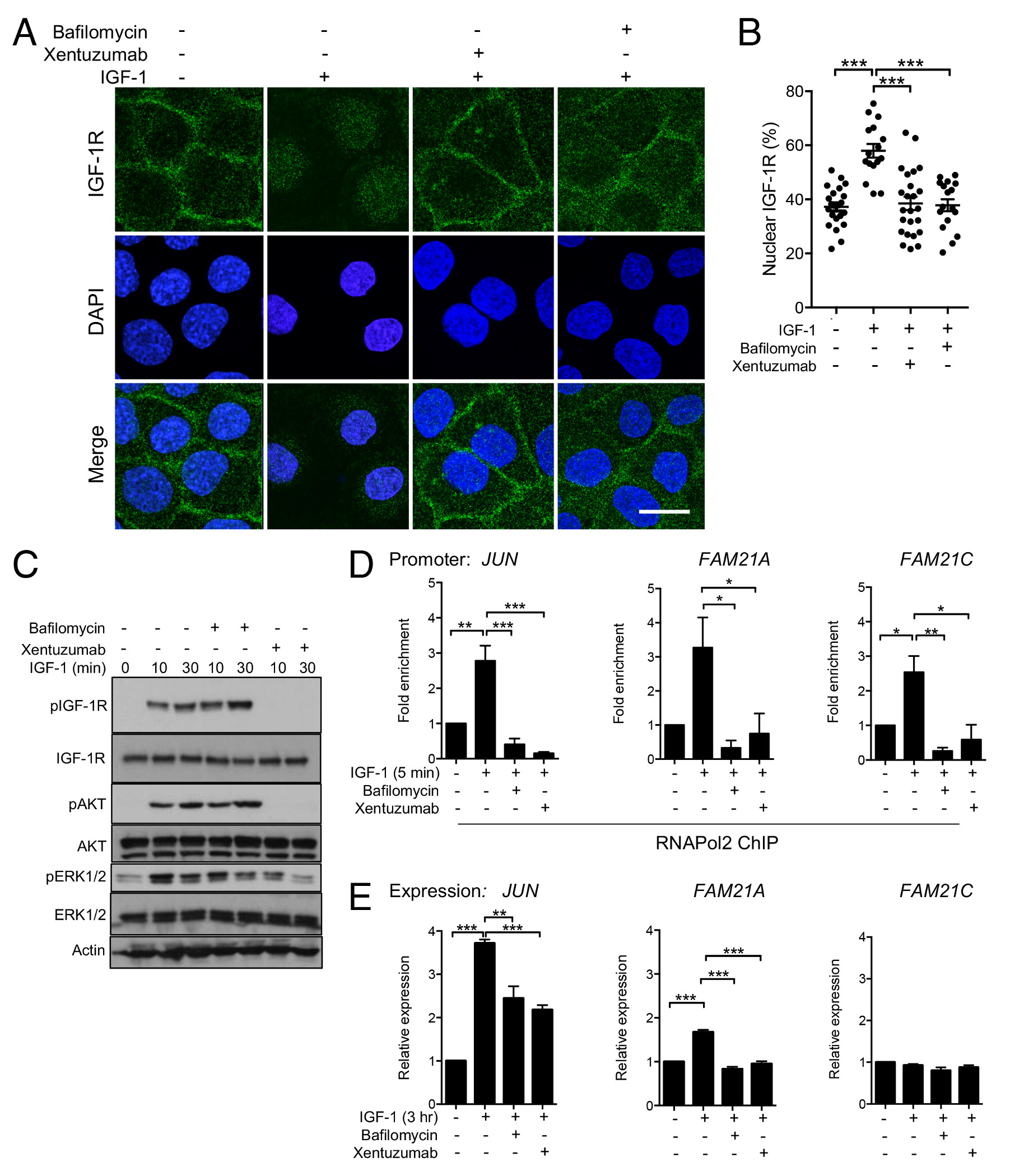
A-C. Serum-starved DU145 cells were incubated with 50nM IGF-1 for 30min alone or with 1hr pre-treatment with 100nM xentuzumab or 50nM BafA1. A: representative IGF-1R immunofluorescence images, scale bar 20μm. B: quantification of mean ± SEM nuclear IGF-1R as % total cellular IGF-1R (***p<0.001). C: western blot to assess IGF-induced activation of IGF-1R, AKT and ERKs; D, E. Serum-starved DU145 cells were treated with IGF-1 alone or with xentuzumab or BafA1. D: ChIP-qPCR: IGF-induced RNAPol2 recruitment to JUN and FAM21A/C promoters was attenuated by xentuzumab and BafA1 (*p<0.05, **p<0.01, ***p<0.001). E. JUN and FAM21A/C expression quantified by qRT-PCR, showing mean ± SEM fold expression corrected for ACTB, relative to serum-starved cells. IGF-induced JUN and FAM21A upregulation was inhibited by both xentuzumab and BafA1 (**p<0.01, ***p<0.001). IGF-1 did not upregulate FAM21C.
To assess the functional significance of JUN and FAM21A upregulation, we tested effects of depleting these proteins (Figure 5A). Consistent with the known pro-tumorigenic role of JUN (29), cell survival was reduced in JUN-depleted, although not FAM21A-depleted prostate cancer cells (Figure 5B). Seeking a FAM21A-associated phenotype, we noted that FAM21 is a component of the Wiskott-Aldrich syndrome protein and SCAR homolog (WASH) complex involved in endosomal trafficking (30). WASH-associated proteins are reportedly required for actin polymerization and cell motility (31), leading us to speculate that FAM21A contributes to this process. Indeed, FAM21A depletion caused delay in cell migration that was significant at 12 and 36hr, while JUN-depleted cells showed a delay only at 12hr (Figure 5C-D, Supplementary Figure S5A-B). To assess more specifically whether JUN and FAM21A contribute to IGF-dependent migration, we performed transwell assays in low serum (0.2% FCS), detecting enhancement of migration towards IGF-1 (1.23 ± 0.03 fold, p<0.001) in control transfectants. This effect was suppressed by both JUN and FAM21A depletion (Figure 5E), supporting the hypothesis that these proteins contribute to pro-migratory effects of IGF-1. In controls, we observed greater enhancement of migration using 10% FCS as stimulus (1.56 ± 0.63 fold, p<0.001), consistent with the presence in serum of pro-migratory factors in addition to IGFs, and this effect was partially suppressed in JUN-depleted but not FAM21A-depleted cells (Supplementary Figure S5C). To assess potentially confounding effects of proliferation we also performed viability assays on parallel siRNA-transfected cultures, finding no differences in proliferation over the 24hr time-course of migration assays (Supplementary Figure S5D).
Figure 5. Genes upregulated following nuclear IGF-1R recruitment contribute to tumor cell survival and IGF-induced motility.

A-B. DU145 cells were siRNA-transfected and the following day were disaggregated and used for A: assessment of JUN and FAM21A expression by qPCR (n=3 assays for JUN, n=5 for FAM21A); B: clonogenic survival assays, showing representative plate and to right, graph of cell survival expressed as % survival of control-transfectants (**p<0.01, ***p<0.001). C-D. Control or FAM21A siRNA-transfected confluent monolayers were scratched and imaged. C: representative images; D: migration expressed as mean ± SEM % defect remaining at: left, 12hr, right: 36hr (*p<0.05, **p<0.01, ***<0.001). E. DU145 cells were transfected with siControl, siJUN_3 or siFAM21A_2, the following day seeded into upper wells of transwell plates in low-serum medium and migration towards 50 nM IGF-1 quantified after 24 hr (*p<0.05, ***<0.001 by 2-way ANOVA).
Nuclear IGF-1R is recruited to gene promoters in clinical cancers and associates with JUN expression
Having identified a transcriptional role for IGF-1R in the nucleus of cultured prostate cancer cells, we used two approaches to investigate the significance of nuclear IGF-1R in clinical cancers. First, we performed ChIP-qPCR on fresh frozen primary prostate cancers, and were able to detect IGF-1R on JUN and FAM21A promoters, with higher signal in tumors with abundant nuclear IGF-1R (Figure 6A), supporting the clinical relevance of IGF-1R ChIP-seq findings in cultured cells. Finally, to further probe the relationship between nuclear IGF-1R and JUN expression, we performed IHC for JUN in adjacent FFPE sections of the radical prostatectomies in which we had evaluated IGF-1R expression and subcellular localization (Figure 1). After scoring JUN signal in the malignant epithelium, it was apparent that JUN showed significant correlation with nuclear IGF-1R (Figure 6B-C, Supplementary Figure S6A). This correlation was not seen for total IGF-1R (Supplementary Figure S6B), supporting the importance of nuclear IGF-1R in upregulating JUN. Taken together, these data highlight an important non-canonical nuclear role for IGF-1R that promotes the properties required to attain advanced tumor stage (Figure 6D).
Figure 6. Nuclear IGF-1R is present on promoters of clinical prostate cancers, and correlates with JUN expression.

A. Fresh frozen prostate cancers underwent IGF-1R or control (IgG) ChIP-qPCR for IGF-1R binding regions of JUN and FAM21A promoters. Upper: relative IGF-1R enrichment; center: representative IGF-1R IHC from adjacent FFPE tumor (scale bar 20μm); lower: Gleason grades and IGF-1R scores in membrane (M), cytoplasm (C), nucleus (N). B. IGF-1R and JUN IHC on adjacent RP sections, showing Gleason 4 pattern gland (scale bar 30μm). C. Graph: total JUN immunoreactive scores in n=80 RPs correlated with nuclear IGF-1R (Spearman coefficient). D. Nuclear IGF-1R binds to DNA and interacts with transcriptional regulators, inducing expression of genes that promote tumor cell survival and migration.
Discussion
The principal findings of our study are that IGF-1R binds directly to DNA, interacts with key transcriptional regulators, contributes to RNAPol2 recruitment and gene expression, and associates with advanced tumor stage. We identified IGF-1R recruitment to regulatory DNA sequences by performing parallel ChIP-seq for RNAPol2 and histone marks of active enhancers and promoters. This strategy allowed us to locate regulatory regions of the genome, and also assess ChIP-seq efficiency. We compared RNAPol2 and H3K4me1/3 peak numbers with those reported by (23), who used ChIP-seq to study transcriptional regulators in LNCaP prostate cancer cells, obtaining 1.36 – 10.22 x 106 uniquely-mapped reads, and reporting 7,028 binding sites for RNAPol2, 25,469 for H3K4me1, 24,921 for H3K4me3. Therefore, we identified more RNAPol2 binding sites and similar numbers of H3K4me1/3 sites, supporting the ability of our ChIP-seq protocol to detect enrichment of regulatory proteins on DNA. We identified far fewer peaks of IGF-1R binding, although two factors support the credibility of the identified peaks and their functional importance. Firstly, we identified IGF-1R binding peaks in common between two cancer cell lines, and we validated IGF-1R binding by ChIP-qPCR. Secondly, two previous studies had performed ChIP-seq using the same IGF-1R antibody, finding relatively few IGF-1R binding sites in genomic DNA. Larsson’s group was the first to use this approach, identifying 568 IGF-1R binding sites in melanoma cells, of which 80% were intergenic and 3.4% (˜20 sites) were ≤20kb of a TSS (6). In immortalized corneal epithelial cells, Wu and colleagues identified nuclear IGF-1R:INSR hybrid receptors, reporting 88 binding peaks for IGF-1R and 86 for INSR, assigned to nearest genes involved in proliferation, cell death, differentiation, cell adhesion, signal transduction, metabolism, and cell communication (32). Thus, there is support for the concept that IGF-1R binds to a limited subset of sites in the human genome. The location of these sites may be cell-type specific, possibly related to differences in nuclear structure and chromatin organization (33), given that the binding sites we identified appear to cluster selectively around the TSS, unlike the majority of sites identified by (6).
In addition to clustering around a TSS, the majority of sites of IGF-1R recruitment we identified were coincident with peaks of RNAPol2 and H3K4me1 enrichment, and 50% coincided with H3K4me3 peaks. This identification of nuclear IGF-1R binding sites at regulatory DNA regions is consistent with a model in which interaction of nuclear IGF-1R with DNA regulates gene transcription, supporting previous reports identifying IGF-1R on the CCDN1 and IGF1R gene promoters (26, 27). The major difference is that we identified specific promoters by ChIP-seq, while these previous reports were guided to the CCDN1 promoter by recognition that nuclear IRS-1 is also present at TCF/LEF sites of this promoter (27), and the IGF1R promoter by interest in regulation of IGF1R gene expression (26). We did not detect IGF-1R on either promoter (Supplementary Figure 3C-D), again suggesting cell type-specific differences.
The concept that IGF-1R binds directly to DNA is supported by our EMSA data using probes corresponding to ChIP-seq-identified IGF-1R binding peaks. In these assays, recombinant IGF-1R protein was the only component added to reactions in which probe mobility was retarded (Figure 2C). Such data have been considered to provide evidence for direct protein:DNA interaction in EMSA characterizing DNA binding of other recombinant or highly-purified proteins (20, 34–36). Our co-IP data (Figure 3D-E) indicate that within intact cells, nuclear IGF-1R exists in protein complexes and may be recruited in this context to chromatin. Importantly, we report a hitherto-unrecognized interaction between nuclear IGF-1R and RNAPol2. The functional implications of this interaction are currently unclear, given the discrepancy between the time-course of the interaction, increasing over 30min (Figure 3C), and more rapid (5min) IGF-induced RNAPol2 recruitment (Figure 3F), which could suggest that this response is independent of nuclear IGF-1R. We considered probing the contribution of nuclear IGF-1R by manipulating its localization, by mutating a nuclear localization sequence (NLS) to block trafficking via importins, or expressing SUMO-site mutant IGF-1R (6). However, recent work detects SUMO-site mutant IGF-1R in the nucleus, possibly via heterodimerization with INSR (37), and furthermore IGF-1R lacks an identifiable NLS, and unlike (8) we cannot detect IGF-1R interaction with importin-beta (7). Therefore, we adopted the strategy of comparing complete pathway blockade by xentuzumab with internalization inhibition using BafA1. This approach generated evidence implicating nuclear IGF-1R, by the suppression of IGF-induced RNAPol2 recruitment by BafA1 (Figure 4D), which blocks receptor internalization but not membrane signalling (Figure 4A-C). Furthermore, IGF-1 did not influence RNAPol2 recruitment to promoters lacking IGF-1R binding sites (Supplementary Figure S4C). The functional significance of IGF-1R:RNAPol2 complex formation could be further explored by identifying and disrupting the IGF-1R domain(s) required for RNAPol2 interaction, using ChIP to test recruitment of IGF-1R and RNAPol2 to JUN and FAM21A promoters.
While nuclear IGF-1R is detectable by immunohistochemistry, immunofluorescence and subcellular fractionation, these techniques also detect abundant cytoplasmic IGF-1R. It is unclear to what extent nuclear IGF-1R functions are related to/dependent on cytoplasmic IGF-1R. Ligand-induced IGF-1R activation promotes IGF-1R internalization into the cytoplasm, but we were unable to detect interaction with RNAPol2 in this subcellular compartment. However, internalized IGF-1R is known to mediate sustained signalling to AKT, and/or reflects IGF-1R that is en route to degradation, recycling to the plasma membrane or trafficking to the nucleus (6, 7, 38, 39). Given these considerations, it is plausible to consider that plasma membrane IGF-1R might be inactive, without pro-tumorigenic function. In future, it will be interesting to assess tissue IGF expression, to determine whether nuclear IGF-1R positivity associates with, and is potentially a response to, high ambient ligand levels.
It is increasingly recognized that multiple components of the IGF-insulin axis undergo nuclear translocation, including IGF and insulin receptors, docking molecules and IGF binding proteins (26, 40–43). Indeed, INSR reportedly undergoes insulin-stimulated recruitment to the promoters of genes already known to be insulin-induced, contributing to glucose homeostasis (41). Similarly, IGF-1 is known to promote tumor growth at least in part by upregulating JUN (44). FAM21 has not previously been linked with IGF signalling, but FAM21A is clearly upregulated here by IGF-1, and IGF-induced JUN and FAM21A transcription is comparably inhibited by IGF neutralizing antibody xentuzumab and by preventing IGF-1R internalization (Figure 4E). The equivalence of these responses suggests that internalized/nuclear IGF-1R contributes to IGF-induced transcription, although we acknowledge that canonical signalling eg via ERKs may also contribute to this transcriptional effect. IGF-1 does not upregulate FAM21C, suggesting either that IGF-1R recruitment has no functional effect at this locus, or is consistent with reports that transcription is initiated but not completed in a large fraction of human genes (45).
Together with our earlier report of association with adverse outcome in renal cancer (7), the finding here that nuclear IGF-1R associates with advanced stage in prostate cancer supports a link with aggressive tumor behaviour. While it is not possible to infer a causative association from these clinical findings, experimental data support functional relevance, linking nuclear IGF-1R with increased IGF-induced proliferation, gefitinib resistance and enhanced tumorigenicity (9, 10, 13). These phenotypes could be mediated at least partly by FAM21, reported to promote chemo-resistance in pancreatic cancer (46), and JUN, which associates with radioresistant prostate cancer in patients and murine models (47, 48). In radiotherapy-treated prostate cancers, we recently reported that IGF-1R upregulation associates with high Gleason grade and risk of metastasis, and cytoplasmic and internalized IGF-1R with biochemical recurrence, although there were no specific associations with nuclear IGF-1R (49). However, nuclear IGF-1R was recently found to interact with PCNA to influence the response to DNA damage (50). Finally, we identify JUN and FAM21 as mediators of IGF-induced migration (Figure 5E). Suppressed migration towards FCS in JUN-depleted but not FAM21A-depleted cells (Supplementary Figure S5C) suggests that JUN mediates migration induced by additional stimuli present in FCS, while FAM21A may more specifically mediate chemotactic response to IGF-1.
Taken together, these data highlight an important non-canonical nuclear role for IGF-1R that associates with advanced tumor stage, and reveal hitherto-unrecognized molecular pathways through which IGFs promote tumor cell survival and motility. Given the reported association of nuclear IGF-1R with clinical response to IGF-1R inhibition (14, 15), and our demonstration that IGF-neutralizing antibody antagonizes nuclear IGF-1R functions, these findings have implications for clinical evaluation of IGF inhibitory drugs.
Supplementary Material
Acknowledgements
We are very grateful to the prostate cancer patients who gave permission for use of tumor tissue in research. We are also grateful for technical advice and support from Simon Engledow (High-Throughput Genomics Group, Wellcome Trust Centre for Human Genetics Oxford), and Graham Brown (Microscopy Core Facility, Department of Oncology Oxford), and for comments on the manuscript from Eric O’Neill and Sovan Sarkar. This study was supported by Prostate Cancer UK (G2011/20, G2012/25), Development Fund of Cancer Research UK Oxford Cancer Research Centre (CRUKDF 0715-VMTA), UCARE-Oxford (TA/VM-2016), National Institute for Health Research (NIHR) Research Capacity Funding (grant AC14/037), Breast Cancer Now (2014NovPR364), The Rosetrees Trust and John Black Charitable Foundation (M330-F1), and support to VMM from the NIHR Oxford Biomedical Research Centre. The ProMPT study is supported by the UK NIHR, Cancer Research UK and the MRC, and the Cambridge and Oxford Biomedical Research Centres. The funding source had no role in the design, conduct of the study, collection, management, analysis and interpretation or preparation, review, or approval of the manuscript.
Footnotes
Conflict of interest statement: UWC, MS and TB are employees of Boehringer Ingelheim. The other authors have declared that there is no conflict of interest.
References
- 1.Travis RC, Appleby PN, Martin RM, Holly JM, Albanes D, Black A, et al. A Meta-analysis of Individual Participant Data Reveals an Association between Circulating Levels of IGF-I and Prostate Cancer Risk. Cancer Res. 2016;76:2288–300. doi: 10.1158/0008-5472.CAN-15-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chitnis MM, Lodhia KA, Aleksic T, Gao S, Protheroe AS, Macaulay VM. IGF-1R inhibition enhances radiosensitivity and delays double-strand break repair by both non-homologous end-joining and homologous recombination. Oncogene. 2014;33:5262–73. doi: 10.1038/onc.2013.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vidal SJ, Rodriguez-Bravo V, Quinn SA, Rodriguez-Barrueco R, Lujambio A, Williams E, et al. A targetable GATA2-IGF2 axis confers aggressiveness in lethal prostate cancer. Cancer Cell. 2015;27:223–39. doi: 10.1016/j.ccell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zu K, Martin NE, Fiorentino M, Flavin R, Lis RT, Sinnott JA, et al. Protein expression of PTEN, insulin-like growth factor I receptor (IGF-IR), and lethal prostate cancer: a prospective study. Cancer Epidemiol Biomarkers Prev. 2013;22:1984–93. doi: 10.1158/1055-9965.EPI-13-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res. 2008;14:6364–70. doi: 10.1158/1078-0432.CCR-07-4879. [DOI] [PubMed] [Google Scholar]
- 6.Sehat B, Tofigh A, Lin Y, Trocme E, Liljedahl U, Lagergren J, et al. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci Signal. 2010;3:ra10. doi: 10.1126/scisignal.2000628. [DOI] [PubMed] [Google Scholar]
- 7.Aleksic T, Chitnis MM, Perestenko OV, Gao S, Thomas PH, Turner GD, et al. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010;70:6412–9. doi: 10.1158/0008-5472.CAN-10-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Packham S, Warsito D, Lin Y, Sadi S, Karlsson R, Sehat B, et al. Nuclear translocation of IGF-1R via p150(Glued) and an importin-beta/RanBP2-dependent pathway in cancer cells. Oncogene. 2015;34:2227–38. doi: 10.1038/onc.2014.165. [DOI] [PubMed] [Google Scholar]
- 9.Bodzin AS, Wei Z, Hurtt R, Gu T, Doria C. Gefitinib resistance in HCC mahlavu cells: upregulation of CD133 expression, activation of IGF-1R signaling pathway, and enhancement of IGF-1R nuclear translocation. J Cell Physiol. 2012;227:2947–52. doi: 10.1002/jcp.23041. [DOI] [PubMed] [Google Scholar]
- 10.Aslam MI, Hettmer S, Abraham J, Latocha D, Soundararajan A, Huang ET, et al. Dynamic and nuclear expression of PDGFRalpha and IGF-1R in alveolar Rhabdomyosarcoma. Mol Cancer Res. 2013;11:1303–13. doi: 10.1158/1541-7786.MCR-12-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Gaal JC, Roeffen MH, Flucke UE, van der Laak JA, van der Heijden G, de Bont ES, et al. Simultaneous targeting of insulin-like growth factor-1 receptor and anaplastic lymphoma kinase in embryonal and alveolar rhabdomyosarcoma: a rational choice. Eur J Cancer. 2013;49:3462–70. doi: 10.1016/j.ejca.2013.06.022. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Huang FF, Wu DS, Li WJ, Zhan HE, Peng MY, et al. SUMOylation of insulin-like growth factor 1 receptor, promotes proliferation in acute myeloid leukemia. Cancer Lett. 2015;357:297–306. doi: 10.1016/j.canlet.2014.11.052. [DOI] [PubMed] [Google Scholar]
- 13.Lin Y, Liu H, Waraky A, Haglund F, Agarwal P, Jernberg-Wiklund H, et al. SUMO-modified Insulin-Like Growth Factor 1 Receptor (IGF-1R) Increases Cell Cycle Progression and Cell Proliferation. J Cell Physiol. 2017 doi: 10.1002/jcp.25818. epub Jan 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asmane I, Watkin E, Alberti L, Duc A, Marec-Berard P, Ray-Coquard I, et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: A predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur J Cancer. 2012;48:3027–35. doi: 10.1016/j.ejca.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Aleksic T, Browning L, Woodward M, Phillips R, Page S, Henderson S, et al. Durable Response of Spinal Chordoma to Combined Inhibition of IGF-1R and EGFR. Front Oncol. 2016;6:98. doi: 10.3389/fonc.2016.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dale OT, Aleksic T, Shah KA, Han C, Mehanna H, Rapozo DC, et al. IGF-1R expression is associated with HPV-negative status and adverse survival in head and neck squamous cell cancer. Carcinogenesis. 2015;36:648–55. doi: 10.1093/carcin/bgv053. [DOI] [PubMed] [Google Scholar]
- 17.Aleksic T, Worrall AR, Verrill C, Turley H, Campo L, Macaulay VM. Improved immunohistochemical detection of type 1 insulin-like growth factor receptor in human tumors. Immunochem Immunopath. 2016;2:114. [Google Scholar]
- 18.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc. 2007;2:1849–61. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rozek D, Pfeifer GP. In vivo protein-DNA interactions at the c-jun promoter: preformed complexes mediate the UV response. Mol Cell Biol. 1993;13:5490–9. doi: 10.1128/mcb.13.9.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hellawell GO, Turner GD, Davies DR, Poulsom R, Brewster SF, Macaulay VM. Expression of the type 1 insulin-like growth factor receptor is up-regulated in primary prostate cancer and commonly persists in metastatic disease. Cancer Res. 2002;62:2942–50. [PubMed] [Google Scholar]
- 23.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–54. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2014;158:673–88. doi: 10.1016/j.cell.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedbichler K, Hofmann MH, Kroez M, Ostermann E, Lamche HR, Koessl C, et al. Pharmacodynamic and antineoplastic activity of BI 836845, a fully human IGF ligand-neutralizing antibody, and mechanistic rationale for combination with rapamycin. Mol Cancer Ther. 2014;13:399–409. doi: 10.1158/1535-7163.MCT-13-0598. [DOI] [PubMed] [Google Scholar]
- 26.Sarfstein R, Pasmanik-Chor M, Yeheskel A, Edry L, Shomron N, Warman N, et al. Insulin-like growth factor-I receptor (IGF-IR) translocates to nucleus and autoregulates IGF-IR gene expression in breast cancer cells. J Biol Chem. 2012;287:2766–76. doi: 10.1074/jbc.M111.281782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warsito D, Sjostrom S, Andersson S, Larsson O, Sehat B. Nuclear IGF1R is a transcriptional co-activator of LEF1/TCF. EMBO Rep. 2012;13:244–50. doi: 10.1038/embor.2011.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huss M, Wieczorek H. Inhibitors of V-ATPases: old and new players. J Exp Biol. 2009;212:341–6. doi: 10.1242/jeb.024067. [DOI] [PubMed] [Google Scholar]
- 29.Eferl R, Ricci R, Kenner L, Zenz R, David JP, Rath M, et al. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003;112:181–92. doi: 10.1016/s0092-8674(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 30.Gomez TS, Billadeau DD. A FAM21-containing WASH complex regulates retromer-dependent sorting. Dev Cell. 2009;17:699–711. doi: 10.1016/j.devcel.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zech T, Calaminus SD, Caswell P, Spence HJ, Carnell M, Insall RH, et al. The Arp2/3 activator WASH regulates alpha5beta1-integrin-mediated invasive migration. J Cell Sci. 2011;124:3753–9. doi: 10.1242/jcs.080986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu YC, Zhu M, Robertson DM. Novel nuclear localization and potential function of insulin-like growth factor-1 receptor/insulin receptor hybrid in corneal epithelial cells. PLoS One. 2012;7:e42483. doi: 10.1371/journal.pone.0042483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy KL, Feinberg AP. Higher order chromatin organization in cancer. Semin Cancer Biol. 2013;23:109–15. doi: 10.1016/j.semcancer.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park OK, Schaefer TS, Nathans D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc Natl Acad Sci U S A. 1996;93:13704–8. doi: 10.1073/pnas.93.24.13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim KH, Yoo S. Sequence-specific interaction between ABD-B homeodomain and castor gene in Drosophila. BMB Rep. 2014;47:92–7. doi: 10.5483/BMBRep.2014.47.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ray S, Maitra A, Biswas A, Panjikar S, Mondal J, Anand R. Functional insights into the mode of DNA and ligand binding of the TetR family regulator TylP from Streptomyces fradiae. J Biol Chem. 2017;292:15301–15311. doi: 10.1074/jbc.M117.788000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warsito D, Lin Y, Gnirck AC, Sehat B, Larsson O. Nuclearly translocated insulin-like growth factor 1 receptor phosphorylates histone H3 at tyrosine 41 and induces SNAI2 expression via Brg1 chromatin remodeling protein. Oncotarget. 2016;7:42288–42302. doi: 10.18632/oncotarget.9785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romanelli RJ, LeBeau AP, Fulmer CG, Lazzarino DA, Hochberg A, Wood TL. Insulin-like growth factor type-I receptor internalization and recycling mediate the sustained phosphorylation of Akt. J Biol Chem. 2007;282:22513–24. doi: 10.1074/jbc.M704309200. [DOI] [PubMed] [Google Scholar]
- 39.Zheng H, Worrall C, Shen H, Issad T, Seregard S, Girnita A, et al. Selective recruitment of G protein-coupled receptor kinases (GRKs) controls signaling of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci U S A. 2012;109:7055–60. doi: 10.1073/pnas.1118359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu A, Chen J, Baserga R. Nuclear insulin receptor substrate-1 activates promoters of cell cycle progression genes. Oncogene. 2008;27:397–403. doi: 10.1038/sj.onc.1210636. [DOI] [PubMed] [Google Scholar]
- 41.Nelson JD, LeBoeuf RC, Bomsztyk K. Direct recruitment of insulin receptor and ERK signaling cascade to insulin-inducible gene loci. Diabetes. 2011;60:127–37. doi: 10.2337/db09-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Azar WJ, Zivkovic S, Werther GA, Russo VC. IGFBP-2 nuclear translocation is mediated by a functional NLS sequence and is essential for its pro-tumorigenic actions in cancer cells. Oncogene. 2014;33:578–88. doi: 10.1038/onc.2012.630. [DOI] [PubMed] [Google Scholar]
- 43.Baxter RC. Nuclear actions of insulin-like growth factor binding protein-3. Gene. 2015;569:7–13. doi: 10.1016/j.gene.2015.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang H, Li TW, Peng J, Mato JM, Lu SC. Insulin-like growth factor 1 activates methionine adenosyltransferase 2A transcription by multiple pathways in human colon cancer cells. Biochem J. 2011;436:507–16. doi: 10.1042/BJ20101754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng ZH, Gomez TS, Osborne DG, Phillips-Krawczak CA, Zhang JS, Billadeau DD. Nuclear FAM21 participates in NF-kappaB-dependent gene regulation in pancreatic cancer cells. J Cell Sci. 2015;128:373–84. doi: 10.1242/jcs.161513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ouyang X, Jessen WJ, Al-Ahmadie H, Serio AM, Lin Y, Shih WJ, et al. Activator protein-1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Res. 2008;68:2132–44. doi: 10.1158/0008-5472.CAN-07-6055. [DOI] [PubMed] [Google Scholar]
- 48.Kajanne R, Miettinen P, Tenhunen M, Leppa S. Transcription factor AP-1 promotes growth and radioresistance in prostate cancer cells. Int J Oncol. 2009;35:1175–82. doi: 10.3892/ijo_00000434. [DOI] [PubMed] [Google Scholar]
- 49.Aleksic T, Verrill C, Bryant RJ, Han C, Worrall AR, Brureau L, et al. IGF-1R associates with adverse outcomes after radical radiotherapy for prostate cancer. Br J Cancer. 2017;117:1600–1606. doi: 10.1038/bjc.2017.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waraky A, Lin Y, Warsito D, Haglund F, Aleem E, Larsson O., Sr Nuclear insulin-like growth factor 1 receptor phosphorylates proliferating cell nuclear antigen and rescues stalled replication forks after DNA damage. J Biol Chem. 2017 doi: 10.1074/jbc.M117.781492. epub Sept 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.